

Abstract
Mice have multiple obstacles to HIV replication, including a block of unspliced and partially spliced viral mRNA nuclear export. In human, Rev binds to the Rev-response element and human (h) Crm1, facilitating nuclear export of RRE-containing viral RNAs. Murine (m) Crm1 is less functional than hCrm1 in this regard. Here we demonstrated that in biochemical experiments mCrm1 failed to interact with HIV Rev whereas hCrm1 did. In genetic experiments in human cells, we observed a modest but significant differential effect between mCrm1 and hCrm1, which was also true of other lentiviral Revs tested. Triple mutant hCrm1 P411T-M412V-F414S behaved similarly to mCrm1, whereas mCrm1 with T411P-V412M-S414F regained some activity, although contribution of additional residues to its function can not be excluded. Similar results were observed in murine cells. This suggests a differential interaction between hCrm1 and mCrm1 and many lentiviral Revs, which may partially explain the HIV replicative defect in mice.
Keywords: Crm1, Rev, Lentiviruses, Virus-host interaction, Nuclear export, HIV
1. Introduction
Since the recognition of AIDS in 1981 more than thirty-five years ago, nearly 70 million individuals have been infected with human immunodeficiency virus type 1 (HIV) and roughly half have died (http://www.unaids.org/en/resources/documents/2016/AIDS-by-the-numbers). Although the introduction of HAART two decades ago has been truly transformative, HIV disease, once invariably fatal, remains incurable. The development and testing of a safe and efficacious prophylactic HIV vaccine, a long-sought goal, has been stymied by the absence of a small animal model. Although chimpanzees can be infected with HIV there is now a moratorium on their experimentation (Kaiser, 2015), and the use of simian-HIV (SHIV) hybrids in rhesus macaques is limited by animal availability and expense. Ideally, a fully permissive small animal model would allow higher throughput testing of candidate vaccines and correlates of immunity, if any. Unfortunately, current humanized mouse models, although highly sophisticated and informative (Denton and Garcia, 2011), do not allow for vaccine testing.
Most rodent species, including the mouse, have multiple blocks to HIV replication, including at the level of viral entry and transcriptional elongation. Even when those obstacles are circumvented using entry factors and human cyclin T1, which allows for high level viral RNA production, very little infectious HIV is produced from murine cells (Coskun et al., 2007, 2006; Elinav et al., 2012; Sherer et al., 2011; Swanson et al., 2010). We and others have pinpointed a major post-integration block in mouse cells at the level of unspliced and partially spliced viral RNA nuclear export (Elinav et al., 2012; Sherer et al., 2011; Swanson et al., 2010).
All retroviruses require nuclear export of intron-containing mRNA for productive, high level replication, and for the lentiviruses this process requires a cis-acting RNA sequence and trans-acting viral and cellular proteins. HIV Rev is an essential regulatory protein that is highly conserved among all viral isolates and clades (Malim et al., 1989a, b, c). It is encoded by a fully spliced mRNA, and is in a different reading frame than Tat, but shares precisely the same major intron. After cytosolic translation, Rev is imported into the nucleus, where it multimerizes on the Rev-response element (RRE, present within that same intron) to allow nuclear export of unspliced and partially spliced viral mRNAs, including genomic RNA (Madore et al., 1994; Malim and Cullen, 1991; Mann et al., 1994; Szilvay et al., 1997; Vercruysse and Daelemans, 2013). Based upon biochemical analysis it is thought that 6–8 Revs cooperatively bind a single RRE (Cook et al., 1991; Daly et al., 1989; Daugherty et al., 2008; Daugherty et al., 2010; Heaphy et al., 1991; Holland et al., 1990; Wingfield et al., 1991); a low resolution structure has demonstrated a Rev dimer binding across a bent RRE (Fang et al., 2013). In the nucleus Rev-RRE complex interacts with host factors chromosome region maintenance 1 (Crm1) and Ran-GTP, and in the cytosol the complex dissociates to be recycled to the nucleus to export additional cargo, leaving RRE-containing viral mRNA in the cytosol. In the absence of Rev multiple viral proteins cannot be synthesized, including Gag, Pol, and Env, and genomic RNA is trapped in the nucleus or spliced. Interestingly there are no cellular homologs of Rev. Only rarely are intron-containing cellular mRNAs exported to the cytosol via an analogous system (Li et al., 2006; Wang et al., 2015), whereas all lentiviruses routinely use the Rev-RRE complex for trafficking of viral mRNAs with retained introns.
We had previously observed a decrease in unspliced, intron-containing HIV mRNA in infected murine cells (Coskun et al., 2007, 2006; Elinav et al., 2012). The presence of human chromosome 2 in murine cells largely reversed this defect and significantly enhanced infectious virus production (Coskun et al., 2006). We and other investigators identified human (h)Crm1 as the likely gene product on chromosome 2 responsible for this effect (Elinav et al., 2012; Nagai-Fukataki et al., 2011; Okada et al., 2009; Sherer et al., 2011). Expression of hCrm1 in murine cells allowed export of intron-containing HIV mRNAs from the nucleus and significantly boosted virus production, whereas murine (m) Crm1 was non-functional. In our hands this defect mapped to HEAT (Huntingtin, elongation factor 3, protein phosphatase 2A, and the yeast kinase TOR1) repeat 9A of hCrm1, specifically to amino acid residues 411, 412, and 414 (Elinav et al., 2012). The effect of hCrm1 was even more pronounced using feline immunodeficiency virus, and hCrm1 acted more than additively with hSRp40, a serine-arginine rich splicing factor, to increase infectious virus production from mouse cells. More recently it was demonstrated that the addition of a second nuclear export signal to HIV Rev allowed for both enhanced HIV Capsid (CA) production and infectious virus release from murine cells (Aligeti et al., 2014), consistent with a fundamental defect in nuclear export of viral mRNAs in rodent cells.
The question remained as to why hCrm1 was functional and mCrm1 was not in terms of Rev and HIV mRNA nuclear export, and previously we proposed three possibilities (Elinav et al., 2012). One was that human cells had a positively acting factor that somehow stabilized the hCrm1 and Rev-RRE complex (but did not do so for mCrm1); a second was that murine cells had a negatively acting factor that disrupted the mCrm1 and Rev-RRE complex (but did not do so for hCrm1). Lastly, we hypothesized that hCrm1 simply interacted more favorably or strongly with Rev-RRE complex, compared to mCrm1. We decided to test the third model by evaluating hCrm1 and mCrm1 interaction with lentiviral Rev proteins, first biochemically and then genetically, the latter using a mammalian two-hybrid system in both human and murine cells.
2. Results
To test whether there is a differential interaction between hCrm1 and mCrm1 and Rev-RRE complex, we first turned to a biochemical method initially developed by Cullen and colleagues (Bogerd et al., 1998), using purified, bacterially expressed HIV Rev-GST and Ran-GTP proteins, along with in vitro transcribed HIV RRE RNA and in vitro translated hCrm1 or mCrm1 proteins. Both Rev-GST and RevM10-GST fusions were purified using glutathione beads as ~45 kD proteins (Fig. 1). When incubated with Ran-GTP and either in vitro translated, HA epitope-tagged hCrm1or mCrm1, only HA-hCrm1 was precipitated, but only in the presence of HIV Rev-GST fusion and RRE RNA (Fig. 1). This is consistent with prior work showing ~2× stronger binding of purified hCrm1 compared to mCrm1 to HIV-1 Rev (Booth et al., 2014). As expected, RevM10-GST fusion failed to precipitate or bind either HA-Crm1 protein, in the presence or absence of RRE.
Fig. 1.
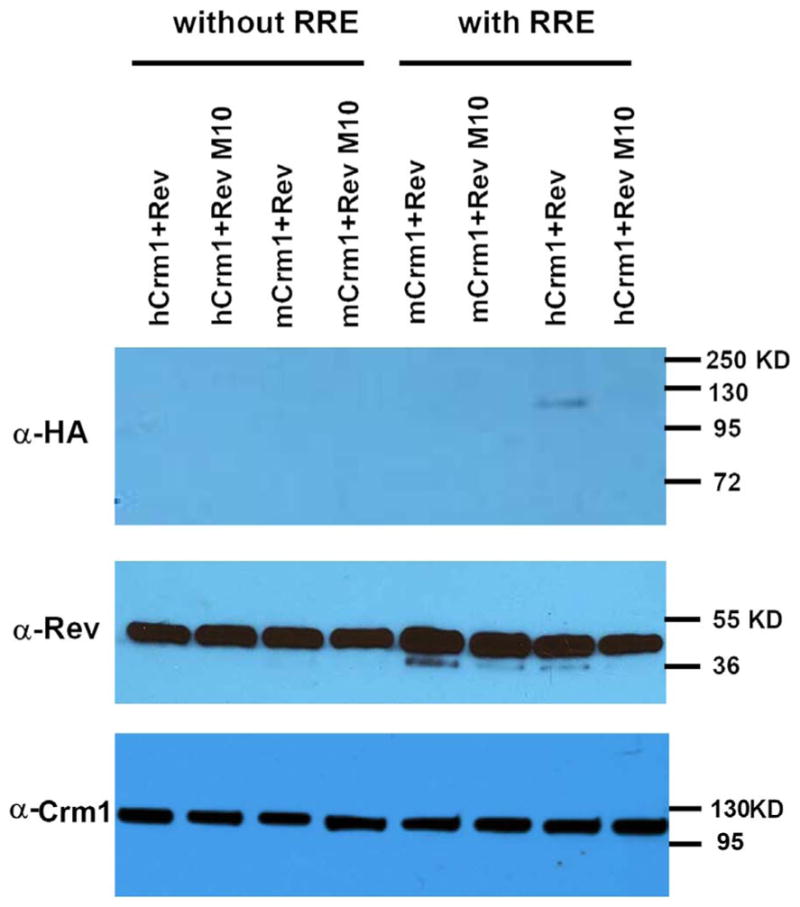
In vitro interaction of human or mouse Crm1 with HIV Rev. Top: GST pull-down assay was performed to compare binding of human vs. mouse Crm1 with HIV1 Rev/RevM10-GST in the presence of RanQ69L-GTP, with or without HIV1 RRE. Middle: input for each reaction probed with anti-Rev antibody; bottom: input for each reaction probed with anti-Crm1 antibody. Molecular mass markers are shown to the right of each panel. This experiment was performed once.
To confirm and extend those findings of a differential interaction between hCrm1 and mCrm1 and Rev-RRE, we first attempted to use the yeast two and three hybrid systems, but failed to observe a detectable interaction between any of the Crm1s and Rev, in the presence or absence of HIV RRE RNA. We then turned to the luciferase complementation system in which protein interaction between amino and carboxy terminal firefly luciferase (FFLUC) fusion proteins results in detectable FFLUC activity by relative light units (RLUs) (Luker et al., 2004). Using this system we were able to detect a strong HIV Rev-Rev genetic interaction but not between Rev and any Crm1 fusion protein. Because the luciferase complementation system may only allow readout when each fusion partner is small enough to permit enzymatic catalysis and full-length Crm1 is greater than 100 kD in size, we next attempted the mammalian two-hybrid system, knowing that we could also separately transfect in HIV RRE RNA in plasmid form, driven by either RNAPII or III promoters.
In the mammalian two-hybrid system one protein fusion is with the Gal4 DNA binding domain (DBD) and the other is with the herpes simplex virus VP16 activation domain (AD), with positive control fusions utilizing cellular p53 and SV40 TAg as known, strong interactors. The reporter we used had five repeats of the 17-mer Gal4 DNA binding motif driving FFLUC. Initially we tested a series of Rev and Crm1 fusion proteins in 293T transient transfection assays to identify a combination that gave the lowest level of autoactivation. Although the Gal4DBD-Crm1 fusions gave some FFLUC readout when transfected in the absence of any VP16 fusion partner, the autoactivation was much lower compared to that of the VP16AD-Crm1 fusions (not shown).
Based on these results, we tested the functionality of the Gal4DBD-Crm1 fusions in a functional assay, transfecting them into murine B78 cells that had an integrated HIV reporter vector encoding both a truncated form of human cyclin T1 and blasticidin resistance (bsd) (Coskun et al., 2007), along with VSV G expression construct and an HIV vector encoding a truncated form of human cyclin T1 and eYFP. The B78 cells express murine Crm1, and this assay allows us to test the functionality of other Crm1 constructs (Fig. 2A). Human cyclin T1 is required since expression of both eYFP and bsd in the vector used are dependent upon the HIV long terminal repeat and the presence of Tat. As anticipated, the non-fused, full-length hCrm1 had the greatest activity in terms of infectious virus release, as measured by the number of blasticidin-resistant colonies on HOS cell targets (Fig. 2B). The Gal4DBD-hCrm1 fusion had ~25% of the activity of the non-fused hCrm1 but was significantly more active than the Gal4DBD-mCrm1 fusion and the Gal4DBD-hCrm1 411–412-414 mutant. As reported previously (Aligeti et al., 2014), the 2xNES-Rev-mCherry expression plasmid gave roughly 50% of the number of colonies, in the absence of hCrm1 (Fig. 2B). As anticipated, similar results were obtained using eYFP as a flow cytometric readout on the HOS targets (not shown).
Fig. 2.

Functionality of Crm1 and HIV Rev fusions. (A) Schematic of how functionality of various Crm1 constructs was tested using B78 cells and titering on HOS. HIV-CIB denotes integrated HIV vector encoding both truncated cyclin T1 and bsdr and HIV-CIY is a similar vector encoding truncated cyclinT1 and eYFP; (B) B78 cells with integrated HIV.CIB were nucleofected in triplicate as in (A) and the indicated plasmids. At 72 h, supernatant was harvested and used to infect HOS cells. After selection in blasticidin-containing medium colonies were enumerated, normalized to the transfection efficiency of the B78 cells based upon eYFP expression. Similar differences in resultant eYFP titers on HOS target cells were observed, as measured by flow cytometry 72 h post-transduction (not shown). (C) Schematic of how functionality of various Rev constructs were tested; HIV-LIB ΔRev denotes an HIV vector that encodes both FFLUC and bsdr with a frameshift in rev; (D) 293Ts were transfected in triplicate with the indicated plasmids and supernatant was titered on HOS targets, with blasticidin-resistant colonies enumerated 7–9 days after selection. (E) As in (D), with FFLUC RLU measured 72 h post-transduction of HOS target cells. Bars indicate mean values +/−1 S.D. * P < 0.001, ** P < 0.01, *** P < 0.05 by student’s t-test. NS: not significant compared to mock transfection. Shown is a representative of three experiments with similar results.
A related, cell-based approach was used to test the functionality of the various HIV Rev fusions, but in a trans-complementation assay using 293T producers transfected with both VSV G and a Rev-deficient HIV reporter vector encoding both FFLUC and bsd (Fig. 2C). As expected, the non-fused, full-length version of NL4-3 Rev had the greatest activity for both readouts (RLU and blasticidin-resistant colonies on susceptible HOS targets), RevM10-GFP fusion had very little to no activity, and the two VP16AD-Rev fusion constructs had between 25% and > 50% of the activity of the non-fused Rev (Fig. 2D and E). Of note, these constructs were all expressed in 293T cells, based upon immunoblotting (Fig. 3A). Based on these results, we decided to test both Gal4DBD-Crm1 fusions and the VP16AD-Rev fusions in the mammalian two hybrid system.
Fig. 3.

Interaction of HIV Rev with human vs. mouse Crm1 detected by Mammalian two-hybrid assay. 293T cells were co-transfected with the indicated expression plasmids along with the pG5LUC reporter and pMax-GFP (Lonza) plasmids, the latter to monitor transfection efficiency. (A) Immunoblotting of indicated proteins in transfected 293T cells. (B) Genetic interaction between indicated Crm1s and HIV Rev proteins was assessed by luciferase activity, normalized to maxGFP expression. Positive control shows interaction of GAL4 DBD-fused mouse p53 protein with VP16 AD-fused SV40 Tag. Empty pVP16 and pM plasmids were used as basal control. Relative luciferase activity is shown by the fold change from the basal control set to 1. Experiments were repeated 3 times with similar results, and a representative one is shown. (C) Results of (B) are shown, but now normalized to the sum of RLU results with the pVP16AD-Rev and the pGal4DBD-Crm1 plasmids each alone (sum set at 1.0). * P < 0.05, ** P < 0.01, *** P < 0.001. Experiments of (B) were repeated at least 3 times with similar results, with the exception that Rev M10 experiments were repeated twice; for (C) values are the average of three experiments, again with the exception of Rev M10 (average of two experiments).
Co-transfection of 293T cells with Gal4DBD-hCrm1 and VP16AD-NL43Rev plasmids along with the Gal4-FFLUC reporter led to an increase in RLU which was greater compared to when either plasmid was transfected alone or when VP16AD-RevM10 plasmid was used (Fig. 3B and C). The amount of RLU was significantly higher when Gal4DBD-hCrm1 was transfected compared to Gal4DBD-mCrm1, and the increase in RLU was observed in the presence and absence of co-transfected HIV RRE (Fig. 3B and C), consistent with the Rev-hCrm1 interaction being independent of the RRE. This may be due to the abundance of cellular RNA to which Rev can bind in a non-specific manner, which would largely be absent in the biochemical in vitro experiments described above (Fig. 1). To confirm that the observed higher level of RLU observed in the presence of Gal4DBD-hCrm1 and VP16AD-Rev was specific, increasing amounts of non-fused, full-length HA-hCrm1 or HA-mCrm1expression plasmid was transfected into 293T cells, and the RLU decreased significantly only in the presence of increasing amounts of hCrm1, not empty vector or mCrm1 (Fig. 4A, B).
Fig. 4.

Interaction specificity of human Crm1 with HIV Rev. 293Ts were co-transfected with pVP16AD-Rev and pGal4DBD-hCrm1, pG5LUC luciferase reporter plasmid and pMax-GFP, with increasing amounts of either pCMV-HA, pCMV-HA-hCrm1, or pCMV-HA-mCrm1, as indicated. (A) Immunoblotting of Crm1 in co-transfected 293T cells using anti-HA 12CA5 antibody, with tubulin serving as loading control. (B) Interaction between pVP16AD-Rev and pGal4DBD-hCrm1 or pGal4DBD-mCrm1, as measured by luciferase activity, normalized to both transfection efficiency and also to resultant RLU for pVP16AD-Rev + pGal4DBD-hCrm1 (set at 100). Positive control as in Fig. 2. Experiments were repeated twice with similar results; representative normalized RLU values are shown. * P < 0.05, **, P < 0.01.
We next turned our attention to other lentiviral Revs, including those of FIV, HIV-2, and EIAV, each of whose function is thought to be dependent on Crm1. Because these Revs are all functional in 293T and not murine cells, with the possible exception of EIAV Rev, we compared the interaction effects of hCrm1 vs mCrm1. In each case the VP16AD-Rev fusion was detected by immunoblotting, although for uncertain reasons the HIV-2 Rev fusion was expressed at lower levels (Fig. 5A). For FIV and HIV-2 Rev fusions with VP16AD, co-transfection with Gal4DBD-hCrm1 resulted in significantly higher RLUs compared to co-transfection with Gal4DBD-mCrm1, in the presence and absence of FIV or HIV-2 RRE, respectively (Fig. 5B and C). On the other hand, for EIAV Rev fusion we never observed a significant increase in RLU when using Gal4DBD-hCrm1 compared to Gal4DBD-mCrm1, in the presence or absence of EIAV RRE (Fig. 5B and C). This is consistent with our prior failure to achieve higher infectious EIAV release from murine cells in the presence of hCrm1 compared to mCrm1 (Elinav et al., 2012).
Fig. 5.

Interaction of human or mouse Crm1 with other lentiviral Revs by mammalian two-hybrid assay. 293Ts were co-transfected the pG5LUC luciferase reporter plasmid, pMax-GFP, either pGal4DBD-hCrm1 or pGal4DBD-mCrm1, and the indicated VP16AD fusions encoding Revs derived from either FIV, EIAV, or HIV-2. (A) Immunoblotting of FIV, EIAV, and HIV-2 Rev-VP16 fusion proteins in transfected 293T cells, with tubulin serving as loading control. (B) Interaction between pGal4DBD-hCrm1 or pGal4DBD-mCrm1 and various VP16AD-Rev fusions was monitored by luciferase activity, normalized to both transfection efficiency and also to resultant RLU from using empty pVP16 and pM plasmids (set at 1). Experiments were repeated 3 times with similar results, and representative normalized RLU are shown. (C) Results of (B) are shown, but now normalized to the sum of RLU results with the pVP16AD-Rev and the pGal4DBD-Crm1 plasmids each alone. * P < 0.05. For (B) and (C) experiments were repeated at least three times with similar results.
We also tested the Gal4DBD-hCrm1-411-412–414 mutant in the above assays. By immunoblotting we confirmed equal expression of that fusion protein compared to Gal4DBD-hCrm1 and Gal4DBD-mCrm1 in 293T cells (Fig. 6A). For all lentivirus Rev fusions tested, there was no increase in RLU in the presence of Gal4DBD-hCrm1-411-412–414 fusion compared to Gal4DBD-hCrm1; in fact typically compared to Gal4DBD-mCrm1 there was a decrease in RLU (Fig. 6B and C). With regards to HIV, HIV-2, and FIV Rev, the presence or absence of the corresponding lentiviral RRE appeared to have little impact on the fold-effect observed with the hCrm1 fusion (Fig. 6B and C). Again, in the presence or absence of EIAV RRE, for EIAV Rev there was no difference in the fold-effect seen, irrespective of Gal4DBD-Crm1 used (Fig. 6C).
Fig. 6.

Interaction of hCrm1 P411-M412-F414 with various Revs. 293T cells were co-transfected with the indicated plasmids, along with pG5LUC luciferase reporter and pMax-GFP to monitor transfection efficiency. (A) Immunoblotting of Gal4DBD-Crm1 fusion constructs along with empty vector (EV) in transfected 293T cells, with tubulin serving as loading control. (B) Interaction of various VP16AD-Rev fusions with Gal4DBD-hCrm1, mCrm1, or hCrm1 P411T-M412V-F414S mutant were measured by RLU using mammalian two-hybrid assay, normalized to GFP expression. (C) Results of (B) are shown, but now normalized to the sum of RLU results with the pVP16AD-Rev and the pGal4DBD-Crm1 plasmids each alone. (D) Immunoblotting using anti-Gal4DBD antibody of indicated Gal4DBD-Crm1 fusion constructs, using four different amounts, in transfected 293T cells, with tubulin again serving as loading control; (E) normalized RLU results with the indicated Gal4DBD-Crm1 plasmids and VP16AD-Rev. Data are mean ± SEM, representative of three different experiments. * P < 0.05, ** P < 0.01, *** P < 0.001. For (B) experiment was done in triplicate; for (C) average data from two independent experiments are shown; for (E) experiment was repeated three times with similar results.
We next made the Gal4DBD-mCrm1 fusion construct with hCrm1 residues at positions 411, 412 and 414 (termed pM-mCrm1h146-444). This construct also has A192S, E284V, G334D, L337I, A346T, and I403V, although these latter residues have not been shown to contribute to HIV Rev activity (Elinav et al., 2012; Sherer et al., 2011). A similar fusion construct but with hCrm1 residuces between amino acids 444 and 805 (termed pM-mCrm1h444-805), the latter to serve as a negative control but also has I474V, K478E, and Q481H. Expression of both of these chimeric fusions in 293T cells was approximately the same by dose titration as the parental Gal4DBD-mCrm1 and Gal4DBD-hCrm1 fusions, allowing for some variance in protein loading (Fig. 6D). In the presence of VP16AD-HIV Rev fusion, there was a significant increase in RLU in the presence of pM-mCrm1h146-444 compared to parental Gal4DBD-mCrm1, although the fold-effect was less than that of Gal4DBD-hCrm1 (Fig. 6E). Construct pM-mCrm1h444-805 trended to slightly higher RLU but was not significant (Fig. 6E). These results suggest those three HEAT repeat 9A amino acid residues are at least partially responsible for the differential effects observed in the mammalian two hybrid assays between hCrm1 and mCrm1 and HIV-1 Rev, consistent with our previous results of infectious virus release from mouse cells (Elinav et al., 2012). We cannot, however, exclude contributions from other downstream residues, as reported previously (Sherer et al., 2011).
The mammalian two-hybrid results described above were all obtained in 293T cells, and thus the higher RLU observed specific to the hCrm1 fusion could be consistent with human cells having an unknown activator that interacts with hCrm1 HEAT repeat 9A and somehow facilitates Crm1-Rev/RRE function. Although this scenario is unlikely based upon the biochemical results of Fig. 1, the mammalian two hybrid experiments were repeated in murine B78 cells using the same Gal4DBD and VP16AD fusion constructs. To achieve adequate transfection efficiency and consequent protein expression levels, B78 cells were transfected using Amaxa nucleofector system with various fusion constructs along with HIV vector FG12, which encodes both an RRE and an eGFP reporter (Qin et al., 2003). Transfection efficiency was ~ 10%, as judged by epifluorescence microcopy of eGFP expressed from FG12 vector. Cells were allowed to recover and proliferate for 4 d and then lysed for FFLUC RLU. B78 cells mock- or transfected with only Crm1 or Rev fusion constructs exhibited low RLU, whereas there was a significant increase in RLU in the presence of both the mCrm1 and Rev fusions (Fig. 7). There was a significantly higher amount of RLU in the presence of both the hCrm1 and Rev fusions compared to each individually or in the presence of mCrm1 and Rev fusions, whereas the highest levels of RLU were observed for the positive control interaction (p53 and SV40 Tag), as expected. Similar results were observed when B78 cells were transfected with cationic lipids (not shown). These results suggest that the more favorable genetic interaction observed between hCrm1 and Rev compared to mCrm1 and Rev is cell-type independent, since it is observed in both human and murine cells.
Fig. 7.
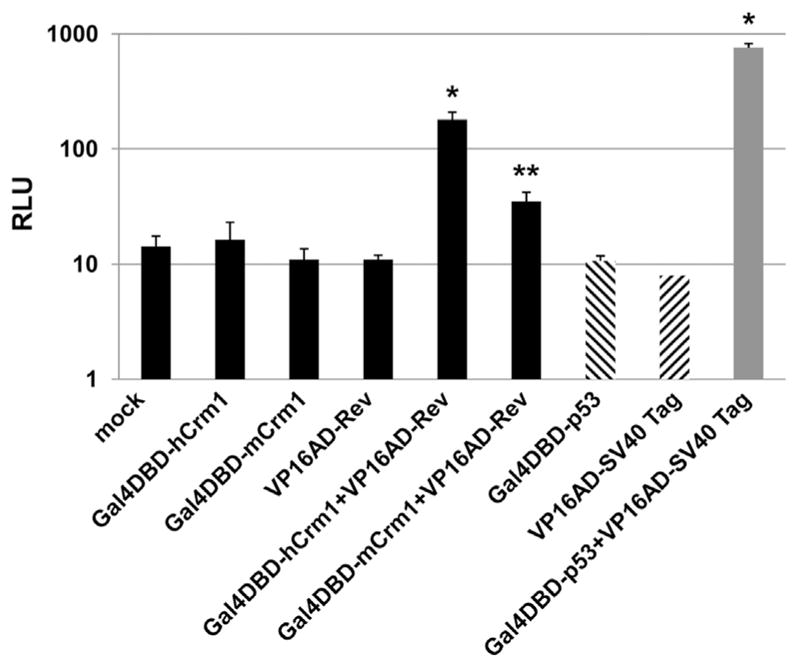
Interaction of HIV Rev with human vs. mouse Crm1 in murine cells. B78 cells were nucleofected with FG12 HIV vector along with the pG5LUC luciferase reporter and the indicated plasmids in triplicate. In each case transfection efficiency was approximately 10%, as judged by eGFP expression from FG12 vector. Cells were lysed at 96 h for luciferase assay. Bars indicate mean values +/− 1 S.D. *P < 0.01, **P > 0.05 by student’s t-test, compared to the sum of RLU values of the respective individual plasmid transfections. Similar results were obtained after Lipofectamine 2000 reagent (Invitrogen) transfection of B78 cells.
3. Discussion
We and others have shown that a major defect to infectious HIV production in murine cells is due to a block at nuclear export of unspliced and partially spliced viral mRNAs (Coskun et al., 2006; Sherer et al., 2011; Swanson et al., 2010). Not only does this result in reduced viral structural protein expression but also decreased levels of cytoplasmic genomic viral mRNA for packaging. This defect was corrected in part by provision of human chromosome 2 (Coskun et al., 2007; Coskun et al., 2006), and more recently it has been demonstrated that at least one of the responsible gene products is Crm1 (Elinav et al., 2012; Nagai-Fukataki et al., 2011; Okada et al., 2009; Sherer et al., 2011), which was known to associate with HIV Rev/RRE complex and facilitate nuclear export of intron-containing viral mRNAs. Murine (m) Crm1, which is precisely the same length at 1071 aa as hCrm1, appears to be a loss-of-function gene, and the important functional differences for HIV infectivity mapped to a cluster or patch of amino acid residues in HEAT repeat 9A, which is distinct and physically distant from the known NES binding domain of Crm1 (Elinav et al., 2012; Sherer et al., 2011). Previously, in vitro experiments had shown that in the presence of HIV Rev two regions of Crm1 were protected against endoprotease treatment—a region encompassing amino acids 800–820 and also an Aspartate at 716, although the functional importance of these regions of Crm1 were not tested (Askjaer et al., 1998).
Rev itself has a single canonical NLS and a leucine rich NES, the latter of which termed the activation domain and is thought to bind to Crm1, but no other Rev sequences or domains have been implicated in Rev binding to Crm1. Rev with two NLS sequences does not have enhanced function in murine cells, whereas Rev with two separated NES sequences (Rev ×2NES) appears to restore function in terms of CA production and infectious virus release in murine cells (Aligeti et al., 2014), such that addition of hCrm1 is unnecessary, which we have confirmed here. This finding is consistent with a fundamental defect in mouse cells being the inability of viral intron-containing mRNAs to be exported from the nucleus. However, the result with Rev ×2NES does not directly address the question as to why mechanistically mCrm1 is less active than hCrm1.
Previously we had proposed three possible models, with one being that hCrm1 fundamentally interacted more favorably or strongly with Rev-RRE complex, compared to mCrm1. We decided to test that model, which was the simplest and most straightforward, by both biochemical and genetic means. By using bacterially-produced Rev-GST and Ran soaked in GTP, along with in vitro transcribed HIV RRE, we were able to demonstrate that in vitro transcribed-translated hCrm1, but not mCrm1, bound to HIV Rev, dependent upon the presence of HIV RRE. As expected, RevM10-GST did not bind either hCrm1 or mCrm1, due to mutations in the NES domain, in the presence or absence of RRE. This result largely confirms the differential binding between mouse and human Crm1 and HIV-1 Rev, as shown by Frankel and colleagues (Booth et al., 2014). To further these results, we tested a number of genetic complementation systems, and the one that gave the most robust and reproducible results was the mammalian two hybrid system. Using that genetic system, in human cells we were able to reproducibly demonstrate a stronger interaction between hCrm1, compared to mCrm1, and HIV Rev, independent of adding HIV RRE. The fold-effect was modest but significant and repeatedly and reproducibly observed, and consistent with fold-effects that we and others have seen on increases in unspliced HIV RNA (Coskun et al., 2006; Elinav et al., 2012; Sherer et al., 2011; Swanson et al., 2010) and the roughly 2-fold higher binding affinity of purifiked hCrm1 compared to mCrm1 to Rev (Booth et al., 2014). Of note, in no genetic system did we detect full-length hCrm1 interacting with itself or mCrm1 or mCrm1 interacting with itself with or without Rev-RRE, observations that have been made by single particle electron microscopy with purified Crm1s in the presence of Rev-RRE and Ran-GTP (Booth et al., 2014). Our negative results, however, may be due to the moderately large size of the Crm1s or relative positioning of the fusion proteins.
Although the RRE-independent interaction between hCrm1 and Rev that we saw in human cells may seem at odds with our in vitro results, Rev could act as a promiscuous RNA binding protein when over-expressed and associate with many non-RRE, cellular RNAs, which may then allow structural conformational changes in Rev, including multimerization and subsequent binding to hCrm1. Because we also observed a stronger, more robust interaction between HIV Rev and hCrm1 compared to mCrm1 in murine cells, this suggests that there is a differential interaction between hCrm1 and mCrm1 and Rev, independent of any other cellular factors in human or mouse cells that could play a role in Rev function. In other words, the third model proposed above is likely to be correct. Because this interaction difference mapped mainly to HEAT repeat 9A of Crm1, distant from the NES domain, this suggests that an unknown region of Rev interacts with HEAT repeat 9A and that mutagenesis of Rev will allow mapping of this binding domain. It may then be possible to identify an altered form of Rev that interacts well with mCrm1 and not hCrm1 to allow infectious particle production from murine cells, in the presence of mCrm1 and absence of hCrm1. Whether mice that are transgenic for hCrm1 and other necessary host factors (e.g., CD4, CCR5, cyclinT1) will permit higher levels of HIV replication is an open question.
Because these results suggest but do not directly demonstrate that HIV Rev interacts with both the NES domain and HEAT repeat 9A of hCrm1, it will be important to perform additional binding studies and biochemical experiments. Previously it had been shown that Crm1 binds Snurportin-1 50-fold greater than Rev (Paraskeva et al., 1999), but it is likely that in those experiments Rev was in monomeric, not multimeric form. Additionally, multiple hCrm1 sites of interaction may be one way that Rev out-competes cellular cargo, such as Snurportin-1, that bind to the NES domain and an adjacent acidic patch of Crm1 (Dong et al., 2009). Enhanced HIV production from murine cells observed with the Rev ×2NES mCherry fusion protein are consistent with these results, suggesting that the additional NES sequence increases the avidity of the Rev fusion for mCrm1, allowing more efficient nuclear export of viral, intron-containing mRNAs. Although tighter binding to mCrm1 may not allow the Rev ×2NES mCherry to recycle back to the nucleus, this may not be relevant in a transient over-expression system in immortalized murine fibroblasts. Our attempts to test the genetic interaction of Rev ×2NES mCherry fusion with various Crm1s failed since the VP16 fusion was poorly expressed for uncertain reasons and did not trans-complement our Rev minus HIV packaging vector system.
Taking into account the modeling study of Booth et al. (2014) suggesting that Crm1 dimerization in the presence of Rev-RRE in vitro depends upon the three key amino acid residues in HEAT Repeat 9A of 411, 412, and 414, and possibly others, a unifying hypothesis is that the interaction of Rev with hCrm1’s NES domain and the dimerization interface is stronger than that of mCrm1. Higher resolution cryoelectron microscopy imaging of the Rev-hCrm1 vs. Rev-mCrm1 complex may be informative in this regard.
Using the mammalian two-hybrid system, in human cells we observed similar fold-effects for hCrm1 but not mCrm1 interaction with FIV and HIV-2 Rev, but not EIAV Rev. Although all of the Rev fusion proteins were at the expected molecular size, we did not perform cell-based complementation assays to determine their functionality. These results are largely consistent with what we observed when transfecting in various lentiviral constructs and over-expressing either hCrm1 or mCrm1 in murine cells (Elinav et al., 2012). One possibility regarding EIAV Rev is that both hCrm1 and mCrm1 are marginally but equally functional and that equine (E. caballus) (e)Crm1 is required for optimal virus production, which we did not test. eCrm1 differs from hCrm1 at aa positions 412 and 414 whereas eCrm1 differs from mCrm1 at aa position 411, the three key residues of HEAT repeat 9A for hCrm1 binding to HIV Rev (Table 1). It should be noted, however, that eCrm1 differs from both hCrm1 and mCrm1 at several other aa positions as well. A prediction would be that provision of eCrm1 to either human or mouse cells would boost production of infectious EIAV. The aa sequences of these three critical aa residues of Crm1 in HEAT repeat 9A, along with three more C-terminal residues in repeat 10 A, of the various mammalian species that have exogenous lentiviruses are summarized in Table 1. That EIAV and FIV behave differently in human and mouse cells suggest that other determinants within Crm1 or other, unknown host factors may be important for EIAV production.
Table 1.
Crm1 heat repeat 9A/10A residues and lentivirus production in various mammalian cells.
| HEAT repeat 9A/10A Crm1 aa Residue | Exogenous lentivirus production | |||||||
|---|---|---|---|---|---|---|---|---|
| Mammalian Species a | 411 | 412 | 414 | 474 | 478 | 481 | Human cells b | Murine cells c |
| Man (Homo sapiens)-HIV | P | M | F | V | E | H | Very high (Elinav et al., 2012) | Very low (Elinav et al., 2012) |
| Goat (Capra hircus)-CAEV | P | V | S | R | E | H | Low (Mselli-Lakhal et al., 2006; Olsen, 2001) | unknown |
| Sheep (Ovis aries)-Visna Maedi virus | P | V | S | R | E | H | low (R.D. Berkowitz et al., 2001) | unknown |
| Horse (Equus caballus)-EIAV | P | V | S | R | E | H | High (Elinav et al., 2012; Olsen, 2001) | Very low (Elinav et al., 2012) |
| Cow (Bos taurus)-BIV | P | V | S | R | E | H | Moderate (R. Berkowitz et al., 2001) | unknown |
| Cow (Bos taurus)-JDV | P | V | S | R | E | H | High (Metharom et al., 2000) | moderate (Metharom et al., 2000) |
| Cat (Felis catus)-FIV | P | V | S | R | E | H | High (Elinav et al., 2012) | Very low (Elinav et al., 2012) |
| Mouse (Mus musculus) | T | V | S | I | K | Q | – | – |
Corresponding exogenous lentivirus also indicated; CAEV denotes Caprine arthritis encephalitis virus, BIV bovine immunodeficiency virus, and JDV Jembrana disease virus.
Very high signifies titers ~107 IU/ml; high ~106 IU/ml; moderate ~105 IU/ml; low ~104 IU/ml.
Very low signifies titers ≤103 IU/ml.
In conclusion, we present both biochemical and genetic evidence of a differential interaction between several lentiviral Revs and hCrm1 and mCrm1, which maps mainly to a triad of aa residues in HEAT repeat 9A of Crm1. Because the effect was observed in both human and murine cells, these data argue against either an additional positive or negative acting ancillary factor in human or mouse cells, respectively. These results also suggest that HIV and other lentiviral Revs have as yet an unidentified domain or region that interacts with Crm1 and that it may be possible to genetically select for altered Revs that interact more strongly with mCrm1, allowing improved HIV replication in murine cells. It may also be possible to identify anti-HIV small molecule inhibitors that reduce Rev avidity or binding for Crm1 and yet do not interfere with essential Crm1 function, by avoiding the NES domain entirely.
4. Materials and methods
4.1. Plasmids & Cells
Full-length, 1071 aa cDNAs of hCrm1, mCrm1, hCrm1 with P411T+M412V+F414S, and mCrm1 with human residues 146–444 and 444–805 were each separately subcloned from previously reported pCMV-HA expression plasmids (Elinav et al., 2012) into expression plasmid pM (Clontech) downstream of Gal4DBD and sequence-verified. Full-length Rev cDNAs of HIV NL4-3, HIV pcv12, HIV-2, FIV, and EIAV were PCR-amplified from respective templates that encoded the respective lentiviral Rev, sequence-verified, and sub-cloned into the expression plasmid pVP16 (Clontech) downstream of VP16AD. Expression of each of these fusion proteins in transfected 293T cells was confirmed by Western blotting using rabbit-derived anti-GAL4 (DBD) (Santa Cruz) or anti-VP16 monoclonal antibody (Sigma). Plasmid encoding hemagglutinin(HA)-tagged hCrm1or mCrm1 was as described elsewhere (Elinav et al., 2012).
HIV transfer vector FG12, FIV transfer vector pGINSIN, EIAV transfer vector pSIN6.1CEGFPW, each driven by CMV IE enhancer-promoter, were as described previously (O’Rourke et al., 2003; Qin et al., 2003; Saenz et al., 2012). HIV-2 RRE from HIV-2 Rod genomic λclone was PCR-amplified and cloned downstream of a U6 promoter. These were used as a source of the corresponding lentiviral RRE.
293T, A9, 3T6, and HOS cell lines were originally obtained from ATCC; B78 was derived from a murine melanoma (Speevak et al., 1995); cell lines were maintained in DMEM with high glucose, 10% fetal calf serum (Invitrogen), 1% penicillin-streptomycin, and 10 μg/ml ciprofloxacin (DMEM complete). Where indicated other antibiotic supplements were added.
4.2. Recombinant protein expression
HIV Rev, HIV Rev M10, and RanQ69L GST fusion plasmids were obtained from Bryan Cullen (Duke University). These were transformed into Escherichia coli strain BL21(DE3) and induced with 1 mM isopropyl-β-d-thiogalactoside for 6 h at 37 °C. Bacteria were pelleted, re-suspended in PBS supplemented with protease inhibitor cocktail (Roche) and sonicated. After centrifugation at 10,000×g for 30 min, supernatant was collected and incubated with GST-Sepharose 4B beads (Pharmacia) for 1 h at room temperature. Bound GST-fusion proteins were washed 3 times with PBS containing 5 mM EDTA and eluted using 10 mM glutathione buffer (50 mM Tris, 10 mM reduced glutathione, pH 8.0). Amounts of purified recombinant protein were estimated using Pierce bicinchoninic acid (BCA) assays (Thermo-Fisher Scientific). GST tag of purified GST-RanQ69L was removed using Factor Xa (Novagen) according to the manufacturers’ instructions. hCrm1-HA and mCrm1-HA proteins were produced by coupled in vitro transcription-translation (Thermo-Fisher Scientific), using previously constructed pCMV-hCrm1-HA or pCMV-mCrm1-HA plasmid as DNA template.
4.3. GST pull-down assay
Purified GST-Rev or GST-Rev M10 protein (0.5 μg of each protein in 100 μl PBS) was used as bait and incubated with equilibrated glutathione-agarose beads (Sigma, 20 μl packed bead volume), with gentle mixing for 1 h at 4 °C. After extensive washing, beads were further incubated with recombinant Ran Q69L protein (1 μg in 100 μl PBS), GTP (2 mM), and HIV RRE (2 μg, prepared by in vitro transcription using a linearized plasmid template and SP6 RNA polymerase), and in vitro transcribed-translated hCrm1-HA or mCrm1-HA protein for 1 h at 4 °Cwith gentle mixing. Beads were extensively washed and then processed for SDS-PAGE electrophoresis followed by immunoblotting with anti-Rev (Abcam) or anti-HA antibody (Santa Cruz).
4.4. Mammalian two-hybrid assay
Intracellular Rev-Crm1 interactions were assessed using the Mammalian Two-Hybrid System according to the manufacturer’s instructions (Clontech), with the modification in which the 0.4 kb Gal4 promoter of the original pG5SEAP plasmid (Clontech) was inserted upstream of FFLUC in the plasmid pGL3-Basic (Promega) to create pG5LUC, with five copies of the Gal4 DNA binding domain upstream of FFLUC. pM-based Crm1 plasmids (1 μg/well for 12-well plate) and pVP16-based Rev plasmids were co-transfected (using the calcium phosphate co-precipitation method), along with the pG5LUC plasmid, into ~100,000 adherent 293T cells and incubated for 37 °Cfor 48 h. Cells were then lysed and assessed for FFLUC activity as described previously (Brasier and Fortin, 2001).
B78 murine cells were maintained in DMEM complete, and at mid-exponential growth were harvested and 5.0 × 106 cells were transfected using 5 μg of each indicated plasmid by nucleofection, using 0.1 ml MEF1 reagent (Amaxa) and program A-024, as previously described (Elinav et al., 2012). Cells were immediately plated in 6-well format, refed the following day, and harvested at 96 h for FFLUC activity after first confirming transfection efficiency by %eGFP+ cells by epifluorescence microscopy.
4.5. Western blotting
293T cells were transfected by calcium phosphate method as described above. Forty-eight h later, where indicated cell lysates were subjected to SDS-PAGE and immunoblotting using anti-tubulin (Cell Signaling Technologies), anti-GAL4 DBD (Santa Cruz Biotechnology), anti-HA (Sigma-Aldrich), anti-Crm1 (clone 17 from BD Transduction Laboratories), anti-Rev (Rev-4: sc-69729 from Santa Cruz Biotechnology), or anti-VP16 (Sigma-Aldrich) antibodies followed by horseradish peroxidase-linked second antibody and enhanced chemiluminescence detection (Pierce) using autoradiographic film (Denville).
4.6. Rev and Crm1 functional testing
For Rev functional testing, a frameshift in the open reading frame of rev was made by filling in at a BamH1 restriction endonuclease site in an HIV packaging vector. This plasmid, pHIV-PVΔRev, was co-transfected using Lipofectamine 2000 (Invitrogen) into 293T cells along VSV G, an HIV transfer vector also deficient in Rev but encoding FFLUC and bsd, and the test Rev construct. Resultant VSV G-pseudotyped vector supernatants were titered on HOS cell targets by assessing both RLU of transduced cells and also by passaging transduced HOS cells into 10 μg/ml blasticidin and enumerating resultant colonies 7–9 d later by methanol-acetic acid fixation and crystal violet staining.
For Crm1 functional testing B78 murine cells with an integrated HIV vector encoding both human cyclinT1 and bsd were transfected using nucleofection (Amaxa) as described above with VSV G, HIV-eYFP, and the test Crm1 construct. Resultant VSV G-pseudotyped vector supernatants were titered on HOS cell targets as described above. Additionally, titer was normalized by transfection efficiency as judged by epifluorescence microscopy of the B78 producers and background colonies (in the absence of any additional Crm1) were subtracted.
4.7. Statistical analysis
Data are presented as mean ± SEM or as mean ± S.D. where indicated. Differences between experimental groups were analyzed for statistical significance by ANOVA followed by Bonferroni’s test for post hoc multiple comparisons, using GraphPad Prism version 4.01, (GraphPad Software Incorporated). Where indicated, student’s t-test was used. A value of p < 0.05 was considered significant.
Acknowledgments
We thank Drs. Nathan Sherer (University of Wisconsin-Madison) and Bryan Cullen (Duke University) for generous reagent gifts. The following reagent was obtained through the NIH AIDS Reagent Program, Division of AIDS, NIAID, NIH: HIV-2ROD phage from Dr. Ronald Desrosiers. This work was supported in part by the Chinese government and NIH/NIDA DP1DA036463. R.E.S. is a NIDA HIV Avant Garde awardee.
References
- Aligeti M, Behrens RT, Pocock GM, Schindelin J, Dietz C, Eliceiri KW, Swanson CM, Malim MH, Ahlquist P, Sherer NM. Cooperativity among Rev-associated nuclear export signals regulates HIV-1 gene expression and is a determinant of virus species tropism. J Virol. 2014;88:14207–14221. doi: 10.1128/JVI.01897-14. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Askjaer P, Jensen TH, Nilsson J, Englmeier L, Kjems J. The specificity of the CRM1-Rev nuclear export signal interaction is mediated by RanGTP. J Biol Chem. 1998;273:33414–33422. doi: 10.1074/jbc.273.50.33414. [DOI] [PubMed] [Google Scholar]
- Berkowitz R, Ilves H, Lin WY, Eckert K, Coward A, Tamaki S, Veres G, Plavec I. Construction and molecular analysis of gene transfer systems derived from bovine immunodeficiency virus. J Virol. 2001;75:3371–3382. doi: 10.1128/JVI.75.7.3371-3382.2001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Berkowitz RD, Ilves H, Plavec I, Veres G. Gene transfer systems derived from Visna virus: analysis of virus production and infectivity. Virology. 2001;279:116–129. doi: 10.1006/viro.2000.0659. [DOI] [PubMed] [Google Scholar]
- Bogerd HP, Echarri A, Ross TM, Cullen BR. Inhibition of human immunodeficiency virus Rev and human T-cell leukemia virus Rex function, but not Mason-Pfizer monkey virus constitutive transport element activity, by a mutant human nucleoporin targeted to Crm1. J Virol. 1998;72:8627–8635. doi: 10.1128/jvi.72.11.8627-8635.1998. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Booth DS, Cheng Y, Frankel AD. The export receptor Crm1 forms a dimer to promote nuclear export of HIV RNA. eLife. 2014;3:e04121. doi: 10.7554/eLife.04121. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Brasier AR, Fortin JJ. Nonisotopic assays for reporter gene activity. Current Protocols in Pharmacology/editorial Board, Enna, S.J. (editor-in-chief) … [et al.] 2001;Chapter 6(Unit 6.5) doi: 10.1002/0471141755.ph0605s05. [DOI] [PubMed] [Google Scholar]
- Cook KS, Fisk GJ, Hauber J, Usman N, Daly TJ, Rusche JR. Characterization of HIV-1 REV protein: binding stoichiometry and minimal RNA substrate. Nucleic Acids Res. 1991;19:1577–1583. doi: 10.1093/nar/19.7.1577. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Coskun AK, van Maanen M, Janka D, Stockton D, Stankiewicsz P, Yatsenko S, Sutton RE. Isolation and characterization of mouse-human microcell hybrid cell clones permissive for infectious HIV particle release. Virology. 2007 doi: 10.1016/j.virol.2006.12.015. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Coskun AK, van Maanen M, Nguyen V, Sutton RE. Human chromosome 2 carries a gene required for production of infectious human immunodeficiency virus type 1. J Virol. 2006;80:3406–3415. doi: 10.1128/JVI.80.7.3406-3415.2006. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Daly TJ, Cook KS, Gray GS, Maione TE, Rusche JR. Specific binding of HIV-1 recombinant Rev protein to the Rev-responsive element in vitro. Nature. 1989;342:816–819. doi: 10.1038/342816a0. [DOI] [PubMed] [Google Scholar]
- Daugherty MD, D’Orso I, Frankel AD. A solution to limited genomic capacity: using adaptable binding surfaces to assemble the functional HIV Rev oligomer on RNA. Mol Cell. 2008;31:824–834. doi: 10.1016/j.molcel.2008.07.016. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Daugherty MD, Liu B, Frankel AD. Structural basis for cooperative RNA binding and export complex assembly by HIV Rev. Nat Struct Mol Biol. 2010;17:1337–1342. doi: 10.1038/nsmb.1902. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Denton PW, Garcia JV. Humanized mouse models of HIV infection. AIDS Rev. 2011;13:135–148. [PMC free article] [PubMed] [Google Scholar]
- Dong X, Biswas A, Suel KE, Jackson LK, Martinez R, Gu H, Chook YM. Structural basis for leucine-rich nuclear export signal recognition by CRM1. Nature. 2009;458:1136–1141. doi: 10.1038/nature07975. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Elinav H, Wu Y, Coskun A, Hryckiewicz K, Kemler I, Hu Y, Rogers H, Hao B, Ben Mamoun C, Poeschla E, Sutton R. Human CRM1 augments production of infectious human and feline immunodeficiency viruses from murine cells. J Virol. 2012;86:12053–12068. doi: 10.1128/JVI.01970-12. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Fang X, Wang J, O’Carroll IP, Mitchell M, Zuo X, Wang Y, Yu P, Liu Y, Rausch JW, Dyba MA, Kjems J, Schwieters CD, Seifert S, Winans RE, Watts NR, Stahl SJ, Wingfield PT, Byrd RA, Le Grice SF, Rein A, Wang YX. An unusual topological structure of the HIV-1 Rev response element. Cell. 2013;155:594–605. doi: 10.1016/j.cell.2013.10.008. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Heaphy S, Finch JT, Gait MJ, Karn J, Singh M. Human immunodeficiency virus type 1 regulator of virion expression, rev, forms nucleoprotein filaments after binding to a purine-rich “bubble” located within the rev-responsive region of viral mRNAs. Proc Natl Acad Sci USA. 1991;88:7366–7370. doi: 10.1073/pnas.88.16.7366. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Holland SM, Ahmad N, Maitra RK, Wingfield P, Venkatesan S. Human immunodeficiency virus rev protein recognizes a target sequence in rev-responsive element RNA within the context of RNA secondary structure. J Virol. 1990;64:5966–5975. doi: 10.1128/jvi.64.12.5966-5975.1990. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kaiser J. Biomedical research. An end to U.S chimp research. Science. 2015;350:1013. doi: 10.1126/science.350.6264.1013. [DOI] [PubMed] [Google Scholar]
- Li Y, Bor YC, Misawa Y, Xue Y, Rekosh D, Hammarskjold ML. An intron with a constitutive transport element is retained in a Tap messenger RNA. Nature. 2006;443:234–237. doi: 10.1038/nature05107. [DOI] [PubMed] [Google Scholar]
- Luker KE, Smith MC, Luker GD, Gammon ST, Piwnica-Worms H, Piwnica-Worms D. Kinetics of regulated protein-protein interactions revealed with firefly luciferase complementation imaging in cells and living animals. Proc Natl Acad Sci USA. 2004;101:12288–12293. doi: 10.1073/pnas.0404041101. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Madore SJ, Tiley LS, Malim MH, Cullen BR. Sequence requirements for Rev multimerization in vivo. Virology. 1994;202:186–194. doi: 10.1006/viro.1994.1334. [DOI] [PubMed] [Google Scholar]
- Malim MH, Bohnlein S, Hauber J, Cullen BR. Functional dissection of the HIV-1 Rev trans-activator–derivation of a trans-dominant repressor of Rev function. Cell. 1989a;58:205–214. doi: 10.1016/0092-8674(89)90416-9. [DOI] [PubMed] [Google Scholar]
- Malim MH, Bohnlein S, Hauber J, Cullen BR. Functional dissection of the HIV-1 Rev trans-activator-derivation of a trans-dominant repressor of Rev function. Cell. 1989b;58:205–214. doi: 10.1016/0092-8674(89)90416-9. [DOI] [PubMed] [Google Scholar]
- Malim MH, Cullen BR. HIV-1 structural gene expression requires the binding of multiple Rev monomers to the viral RRE: implications for HIV-1 latency. Cell. 1991;65:241–248. doi: 10.1016/0092-8674(91)90158-u. [DOI] [PubMed] [Google Scholar]
- Malim MH, Hauber J, Le SY, Maizel JV, Cullen BR. The HIV-1 rev trans-activator acts through a structured target sequence to activate nuclear export of unspliced viral mRNA. Nature. 1989c;338:254–257. doi: 10.1038/338254a0. [DOI] [PubMed] [Google Scholar]
- Mann DA, Mikaelian I, Zemmel RW, Green SM, Lowe AD, Kimura T, Singh M, Butler PJ, Gait MJ, Karn J. A molecular rheostat. Co-operative rev binding to stem I of the rev-response element modulates human immunodeficiency virus type-1 late gene expression. J Mol Biol. 1994;241:193–207. doi: 10.1006/jmbi.1994.1488. [DOI] [PubMed] [Google Scholar]
- Metharom P, Takyar S, Xia HH, Ellem KA, Macmillan J, Shepherd RW, Wilcox GE, Wei MQ. Novel bovine lentiviral vectors based on Jembrana disease virus. J Gene Med. 2000;2:176–185. doi: 10.1002/(SICI)1521-2254(200005/06)2:3<176::AID-JGM106>3.0.CO;2-Q. [DOI] [PubMed] [Google Scholar]
- Mselli-Lakhal L, Guiguen F, Greenland T, Mornex JF, Chebloune Y. Gene transfer system derived from the caprine arthritisencephalitis lentivirus. J Virol Methods. 2006;136:177–184. doi: 10.1016/j.jviromet.2006.05.006. [DOI] [PubMed] [Google Scholar]
- Nagai-Fukataki M, Ohashi T, Hashimoto I, Kimura T, Hakata Y, Shida H. Nuclear and cytoplasmic effects of human CRM1 on HIV-1 production in rat cells. Genes Cells. 2011;16:203–216. doi: 10.1111/j.1365-2443.2010.01476.x. [DOI] [PubMed] [Google Scholar]
- O’Rourke JP, Hiraragi H, Urban K, Patel M, Olsen JC, Bunnell BA. Analysis of gene transfer and expression in skeletal muscle using enhanced EIAV lentivirus vectors. Mol Ther. 2003;7:632–639. doi: 10.1016/s1525-0016(03)00074-1. [DOI] [PubMed] [Google Scholar]
- Okada H, Zhang X, Ben Fofana I, Nagai M, Suzuki H, Ohashi T, Shida H. Synergistic effect of human CycT1 and CRM1 on HIV-1 propagation in rat T cells and macrophages. Retrovirology. 2009;6:43. doi: 10.1186/1742-4690-6-43. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Olsen JC. EIAV, CAEV and other lentivirus vector systems. Somatic Cell Mol Genet. 2001;26:131–145. doi: 10.1023/a:1021030830943. [DOI] [PubMed] [Google Scholar]
- Paraskeva E, Izaurralde E, Bischoff FR, Huber J, Kutay U, Hartmann E, Luhrmann R, Gorlich D. CRM1-mediated recycling of snurportin 1 to the cytoplasm. J Cell Biol. 1999;145:255–264. doi: 10.1083/jcb.145.2.255. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Qin XF, An DS, Chen IS, Baltimore D. Inhibiting HIV-1 infection in human T cells by lentiviral-mediated delivery of small interfering RNA against CCR5. Proc Natl Acad Sci USA. 2003;100:183–188. doi: 10.1073/pnas.232688199. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Saenz DT, Barraza R, Loewen N, Teo W, Poeschla EM. Feline immunodeficiency virus-based lentiviral vectors. Cold Spring Harbor Protoc. 2012;2012:71–76. doi: 10.1101/pdb.ip067579. [DOI] [PubMed] [Google Scholar]
- Sherer NM, Swanson CM, Hue S, Roberts RG, Bergeron JR, Malim MH. Evolution of a species-specific determinant within human CRM1 that regulates the post-transcriptional phases of HIV-1 replication. PLoS Pathog. 2011;7:e1002395. doi: 10.1371/journal.ppat.1002395. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Speevak MD, Berube NG, McGowan-Jordan IJ, Bisson C, Lupton SD, Chevrette M. Construction and analysis of microcell hybrids containing dual selectable tagged human chromosomes. Cytogenet Cell Genet. 1995;69:63–65. doi: 10.1159/000133939. [DOI] [PubMed] [Google Scholar]
- Swanson CM, Sherer NM, Malim MH. SRp40 and SRp55 promote the translation of unspliced human immunodeficiency virus type 1 RNA. J Virol. 2010;84:6748–6759. doi: 10.1128/JVI.02526-09. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Szilvay AM, Brokstad KA, Boe SO, Haukenes G, Kalland KH. Oligomerization of HIV-1 Rev mutants in the cytoplasm and during nuclear import. Virology. 1997;235:73–81. doi: 10.1006/viro.1997.8671. [DOI] [PubMed] [Google Scholar]
- Vercruysse T, Daelemans D. HIV-1 Rev multimerization: mechanism and insights. Curr HIV Res. 2013;11:623–634. doi: 10.2174/1570162x12666140307094603. [DOI] [PubMed] [Google Scholar]
- Wang B, Rekosh D, Hammarskjold ML. Evolutionary conservation of a molecular machinery for export and expression of mRNAs with retained introns. RNA (New York, NY) 2015;21:426–437. doi: 10.1261/rna.048520.114. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wingfield PT, Stahl SJ, Payton MA, Venkatesan S, Misra M, Steven AC. HIV-1 Rev expressed in recombinant Escherichia coli: purification, polymerization, and conformational properties. Biochemistry. 1991;30:7527–7534. doi: 10.1021/bi00244a023. [DOI] [PubMed] [Google Scholar]