

Abstract
Replication and maintenance of mitochondrial DNA (mtDNA) is essential for cellular function, yet few DNA polymerases are known to function in mitochondria. Here, we conclusively demonstrate that DNA polymerase θ (Polθ) localizes to mitochondria and explore whether this protein is overexpressed in patient-derived cells and tumors. Polθ appears to play an important role in facilitating mtDNA replication under conditions of oxidative stress, and this error-prone polymerase was found to introduce mutations into mtDNA. In patient-derived cells bearing a pathogenic mtDNA mutation, Polθ expression levels were increased, indicating that the oxidative conditions in these cells promote higher expression levels for Polθ. Heightened Polθ expression levels were also associated with elevated mtDNA mutation rates in a selected panel of human tumor tissues, suggesting that this protein can influence mutational frequencies in tumors. The results reported indicate that the mitochondrial function of Polθ may have relevance to human disease.
Mitochondria are critical, energy-producing organelles within eukaryotic cells that contain a small amount of their own genetic material (mtDNA). In humans, this circular, 16 kb genome encodes 13 essential subunits of the electron transport chain (ETC), a set of protein complexes essential for enabling mitochondrial respiration and ATP synthesis. Mitochondrial DNA must be properly replicated and protected from damage, making DNA repair and damage tolerance pathways critical to mitochondrial function.1−3 While mitochondria were long thought to possess limited capacity for DNA repair, more recent research has indicated a more expansive role for such mechanisms within the organelle.4,5 In particular, the longstanding dogma that only a single mitochondrial DNA polymerase enzyme, POLγ, functions in mtDNA synthesis has recently come into question.2 Discovery of mitochondrial localization for the polymerase enzymes PrimPol6 and Rev3,7 for example, has indicated that mtDNA replication may be significantly more sophisticated than previously believed. Nevertheless, the complement of proteins involved in catalyzing mtDNA replication remains poorly defined relative to the nuclear genome, where over 16 distinct DNA polymerases have been characterized in mammalian cells.8−10
In a prior study exploring a new chemical probe-based approach to high-throughput screening for mitochondrial DNA maintenance factors, DNA Polymerase θ (Polθ) appeared in a panel of hits and was proposed as a potential mitochondrially localized factor.11 Using a family of mitochondria-targeted chemical probes12−14 in conjunction with a genetic screening approach, a series of DNA maintenance proteins was identified, with Polθ being one of the strongest hits observed. Along with the screening effort, we presented a limited set of preliminary findings suggesting that genetic ablation of DNA polymerase θ (Polθ) expression sensitized cells to targeted mtDNA damage, implying a novel mitochondrial role for this protein.11
Polθ is a DNA polymerase enzyme that has been implicated in a variety of DNA repair processes in the nucleus, including DNA replication timing, dsDNA break repair, and translesion bypass synthesis.15−17 The primary nuclear function of Polθ appears to relate to a noncanonical pathway of dsDNA break repair that is essential for maintaining nuclear genomic stability.18 Prior to our work, no mitochondrial function had been suggested for Polθ. While this earlier study presented initial results indicating that Polθ localizes to mitochondria and that genetic knockout of Polθ impaired mitochondrial function, several questions regarding the role of this protein in mitochondria remained. Most importantly, the submitochondrial localization of Polθ, the mechanism by which it is targeted to mitochondria, and the nature of its activity in the organelle all required further exploration before Polθ could be definitively categorized as a functional mtDNA polymerase. Here, we present conclusive evidence that Polθ is in fact an mtDNA polymerase that is directly involved in maintaining mtDNA replication under conditions of oxidative stress. We also present results showing that Polθ is overexpressed in patient-derived cells corresponding to a genetic mitochondrial pathology, and that Polθ expression is correlated with mtDNA mutational frequency in a subset of tumors. The results presented indicate that this DNA polymerase may play a role in disease-related cellular dysfunction.
Polθ Localizes to Mitochondria
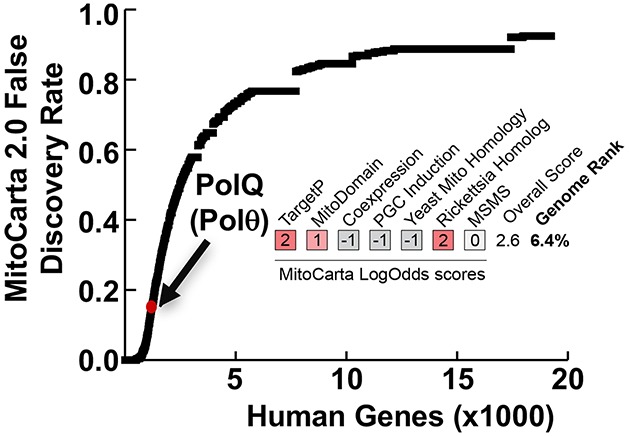
In assessing the likelihood that Polθ functions in mitochondria, we consulted the MitoCarta2.0 list of mammalian mitochondrial proteins.19 While MitoCarta does not cite Polθ as a mitochondrial protein, it is noteworthy that this database ranks the probability that each gene in the mouse or human genomes encodes a protein whose primary residence is in the mitochondrion, and its scoring system thus penalizes “moonlighting” proteins that are predominantly found in other cellular locations, such as polymerases functioning in the nucleus.20 However, Polθ still ranks in the top 7% of all genes on the human MitoCarta2.0 list based on estimated false discovery rate (FDR), driven by its favorable scores for mitochondrial import sequence prediction (TargetP), protein domain structure (MitoDomain), and homology to a protein encoded in the Rickettsia prowazekii genome (Rickettsia Homologue; Figure 1a). Indeed, 137 human genes with higher FDRs are included in the MitoCarta2.0 list based on direct evidence in the literature.19 As such, the MitoCarta analyses are consistent with Polθ being a candidate moonlighting mitochondrial protein.
Figure 1.

POLθ localizes to mitochondria. (a) All genes in the human genome ranked by their estimated false discovery rate in the MitoCarta2.0 study. POLθ is highlighted in red, and its individual LogOdds scores for each MitoCarta2.0 feature are listed. (b) POLθ is present in mitochondrial fractions. PCNA was used as a control for nuclear contamination, HSP70 as a control for cytosolic contamination. Vec = mock-transfected, Polθ-FLAG = pCDH-POLQ-WT-HA-FLAG transfected. (c) POLθ is protected from proteinase K digestion of isolated mitochondria, indicating localization within the mitochondrial matrix. Bcl-XL is a mitochondrial outer membrane protein; TFAM is a mitochondrial matrix protein. (d) POLθ colocalizes with the mitochondrial network in cells by confocal fluorescence microscopy. Pearson correlation coefficient = 0.39 ± 0.09. Image of cell is representative of all cells analyzed.
In previous studies, we assessed the mitochondrial localization of Polθ using an antibody directed against the endogenous Polθ protein. While the data generated previously supported mitochondrial localization, the low native expression levels of Polθ in mammalian cells made it difficult to observe Polθ’s subcellular localization with high resolution.11 Given the importance of unequivocally establishing the mitochondrial localization of this protein, we cloned and expressed a Polθ construct featuring a C-terminal FLAG tag in human cells. Stringent isolation of highly purified mitochondria from cells, followed by Western blot detection of Polθ using an anti-FLAG antibody, revealed the presence of a significant fraction of Polθ protein within mitochondria (Figure 1b). When intact isolated mitochondria were exposed to digestion with proteinase K, Polθ was protected from degradation, indicating its localization within the mitochondrial matrix (Figure 1c).
Immunofluorescence-based detection of Polθ in human cells showed punctate extranuclear localization for the protein colocalizing extensively with a mitochondria-specific marker (MTCO2; Figure 1d), also indicating mitochondrial localization. Quantification of the association between these two stains over a number of images containing different cells revealed a Pearson’s correlation coefficient of 0.39 ± 0.09, which compares favorably to the degree of colocalization observed for previously studied known mitochondrial proteins and dyes.11,21 To more closely visualize the submitochondrial localization of Polθ and to confirm localization with an independent method, we also performed immunogold-labeling assays to detect Polθ by transmission electron microscopy. Minimal background staining was detected upon labeling of untransfected cells (Supporting Information Figure 1a). In contrast, significant gold labeling was detected in both the nucleus and within mitochondria of Polθ-transfected cells (Supporting Information Figure 1b). Mitochondrial staining was mainly confined to the mitochondrial matrix (Supporting Information Figure 1b), indicating the localization of Polθ to this DNA-containing mitochondrial compartment. These experiments provide important data that support the prior identification of Polθ as a DNA polymerase with mitochondrial function.11
Polθ Contains a Mitochondria-Targeting Sequence
Proteins are typically targeted to mitochondria with a mitochondrial targeting sequence (MTS) containing a N-terminal amphipathic α-helix via the protein import machinery located on the mitochondrial outer membrane.22 Our analysis of the appearance of Polθ in the MitoCarta database indicated that this protein had a putative MTS. Computational analysis of Polθ cDNA using the MitoProt program revealed a predicted MTS at the N-terminus of the protein with the sequence MNLLRRSGKRRRS.23 This sequence was fused upstream of a cDNA encoding eGFP to create a reporter construct (MTS-eGFP) that allowed us to assess its mitochondria-targeting capacity. This reporter construct displayed significant mitochondrial localization when expressed in cells, confirming that this N-terminal sequence of Polθ can act as an MTS (Figure 2a). The untagged eGFP vector, in contrast, showed mainly cytosolic and nuclear staining (Figure 2b).
Figure 2.
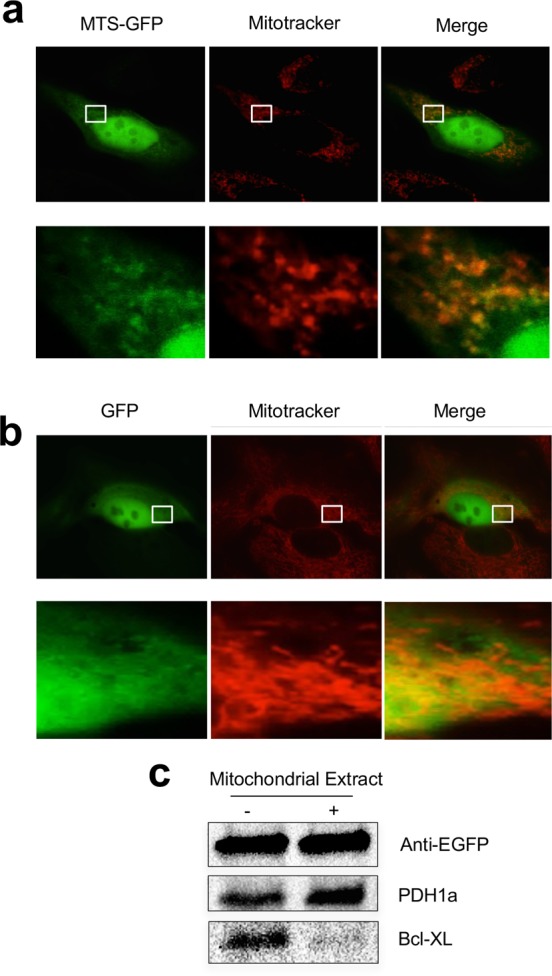
POLθ contains a mitochondria-targeting sequence. (a) MTS-GFP construct colocalizes with mitochondria by fluorescence microscopy when transfected into cells. Nuclear staining is also seen, as is characteristic of tagged GFP-constructs. Images are at 63× magnification. (b) An untagged GFP construct displays mainly cytosolic localization by fluorescence microscopy when expressed in cells. (c) MTS-GFP localizes to the mitochondrial matrix as indicated by proteinase K digestion of isolated mitochondria. Bcl-XL is a mitochondrial outer membrane protein; PDH1a is a mitochondrial matrix protein.
Mitochondrial localization of MTS-eGFP was further confirmed by biochemical isolation of mitochondria and detection of eGFP with an anti-eGFP antibody. MTS-eGFP protein was protected from degradation following proteinase K digestion of isolated mitochondria, indicating that the MTS of Polθ directs localization to the mitochondrial matrix (Figure 2c). Significant nuclear localization was also observed for the MTS-eGFP construct. It should be noted that the same leader sequence of Polθ also contains stretches of positively charged amino acids that are characteristic of nuclear-localization sequences. It is possible that the dual localization of Polθ to both mitochondria and nuclei we observe is mediated by competition between these adjacent localization sequences for interaction with different protein trafficking machineries in the cell, a mechanism that has been observed previously for other dual-targeted proteins.22 Indeed, deletion of this leader sequence caused Polθ to show diffuse cytosolic localization when expressed in cells, eliminating both the nuclear and mitochondrial localization of the protein (Supporting Information Figure 2).
Polθ Associates with the Mitochondrial Genome
We next evaluated the association of Polθ with mtDNA nucleoids, which are protein–DNA complexes in mitochondria housing the majority of mtDNA repair and replication factors.24,25 These nucleoids were stained with an anti-dsDNA antibody and visualized by confocal immunofluorescence microscopy. An anti-dsDNA antibody was used as it has been found that DNA chemical stains like DAPI do not stain mitochondrial nucleoids strongly enough to observe mtDNA localization effectively.26 In our hands, immunological detection of dsDNA has been found to produce a strong mtDNA signal (colocalizing with mitochondrial protein markers) while only weakly staining chromosomal DNA around the nuclear envelope, providing ideal levels of contrast for visualizing these nucleoids.11 Polθ was found to colocalize with these mitochondrial structures to a significant extent, providing evidence for the association of Polθ with mtDNA in cells (Figure 3a).
Figure 3.
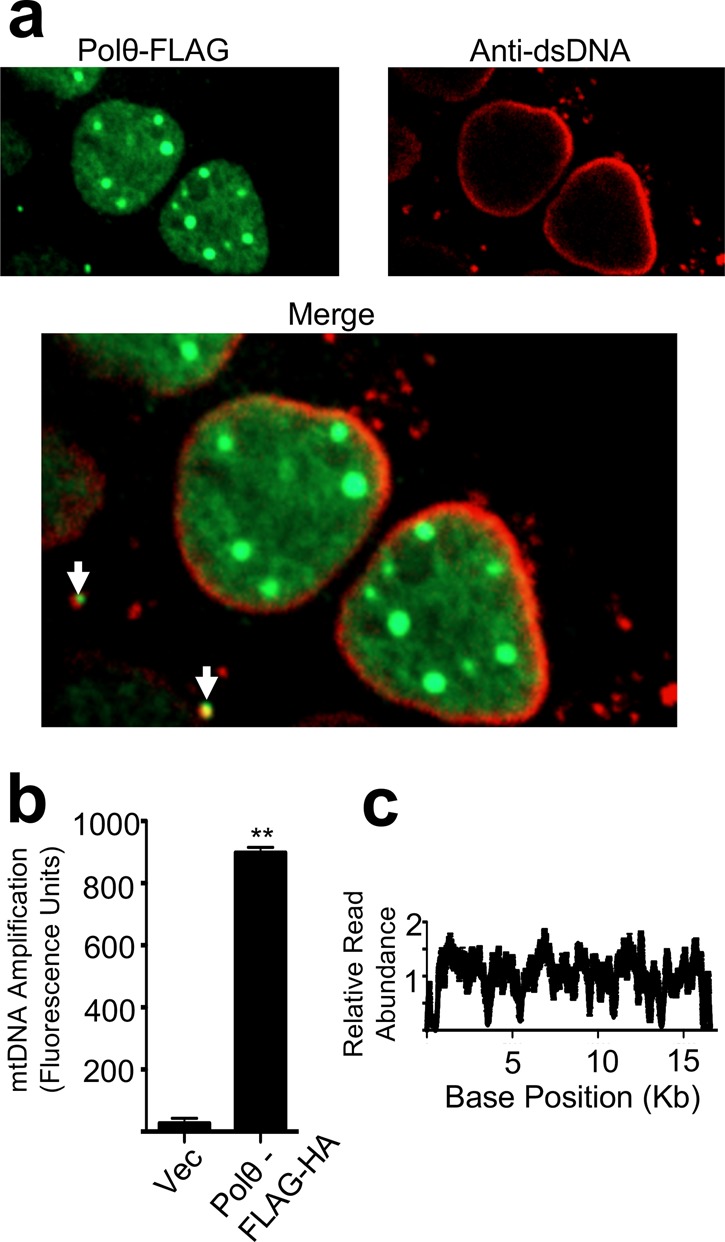
Polθ associates with the mitochondrial genome. (a) Polθ associates with mtDNA nucleoids in cells as indicated by confocal fluorescence microscopy. Nucleoids were stained with an anti-dsDNA antibody and are indicated by arrows. (b) HA- tagged Polθ associates with mtDNA in cells as indicated by a ChIP-PCR using an anti-HA antibody. n = 3, error bars indicate SEM. (c) Sequencing of Polθ-associated mtDNA fragments indicates Polθ binds widely at many positions in the mitochondrial genome.
The association of Polθ with mtDNA was further confirmed using a mitochondrial DNA immonoprecipitation assay. This assay directly probes whether there is a direct physical interaction between the protein and mtDNA. Cells were transfected with a HA-tagged Polθ construct and chemically cross-linked to irreversibly link Polθ to DNA bound by the protein. Immunoprecipitation of Polθ was then performed to isolate fragments of bound DNA. Selective amplification of a mitochondrial genomic region from this isolate was successful specifically in Polθ-HA transfected cells, demonstrating the association of Polθ with mtDNA (Figure 3b). Sequencing of the associated DNA fragments showed a binding of Polθ across all regions of the mitochondrial genome, indicating a broad role for Polθ in mediating mtDNA synthesis (Figure 3c).
Polθ Sustains mtDNA Replication Following Oxidative mtDNA Damage
We next investigated the mitochondrial role of Polθ by assessing how mtDNA replication is altered in cells with a CRISPR-engineered genetic knockout in the Polθ gene. Polθ has been posited to have numerous functions in nuclear DNA repair, including in the repair of both oxidative DNA damage and dsDNA breaks.16,17 Our previous studies, however, showed that Polθ knockdown sensitizes cells specifically to oxidative mtDNA damage and not to other lesion-inducing agents, indicating a specific role in the response to mitochondrial oxidative stress.11 Additionally, we had previously observed that knockout of Polθ is associated with depletion of cellular mtDNA content following oxidative stress; the biochemical mechanism underlying this phenotype, however, is unclear.11 Previous measurements were obtained by analyzing mtDNA content in cells, but the direct assessment of replication efficiency was not probed.
To assess the role of Polθ in maintaining mtDNA replication following mtDNA oxidation, WT and Polθ KO cells were incubated with the nucleotide analogue EdU, which is selectively incorporated into actively replicating DNA. EdU incorporation was then detected postfixation by a click-chemistry-based reaction, and EdU puncta colocalizing with the mitochondrial nucleoid protein TFAM were visualized by confocal fluorescence microscopy (Figure 4a).27 The number of EdU puncta/cell was quantitated over a series of images to assess the rate of mtDNA replication in the Polθ deficient cell line. Polθ KO cells displayed significantly fewer actively replicating nucleoids when compared to WT cells, indicating significant defects in mtDNA replication (Figure 4a). Notably, Polθ KO cells were also more sensitive to mtDNA replication arrest following treatment with the oxidant H202, indicating a potential specialized role for Polθ in tolerating oxidative mtDNA lesions (Figure 4a).11
Figure 4.

Polθ protects cells from oxidative mtDNA damage by facilitating mtDNA replication. (a) Polθ knockout reduces mtDNA replication rates as measured by EdU staining under conditions of oxidative stress. Arrows indicate EdU puncta that colocalize with the mitochondrial marker TFAM. Nuclear signal is overexposed so as to visualize weakly staining nucleoids, n = 3. (b) Polθ KO cells have reduced expression of a mitochondria-encoded ETC subunit. MTCO2 is a mitochondria-encoded protein. VDAC1 is a nuclear-encoded mitochondrial protein. HSP70 is a cytosolic protein used as a loading control. (c) Treatment of Polθ KO cells with a mitochondria-targeted DNA damaging agent results in a decrease in mitochondrial mass as assessed by MitoTracker Green staining, n = 3. (d) Expression of Polθ rescues cells from H202 induced mtDNA replication arrest, while Polθ-NLS, a mutant lacking mitochondrial localization, does not, n = 3. (e) Treatment of LentiX cells with the indicated concentrations of mtOx for 24 h induces an increase in expression of Polθ mRNA as measured by qPCR, n ≥ 3. (f) Cultured cells from a patient with MELAS caused by an A3243G mutation in mtDNA express Polθ at higher levels than three control cell lines, n = 3. Mean values indicated in all cases; error bars indicate SEM. *p < 0.05, **p < 0.01. ***p < 0.0001.
To connect this replication defect with alterations in mitochondrial gene expression, we questioned how these mtDNA replication defects affect expression of mtDNA-encoded genes. Polθ KO cells were observed to have significantly reduced expression of the mitochondria-encoded ETC subunit MTCO2, possibly reflecting a reduction in mtDNA abundance in this cell line (Figure 4b). Expression of VDAC1, a nuclear-encoded mitochondrial protein, was only moderately affected. We also gauged the impact of Polθ knockout on mitochondrial biogenesis by staining cells with MitoTracker Green, a dye that localizes to mitochondria in a membrane-potential independent manner. While mitochondrial mass was equivalent in WT and Polθ KO cells under normal growth conditions, mitochondrial content was significantly reduced in Polθ KO cells that had been treated for 24 h with mt-Ox, a mitochondria-localizing compound that has been shown in previous work to induce significant oxidative damage specifically to mtDNA (Figure 4c).11 Mitochondrial mass in WT cells was unaffected by this treatment, underscoring that the mtDNA replication activity of Polθ is essential to maintaining mitochondrial abundance in the context of oxidative stress.
The role of this DNA polymerase in the oxidative stress response was probed further by assessing whether heightened expression of Polθ conferred increased tolerance of cells to mtDNA damage. We found that treatment of cells with H2O2 induced a decrease in active mtDNA replication by EdU staining (Figure 4d). Cells transfected to express WT Polθ before treatment with H2O2 were somewhat protected from this arrest in mtDNA replication (Figure 4d). In order to confirm that this protective effect of Polθ expression arises from activity specifically within mitochondria, we generated a new Polθ construct where the N-terminal MTS region is replaced by the known nuclear localization sequence PKKKRV.28 Expression of this construct in cells caused the Polθ protein to localize exclusively to the nucleus, with no mitochondrial localization observable (Supporting Information Figure 3). Expression of Polθ-NLS, additionally, did not increase rates of mtDNA replication following mtDNA oxidation, indicating that Polθ’s activity is mediated through direct action on the mitochondrial genome (Figure 4d).
Given the apparent importance of Polθ in mediating an effective response to mtDNA damage, we also evaluated whether expression of Polθ is naturally elevated in cells undergoing mtDNA-damaging stress. Gene expression analysis by qPCR revealed that steady-state levels of Polθ mRNA increased significantly 24 h after treatment of cells with mt-Ox (Figure 4e). This observation indicates that cellular upregulation of Polθ is part of the response to oxidative stress.
We probed whether Polθ expression was perturbed in a patient-derived cell line harboring a mtDNA mutation causing MELAS, a mitochondrial disease associated with severe mitochondrial dysfunction and oxidative cellular damage (Figure 4f).29 A significant upregulation of the Polθ mRNA was observed in these cells. Taken together, these results imply that Polθ is a key DNA damage response gene that regulates tolerance of cells to oxidative mtDNA damage.
Polθ Modulates mtDNA Mutagenesis in Both Cultured Cells and Clinical Samples
Finally, we wished to more closely examine how Polθ activity affects the integrity of the mitochondrial genome. Prior work had indicated that Polθ knockout alters mtDNA mutation rates.11 Additionally, experiments in vitro have shown that Polθ is a relatively error-prone polymerase, raising the possibility that mitochondrial Polθ may be mutagenic in living cells.30 Indeed, deep sequencing of mtDNA from cells transfected with WT Polθ showed an increase in the frequency of heteroplasmic point mutations (mutations making up greater than 0.5% of the overall mtDNA content at a given base position) in mtDNA (Figure 5a). This effect was not observed in cells expressing a catalytically inactive form of Polθ (Figure 5a), strongly supporting Polθ’s direct action on the mitochondrial genome. Interestingly, while Polθ has been suggested to play a role in dsDNA break repair in the nucleus, the rate of insertions (Figure 5b) or deletions (Figure 5c) in mtDNA was unaffected by Polθ overexpression. Read depth patterns over the mitochondrial genome were also unaffected, indicating the absence of any large-scale genomic rearrangements caused by heightened Polθ activity (Figure 5d). These results suggest a distinct role for Polθ in mitochondria versus the nucleus.
Figure 5.
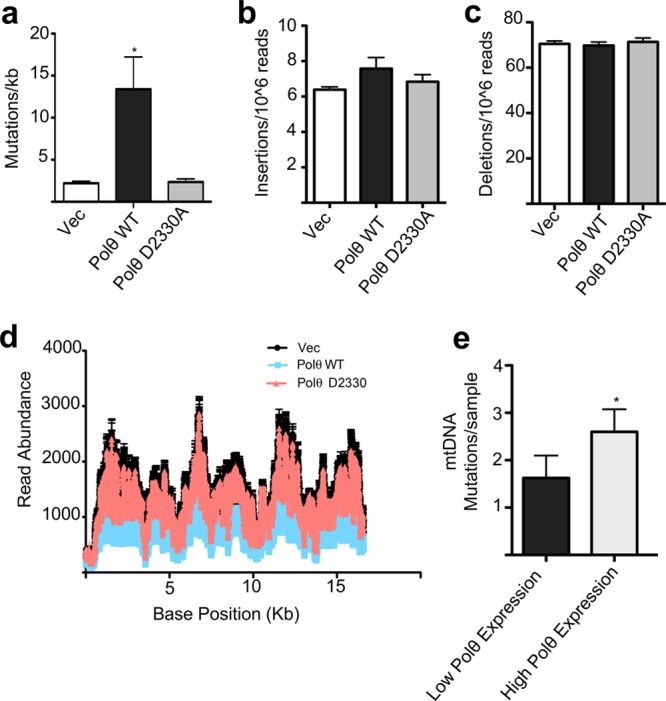
Polθ specifically induces mtDNA mutagenesis. (a) Polθ overexpression increases mtDNA point mutations. Increase in mtDNA mutagenesis is not seen with Polθ D2330A, a catalytic mutant lacking DNA polymerase activity, n = 3. (b) Polθ overexpression does not affect the rate of mtDNA insertion mutations, n = 3. (c) Polθ overexpression does not affect the frequency of small mtDNA deletions, n = 3. (d) Polθ overexpression does not change read-depth patterns over the mitochondrial genome, indicating the absence of large genomic rearrangements, n = 3. (e) Hepatocellular tumor samples with high Polθ mRNA expression are more likely to have somatic mutations in their mtDNA than tumor samples with low measured Polθ mRNA expression, n = 31. Mean values indicated; all error bars indicate SEM, *p < 0.05.
Polθ has been reported to be transcriptionally upregulated in a variety of human cancers.31,32 As our studies indicate that Polθ overexpression modulates rates of mutation in mtDNA, we next sought to determine whether mtDNA mutation rates are altered in Polθ-overexpressing tumors. We acquired whole-exome sequencing data from the Cancer Genome Atlas for 47 hepatocellular tumor samples and identified a set of tumors with both high (mRNA expression Z score > 0.5) and low (Z < −0.5) Polθ expression. We then quantitated the number somatic mtDNA mutations in these same tumors identified by previously reported deep sequencing analysis.33 High Polθ-expression tumors were found to harbor significantly larger numbers of mtDNA point mutations when compared to tumors with low Polθ expression (Figure 5e). These results show that high Polθ expression levels may drive mitochondrial mutagenic activity in a clinically relevant context and raise interesting questions about how mitochondrial function may be altered in Polθ-overexpressing tumors.
DNA Polymerase θ is a Mitochondrial Protein that Modulates Replication Error
While mitochondrial DNA repair and replication had long been thought to be governed by simpler and less sophisticated biochemical processes than those that exist in the nucleus, a growing body of research is convincingly showing that mitochondria do in fact possess advanced pathways of DNA damage tolerance.6,7 Here, we provide critical support to the initial discovery that DNA Polymerase θ is a central factor in mtDNA replication and repair, localizing to and mediating DNA synthesis within the organelle. We report a series of new experiments and insights that provide strong evidence that Polθ is a mitochondrial protein and, importantly, explore the physiological relevance of this finding.
We observed that knockout of Polθ is associated with decreased mtDNA replication when cells are exposed to oxidative stress. Polθ expression increases substantially following oxidative mtDNA damage, facilitating efficient replication of the mitochondrial genomic material. These findings elucidate a novel pathway by which mitochondria maintain their mtDNA levels under oxidative DNA-damaging stress conditions. Interestingly, this mitochondrial function for Polθ is distinct from this protein’s roles in the nucleus, where it appears to act in error-prone repair of dsDNA breaks as well as DNA replication timing and regulation of G-quadraplex structures.15,34−37 Polθ’s many diverse roles in different compartments of the cell thus mark it as a unique multifunctional enzyme that deserves significant further study and research.
Our previous work had already provided preliminary support for Polθ’s mitochondrial localization, but functional characterization of mitochondrial proteins can always be prone to artifacts and misleading results. This is especially true when available antibodies against a protein of interest are of mixed quality and a protein has multiple cellular roles that could be indirectly impacting mitochondrial function. In this study, we were able to confirm our results indicating Polθ’s mitochondrial localization with much higher specificity and resolution by using a controlled gene expression system to detect tagged Polθ protein. We found that Polθ localizes to the mitochondrial matrix and directly associates with mtDNA, answering questions not addressed in prior work. A putative MTS of Polθ is also identified for the first time. Similarly, our functional studies clearly demonstrate a novel role for Polθ in mtDNA replication and show conclusively that this activity relates to Polθ’s direct action within mitochondria. All of these key findings were not addressed by our previous study.
Finally, we present intriguing new results confirming that the replicative function of Polθ might have significant consequences for the long-term maintenance of the mitochondrial genetic material. High expression of Polθ appears to affect the mitochondrial genome by increasing the frequency of mtDNA point mutations; this effect is also observed in a panel of human tumor tissues reported to express Polθ at high levels. While our previous study had indicated a link between Polθ function and mtDNA mutagenesis in cultured cells, this is the first demonstration that this activity might be relevant in vivo. The finding that Polθ could modulate mtDNA mutation rates in cancer is highly significant, as Polθ is frequently overexpressed across a wide range of tumor subtypes.31 Mitochondrial DNA mutations, additionally, have been found to drive increased cancer growth and metastasis in a number of different studies.38 The work we present here suggests a potentially novel mechanism linking these two pathophysiological processes, implying that Polθ’s mitochondrial activity may have significant relevance in human health and disease.
Materials and Methods
Cell Culture
LentiX-293T cells were obtained from ATCC and cultured in DMEM containing 10% FBS. HeLa cells were also obtained from ATCC and cultured in MEM-α containing 10% FBS. HAP1 WT and POLQ KO cells were obtained from Horizon Genomics and cultured in IMDM containing 10% FBS. In all cases, cells were maintained in a 37 °C incubator with 5% CO2.
Mitochondrial DNA Immunoprecipitation Assays
LentiX cells were seeded in 150 mm dishes at a density of 3 × 106 cells/dish and allowed to attach overnight. Cells were then transfected using 24 μg of DNA and 60 μL of Lipofectamine 2000 according to the manufacturer’s instructions. After 48 h, cells were cross-linked by the addition of 16% PFA directly to the cell culture media to achieve a final 1% PFA concentration. After 10 min, the cross-linking reaction was quenched by the addition of 1 M glycine to a final concentration of 0.125 M. Cells were washed once with dPBS and transferred using a cell scraper into a 15 mL falcon tube. Cells were then spun down at 600g for 5 min, resuspended in lysis buffer (20 mM Tris, 125 mM NaCl 0.2% Triton-X-100, 1 mM EDTA), and sonicated in a VWR Symphony sonicating bath for 30 min to shear genomic DNA. The insoluble protein fraction was removed by centrifugation at 15 000g for 15 min. Protein concentration in the resultant soluble fraction was quantitated by BCA assay. A total of 200 μg of protein was diluted to 500 μL in PBS and incubated for 1 h with rotation with 25 μL of Protein G Dynabeads (NEB) to preclear nonspecific interactions. Precleared lysate was then incubated overnight with 2 μg of anti-HA antibody, followed by a 1 h incubation with 25 μL of Protein G Dynabeads. Beads were isolated using a magnetic bar and washed four times with lysis buffer and four times with TE buffer. To reverse cross-links and elute DNA, beads were resuspended in a pH 11 TE buffer containing 1 mg mL–1 proteinase K, incubated at 55 °C for 1 h and then at 95 °C for 15 min. A PCR was then performed to detect mtDNA as described previously.11 Alternatively, isolated DNA was also sequenced as described below.
Confocal Immunofluorescence Microscopy
LentiX cells were seeded in six well plates containing 25 mm glass coverslips at a density of 150 000 cells/well. After overnight attachment, they were transfected using 2 μg of DNA and 5 μL of Lipofectamine 2000 according to the manufacturer’s instructions. A total of 48 h following transfection, cells were fixed for 10 min by incubation with 4% PFA in PBS at RT. Cells were washed with PBS twice for 10 min each, then permeabilized with 0.4% Triton-X-100 for 20 min. Cells were washed twice with PBS for 5 min each, then incubated for 1 h with blocking buffer containing 0.1% Triton-X-100 and 5% Goat Serum in PBS. Primary antibodies were diluted in blocking buffer at the following dilutions: 1:250 rabbit anti-FLAG (Sigma F7425), 1:250 rabbit anti-HA (Abcam ab9110), 1:200 mouse anti-MTC02 (Abcam, ab110258), 1:200 anti-dsDNA (Abcam, ab27156), and 1:200 TFAM (Abcam ab131607). Antibodies were applied overnight. Following incubation; cells were washed twice with PBS and incubated for 1 h with either AlexaFluor 488 goat anti-rabbit antibody (Life Technologies, A11008), AlexaFluor 568 goat anti-mouse (Life Technologies, A11004), or AlexaFluor 647 goat antirabbit (ab150155) diluted 1:500 in PBS. Where applicable, Hoechst 33332 (Sigma) was used to stain nuclei at a concentration of 1:1000. Cells were then washed twice with PBS and imaged using a Quorum spinning disk confocal microscope.
Live Cell Fluorescence Microscopy
HeLa cells were seeded in eight well ibidi μ-slides at a density of 16 000 cells/well and allowed to attach overnight. Cells were then transfected with indicated eGFP constructs using 100 ng of DNA and 0.25 μL of Lipofectamine 2000 according to he manufacturer’s instructions. After 24 h, media were removed and cells were stained with 100 nM Mitotracker Deep Red in Opti-MEM for 15 min. Cells were washed twice with Opti-MEM and imaged using a Zeiss Observer microscope.
Mitochondrial Isolation
LentiX cells were seeded in T175 flasks at a density of 3 × 106 cells/dish and allowed to attach overnight. Cells were then transfected using 24 μg of DNA and 60 μL of Lipofectamine 2000 according to the manufacturer’s instructions. After 48 h, cells were isolated by trypsinization and centrifugation at 600g for 10 min. Sucrose gradient isolation of mitochondria was performed as described previously.11
Proteinase K Digestion Assays
Mitochondria were isolated from POLQ-HA-FLAG or MTS-eGFP transfected LentiX cells as described above. Mitochondria were resuspended in buffer containing 10 mM Tris, 1 mM EDTA, and 250 mM sucrose at pH 7.5. Protein content was quantitated by BCA. Subsequently, 80 μg of mitochondrial extract was incubated either with or without 100 μg/mL Proteinase K for 20 min on ice. Protein was precipitated by addition of two volumes of 20% ice-cold TCA and isolated by centrifugation at 3000g. The protein pellet was washed with acetone and dissolved directly in two times Laemmli buffer with 100 mM DTT for subsequent analysis by SDS-PAGE. An antibody against Bcl-XL (Abcam, ab31396) was used to monitor degradation of a mitochondrial outer membrane protein by proteinase K. Antibodies against TFAM (ab131607) or PDH1a (sc-292543) were used to monitor degradation of a mitochondrial matrix protein by proteinase K, with antibody choice determined by protein size so as to maximize efficient use of a given protein extract in Western blotting.
EdU Staining
HAP1 WT and POLQ KO cells were seeded in six well plates containing 25 mm glass coverslips at a density of 250 000 cells/well and allowed to attach overnight. LentiX cells were seeded in six well plates containing 25 mm glass coverslips at a density of 150 000 cells/well and subsequently transfected for 24 h with pCDH-POLQ-WT-HA-FLAG and pCDH-POLQ-D2330A-HA-FLAG constructs as described above. Cells were subsequently treated with media containing 10 μM EdU and the indicated concentrations of H2O2 for 24 h. Concentrations of H2O2 were chosen based on observed sensitivity of different cell lines to toxic effects of H2O2; LentiX cells were found to be more H2O2-resistant than HAP1 cells, and so higher absolute concentrations were used. Following incubation, cells were fixed in 4% PFA in PBS for 10 min at RT. Cells were then washed twice with 3% BSA in PBS for 5 min each. Cells were permeabilized at RT using 0.5% Triton-X-100 in PBS and subsequently washed twice with PBS for 5 min each. EdU labeling was performed using the Click-iT EdU AlexaFluor 488 Imaging Kit using manufacturer’s instructions. Cells were imaged using a Quorum spinning disk confocal microscope. Ten images covering at least 30 cells were acquired for each experimental condition. Extranuclear puncta corresponding to mitochondrial nucleoids were counted by eye over each image set. H2O2 was used as an oxidizing agent for these studies because mt-Ox is fluorescent and interferes with multicolor imaging experiments.39
mtDNA Sequencing
LentiX cells were seeded in T175 flasks at a density of 3 × 106 cells/dish and allowed to attach overnight. Cells were then transfected using 24 μg of DNA and 60 μL of Lipofectamine 2000 according to the manufacturer’s instructions. Isolation of mitochondria was performed as described previously.11 Mitochondrial DNA was extracted from the resulting mitochondrial extracts using the GenElute Mammalian DNA Miniprep Kit (Sigma) using manufacturer’s directions. Paired-end sequencing was performed on an Illumina MiSeq platform with a Nextera XT library preparation. Reads were aligned to a mitochondrial DNA consensus sequence using Bowtie2,40 and point mutations were analyzed using Samtools.41 Mutations were defined as deviations from the consensus mitochondrial genome sequence. In order to define single nucleotide mutations, quality settings were applied such that a mutation must represent a number of reads corresponding to 0.5% of the average read depth for the given sample. These parameters allow for normalization of differences in sequence coverage between cell lines and biological replicates. Insertions and deletions, being much rarer events, were simply counted and normalized to the number of reads for each sample. Some SNPs corresponding to variations in native mtDNA sequence between our cell lines and the mtDNA reference sequence (GRCh38 build) were observed and excluded from global mutation analysis.
Electron Microscopy
LentiX cells were transfected as described above with POLQ-FLAG-HA WT DNA. After 48 h, cells were pelleted at 1500g and fixed in 0.1 M phosphate buffer (pH 7.2) containing 4% paraformaldehyde and 0.2% glutaraldehyde for 2 h at RT. Samples were then washed 3 times for 15 min each with 0.1 M phosphate buffer (pH 7.2) and dehydrated through a series of graded ethanol solutions. Samples were then embedded in resin, polymerized at −20 °C with UV light using a 0.1% benzol UV catalyst for 24 h and then moved to 60 °C for 48 h with a 5 ppm hydroquinone thermal catalyst. Ultrathin sections were collected on nickel grids and floated for 30 min at RT under conditions with TBS (1 mM CaCl2, 1 mM MgCl2, 4% fish gelatin, 50 mM Tris, 150 mM NaCl). Grids were then floated on drops containing Anti-FLAG M2 antibody (Sigma F1804) diluted 1:50 in conditioned TBS for 1 h. Grids were washed by floating on drops of conditioned TBS five times for 5 min each. Subsequently, grids were stained by floating on drops containing goat anti-mouse IgG-10 nm gold antibody conjugate diluted 1:50 in conditioned TBS for 30 min. Grids were again washed five times for 5 min each as above. Finally, grids were contrast-stained with 5% uranyl acetate for 10 min at RT and imaged on a Hitachi H-7000 transmission electron microscope.
Mitotracker Green Staining
WT and Polθ KO cells were seeded in 12 well plates at a concentration of 150 000 cells/well and allowed to attach overnight. The next day, cell growth media were replaced with media containing 4 μM mtOx. After 24 h of treatment, cell growth media were removed and replaced with media containing 250 nM Mitotracker Green. Cells were subsequently incubated for 20 min. Media were then removed, and cells were harvested by trypsinization. Cells were spun down at 600g, washed once with PBS, and subsequently resuspended in PBS containing 5 nM Sytox Red to allow for identification of dead cells. Cells were then analyzed by flow cytometry using a BD FACSCanto flow cytometer. Average fluorescence in the FITC channel was measured and taken to be a measure of mitochondrial mass.
Western Blotting
Unless otherwise indicated, cell lysates were extracted by resuspension of cells in RIPA buffer containing 1 mM PMSF. TGX Mini-Protean Tris Glycine 4–15% gradient gels (BioRad) were used for SDS-PAGE. Transfer was performed to the PVDF membrane over 1 h using Novex Tris-Glycine transfer buffer (Life Technologies) containing 20% methanol. Blocking was performed in Tris-buffered glycine buffer containing 0.1% Tween and 5% skim milk for 1 h at RT. Primary antibodies against MTCO2 (Abcam, ab110258), HSP70 (Abcam, ab2787), and VDAC1 (Abcam, ab15895) were all diluted 1:500 in TBST and incubated overnight. Secondary anti-mouse (Cell Signaling, 7076S) and anti-rabbit (Cell Signaling, 7074S) HRP conjugate antibodies were applied for 1 h at RT. Membranes were washed four times for 5 min each with TBST following each antibody application. Blots were exposed using a ChemiDoc imager (BioRad).
mRNA Expression Experiments
LentiX cells were seeded in 12 well plates at a concentration of 150 000 cells/well and allowed to attach overnight. The next day, cell growth media were replaced with media containing 4 μM mtOx or 8 μM mtOx. RNA was extracted using TRIzol Reagent (Life Technologies) according to the manufacturer’s instructions after 24 h. The extracted RNA was treated with TURBO DNase (Life Technologies, 2U/μL) then quenched and purified through a subsequent TRIzol isolation. RNA was quantified via NanoDrop2000 (ThermoFisher Scientific). RT-qPCR was performed using a TaqMan RNA-to-CT 1-Step Kit (Life Technologies) according to the manufacturer’s directions for a 96-well Applied Biosystems 7500 Real-Time PCR System (50 μL/reaction). Predesigned TaqMan Gene Expression Assay primer/probe sets were used for detection of GAPDH (Life Technologies, Hs02786624_g1) and POLQ (Life Technologies, Hs00981375_m1). Fold change values for POLQ were calculated based on relative quantitation using comparative CT values corrected by GAPDH endogenous control.
Bioinformatic Studies
mRNA expression data for the POLQ gene was obtained for a list of 47 hepatocellular cancers from the TCGA provisional data set using cBioPortal.42,43 Information on somatic mtDNA mutation rates was obtained from a separate study that included the same tumor specimens.33 Hepatocellular tumors were chosen for this study because of the wide spread in POLQ expression levels observed in this data set as well as the availability of mtDNA sequencing data.
Molecular Cloning
pCDH-POLQ-WT and pCDH-POLQ-D2330A plasmids were generously donated by Rick Wood’s lab (MD Anderson Cancer Centre). The pCDH-POLQ-NLS and pCDH-POLQ-delMTS construct was generated by de novo synthesis of a POLQ gene fragment featuring the desired sequence (ACGT), followed by standard subcloning protocols.
Statistical Analysis
Sample sizes for each given experiment are detailed in corresponding figure legends. All p values were determined by the student’s two-tailed t test.
Acknowledgments
We would like to extend thanks to R. Wood (University of Texas, MD Anderson Cancer Centre) for kindly sharing pCDH-POLQ-WT-HA-FLAG and pCDH-POLQ-D2330A-HA-FLAG plasmids for these experiments. We would also like to thank N. Sondheimer for generously donating A3243G MELAS cells for our study. This work was funded with National Institutes of Health grants R01GM115591 (to D.J.P.) and R01GM116886 (to S.O.K.)
Supporting Information Available
The Supporting Information is available free of charge on the ACS Publications website at DOI: 10.1021/acschembio.8b00072.
Additional localization experiments for Pol θ and modified constructs (PDF)
The authors declare no competing financial interest.
Supplementary Material
References
- Kazak L.; Reyes A.; Holt I. J. (2012) Minimizing the damage: repair pathways keep mitochondrial DNA intact. Nat. Rev. Mol. Cell Biol. 13, 659–671. 10.1038/nrm3439. [DOI] [PubMed] [Google Scholar]
- Holt I. J.; Reyes A. (2012) Human mitochondrial DNA replication. Cold Spring Harbor Perspect. Biol. 4, a012971–a012971. 10.1101/cshperspect.a012971. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Alexeyev M.; Shokolenko I.; Wilson G.; LeDoux S. (2013) The maintenance of mitochondrial DNA integrity—critical analysis and update. Cold Spring Harbor Perspect. Biol. 5, a012641. 10.1101/cshperspect.a012641. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Quiros P. M.; Mottis A.; Auwerx J. (2016) Mitonuclear communication in homeostasis and stress. Nat. Rev. Mol. Cell Biol. 17, 213–226. 10.1038/nrm.2016.23. [DOI] [PubMed] [Google Scholar]
- Guilliam T. A.; Doherty A. J. (2017) PrimPol—prime time to reprime. Genes 8, 20. 10.3390/genes8010020. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Garcia-Gomez S.; Reyes A.; Martinez-Jimenez M. I.; Chocron E. S.; Mouron S.; Terrados G.; Powell C.; Salido E.; Mendez J.; Holt I. J.; Blanco L. (2013) PrimPol, an archaic primase/polymerase operating in human cells. Mol. Cell 52, 541–553. 10.1016/j.molcel.2013.09.025. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Singh B.; Li X.; Owens K. M.; Vanniarajan A.; Liang P.; Singh K. K. (2015) Human REV3 DNA polymerase zeta localizes to mitochondria and protects the mitochondrial genome. PLoS One 10, e0140409. 10.1371/journal.pone.0140409. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lange S. S.; Takata K.-i.; Wood R. D. (2011) DNA polymerases and cancer. Nat. Rev. Cancer 11, 96–110. 10.1038/nrc2998. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Krasich R.; Copeland W. C. (2017) DNA polymerases in the mitochondria: A critical review of the evidence. Front. Biosci., Landmark Ed. 22, 692–709. 10.2741/4510. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bailey L. J.; Doherty A. J. (2017) Mitochondrial DNA replication: a PrimPol perspective. Biochem. Soc. Trans. 45, 513–529. 10.1042/BST20160162. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wisnovsky S.; Jean S. R.; Kelley S. O. (2016) Mitochondrial DNA repair and replication proteins revealed by targeted chemical probes. Nat. Chem. Biol. 12, 567–573. 10.1038/nchembio.2102. [DOI] [PubMed] [Google Scholar]
- Rin Jean S.; Tulumello D. V.; Wisnovsky S. P.; Lei E. K.; Pereira M. P.; Kelley S. O. (2014) Molecular vehicles for mitochondrial chemical biology and drug delivery. ACS Chem. Biol. 9, 323–333. 10.1021/cb400821p. [DOI] [PubMed] [Google Scholar]
- Fonseca S. B.; Pereira M. P.; Mourtada R.; Gronda M.; Horton K. L.; Hurren R.; Minden M. D.; Schimmer A. D.; Kelley S. O. (2011) Rerouting chlorambucil to mitochondria combats drug deactivation and resistance in cancer cells. Chem. Biol. 18, 445–453. 10.1016/j.chembiol.2011.02.010. [DOI] [PubMed] [Google Scholar]
- Chamberlain G. R.; Tulumello D. V.; Kelley S. O. (2013) Targeted delivery of doxorubicin to mitochondria. ACS Chem. Biol. 8, 1389–1395. 10.1021/cb400095v. [DOI] [PubMed] [Google Scholar]
- Fernandez-Vidal A.; Guitton-Sert L.; Cadoret J.-C.; Drac M.; Schwob E.; Baldacci G.; Cazaux C.; Hoffmann J. S. (2014) A role for DNA polymerase θ in the timing of DNA replication. Nat. Commun. 5, 1–10. 10.1038/ncomms5285. [DOI] [PubMed] [Google Scholar]
- Mateos-Gomez P. A.; Gong F.; Nair N.; Miller K. M.; Lazzerini-Denchi E.; Sfeir A. (2015) Mammalian polymerase θ promotes alternative NHEJ and suppresses recombination. Nature 518, 254–257. 10.1038/nature14157. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Yoon J. H.; Roy Choudhury J.; Park J.; Prakash S.; Prakash L. (2014) A role for DNA polymerase θ in promoting replication through oxidative DNA lesion, thymine glycol, in human cells. J. Biol. Chem. 289, 13177–13185. 10.1074/jbc.M114.556977. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wyatt D. W.; Feng W.; Conlin M. P.; Yousefzadeh M. J.; Roberts S. A.; Mieczkowski P.; Wood R. D.; Gupta G. P.; Ramsden D. A. (2016) Essential roles for polymerase theta-mediated end joining in the repair of chromosome breaks. Mol. Cell 63, 662–673. 10.1016/j.molcel.2016.06.020. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Calvo S. E.; Clauser K. R.; Mootha V. K. (2016) MitoCarta2.0: an updated inventory of mammalian mitochondrial proteins. Nucleic Acids Res. 44, 1251–1257. 10.1093/nar/gkv1003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Pagliarini D. J.; Calvo S. E.; Chang B.; Sheth S. A.; Vafai S. B.; Ong S. E.; Walford G. A.; Sugiana C.; Boneh A.; Chen W. K.; Hill D. E.; Vidal M.; Evans J. G.; Thorburn D. R.; Carr S. A.; Mootha V. K. (2008) A mitochondrial protein compendium elucidates complex I disease biology. Cell 134, 112–123. 10.1016/j.cell.2008.06.016. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Horton K. L.; Stewart K. M.; Fonseca S. B.; Guo Q.; Kelley S. O. (2008) Mitochondria-penetrating peptides. Chem. Biol. 15, 375–382. 10.1016/j.chembiol.2008.03.015. [DOI] [PubMed] [Google Scholar]
- Yogev O.; Pines O. (2011) Dual targeting of mitochondrial proteins: mechanism, regulation and function. Biochim. Biophys. Acta, Biomembr. 1808, 1012–1020. 10.1016/j.bbamem.2010.07.004. [DOI] [PubMed] [Google Scholar]
- Claros M. G. (1995) MitoProt, a Macintosh application for studying mitochondrial proteins. Bioinformatics 11, 441–447. 10.1093/bioinformatics/11.4.441. [DOI] [PubMed] [Google Scholar]
- Bogenhagen D. F. (2012) Mitochondrial DNA nucleoid structure. Biochim. Biophys. Acta, Gene Regul. Mech. 1819, 914–920. 10.1016/j.bbagrm.2011.11.005. [DOI] [PubMed] [Google Scholar]
- Gilkerson R.; Bravo L.; Garcia I.; Gaytan N.; Herrera A.; Maldonado A.; Quintanilla B. (2013) The mitochondrial nucleoid: integrating mitochondrial DNA into cellular homeostasis. Cold Spring Harbor Perspect. Biol. 5, a011080. 10.1101/cshperspect.a011080. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ashley N.; Harris D.; Poulton J. (2005) Detection of mitochondrial DNA depletion in living human cells using PicoGreen staining. Exp. Cell Res. 303, 432–446. 10.1016/j.yexcr.2004.10.013. [DOI] [PubMed] [Google Scholar]
- Lentz S. I.; Edwards J. L.; Backus C.; McLean L. L.; Haines K. M.; Feldman E. L. (2010) Mitochondrial DNA (mtDNA) biogenesis: visualization and duel incorporation of BrdU and EdU into newly synthesized mtDNA In Vitro. J. Histochem. Cytochem. 58, 207–218. 10.1369/jhc.2009.954701. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kalderon D.; Roberts B. L.; Richardson W. D.; Smith A. E. (1984) A short amino acid sequence able to specify nuclear location. Cell 39, 499–509. 10.1016/0092-8674(84)90457-4. [DOI] [PubMed] [Google Scholar]
- Taylor R. W.; Turnbull D. M. (2005) Mitochondrial DNA mutations in human disease. Nat. Rev. Genet. 6, 389–402. 10.1038/nrg1606. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Seki M.; Marini F.; Wood R. D. (2003) POLQ (Pol θ), a DNA polymerase and DNA-dependent ATPase in human cells. Nucleic Acids Res. 31, 6117–6126. 10.1093/nar/gkg814. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kawamura K.; Bahar R.; Seimiya M.; Chiyo M.; Wada A.; Okada S.; Hatano M.; Tokuhisa T.; Kimura H.; Watanabe S.; Honda I.; Sakiyama S.; Tagawa M.; O-Wang J. (2004) DNA polymerase θ is preferentially expressed in lymphoid tissues and upregulated in human cancers. Int. J. Cancer 109, 9–16. 10.1002/ijc.11666. [DOI] [PubMed] [Google Scholar]
- Ceccaldi R.; Liu J. C.; Amunugama R.; Hajdu I.; Primack B.; Petalcorin M. I.; O’Connor K. W.; Konstantinopoulos P. A.; Elledge S. J.; Boulton S. J.; Yusufzai T.; D’Andrea A. D. (2015) Homologous-recombination-deficient tumours are dependent on Polθ-mediated repair. Nature 518, 258–262. 10.1038/nature14184. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ju Y. S.; Alexandrov L. B.; Gerstung M.; Martincorena I.; Nik-Zainal S.; Ramakrishna M.; Davies H. R.; Papaemmanuil E.; Gundem G.; Shlien A.; Bolli N.; Behjati S.; Tarpey P. S.; Nangalia J.; Massie C. E.; Butler A. P.; Teague J. W.; Vassiliou G. S.; Green A. R.; Du M. Q.; Unnikrishnan A.; Pimanda J. E.; Teh B. T.; Munshi N.; Greaves M.; Vyas P.; El-Naggar A. K.; Santarius T.; Collins V. P.; Grundy R.; Taylor J. A.; Hayes D. N.; Malkin D.; Foster C. S.; Warren A. Y.; Whitaker H. C.; Brewer D.; Eeles R.; Cooper C.; Neal D.; Visakorpi T.; Isaacs W. B.; Bova G. S.; Flanagan A. M.; Futreal P. A.; Lynch A. G.; Chinnery P. F.; McDermott U.; Stratton M. R.; Campbell P. J. (2014) Origins and functional consequences of somatic mitochondrial DNA mutations in human cancer. eLife 3, e02935. 10.7554/eLife.02935. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ceccaldi R.; Liu J. C.; Amunugama R.; Hajdu I.; Primack B.; Petalcorin M. I.; O’Connor K. W.; Konstantinopoulos P. A.; Elledge S. J.; Boulton S. J.; Yusufzai T.; D’Andrea A. D. (2015) Homologous-recombination-deficient tumours are dependent on Poltheta-mediated repair. Nature 518, 258–262. 10.1038/nature14184. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Koole W.; van Schendel R.; Karambelas A. E.; van Heteren J. T.; Okihara K. L.; Tijsterman M. (2014) A Polymerase Theta-dependent repair pathway suppresses extensive genomic instability at endogenous G4 DNA sites. Nat. Commun. 5, 3216. 10.1038/ncomms4216. [DOI] [PubMed] [Google Scholar]
- Black S. J.; Kashkina E.; Kent T.; Pomerantz R. T. (2016) DNA polymerase θ: A unique multifunctional end-joining machine. Genes 7, 67. 10.3390/genes7090067. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wood R. D.; Doublié S. (2016) DNA polymerase θ (POLQ), double-strand break repair, and cancer. DNA Repair 44, 22–32. 10.1016/j.dnarep.2016.05.003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ishikawa K.; Takenaga K.; Akimoto M.; Koshikawa N.; Yamaguchi A.; Imanishi H.; Nakada K.; Honma Y.; Hayashi J. I. (2008) ROS-generating mitochondrial DNA mutations can regulate tumor cell metastasis. Science 320, 661–664. 10.1126/science.1156906. [DOI] [PubMed] [Google Scholar]
- Mahon K. P.; Potocky T. B.; Blair D.; Roy M. D.; Stewart K. M.; Chiles T. C.; Kelley S. O. (2007) Deconvolution of the cellular oxidative stress response with organelle-specific peptide conjugates. Chem. Biol. 14, 923–930. 10.1016/j.chembiol.2007.07.011. [DOI] [PubMed] [Google Scholar]
- Langmead B.; Trapnell C.; Pop M.; Salzberg S. L. (2009) Ultrafast and memory-efficient alignment of short DNA sequences to the human genome. Genome Biol. 10, R25. 10.1186/gb-2009-10-3-r25. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Li H.; Handsaker B.; Wysoker A.; Fennell T.; Ruan J.; Homer N.; Marth G.; Abecasis G.; Durbin R. (2009) The sequence alignment/map format and SAMtools. Bioinformatics 25, 2078–2079. 10.1093/bioinformatics/btp352. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gao J.; Aksoy B. A.; Dogrusoz U.; Dresdner G.; Gross B.; Sumer S. O.; Sun Y.; Jacobsen A.; Sinha R.; Larsson E.; Cerami E.; Sander C.; Schultz N. (2013) Integrative analysis of complex cancer genomics and clinical profiles using the cBioPortal. Sci. Signaling 6, pl1. 10.1126/scisignal.2004088. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Cerami E.; Gao J.; Dogrusoz U.; Gross B. E.; Sumer S. O.; Aksoy B. A.; Jacobsen A.; Byrne C. J.; Heuer M. L.; Larsson E.; Antipin Y.; Reva B.; Goldberg A. P.; Sander C.; Schultz N. (2012) The cBio cancer genomics portal: an open Platform for exploring multidimensional cancer genomics data. Cancer Discovery 2, 401. 10.1158/2159-8290.CD-12-0095. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.