

Abstract
Asymmetric cell division results in two distinctly fated daughter cells. A molecular hallmark of asymmetric division is the unequal partitioning of cell fate determinants. We have previously established that growth factor signaling promotes protein depalmitoylation to foster polarized protein localization, which in turns drives migration and metastasis. Here, we report protein palmitoylation as a key mechanism for the asymmetric partitioning of the cell fate determinants Numb and β-catenin through the activity of the depalmitoylating enzyme APT1. Using point mutations, we show specific palmitoylated residues on Numb are required for asymmetric localization. By live-cell imaging, we show that reciprocal interactions between APT1 and the Rho-family GTPase CDC42 promote the asymmetric localization of Numb and β-catenin to the plasma membrane. This in turn restricts Notch- and Wnt-responsive transcriptional activity to one daughter cell. Moreover, we show altering APT1 expression changes the transcriptional signatures of MDA-MB-231 triple receptor–negative breast cancer cells similarly to changes in Notch and β-catenin–mediated Wnt signaling. We also show that loss of APT1 depletes a specific subpopulation of tumorigenic cells in colony formation assays. Together, our findings demonstrate that APT1-mediated depalmitoylation is a major mechanism of asymmetric cell division maintaining Notch and Wnt-associated protein dynamics, gene expression, and cellular functions.
Introduction
Asymmetric cell division yields two morphologically and functionally distinct daughter cells and serves as a major contributor to cellular heterogeneity during development and tissue homeostasis (1). In dividing stem and progenitor cells, cell fate determinant proteins are unequally segregated along the division axis and inherited by one cell, resulting in the differential activation of transcriptional networks that establish non-identical daughter cells (2–5). For example, Drosophila melanogaster neuroblasts divide asymmetrically to produce a self-renewing neuroblast and a differentiating cell (6, 7). Similarly, transformed cells can also exhibit cellular heterogeneity with variations in properties such as signaling activity, tumorigenicity, and drug resistance exhibited by distinct cell populations (8–10). The cause of tumor heterogeneity has generally been attributed to genomic instability, epigenetic alterations, or interactions with the tumor microenvironment (11–13), though it is possible that asymmetric cell division may also play a role.
The molecular mechanisms driving and maintaining asymmetric divisions are poorly understood, but developmental signaling pathways such as Notch and Wnt have been shown to be key factors. In Drosophila neuroblasts, polarized cell division results in asymmetric Numb (the Notch antagonist) localization at the plasma membrane, resulting in unequal inheritance by cells fated to differentiate into neurons (6, 7, 14–16). Numb is also partitioned asymmetrically in dividing mammalian cells such as mammary epithelial precursors, hematopoietic stem and progenitor cells, and T-lymphocyte precursors (2, 17, 18). Likewise, directionally applied Wnt signals restrict β-catenin (a critical intracellular mediator of canonical Wnt signaling) in a polarized manner to mouse embryonic stem cells and C.elegans seam cells that are fated to remain as progenitor cells rather than differentiate (4, 19). Differential spatial organization of proteins and the resulting cell polarity is established by the evolutionarily conserved Par-aPKC (atypical protein kinase C)-CDC42 complex (20–22). CDC42 is a small Rho-family GTPase that mediates polarized processes such as vesicle budding, trafficking, and directional cell migration by remodeling the actin and microtubule cytoskeleton networks (23). Although the CDC42 polarity complex is involved during asymmetric cell division, the mechanisms directing the asymmetric recruitment and retention of cell fate determinant proteins at the plasma membrane during cell division remain unclear.
Posttranslational lipid modification is a prevalent mechanism of targeting proteins to membranes that alters molecular conformations and increases protein affinity for hydrophobic environments (24). Lipid modifications are largely non-reversible and maintain protein enzymatic function and stability, protein-protein interactions, and intracellular trafficking (25). Palmitoylation, in contrast, is a reversible lipid modification that modulates subcellular polarity and signaling cascade activity, such as that of the epidermal growth factor receptor (EGFR) and the small GTPase Ras, by rapid protein shuttling between the cytosol and lipid rafts within the plasma membrane (26–32). Palmitoylation is modulated by two classes of enzymes: palmitoyltransferases that add palmitate to cysteine residues, and depalmitoyltransferases that remove palmitate at the membrane and promote protein localization to the cytosol (33). Thus, palmitoylation is an attractive, but untested, candidate mechanism for the dynamic asymmetric targeting of proteins to the plasma membrane during cell division to drive cellular heterogeneity in embryogenesis, development, and tumorigenesis.
In this study, we investigated the role of palmitoylation on the polarized partitioning of cell fate determinants during asymmetric cell division and clarified how palmitoylation contributes to cellular heterogeneity. Using human cancer cell lines derived from malignant tumors that exhibit high cellular heterogeneity, such as triple receptor–negative breast cancers and osteosarcomas (34–38), we found that the depalmitoylating enzyme acyl-protein thioesterase 1 (APT1) (39–41), directs the asymmetric localization of Numb and β-catenin. Additionally, we found that APT1 activity during mitosis restricted Notch-, Wnt-, and Sox2-dependent transcription to one daughter cell. APT1 also maintained the gene expression signature and colony-forming potential of transformed cells. Moreover, our observations indicate that APT1 was required for the transcriptional output of different signaling pathways in cancer cells grown in serial colony formation assays, which correlates with cellular heterogeneity and tumorigenic potential. With these findings, we identify a palmitoylation-mediated mechanism of asymmetric cell division that promotes the generation of functionally heterogeneous cells in tumors.
Results
Activity of the depalmitoylating enzyme APT1 is required for asymmetric localization of Numb and β-catenin
We have previously shown that APT1 promotes the transient and asymmetric localization of cell adhesion molecules during interphase in response to extracellular signals (26). To test the hypothesis that APT1 directs the asymmetric localization of Notch and Wnt signaling-associated cell fate determinants during cell division, we examined the spatial organization of Numb and β-catenin in fixed cells. Using the MDA-MB-231 human triple receptor–negative breast cancer cell line, dividing cells were identified by immunostaining for acetylated tubulin, a marker of stabilized tubulin structures such as the mitotic spindle and cytokinetic midbody (42), and counterstained for Numb or β-catenin. In the absence of exogenous stimuli, we observed symmetric localization of both proteins (Fig. 1A, B), as assessed by measuring the percentage difference in the mean fluorescence pixel intensity of Numb or β-catenin across dividing cells (fig. S1A). The distribution of the percentage differences of all quantified cells was plotted, and cells with a percentage difference of 20 or greater were scored as asymmetric (Fig. 1C, D). Treating cells with Palmostatin B (PalmB), a pharmacological inhibitor of APT enzymes (43), reduced the asymmetric localization of Numb by 3.0-fold (29.4% vs. 9.9%) and β-catenin by 3.3-fold (26.1% vs. 8.0%) (Fig.1 E, F). These findings suggest that APT enzymes are required for establishing asymmetric localizations of Numb and β-catenin in dividing cells.
Fig. 1. Activity of the depalmitoylating enzyme APT1 is required for asymmetric localization of Numb and β-catenin.
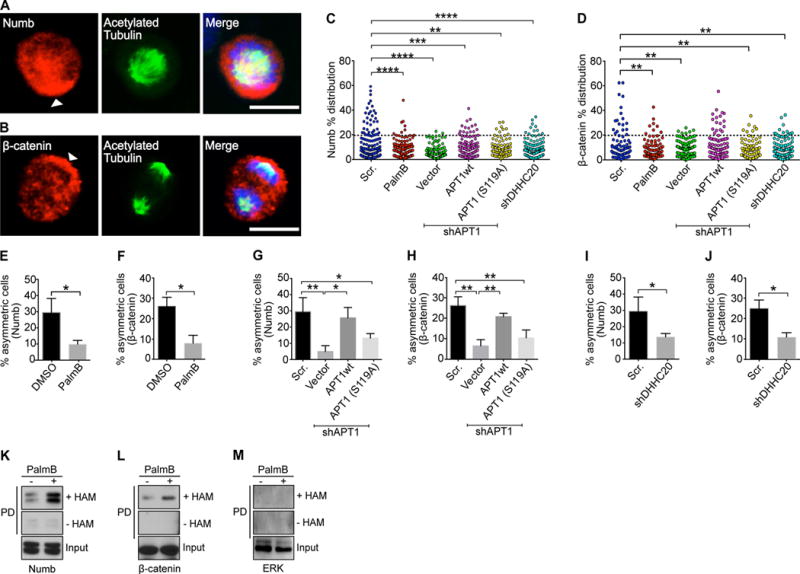
(A and B) Images of dividing MDA-MB-231 cells stained to show endogenous Numb (A) and β-catenin (B) in red, acetylated tubulin in green, and nuclei in blue. Arrowheads indicate asymmetric localization of Numb and β-catenin. Scale bars, 15 μm. (C and D) Distribution dot plots showing the difference in mean fluorescence pixel intensity of endogenous Numb (C) and β-catenin (D) across dividing MDA-MB-231 cells. The distribution of the percentage differences of all quantified cells was plotted, and cells with a difference of >20% (black dotted line) were scored as asymmetric. n= 508-582 cells scored for each experimental group. Each dot represents a single cell. Asterisks indicate statistically significant differences between the indicated groups. (E and F) Quantification of dividing MDA-MB-231 cells showing asymmetric Numb (E) and β-catenin (F) localization after treatment with PalmB or DMSO. (G and H) Quantification of the number of dividing MDA-MB-231cells showing asymmetric Numb (G) and β-catenin (H) localization when APT1 was knocked down with shAPT1, and when wild-type APT1 (APT1WT) or the catalytically inactive APT1S119A mutant was coexpressed with shAPT1. Cells expressing a scrambled (Scr) shRNA sequence were used as a negative control for APT1 knockdown, and cells expressing the empty vector were used as the negative control for the APT1 rescue experiments. (I and J) Quantification of the number of dividing MDA-MB-231 cells showing asymmetric Numb (I) and β-catenin (J) localization when DHHC20 was knocked down with shDHHC20. (K to M) Immunoblots showing biotin-labeled Numb (K), β-catenin (L), and Erk (M) in MDA-MB-231 cell lysates following acyl-biotin exchange (ABE) assays and pulldown on streptavidin beads (PD). Cells were grown in either in the presence either PalmB or vehicle control (DMSO). Input lanes show cell lysates prior to pulldown. Samples without hydroxylamine (-HAM) were negative controls for the ABE reactions. *P < 0.05; **P < 0.01 T-test (E and F) or ANOVA (G to J). Error bars indicate standard deviation (SD).
To address a specific requirement for APT1, we knocked down APT1 with a short hairpin RNA (shRNA) (fig. S1B). In APT1 knockdown cells, asymmetric localization of Numb was reduced by 6-fold (29.4% vs. 4.9%), and asymmetric localization of β-catenin was reduced by 4.1-fold (26.1% vs. 6.4%). Ectopic expression of wild-type human APT1 (APT1WT) from a plasmid restored asymmetric localization of Numb and β-catenin to baseline control conditions (Fig. 1G, H). However, ectopic expression of a catalytically inactive mutant form of APT1, in Ser119 in the catalytic domain is mutated to Ala (APT1S119A) (40, 41), failed to rescue asymmetric Numb and β-catenin localization (fig. S1B; Fig. 1G, H). This indicates that the catalytic activity of APT1 is critical for asymmetric Numb and β-catenin localization.
Of the 23 DHHC palmitoyltransferases found in mammalian cells, DHHC20 is one of three that is localized to the plasma membrane where we would expect it to be available to palmitoylate proteins like Numb and β-catenin (44). DHHC20 is also known to be expressed in MDA-MB-231 cells, where it palmitoylates EGFR and attenuates EGFR signaling (28). We asked whether loss of DHHC20 would affect protein localization. Knockdown of DHHC20 reduced asymmetric localization of Numb and β-catenin in a manner similar to knockdown of APT1 (fig. S1C, D; Fig. 1I, J).
The dependence of Numb and β-catenin asymmetric localization on APT1 suggests that Numb and β-catenin may be palmitoylated. To evaluate protein palmitoylation, we used acyl-biotin exchange (ABE) assays (45). In the ABE assay, purified proteins are treated with N-ethylmaleimide to block free thiol groups, then the Cys-palmitoyl thioester linkages are cleaved with hydroxylamine (HAM), and the newly exposed thiol groups are coupled to biotin. The modified proteins are then purified with streptavidin. Results of ABE assays were consistent with Numb and β-catenin being palmitoylated under normal growth conditions (Fig. 1K-L). Treating cells with PalmB increased the total amounts of palmitoylated Numb and β-catenin (Fig. 1K, L), indicating that palmitoylation of these proteins was inhibited in part by APT enzymes. As a negative control, ABE assays did not detect palmitoylation of the extracellular signal–regulated kinase (ERK), which is not palmitoylated (Fig. 1M).
To determine whether palmityolation-dependent asymmetric localization was specific to Numb and β-catenin, dividing cells were immunostained for CD44 and RhoB, both of which are palmitoylated and unrelated to Wnt or Notch signaling (46–49), as well as for GFP in cells expressing a control GFP plasmid. The percentage of cells with asymmetrically localized GFP, CD44, or RhoB were not higher than the background percentages observed for β-catenin and Numb (approximately 8%), and were unaffected by PalmB treatment (fig. S1E-J). Together, these findings uncover a direct role for APT1 in the spatial distribution of the palmitoylated cell fate determinants Numb and β-catenin.
Asymmetric localization of Numb requires palmitoylation of the phosphotyrosine binding domain
Although we found β-catenin to be palmitoylated, β-catenin cortical localization is mediated through association with cadherins at tight junctions (50, 51). Thus, the necessity of palmitoylation for the membrane localization of β-catenin is unclear. However, the conserved phosphotyrosine binding (PTB) domain of Numb is required for association of Numb with the plasma membrane and for asymmetric localization of Numb in Drosophila through mechanisms that are still unknown (52). We sought to directly test whether palmitoylation of Numb is required for its asymmetric localization. Using the palmitoylation prediction algorithm CSS-Palm (53) and through identification of solvent-exposed cysteine residues within the PTB domain crystal structure (54), three conserved and potentially palmitoylated cysteine residues (Cys37, Cys160, and Cys165) were identified and mutated to Ala (Fig. 2A). The Numb triple Cys-to-Ala mutant (NumbAAA) showed reduced palmitoylation as measured by metabolic labeling of cells with palmitic acid azide (Fig. 2B). Endogenous β-catenin was also metabolically labeled with palmitic acid azide, confirming the efficiency of labeling in all reactions (Fig. 2B). These results demonstrate that Numb and β-catenin are continuously palmitoylated in MDA-MB-231 cells.
Fig. 2. Palmitoylation and APT1 activity drive Numb localization.
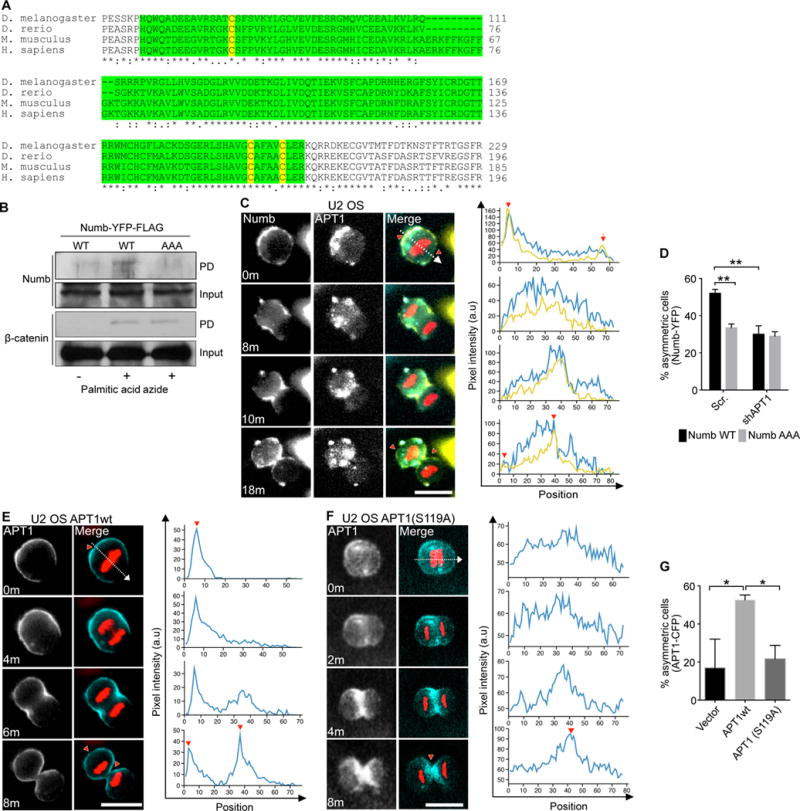
(A) Sequence comparison of the N-terminus of Numb from fruit fly (D. melanogaster), zebrafish (D. rerio), mouse (M. musculus), and human (H. sapiens). The phosphotyrosine domain (PTB) is highlighted in green, and the putative palmitoylated cysteines are highlighted in yellow. Conserved residues indicated by asterisk (*). (B) Immunoblot showing transgenically-expressed wild-type Numb (NumbWT) or the NumbAAA mutant and endogenous β-catenin in U2 OS cell lysates following purification of palmitoylated proteins. Cells were metabolically labelled with palmitic acid azide or treated with DMSO (vehicle control), then lysates were subjected to click chemistry to convert the palmitic acid moiety to biotin, pulled down on streptavidin beads, and used for immunoblotting. Input was taken from cell lysates prior to pulldown. (C) Time-lapse images of dividing U2 OS cells co-expressing NumbWT-YFP (yellow), APT1WT-CFP (blue), and mCherry-Histone H2B (red). Fluorescence pixel intensity was measured along the division axis (dashed line) and the corresponding pixel values of Numb (yellow line) and APT1 (blue line) along the division axis were plotted on graphs. Red arrowheads on images and graphs indicate the peak Numb and APT1 pixel intensity at the membrane or cytokinetic midbody. Time shown in minutes, m. Arbitrary units, a.u. Scale bar, 15 μm. (D) Quantification of the number of dividing U2 OS cells showing asymmetric localization of NumbWT-YFP (black bar) and NumbAAA-YFP (grey bar) when each was coexpressed with shAPT1. Cells expressing a scrambled (Scr) shRNA sequence were used as a negative control for APT1 knockdown. (E and F) Time-lapse images of dividing U2 OS cells coexpressing either APT1WT-CFP (E) or APT1S119A-CFP (F) with mCherry-Histone H2B (red). Fluorescence pixel intensity was quantified as in (C). Time shown in minutes, m. Arbitrary units, a.u. Scale bars, 15 μm. (G) Quantification of the number of dividing U2 OS cells showing asymmetric APT1WT-CFP or APT1S119A-CFP localization. Cells expressing an empty GFP plasmid (Vector) were used as a negative control. n= 102-143 cells scored for each group from three independent experiments. *P< 0.05; **P < 0.01 T-test and ANOVA. Error bars indicate standard deviation (SD).
Next, we visualized the dynamic localization and segregation of fluorescently labeled Numb and APT1 during cell division by live-cell imaging. Because MDA-MB-231 cells shifted from a flat, spread morphology to a raised, rounded morphology out of the imaging plane during cell division, we instead utilized U2 OS human osteosarcoma cells, which maintained a consistent rounded morphology within the imaging plane during division. U2 OS cells were transduced to stably express an mCherry-Histone B (H2B) plasmid, allowing for the unambiguous identification of cells undergoing division in real time. Over the course of cell division, cyan fluorescent protein (CFP)-tagged APT1WT (APT1WT-CFP) and yellow fluorescent protein (YFP)-tagged NumbWT (NumbWT- YFP) exhibited highly dynamic asymmetric localization (Fig. 2C). At the beginning of the cell division cycle, NumbWT was concentrated at one end of the cell at the plasma membrane, but as daughter cells formed, the localization of Numb shifted to membrane regions at and near the cleavage furrow. Finally, as daughter cells separated, Numb was partitioned to the plasma membrane of the cell that emerged from the same side of the mother cell to which Numb was initially concentrated. APT1WT co-segregated with Numb to membrane regions and was retained in daughter cells with high Numb signal as indicated by line-scan analysis of YFP and CFP pixel intensity along the division axis (Fig. 2C, Movie S1–3, red arrows). This suggests that APT1 either responds to the same spatial cues as Numb. Alternatively, APT1 may direct Numb localization or vice versa. YFP-tagged NumbAAA (NumbAAA-YFP) was live-imaged to determine the contribution of Numb palmitoylation on its localization. NumbAAA showed a 1.5-fold reduced asymmetric localization in dividing cells as compared to NumbWT (Fig. 2D). Finally, knocking down APT1 reduced asymmetric NumbWT localization but did not further reduce the asymmetry of NumbAAA (Fig. 2D). Our results show that the asymmetric partitioning of Numb is actively maintained by a mechanism that requires both APT1-mediated depalmitoylation and palmitoylation of Cys37, Cys160, and/or Cys165 within the PTB domain.
Asymmetric localization of APT1 during cell division requires APT1 catalytic activity
Having determined that APT1 activity is essential for asymmetric localization of Numb and β-catenin, we next examined whether the catalytic activity of APT1 is required for its own asymmetric localization. Immunostaining fixed cells showed that endogenous APT1 was asymmetrically localized in 22.3% of control cells. Expressing the catalytically inactive form APT1S119A reduced this asymmetric localization by 2.4-fold, to 9.4%, whereas expressing APT1WT had no significant effect (fig. S2A-C). Knocking down DHHC20 also reduced asymmetric APT1 partitioning, suggesting that DHHC20 promotes asymmetric localization of APT1 (fig. S2B, D).
To gain detailed insights into how APT1 activity promotes and maintains the dynamics of its own asymmetric localization, we compared the spatiotemporal distribution of ectopically expressed APT1WT-CFP versus APT1S119A-CFP by live-cell imaging (Fig. 2E-G; Movies S4–7). In 52.6% of dividing cells, APT1WT-CFP asymmetry was maintained at the plasma membrane through cytokinesis (Fig. 2E,G; Movies S4–5). The asymmetric localization of APT1S119A-CFP was significantly reduced to 20.6% of cells, similar to the asymmetric localization of GFP-vector (16.9%) (Fig. 2F,G; Movies S6–7). Additionally, the catalytically inactive mutant APT1S119A-CFP was not discretely localized to the plasma membrane at the start of division and appeared to be stuck at the cytokinetic midbody during cytokinesis (Fig. 2F, Movie S6–7). The data up to this point demonstrate that asymmetric localization of APT1 requires its catalytic activity and suggest a role for protein depalmitoylation activity at the site of asymmetric protein localization.
Palmitoylating and depalmitoylating enzymes localize asymmetrically with palmitoylated proteins during cell division
To clarify the purpose of APT1 accumulation at sites of asymmetrically localized proteins, we hypothesized that APT1 localizes to regions of high protein palmitoylation. Fixed MDA-MB-231 cells were immunostained for DHHC20, which was asymmetrically partitioned to the same region of a dividing cell as APT1 (Fig. 3A). Knocking down DHHC20 reduced asymmetric APT1 partitioning (fig. S2D). Additionally, the distribution of APT1 and DHHC20 overlapped with that of caveolin, a palmitoylated protein (55), at the plasma membrane (Fig. 3B, C). This would suggest DHHC20-mediated palmitoylation of substrates could recruit APT1 to the membrane.
Fig. 3. Palmitoylating enzymes and depalmitoylating enzymes are asymmetrically partitioned during cell division.
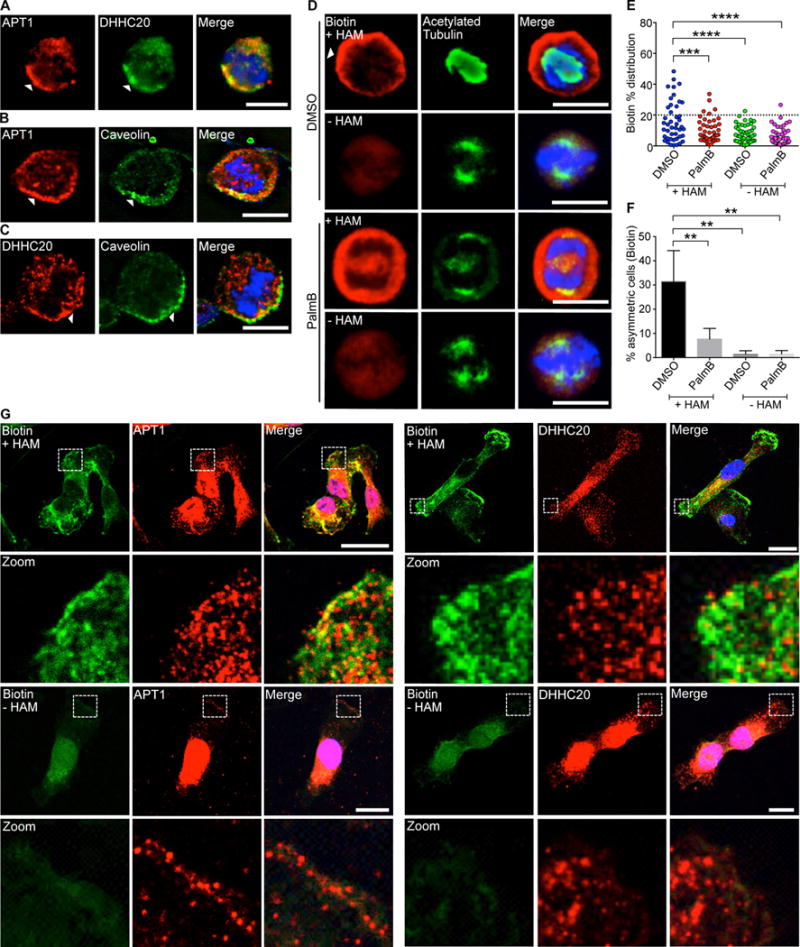
(A) Images of dividing MDA-MB-231 cells stained to show endogenous APT1 (red), DHHC20 (green), and nuclei (blue). (B and C) Images of dividing MDA-MB-231 cells stained to show endogenous APT1 (B) or DHHC20 (C), caveolin, and nuclei. Asymmetric localization is indicated by arrowheads (A to C). (D) Images of dividing MDA-MB-231 cells treated with PalmB or DMSO and stained to show biotin-labeled palmitoylated proteins (red), acetylated tubulin (green), and nuclei (blue) by ABE immunofluorescence. Samples without hydroxylamine (-HAM) were negative controls for the ABE reaction. (E) Distribution dot plots showing the difference in mean fluorescence pixel intensity of biotin-labeled palmitoylated proteins. The distribution of the percentage differences of all quantified cells was plotted, and cells with a difference of >20% (dotted line) were scored as asymmetric. n = 91-101 cells scored for each experimental group. Each dot represents a single cell. Asterisks indicate statistically significant differences between the indicated groups. (F) Quantification of the number of dividing MDA-MB-231 cells showing asymmetric palmitoylated proteins after treatment with PalmB or DMSO control. (G) Confocal images of ABE immunofluorescence in non-dividing MDA-MB-231 cells showing all palmitoylated proteins (green), APT1 or DHHC20 (red), and nuclei (blue) by ABE immunofluorescence. White dotted boxes indicated magnified areas shown directly below (Zoom). Samples without hydroxylamine (-HAM) were negative controls for the ABE reaction. Scale bars (including zoom), 15μm. *P < 0.05 T-test. Error bars indicate standard deviation (SD).
The presence of both the depalmitoylating enzyme APT1 and palmitoylating enzyme DHHC20 in plasma membrane–associated domains led us to test whether these regions were also enriched for palmitoylated proteins. Because there are currently no palmitoylation-specific antibodies to visualize the localization of palmitoylated proteins, we modified the ABE assay for immunofluorescence and detected asymmetric palmitoylated protein localization at the cortex of dividing cells (Fig. 3D). This asymmetric localization depended on the activity of APT1, because PalmB treatment decreased the enrichment of palmitoylated proteins at the cortex and reduced the percentage of cells showing asymmetric localization of palmitoylated proteins by 4.1-fold (31.4% vs. 7.6%). The immunofluorescence signal generated by the modified ABE assay was specific to palmitoylated proteins; negative control staining of samples in which HAM was omitted from the ABE reaction showed greatly reduced signal and symmetric localization (Fig. 3D-F). To determine the localization of palmitoylated proteins relative to APT1 and DHHC20 and whether this asymmetric localization was only observed during mitosis, we performed the modified ABE immunofluorescence assay on non-dividing cells. Both APT1 and DHHC20 puncta were localized to regions enriched in palmitoylated proteins at membrane ruffles as assessed by confocal microscopy (Fig. 3G). The data thus far indicate that both APT1 and DHHC20 localize to regions enriched in palmitoylated proteins.
A constitutively active CDC42 mutant promotes asymmetric localization of APT1, Numb, and β-catenin during cell division
We next examined whether APT1-mediated asymmetric protein partitioning functions independently of known polarity-establishing mechanisms. The Par-aPKC-CDC42 polarity complex promotes the asymmetric subcellular distribution of cell fate determinants (15, 56). We knocked down CDC42 and PARD3 (the mammalian homolog of Par3) to assess the requirement of these factors for APT1, Numb, and β-catenin localization (fig. S3A, B). Asymmetric localization of endogenous APT1 was reduced by 2.6-fold (22.3% vs. 8.5%) in CDC42 knockdown cells, and by 4.3-fold (22.3% vs. 5.2%) in PARD3 knockdown cells (Fig. 4A, B; fig. S4C). PARD3 knockdown reduced the asymmetric localization of β-catenin, but not that of Numb (fig. S3D-G). We next asked if CDC42 and APT1 double knockdown would completely abolish asymmetric Numb and β-catenin partitioning. The percentage of cells showing asymmetric partitioning of Numb and β-catenin in the double knockdown condition did not further decrease as compared to APT1 or CDC42 single knockdown (fig. S3D, E, H, I). This suggests there might be a basal level of asymmetric distribution for both of these proteins that is independent of both APT1 and CDC42.
Fig. 4. The reciprocal interaction between APT1 and CDC42 establishes and maintains asymmetric protein partitioning during cell division.
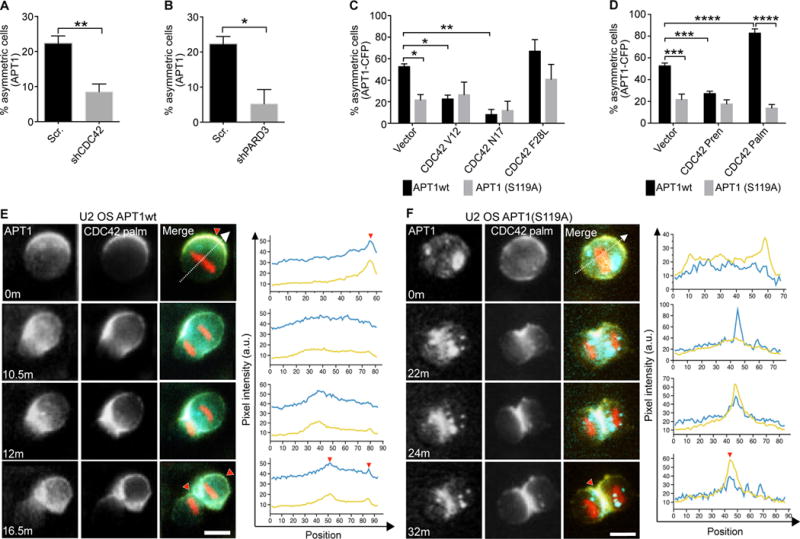
(A and B) Quantification of the number of dividing MDA-MB-231 cells showing asymmetric localization of APT1 when CDC42 was knocked down with shCDC42 (A) or PARD3 was knocked down with shPARD3 (B). Cells expressing a scrambled (Scr) shRNA sequence were used as a negative control for knockdown. (C and D) Quantification of the number of dividing U2 OS cells showing asymmetric APT1WT-CFP or APT1S119A-CFP in cells expressing constitutively active (CDC42V12 and CDC42F28L) or dominant negative (CDC42N17) forms of CDC42 (C), or expressing isoforms of CDC42 that are prenylated (CDC42Pren) or both palmitoylated and prenylated (CDC42Palm) (D). (E and F) Time-lapse images of dividing U2 OS cells co-expressing CDC42Palm-YFP (yellow) and either APT1WT-CFP (E) or APT1S119A-CFP (F) (blue), and mCherry-Histone H2B (red). Overlapping CFP and YFP signal in the merge (green). Fluorescence pixel intensity was measured along the division axis (dashed line) and the corresponding pixel values of CDC42 (yellow line) and APT1 (blue line) along the division axis were plotted. Red arrowheads on images and corresponding graphs mark the peak asymmetric CDC42 and APT1 accumulation at the membrane or cytokinetic midbody. Time shown in minutes, m. Arbitrary units, a.u. Scale bars, 15 μm. n= 152-288 cells scored for each group from 4 independent experiments. *P < 0.05; **P < 0.01 T-test (A–D) and ANOVA (C, D). Error bars indicate standard deviation (SD).
We next investigated whether APT1 asymmetric localization depended on CDC42 by altering CDC42 activity. Work from other groups has shown that expression of either GTP-locked or GDP-locked mutant forms of CDC42 can interfere with CDC42-dependent cellular processes (20, 23, 57–60). We used live-cell imaging of APT1WT-CFP and YFP-tagged CDC42 mutants to test the function of CDC42 activity on APT1 localization. Cells expressing a GTP-locked, constitutively active CDC42 mutant (CDC42V12) or a GDP-locked, inactive CDC42 mutant (CDC42N17) showed reduced asymmetry of APT1WT during cell division as compared to cells expressing a control GFP vector (Fig. 4C). In fixed cells, knocking down CDC42 and expressing shRNA-resistant CDC42V12 or CDC42N17 also significantly reduced asymmetric APT1 and β-catenin localization. Compared to CDC42 knockdown, which had minimal effect, expressing CDC42V12 or CDC42N17 resulted in a strong reduction of asymmetric Numb localization (fig. S4A-G).
To demonstrate the requirement of CDC42 cycling activity on APT1 localization in cells, we expressed a YFP-tagged constitutively active CDC42 mutant that retains GTP-GDP cycling (CDC42F28L). CDC42F28L was previously shown by other groups to mimic CDC42-mediated effector binding and subcellular localization (57). Expressing shRNA-resistant CDC42F28L in CDC42 knockdown cells was sufficient to rescue β-catenin and APT1 asymmetric localization, but only partially rescued asymmetric Numb localization in CDC42 knockdown cells (fig S4A-G). Additionally, expression of CDC42V12, CDC42N17, or CDC42F28L did not significantly alter the low percentage of asymmetrically localized catalytically inactive APT1S119A as compared to APT1WT by live-cell imaging (Fig. 4C). These results demonstrate a requirement for the polarity complex and, specifically, CDC42 activity in promoting the asymmetric partitioning of APT1 and β-catenin to the membrane. However, although Numb asymmetric localization required CDC42 activity, overall it appears to be less dependent than APT1 and β-catenin on the canonical Par-aPKC-CDC42 polarity complex.
The reciprocal interaction between APT1 and palmitoylated CDC42 is sufficient to promote asymmetric protein partitioning during cell division
In addition to cycling between GTP and GDP, CDC42 function is also maintained in part by membrane association though a polybasic region and lipid modification of the C-terminal tail (23, 59). There are two known naturally occurring exon splice variants of CDC42 with distinct lipid modifications– a solely prenylated splice variant and a dually palmitoylated and prenylated splice variant (61). We asked whether either CDC42 splice variant could rescue asymmetric protein localization in CDC42 knockdown cells during division by expressing shRNA-resistant constructs of the dually lipid modified isoform (CDC42Palm) or the solely prenylated CDC42 (CDC42Pren) splice variants (fig. S4A) and examining the localization of Numb, β-catenin, and APT1 in fixed cells. In CDC42 knockdown cells, expressing CDC42Palm was sufficient to fully rescue APT1 asymmetry, whereas expression of CDC42Pren appeared to inhibit asymmetric APT1 localization. CDC42Palm and CDC42Pren had little effect on asymmetric Numb localization in CDC42 knockdown cells. CDC42Palm rescued asymmetric β-catenin localization to baseline control conditions, but CDC42Pren had no observable effect (fig. S4B-D, H-J). These results suggest that palmitoylated CDC42 promotes asymmetric localization of specific downstream proteins, and APT1 may reciprocally promote asymmetric localization of palmitoylated CDC42 during cell division.
To spatiotemporally visualize CDC42 and APT1 localization during cell division, we measured the distribution of APT1WT-CFP and CDC42Pren-YFP or CDC42Palm-YFP by live-cell imaging. CDC42Palm expression increased the percentage of asymmetric APT1WT-CFP in dividing cells by 1.5-fold (52.6% vs. 82.7%) whereas CDC42Pren suppressed asymmetric partitioning of APT1WT-CFP by 1.9-fold (52.6% vs. 27.2%) (Fig. 4D). In contrast, neither isoform sufficiently altered asymmetric partitioning of catalytically inactive APT1S119A (Fig. 4D). This led us to ask whether APT1 was sufficient to promote asymmetric CDC42Palm localization. Both APT1WT-CFP and CDC42Palm-YFP were asymmetric at the plasma membrane early during division, and asymmetrically redistributed to the membrane of one daughter cell upon cytokinesis (Fig. 4E, Movies S8–10). As expected, expression of APT1S119A inhibited asymmetric redistribution of CDC42Palm-YFP and retained CDC42Palm-YFP at the cytokinetic midbody (Fig. 4F). We next asked whether CDC42Palm was endogenously expressed in MDA-MB-231 cells using exon-spanning primers. We detected the endogenous transcript for this palmitoylated CDC42 splice variant by PCR (fig. S4K, L). Furthermore, we detected CDC42 palmitoylation in U2 OS cells by the ABE assay and demonstrated that treatment with PalmB increased the amount of palmitoylated CDC42, whereas treatment with the palmitoyltransferase inhibitor 2-bromopalmitate (2BP) decreased palmitoylation (fig. S4M). These results suggest that palmitoylated CDC42 could function with APT1 in these cells either at a basal level or in a subset of cells to promote asymmetric protein localization.
APT1 restricts Wnt and Notch transcriptional activity to one daughter cell
One downstream effect of asymmetrically partitioning cell fate determinants during cell division is the activation of different transcriptional networks in the two daughter cells, resulting in cells with unique transcriptional profiles (3, 4, 6). We hypothesized that APT1 could also mediate the partitioning of asymmetric transcriptional activity of the Notch and Wnt-β-catenin signaling pathways to one daughter cell. We expressed GFP reporter transgenes containing Notch-activated RBPJ (recombination signal binding protein for immunoglobulin kappa J region) binding sites (pGF1-Notch) (62, 63) or Wnt-activated TCF and Lef1 binding sites (pGF1-TCF/Lef1) in MDA-MB-231 cells, which were then immunostained for GFP and acetylated tubulin in daughter cells (Fig. 5A, B). After plotting the distribution of asymmetric divisions as described in fig. S1, we observed asymmetric Notch and TCF/Lef1 reporter signal in 22.7% and 31.7% of daughter cells, respectively. A control GFP reporter containing a minimal CMV promoter (mCMV) lacking pathway-specific promoter elements showed symmetric signal in most cells and confirmed that the observed asymmetries were dependent on the TCF/Lef1 and Notch enhancer elements (Fig. 5C-H).
Fig. 5. APT1 restricts Wnt and Notch transcriptional activity to one daughter cell.
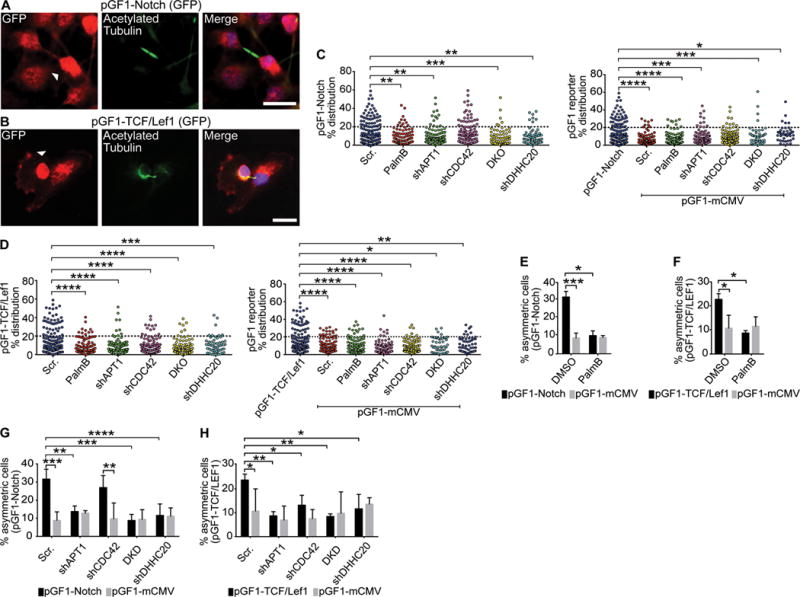
(A and B) Images of cytokinetic MDA-MB-231 cells stained to show expression of the pGF1-Notch GFP reporter (A) or pGF-1 TCF/Lef1 GFP reporter (B) (red), acetylated tubulin (green), and nuclei (blue). Arrowhead indicate asymmetric localization. Scale bars, 15μm. (C and D) Distribution dot plots showing the difference in mean fluorescence pixel intensity of pGF1-Notch reporter (C) or pGF1-TCF/Lef1 reporter (D) across dividing cells. Cells expressing an empty pGF1-mCMV GFP reporter were used as a negative control for the reporters. The distribution of the percentage differences of all quantified cells was plotted, and cells with a difference of >20% (dotted line) were scored as asymmetric. n= 784-822 cells scored for each experimental group. Each dot represents a single cell. Asterisks indicate statistically significant differences between the indicated groups. (E and F) Quantification of dividing MDA-MB-231 cells showing asymmetric localization of pGF1-Notch GFP reporter (E) or pGF1-TCF/Lef1 GFP reporter (F) (black bars) following treatment with PalmB or DMSO vehicle control. Cells expressing an empty pGF1-mCMV reporter (grey bars) were used as a negative control for reporter expression. (G and H) Quantification of the number of dividing MDA-MB-231 cells showing asymmetric localization of the pGF1-Notch GFP reporter (G) or pGF1-TCF/Lef1 GFP reporter (H) (black bars) when coexpressed with shAPT1, shCDC42, both shAPT1 and shCDC42 (DKD), and shDHHC20. Cells expressing an empty pGF1-mCMV GFP reporter (grey bars) were used as a negative control for reporter expression, and cells expressing a scrambled (Scr) shRNA sequence were used as a negative control for knockdown. *P < 0.05; **P < 0.01; ***P < 0.001; ****P < 0.001 T-test (between reporters and pGF1-mCMV) or ANOVA (C to H). Error bars indicate standard deviation (SD).
Pharmacologically inhibiting APT1 with PalmB treatment reduced asymmetric Notch reporter signal by 3.1-fold (31.7% vs. 10.2%), and Wnt reporter signal by 2.6-fold (22.7% vs. 8.7%), respectively (Fig. 5E, F). Knocking down APT1 or DHHC20 also reduced asymmetric Notch and Wnt reporter signal, indicating that palmitoylation can restrict Notch- and Wnt-dependent transcription to one daughter cell. Furthermore, knocking down CDC42 significantly disrupted asymmetric Wnt reporter signal, whereas the Notch reporter signal was unchanged, consistent with observations of asymmetric Numb localization (fig. S4). Additionally, CDC42 and APT1 may function in the same pathway that stimulates signal-activated transcription because double knockdown of APT1 and DHHC20 did not appreciably decrease asymmetric Wnt or Notch reporter signal over APT1 or CDC42 single knockdown (Fig. 5G, H). Together these findings suggest palmitoylation directs the localization of a Wnt activating factor and a Notch inhibitory factor to restrict transcription to one daughter cell.
APT1 induces Wnt, Notch, and mammary stem cell transcriptional signatures in MDA-MB-231 triple receptor–negative breast cancer cells
To date, APT1 has not been shown to drive transcriptional changes in the contexts of development or disease. To further investigate the observed influence of APT1 on asymmetric Notch and Wnt reporter activity, we asked whether APT1 influences a Notch or Wnt gene signature in cells. We performed RNA-seq and gene set enrichment analysis (GSEA) on wild-type control, APT1 knockdown, or APT1WT-expressing MDA-MB-231 cells (table S1). We performed GSEA using a ranked list of differentially expressed genes and compared APT1WT-expressing cells vs. control cells, APT1 knockdown cells vs. control cells, and APT1WT-expressing cells vs. APT1 knockdown cells. In APT1 knockdown cells compared to control cells, we identified a high scoring signature [NES (normalized enrichment score) of 1.46 and a false discovery rate (FDR) q-value of 0.073] that positively correlated with genes reported to increase in cells overexpressing active β-catenin (BCAT_UP.V1_UP), suggesting that APT1 depletion promotes β-catenin signaling (Fig. 6A; fig. S5A). When comparing APT1WT-expressing cells to control cells, we identified a negative correlation (NES 1.25 and FDR q-value 0.302), with a gene signature that includes genes reported to decrease in cells treated with a Notch inhibitor (NOTCH_DN.V1_DN), suggesting APT1 suppresses Notch signaling (Fig. 6B, fig. S5A). When APT1WT-expressing cells were compared to APT1 knockdown cells, we identified a high scoring signature (NES 2.34, FDR q-value=0.0001) for genes reported to increase in mammary stem cells (PECE_MAMMARY_STEM_CELL_UP) (Fig. 6C). Visualization of the leading-edge genes from the mammary stem cell signature also indicated a strong decrease in expression with APT1 knockdown when normalized to control wild-type cells, (Fig. 6D).
Fig. 6. Altering APT1 expression changes β-catenin and Notch gene signatures in MDA-MB-231 cells.
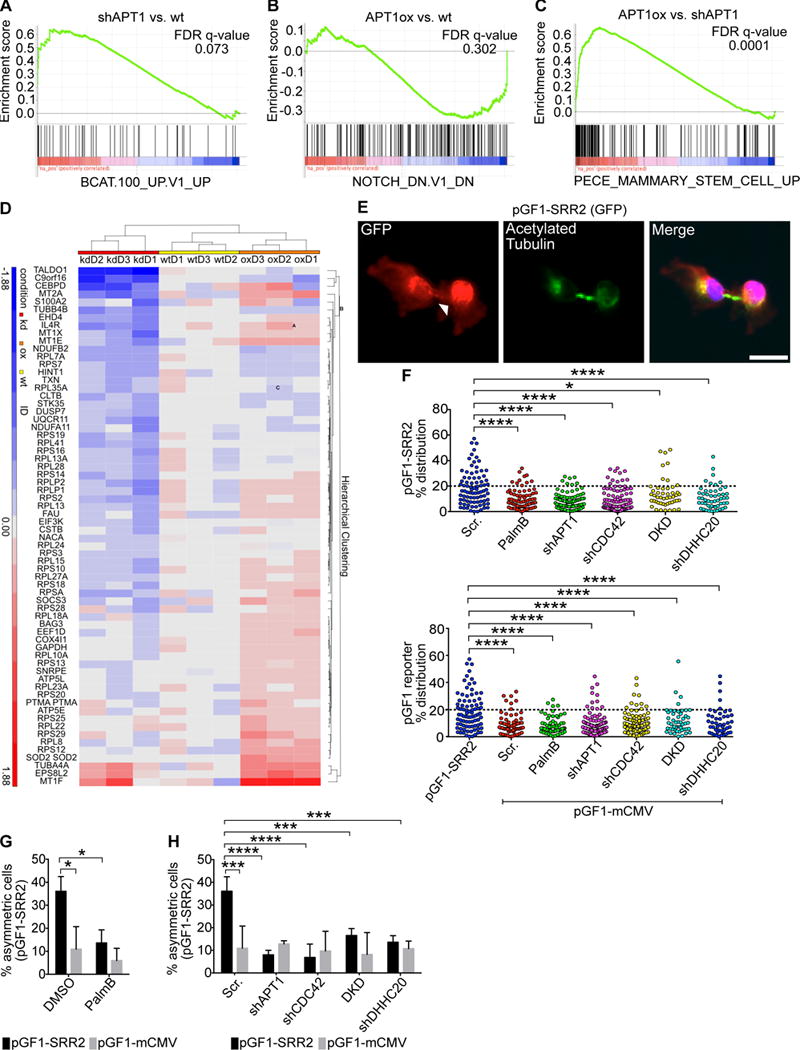
Gene set enrichment analysis (GSEA) of (A) a top-scoring β-catenin overexpression transcriptional signature in MDA-MB-231 cells when APT1 was knocked down and compared against control cells (FDR q-value= 0.073), (B) a Notch inhibition signature when APT1WT was overexpressed and compared against control cells (FDR q-value= 0.302) (B), and (C) a mammary stem cell signature when APT1WT was overexpressed and compared against shAPT1 cells (FDR q-value= 0.0001). (D) Heat map of leading edge genes obtained from the mammary stem cell signature shown in (C). Data is grouped by APT1 knockdown (kd) cells, APT1WT-overexpressing (oe) cells, and control (wt) cells. (E) Images of cytokinetic MDA-MB-231 cells stained to show the pGF1-SRR2 GFP reporter (red), acetylated tubulin (green), and nuclei (blue). Arrowheads indicate asymmetric localization of pGF1-SRR2. Scale bar, 15μm. (F) Distribution dot plots showing the difference in mean fluorescence pixel intensity of the pGF1-SRR2 reporter across dividing MDA-MB-231 cells. Cells expressing an empty pGF1-mCMV GFP reporter were used as a negative control for the reporters. The distribution of the percentage differences of all quantified cells was plotted, and cells with a difference of >20% (dotted line) were scored as asymmetric. n = 959 cells scored for the experimental group. Each dot represents a single cell. Asterisks indicate statistically significant differences between the indicated groups. (G) Quantification of dividing MDA-MB-231 cells showing asymmetric localization of the pGF1-SRR2 GFP reporter after treatment with PalmB or DMSO control (black bars). Cells expressing an empty pGF1-mCMV reporter (grey bars) were used as a negative control for reporter expression. (H) Quantification of the number of dividing MDA-MB-231 cells showing asymmetric localization of pGF1-SRR2 GFP reporter (black bars) when coexpressed with shAPT1, shCDC42, both shAPT1 and shCDC42 (double knockdown, DKD), or shDHHC20. Cells expressing an empty pGF1-mCMV GFP reporter (grey bars) were used as a negative control for reporter expression, and cells expressing a scrambled (Scr) shRNA sequence were used as a negative control for knockdown conditions. **P < 0.01; ****P < 0.0001 T-test (between reporters and pGF1-mCMV) or ANOVA (H). Error bars: SD.
Two other high scoring signatures found in APT1WT-overexpressing cells compared to APT1 knockdown cells were signatures for Myc and Cyclin D1 overexpression, both of which are maintained by Wnt and Notch signaling (NES 2.08, FDR q-value 0.0001; NES 1.69, FDR q-value 0.014) (fig. S5B, C). Target genes with high log2FC (fold change) values were validated by qRT-PCR. Notable APT1-driven genes included DKK1, BMP4, GATA6, and KLF5 (64–66) (fig. S5D, E; table S1).
Based on the high-scoring gene expression signature for mammary stem cells observed in APTWT-overexpressing cells, it is possible APT1 asymmetrically restricts signaling pathways that are activated in stem cells. The transcription factor Sox2 has been implicated in mammary stem cell function (67). Expression of a Sox2-responsive reporter (pGF1-SRR2) in MDA-MB-231 cells revealed that its asymmetric signal was reduced with APT1 knockdown or PalmB treatment. DHHC20, CDC42, or DHHC20 and CDC42 double knockdown reduced the SRR2 asymmetric signal (Fig. 6E-H). We also observed asymmetric APT1 localization in dividing mouse embryonic stem cells expressing an APT1WT plasmid (fig. S6). These results implicate a role for palmitoylation in promoting cell fate–related transcriptional signatures and maintaining asymmetric cell division in progenitor cells.
APT1 and CDC42 maintain unique cell populations in MDA-MB-231 colonies
Tumors are heterogeneous populations of cells that have various degrees of proliferative and self-renewal potential, and these properties contribute to tumorigenicity (35). Triple receptor–negative breast tumors, like that from which MDA-MB-231 cells were derived, contain subpopulations of cells that vary in proliferative and self-renewing, tumor-initiating potential (17, 34, 36). Whether asymmetric cell division contributes to the generation of functionally heterogeneous cells in tumors is unclear. We utilized the colony formation assay, which measures the growth of transformed cells in anchorage-independent conditions and correlates with tumorigenicity. Because only a subset of transformed cells can form colonies, this assay may also indicate the degree of heterogeneity within a cell population (17, 68). Knocking down APT1 reduced colony-forming potential of MDA-MB-231 cells by 2.3-fold (average colonies counted 39 vs. 90), whereas expressing APT1WT increased colony numbers by 1.7-fold (average colonies counted 152 vs. 90), suggesting that APT1 maintains anchorage-independent growth and tumorigenic potential of transformed cells (fig. S7A).
Self-renewing cells within the colony are expected to form new colonies upon serial dissociation and replating, indefinitely. To test whether APT1-mediated asymmetry maintains a self-renewing population within colonies, we examined the self-renewal potential of APT1 knockdown or CDC42 knockdown MDA-MB-231 cells in colonies over 3 rounds of dissociation and replating. APT1 knockdown significantly reduced the number of colonies on the second and third plating, suggesting depletion of colony-initiating cells (Fig. 7A). Unexpectedly, CDC42 knockdown increased the number of colonies formed with each plating compared to control cells, suggesting an expansion of a colony-initiating cell population (Fig. 7B). Under normal two-dimensional culture conditions, we observed a slight lag in proliferation for APT1 knockdown, CDC42 knockdown, and double knockdown cells. Because we did not see a significant impairment of proliferation, this result would suggest that the reduced colony-forming potential is not due decreased proliferation under normal growth conditions (fig. S7B-D). Colonies from APT1 and CDC42 double knockdown cells showed reduced replating ability (fig. S7E), again demonstrating that the expansion of this colony-forming population is dependent on APT1.
Fig. 7. APT1 and CDC42 maintain tumorigenic cell populations in MDA-MB-231 colonies.
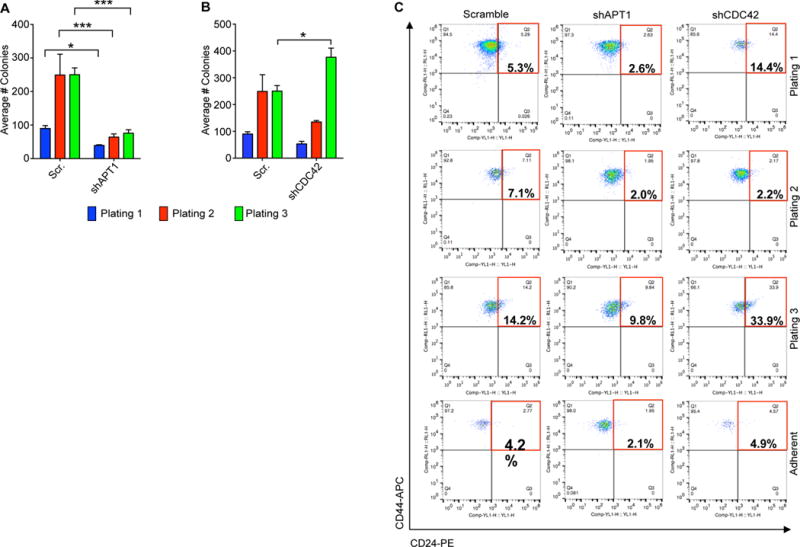
(A and B) Quantification of the average number of colonies formed from MDA-MB-231 shAPT1 cells (A) or shCDC42 cells (B) over 3 serial replatings. Cells expressing a scrambled (Scr) shRNA sequence were used as a negative control for knockdown. Each graph shows means taken from three independent experiments. *P < 0.05; ***P < 0.0001 as measured by T-test. Error bars indicate standard error of the mean (SEM). (C) Representative flow cytometry analysis showing gating strategy of CD44+/CD24lo cells (red box) in cells dissociated from colonies or adherent. The population was gated off of ALDH+ cells shown in fig. S7. The percentages inside the red box indicate the relative proportion of the CD44+/CD24lo cell population. Cells were stained with phycoerythrin (PE)-conjugated anti-CD24 (CD24-PE) (x-axis) and allophycocyanin (APC)-conjugated CD44-APC (y-axis). Flow cytometry plots are representative of results from six independent experiments.
Because the increase in colony-forming potential of CDC42 knock down cells was unexpected, we hypothesized this could be caused by increased symmetric divisions of colony-initiating cells. A subset of highly tumorigenic cells in basal breast cancers have been shown to reactivate developmental signaling pathways such as those dependent on Notch, Wnt, or Sox2 (34, 69–71). Pharmacologic inhibition of Wnt (with the Porcupine inhibitor IWP2) or Notch (with the γ-secretase inhibitor compound E) reduced the self-renewal potential of colonies similarly to APT1 knockdown or APT1 and CDC42 double knockdown (fig. S7E). Furthermore, APT1 knockdown increased Wnt reporter signal and decreased Notch and Sox2 reporter signals in colonies (fig. S7F). This is consistent with our GSEA analysis that indicated APT1 knockdown suppresses Notch signaling and promotes β-catenin signaling and pathways active in mammary stem cells.
Knocking down CDC42 reduced the Notch reporter signal, but increased the Sox2 reporter signal in most of the cells within the colony (fig. S7F). The Sox2 reporter has been previously shown to correlate with increased tumorigenicity (71); these results therefore suggest that the increase in colony formation caused by CDC42 knockdown is due to an increase in Sox2 transcriptionally-active cells.
To address the possibility that APT1 maintains a specific subpopulation of tumorigenic cells, we dissociated colonies and sorted the cells by flow cytometry to test for the cell surface marker profile that is associated with highly tumorigenic cells in breast cancers: CD44 high, CD24 low, and Aldehyde dehydrogenase high (CD44+/ CD24lo/ ALDH+) (72). We first examined the size of the ALDH+ population, which was higher in APT1 knockdown cells (16.7%) as compared to control (7.3%) or CDC42 knockdown (7.8%) cells on the first replating (fig. S8). Further gating the ALDH+ population for CD44+/CD24lo cells revealed that knocking down APT1 reduced CD44+/CD24lo cells (2.6%), whereas knocking down CDC42 increased this population (14.4%), as compared to control cells (5.3%) (Fig. 7C). The trend of the number of CD44+/CD24lo/ALDH+ cells being reduced in the APT1 knockdown condition and increased in the CDC42 knockdown condition was sustained on second and third platings (Fig. 7C). This pattern was observed to a lesser extent in adherent cells (fig. 7C; fig. S8). These results are consistent with the observed colony phenotypes and may explain why CDC42 knockdown cells form many colonies, whereas APT1 knockdown cells are unable to do so. Taken together, these findings demonstrate that in an anchorage-independent setting, an APT1-CDC42 axis maintains the expansion of a self-renewing, tumorigenic cell population and activation of transcriptional profiles required to maintain this population.
Discussion
Our results uncover a molecular mechanism through which palmitoylation restricts the localization of the cell fate determinants β-catenin and Numb and the downstream transcriptional responses to Wnt and Notch signaling to one daughter cell during cell division. Furthermore, palmitoylation directly promotes asymmetric Numb localization, because point mutations in conserved cysteine residues in Numb inhibit not only its palmitoylation but also its asymmetric localization. Inhibition of APT1 reduces the percentage of cells with asymmetrically localized cell fate determinants indicating it facilitates a controlled, rather than a stochastic, process. The observation that both decreased palmitoylation of the NumbAAA mutant or increased palmitoylation of Numb upon APT1 knockdown inhibits asymmetric localization would suggest it is the dynamic association with the plasma membrane that promotes asymmetry. The enrichment of both the palmitoyltransferase DHHC20 and the depalmitoylating enzyme APT1 to a discrete membrane domain could reinforce this process. APT1 inhibition did not completely block the asymmetric localization of β-catenin and Numb, but resulted in a residual asymmetry in about 8% of cells. This is similar to the percentage of cells with asymmetrically localized GFP that does not respond to APT1 inhibition, and is most likely independent of palmitoylation. This palmitoylation-independent asymmetric localization could be the effect of previously reported asymmetric partitioning of machinery that maintains protein abundance such as the proteasome (3, 73) or protein translation machinery (74, 75), which could result in localized differences in degradation or synthesis of proteins during cell division.
Our data show an interdependence of APT1 and CDC42 activity for their own asymmetric localization. This interdependence could occur through association of two distinct compartments of the plasma membrane: the lipid bilayer and the membrane-associated, cortical actin cytoskeleton. CDC42 establishes polarity in cells by spatially reorganizing actin to direct vesicle traffic to and fusion with the plasma membrane (23, 76, 77). Because active CDC42 is membrane-associated, APT1 could promote the recruitment of palmitoylated proteins that also stimulate CDC42 function, such as the CDC42 activator Intersectin, which is also activated by Numb (78), to the plasma membrane. This is consistent with current models of diffusible, membrane-bound molecules establishing distinct polarized domains at the plasma membrane in a feedforward manner (79, 80). It has also been hypothesized that the formation of polarized domains can be inhibited if the association rate outpaces the dissociation rate or vice versa (79, 81). This could explain why polarized membrane domains are sensitive to changes in CDC42 protein abundance, activity, and lipid-modification, as demonstrated by our findings.
Whereas the asymmetric distributions of CDC42 and APT1 appear to be interdependent, the depletion of either protein individually has distinct effects on the Wnt and Notch pathways. Knocking down CDC42 significantly reduces both APT1 and β-catenin asymmetric localization, without affecting the localization of Numb. This would suggest that although asymmetric β-catenin and Numb localization require APT1 activity, only the asymmetric localization of β-catenin requires asymmetric localization of APT1. CDC42 is known to promote canonical Wnt signaling activity by sequestering adenomatosis polyposis coli (APC), a component of the β-catenin destruction complex, to the plasma membrane in an actin-dependent manner (82, 83). Thus, maintenance of the actin cytoskeleton by CDC42 may be required for asymmetric β-catenin localization, but not for Numb. As such, the requirement for CDC42 may be necessitated by unique mechanisms of Wnt signal activation distinct from a requirement for APT1 activity.
By manipulating APT1, we demonstrate that we can alter the asymmetric activation of Notch, Wnt, and Sox2 transcriptional reporters without directly altering the expression of transcription factors or upstream signaling factors. The fact that β-catenin and Notch gene signatures were not as high-scoring as compared to those of mammary stem cells, Myc, and Cyclin D1 signatures may indicate that APT1 directs combinatorial signaling to determine cell identity. Myc and Cyclin D1 are canonical downstream transcriptional targets of both Wnt and Notch and are also involved in determining mammary stem cell identity (84–86). Within the mammary stem cell signature is a notable abundance of ribosomal proteins. A similar abundance in ribosomal proteins is also present in the Myc signature, consistent with its role in driving ribosomal biogenesis and protein translation in the mammary gland, among other tissues, and in cancer (87, 88). Because Myc is a critical factor for mammary stem cell identity and function (85), our results would suggest APT1 has a role in translation in mammary stem cells. What remains unclear is whether altering APT1 protein amounts induces the expansion of a population of cells expressing a mammary stem cell transcriptional signature or induces a de novo transcriptional signature in most cells. We also have evidence of asymmetric APT1 localization in dividing mouse embryonic stem cells expressing APT1WT, suggesting that this mechanism may also have a broader and conserved role in development. Future experiments, including single-cell RNAseq, may address these questions.
Consistent with the concept of changing cell populations, our results suggest that APT1 is required to maintain a tumorigenic population of breast cancer cells whereas CDC42 appears to restrict the size of this population. Our findings indicate that APT1 may contribute to the activation of transcriptional programs that promote colony-forming potential, whereas CDC42 could restrict APT1 activity to one daughter during asymmetric division. This would be consistent with the increase in the CD44+/ CD24lo/ALDH+ population in CDC42 knockdown colonies. The implications of this study may be relevant to human disease because APT1 is amplified in various cancers, and this amplification is associated with poor patient prognosis, suggesting APT1 could function in the development and progression of human cancers (89, 90). Although the importance of self-renewing, tumor-initiating or stem-like cells in cancer is still unclear, the question of how tumors maintain and generate cells with diverse properties such as metastatic potential, dormancy, and drug resistance is still a critical one. Further exploration of the mechanisms that establish and direct asymmetric cell division and promote cellular heterogeneity may help us understand tumor development and progression.
Materials and Methods
Cell Culture
MDA-MB-231 and U2OS cells were cultured in DMEM + glutamax (Thermo Fisher; Cat. No. 10566-016) and 10% fetal bovine serum. For drug treatment, cells were treated with DMSO (Sigma; Cat. No. D2650), 10μM Palmostatin B (EMD Millipore; Cat. No. 178501) prepared in DMSO, or 5 μM 2-bromopalmitate (Sigma; Cat. No. 21604-1G) prepared in DMSO for 16 hours before staining or harvesting for cell lysates. Cells were treated with 0.5ug/mL puromycin for selection. E14 ESCs were cultured in DMEM Knockout (ThermoFisher. Cat. 10829-018), 15% fetal bovine serum, 1% L-glutamine, 1% Pen Strep, 1% Non-essential amino acids, 0.1 mM 2 mercaptoethanol, 1000 units/mL Leukemia inhibitory Factor (Sigma, Cat. L5158), 1 μM MEK I/II Inhibitor (Millipore, Cat. 444966), 3μM GSK3 Inhibitor XVI (Millipore, Cat. 361559) on gelatin coated tissue culture plates.
Stable cell lines
HEK cells were transfected with 0.69 μg/μL of a GAG, Rev, and Vsvg mix and 1.42 μg/μL of the following plasmids: Scramble control, GFP-PRRL, APT1WT -CFP-Flag-PRRL, APT1 (S119)-CFP-Flag-PRRL, FLAG- CDC 42 Pren -PRRL, FLAG-CDC42-V12-PRRL, FLAG-CDC42-N17-PRRL, FLAG-CDC42-F28-PRRL, FLAG-CDC42 Palm-PRRL, pGF-SRR2-mCMV-GFP-puro (System Biosciences; Cat. No. SR20071-PA-P), pGF-Notch-mCMV-GFP-puro (System Biosciences; Cat. No. TR020PA-P), pGF-TCF/Lef-mCMV-GFP-puro (System Biosciences; Cat. No.TR013PA-P), or pGF-mCMV-GFP (System Biosciences; Cat. No. TR011PA-1), for 24 hours with LT-1 transfection reagent (Mirus Bio. Cat. MIR2300). The aforementioned APT1 and CDC42 plasmids were designed with short hairpin resistant sequences for rescue studies. Virus was collected 72 hours after infection with 0.5-1mL virus used for stable cell line generation. U2OS cells were infected with APT1WT -CFP-FLAG or APT1(S119)-CFP-FLAG lentivirus for 24 hours, then recovered in complete DMEM for 48 hours prior to cell culture. E14 ESCs were infected with APT1WT -CFP-FLAG lentivirus for 24 hours, then recovered in complete DMEM-knockout for 48 hours prior to cell culture. E14 ESCs were infected with APT1WT -CFP-FLAG lentivirus for 24 hours, and then recovered in complete DMEM for 48 hours prior to cell culture. MDA-MB-231 were infected with the aforementioned lentivirus for 24 hours and recovered in complete DMEM for 48 hours prior to cell culture.
Short hairpin design
shRNA for PARD3
F: 5′-CCGGGCCATCGACAAATCTTATGATCTCGAGATCATAAGATTTGTCGATGGCTTTTTG-3′
R: 5′-AATTCAAAAAGCCATCGACAAATCTTATGATCTCGAGATCATAAGATTTGTCGATGGC-3′
F: 5′-CCGGGCCATCGACAAATCTTATGATCTCGAGATCATAAGATTTGTCGATGGCTTTTTG-3′
R: 5′-AATTCAAAAAGCCATCGACAAATCTTATGATCTCGAGATCATAAGATTTGTCGATGGC-3′
F: 5′-CCGGAGTCAATTGGATTTCGTTAAACTCGAGTTTAACGAAATCCAATTGACTTTTTTG-3′
R: 5′-AATTCAAAAAAGTCAATTGGATTTCGTTAAACTCGAGTTTAACGAAATCCAATTGACT-3′
shRNA for CDC42
F: 5′-CCGGCGGAATATGTACCGACTGTTTCTCGAGAAACAGTCGGTACATATTCCGTTTTTG-3′
R: 5′-AATTCAAAAACGGAATATGTACCGACTGTTTCTCGAGAAACAGTCGGTACATATTCCG-3′
F: 5′-CCGGTGCTTGTTGGGACTCAAATTGCTCGAGCAATTTGAGTCCCAACAAGCATTTTTG-3′
R: 5′-AATTCAAAAATGCTTGTTGGGACTCAAATTGCTCGAGCAATTTGAGTCCCAACAAGCA-3′
F: 5′-CCGGAGATTACGACCGCTGAGTTATCTCGAGATAACTCAGCGGTCGTAATCTTTTTTG-3′
R: 5′AATTCAAAAAAGATTACGACCGCTGAGTTATCTCGAGATAACTCAGCGGTCGTAATCT-3′
shRNA for APT1
F 5′- CCGGTAGGCCTGTTACATTAAATATCTCGAGATATTTAATGTAACAGGCCTATTTTTG-3′
R 5′- AATTCAAAAATAGGCCTGTTACATTAAATATCTCGAGATATTTAATGTAACAGGCCTA-3′
Alignment of Numb
Numb sequences from D. melanogaster (P16554), mouse (Q9QZS3), zebrafish (Q5FBC1), and human (P49757) were chosen from UniProt canonical sequences. Alignment was performed with Clustal Omega. Conserved domains were identified by the Conserved Domain Database (NCBI).
Mutagenesis
Site-directed mutagenesis of Numb C37, C160, and C165 to alanine, and APT1 (S119) to alanine were performed using QuikChange multisite-directed mutagenesis kit (Agilent; Cat. No. 210515). Potential palmitoylated cysteines on Numb were identified using CSS-Palm 3.0 developed by Zhou. et. al. (53) and analysis of the PTB domain crystal structure. Mutants were sequence verified by the DNA Sequencing Facility at the Perelman School of Medicine, University of Pennsylvania.
Live-cell imaging
MDA-MB-231 cells were not conducive to studying the dynamics of asymmetric localization at the plasma membrane by live-imaging, as these cells frequently divided out of the focal imaging plane. Thus, U2 OS cells stably expressing mCherry-Histone H2B facilitated protein tracking over time and allowed us to study the dynamics of asymmetric localization at the plasma membrane over the course of cell division. U2 OS cells stably expressing APT1WT -CFP-FLAG-PRRL or APT1(S119)-CFP-FLAG-PRRL were transfected with 2 μg of YFP-CDC42 Pren-PCDNA3.1, YFP-CDC42 Palm-PCDNA3.1, YFP-CDC42 V12-PCDNA3.1, YFP-CDC42 N17-PCDNA3.1, YFP-CDC42 F28-PCDNA3.1, or NUMB-YFP-FLAG- PRRL wild-type or mutant for 24 hours using LT-1 (Mirus Bio; Cat. No. MIR2300) according to manufacturer’s protocol. Cells were imaged 48 hours after transfection in HBSS (Life Technologies; Cat. No. 14175079) containing 2% fetal bovine serum, 1 mg/mL glutamine, and 20mM HEPES pH 7.4 at 37°C. Cells were imaged using the Leica DMI6000 B inverted microscope.
Immunofluorescence
MDA-MB-231 cells were plated on glass coverslips and treated as described. Cells were fixed in 10% formalin, blocked in 5% BSA in TBS containing 0.1% Triton-X (Roche), incubated in 1:500 primary antibody (β-catenin, Cell Signaling Technologies; Cat. No. 9581S), (PARD3 (ab64646)/ Numb (ab14140)/ APT1 (ab91606)/ GFP (ab290)/ Caveolin (ab17052), Abcam), (DHHC20, Sigma; Cat. No. HPA014483), (Acetylated tubulin, Santa Cruz; Cat. No. sc23950), 1:1000), for 1-2 hours at room temperature, incubated in secondary antibody (Alexafluor 488 goat anti-mouse (A11001)/ Alexafluor 594 goat anti-rabbit (A11012), Life Technologies; 1:1000) for 1 hour at room temperature, and mounted in DAPI-mount (Southern Biotech; Cat. No. 0100-20). ESCs were cultured without LIF, MEK or GSK inhibitors for 24 hours before staining with 1:1000 anti-GFP (ab290) as described. Cells were imaged using the Leica DMI6000 B inverted microscope on 40X magnification and colonies were imaged on 20X magnification.
Western Blotting
200,000 MDA-MB-231 cells were plated on 60mm tissue culture dishes and were lysed in Tris lysis buffer (50mM Tris pH 7.4 buffer, 150mM NaCl, 2% Triton-X, 1μg/ml leupeptin, 1μg/ml aprotinin, 2μg/ml pepstatin A). Proteins were run out on 10% acrylamide gel and probed with PARD3 1:1000, CDC42 1:500 (Cytoskeleton; Cat. No. ACD03), Flag 1:500 (Sigma; Cat. No. F3165), APT1 1:500, and DHHC20 1:1000, at 4°C, overnight, then incubated in secondary anti-rabbit HRP (Jackson Immunoresearch; Cat. No. 211-032-171) or anti-mouse HRP (Jackson Immunoresearch; Cat. No. 115-035-003) for 1 hour at room temperature. Blots were developed in Piere ECL Chemilluminescence solution (Thermo Fisher; Cat. No. 32106).
Acyl-Biotin Exchange (ABE) Assay
The protocol is adapted from Wan et al., 2007. : 200,000 MDA-MB-231 cells were plated on 60mm tissue culture dishes and were harvested by scraping in ABE lysis buffer (50mM HEPES pH 7.4, 1% Triton X-100, 150mM NaCl, 5mM EDTA, 50mM N-ethyl-maleimide (NEM), 1μg/ml leupeptin, 1μg/ml aprotinin, 2μg/ml pepstatin A). Lysates were clarified by centrifugation at 15,000 RPM for 10 minutes, and incubated with NEM overnight at 4°C. The samples were m/c precipitated twice then resuspended in 80μL 4%SDS buffer. The samples were split in half and 160μL of hydroxylamine buffer (0.7M hydroxylamine pH 7.4, 50mM HEPES pH 7.4, 0.2% Triton X-100, 150mM NaCl, 5M EDTA) was added to one half of the sample and control 0.2% Triton X-100 buffer (50mM HEPES pH 7.4, 0.2% Triton X-100, 150mM NaCl, 5mM EDTA) was added to the remaining sample and incubated at room temperature for 1hour. The samples were m/c precipitated and resuspended in 40μL4%SDS buffer containing 10μM Biotin-HPDP (Thermo Fisher; Cat. No. 21341). 160μLof 0.2% Triton X-100 buffer +10μM Biotin-HPDP was added and incubated at RT for 1hour. The samples were m/c precipitated and resuspended in 20μL of 4%SDS buffer followed by addition of 800μL of 1% Triton X-100 buffer (50μL removed for analysis as “input”). 30μL of streptavidin agarose buffer (50mM HEPES pH 7.4, 0.2% Triton X-100, 150mM NaCl, 5mM EDTA) was added to the remaining sample and incubated at room temperature for 1hour. The samples were m/c precipitated and resuspended in 40μL4%SDS buffer containing 10μM Biotin-HPDP (Pierce). 160μLof 0.2% Triton X-100 buffer +10μM Biotin-HPDP was added and incubated at RT for 1hour. The samples were m/c precipitated and resuspended in 20μLof 4%SDS buffer followed by addition of 800μL of 1% Triton X-100 buffer (50μLremoved for analysis as “input”). 30μLof streptavidin agarose beads (Thermo Scientific; Cat. No. 20349) were added to the samples and incubated 20 overnight at 4°C rotating. The samples were washed in 1% Triton-X100 buffer and analyzed by SDS PAGE.
ABE Assay for Immunofluorescence
Washes were performed in 1X ABE buffer + 0.2% Triton-X + 0.1% SDS. Cells were seeded onto coverslips. Cells were fixed in 10% formalin + 50mM NEM for 10 minutes at room temperature and washed once before being incubated overnight in 1X ABE buffer + 0.2% Triton-X + 0.1% SDS* + 50mM NEM at 4°C. The following day, 3 × 15 minute washes were performed. Cells were incubated in HA+ or HA- buffer for 2 hours at room temperature, then washed 3 × 15minutes. Cells were incubated in Biotin-HPDP buffer for 1 hour at room temperature and washed for 3 × 20 minutes. Cells were Incubated in primary antibody (Biotin, 1:500; Abcam; Cat. No. ab53494), at 4°C overnight, and immunofluorescence was performed as described above.
Click Chemistry Assay for Palmitoylation
200,000 U2 OS cells were plated on 60mm tissue culture dishes and were transfected with 2μg Numb wt or NumbAAA YFP-Flag for 24 hours, labeled with 100μM palmitic acid azide (Life Technologies; Cat. No. C10265). Cells were prepared using the Click-IT protein reaction buffer (Life Technologies; Cat. No. C10276) according to manufacturer’s protocol and analyzed with Western blot as described above.
Quantification and linescan analysis
Cells were quantified by drawing around the mitotic spindle poles, or around each daughter cell in cytokinesis using the Leica LAS AF software as shown in Fig. S1A. Percent difference was calculated from the Mean Gray Values generated in Leica LAS AF and calculated as described in Fig. S1A. The distribution of acquired percentage differences for each experimental condition were plotted as dot plots. Cells with a percentage difference of 20 or greater were counted as asymmetric and plotted in a bar graph. All graphs were generated with Prism software. Linescan analysis of pixel intensity was performed on still frames from live-cell movies using ImageJ software.
Colony Assays
MDA-MB-231 cells were plated on 6-well, low adhesion plates (Corning; Cat. No. 3471) in 2mL of WIT-P plus serum-free supplement growth media (Cellaria; Cat. No. 00-0045-500) containing 20ng/mL FGF (Life Technologies; Cat. No. PHG0024), 20ng/mL EGF (Life Technologies; Cat. No. PHG0311), and 10 μg /mL heparin (StemCell Technologies; Cat. No. 07980). Each well contained 4,000 cells. Cells were treated with 1 μM GSI (Compound E; Millipore; Cat. No. 565790), 5 μM IWP2 (StemCell Technologies; Cat. No. 72124), or DMSO (Sigma; Cat. No. D2650) and grown for 7 days before counting. For replating, cells were grown into colonies as described. On Day 7, colonies were spun down at 1000rpm × 5min, resuspended in 0.05% Trypsin-EDTA, reconstituted in growth media, and plated as described above. Process was repeated for three replatings in duplicate.
Proliferation Assays
15,000 MDA-MB-231 cells were plated on a 12-well dish. At 24, 48, and 72 hours, cells were washed with 1X PBS, trypsinized, and spun at 1000rpm × 5 minutes. Pellets were resuspended in DMEM and counted by a hemacytometer.
RNA Isolation and Quantitative Real-Time PCR
Total RNA was isolated from cells using RNeasy Mini Kit (Qiagen; Cat. No. 74104) and cDNA was synthesized from 2,000ng total RNA using SuperScript III First-Strand Synthesis SuperMix (Life Technologies; Cat. No. 18080400). qRT-PCRs were performed in triplicate using standard SYBR green reagents and protocols on a StepOnePlus Real-Time PCR system (Applied Biosystems). Target genes with high log2FC values were chosen for validation. The target mRNA expression was quantified using ΔΔCt method and normalized to GAPDH expression. The following primers were used for validation:
hDKK1
F: 5′- CAGGCGTGCAAATCTGTCT – 3′
R: 5′- AATGATTTTGATCAGAAGACACACATA – 3′
hFGF5
F: 5′- CCCAGAATCAGCCCTACAAG – 3′
R: 5′- GAGGAGGAAGGACAAGCTCA – 3′
hGATA6
F: 5′- GCAAAAATACTTCCCCCACA – 3′
R: 5′- TCTCCCGCACCAGTCATC – 3′
hKLF5
F: 5′- CTGCCTCCAGAGGACCTG – 3′
R: 5′- TCGTCTATACTTTTTATGCTCTGGAAT – 3′
hITGB4
F: 5′- TCAGCCTCTCTGGGACCTT – 3′
R: 5′- TCCTTATCCACACGGACACA – 3′
hBMP4
F: 5′- TCCACAGCACTGGTCTTGAG – 3′
R: 5′- TGGGATGTTCTCCAGATGTTCT – 3′
hPTK7
F: 5′- CAGAGGACTCACGGTTCGAG – 3′
R: 5′- TACCAGGGTCTCTGCCACTC – 3′
hGAPDH
F: 5′- ACA CCA TGG GGA AGG TGA AG-3′
R: 5′-AAG GGG TCA TTG ATG GCA AC -3′
hCDC42 palm
F: 5′-TGGAGTGTTCTGCACTTACA-3;
R: 5′-GAATATACAGCACTTCCTTTTGGG-3′
hCDC42 prenyl
F: 5′-AGGCTGTCAAGTATGTGG-3′
R: 5′-TAGCAGCACACACCTGCG-3′
RNA Isolation and analysis for RNA-seq
Total RNA was harvested from MDA-MB-231 control, APT1WT-overexpressing or shAPT1 cells using the RNeasy RNA isolation kit (Qiagen; Cat. No. 74104). 100ng of RNA was used to generate cDNA libraries with the Illumina TruSeq mRNA Library Prep Kit for NeoPrep library preparation system (Illumina; Cat. No. NP-202-1001) and analyzed on a NextSeq500 sequencer. Analysis was prepared by the DNA Sequencing Facility at the Perelman School of Medicine, University of Pennsylvania as follows: estimated transcript levels were ranked with Salmon, TX Import was used to condense transcript levels to gene intensity, and Deseq2 was used to calculate statistical levels for each condition. The scaled values (determined by DeSeq2) were input to Gene Set Enrichment Analysis software (Broad Institute) and analyzed against the C2 Chemical and Genetic perturbations, C6 Oncogenic, and Hallmark signatures gene matrix applying classic enrichment statistic. Heatmaps were generated by taking the log2 values with an offset of 1 for all conditions and targets were chosen by taking the leading-edge targets from GSEA BCAT, NOTCH, and mammary stem cell sets. The average of the 3 control wild-type values were subtracted from each individual value. Values were clustered for samples and genes using Euclidian similarity measure with average linkage.
PCR
To confirm the expression of FLAG-CDC42 plasmids in shCDC42 MDA-MB-231 cells, we isolated RNA and prepared cDNA from FLAG-prenylated CDC42, FLAG-CDC42-V12, FLAG-CDC42-N17, FLAG-CDC42-F28, FLAG-palmitoylated CDC42 cell lines. PCR reaction was performed using 2ug of cDNA, 10X PCR buffer (Sigma; Cat. No. P2192), 10mM dNTPs (Life Technologies; Cat. no. 18252015), Taq polymerase (Sigma; Cat. No D1806) and 10μM of hCDC42-palm and hCDC42-prenyl primers mentioned above, which are designed to span the alternatively spliced exon. The reaction was run out on a 1.4% agarose gel.
Flow cytometry
Adherent non-confluent MDA-MB-231 cells or colony-dissociated cells were treated to detect ALDH activity using the ALDEFLUOR assay (Stem Cell Technologies; Cat. No. 01702) according to manufacturer’s protocol. Diethylaminobenzaldehyde (DEAB) was used as a negative control to set ALDH + gates. Cells were stained for surface markers with CD44-APC (BD Biosciences; Cat. No. 560890), CD24-PE (BD Biosciences; Cat. No. 560991), and Live/Dead Violet (Thermo Fisher; Cat. No. L34963) for viability. Compensation was performed using Ultra Comp beads (Thermo Fisher; Cat. No. 01-2222-41). Experiments were run on the Attune NxT flow cytometer system (Life Technologies) and analysed with FlowJo software. ALDH+ cells (Fig. S7) were gated for CD44+/CD24lo (Fig. 7C) for comparison.
Supplementary Material
Movie S1. Single channel of a U2 OS cell ectopically expressing APT1WT-CFP (blue).
Movie S10. Merge of Movie S1 of a U2 OS cell ectopically expressing APT1WT-CFP (blue), YFP-CDC42 Palm (yellow), and mCherry-Histone-H2B (red).
Movie S2. Single channel of a U2 OS cell from Movie S1 ectopically expressing NumbWT-YFP (yellow).
Movie S3. Merge of Movie S1 of a U2 OS cell ectopically expressing APT1WT-CFP (blue), NumbWT-YFP (yellow), and mCherry-Histone-H2B (red).
Movie S4. Single channel of a U2 OS cell ectopically expressing APT1WT-CFP (blue).
Movie S5. Merge of Movie S4 of a U2 OS cell ectopically expressing APT1WT-CFP (blue) and mCherry-Histone-H2B (red).
Movie S6. Single channel of a U2 OS cell ectopically expressing APT1S119A-CFP (blue).
Movie S7. Merge of Movie S6 of a U2 OS cell ectopically expressing APT1S119A-CFP (blue) and mCherry-Histone-H2B (red).
Movie S8. Single channel of a U2 OS cell ectopically expressing APT1WT-CFP (blue).
Movie S9. Single channel of a U2 OS cell ectopically expressing YFP-CDC42 Palm (yellow).
Fig. S1. Scoring method for determining asymmetric divisions.
Fig. S2. DHHC20 and APT1 localization in MDA-MB-231 cells.
Fig. S3. Effect of CDC42 and PARD3 knockdown on asymmetric Numb and β-catenin localization.
Fig. S4. CDC42 activity and lipidation promote asymmetric APT1, Numb, and β-catenin localization.
Fig. S5. Validation of RNA-seq and additional GSEA analyses relating to Figure 6.
Fig. S6. Staining of asymmetric APT1 in mouse embryonic stem cell.
Fig. S7. Colony counts, growth curves, and reporter expression relating to Figure 7.
Fig. S8. Gating scheme for ALDH+ cells on dissociated colonies or adherent cells.
Table S1. Excel spreadsheet of RNA-seq annotated genes.
One-Sentence Summary.
Palmitoylation-dependent asymmetric partitioning of cell fate determinants correlates with tumor cell heterogeneity.
Editor’s Summary.
Lipid modification promotes cancer cell heterogeneity
Asymmetric cell division generates daughter cells that have distinct fates and is accomplished through the unequal distribution of cell fate determinants or signaling pathway components. Stypulkowski et al. found that the depalmitoylase APT1 was asymmetrically localized in dividing breast cancer and osteosarcoma cells and required for the asymmetric localization of the Notch antagonist Numb and the Wnt signaling mediator β-catenin, both of which are palmitoylated. The catalytic activity of APT1 was required for asymmetric localization of APT1, Numb, and β-catenin, and the polarity complex component CDC42 was required for the asymmetric localization of both APT1 and β-catenin. APT1-mediated asymmetric partitioning of Numb and β-catenin restricted Notch and Wnt signaling to one daughter cell or the other and induced a mammary stem cell transcriptional signature in breast cancer cells. The absence of APT1 or CDC42 reduced heterogeneity and self-renewal capacity in breast cancer cell populations. These results identify palmitoylation-dependent asymmetric partitioning of cell fate determinants as a potential driver of tumor cell heterogeneity, which has been associated with tumor progression and metastatic potential.
Acknowledgments
We would like to thank Christopher J. Lengner and Donita C. Brady for their insightful comments on this manuscript. We also thank John Tobias and the Molecular Profiling Core at the University of Pennsylvania for analysis of the RNAseq data and generation of heat maps and GSEA signatures. The results shown here are in whole or part based upon data generated by the TCGA Research Network: http://cancergenome.nih.gov/.
Funding: This work was supported by NIH Grant R01CA181633, ACS Grants RSG-15-027-01, IRG –78-002-34.
Footnotes
Author contributions: E.S. designed and conducted the experiments. I.A. performed the RNA-seq experiment. E.S. and E.S.W wrote the manuscript.
Competing interests: The authors declare that they have no competing interests.
Data and materials availability: RNA sequencing data is available at GEO (ID: GSE105486).
References and Notes
- 1.Neumüller RA, Knoblich JA. Dividing cellular asymmetry: Asymmetric cell division and its implications for stem cells and cancer. Genes Dev. 2009;23:2675–2699. doi: 10.1101/gad.1850809. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Chang JT, Palanivel VR, Kinjyo I, Schambach F, Intlekofer AM, Banerjee A, Longworth Sa, Vinup KE, Mrass P, Oliaro J, Killeen N, Orange JS, Russell SM, Weninger W, Reiner SL. Asymmetric T lymphocyte division in the initiation of adaptive immune responses. Science. 2007;315:1687–1691. doi: 10.1126/science.1139393. [DOI] [PubMed] [Google Scholar]
- 3.Chang JT, Ciocca ML, Kinjyo I, Palanivel VR, McClurkin CE, DeJong CS, Mooney EC, Kim JS, Steinel NC, Oliaro J, Yin CC, Florea BI, Overkleeft HS, Berg LJ, Russell SM, Koretzky GA, Jordan MS, Reiner SL. Asymmetric Proteasome Segregation as a Mechanism for Unequal Partitioning of the Transcription Factor T-bet during T Lymphocyte Division. Immunity. 2011;34:492–504. doi: 10.1016/j.immuni.2011.03.017. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Mizumoto K, Sawa H. Cortical ??-Catenin and APC Regulate Asymmetric Nuclear ??-Catenin Localization during Asymmetric Cell Division in C. elegans. Dev Cell. 2007;12:287–299. doi: 10.1016/j.devcel.2007.01.004. [DOI] [PubMed] [Google Scholar]
- 5.Katajisto P, Döhla J, Chaffer CL, Pentinmikko N, Marjanovic N, Iqbal S, Zoncu R, Chen W, Weinberg RA, Sabatini DM. Stem cells. Asymmetric apportioning of aged mitochondria between daughter cells is required for stemness. Science. 2015;348:340–3. doi: 10.1126/science.1260384. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Rhyu MS, Jan LY, Jan YN. Asymmetric distribution of numb protein during division of the sensory organ precursor cell confers distinct fates to daughter cells. Cell. 1994;76:477–491. doi: 10.1016/0092-8674(94)90112-0. [DOI] [PubMed] [Google Scholar]
- 7.Zhong W, Feder JN, Jiang MM, Jan LY, Jan YN, Artavanis-Tsakonas S, Matsuno K, Fortini M, Blaikie P, Immanuel D, Wu J, Li N, Yajnik V, Margolis B, Blochlinger K, Bodmer R, Jack J, Jan L, Jan Y, Blochlinger K, Jan L, Jan Y, Bork P, Margolis B, Brand A, Perrimon N, Brand M, Jarman A, Jan L, Jan Y, Caviness V, Takahashi T, Nowakowski R, Chenn A, McConnell S, Church G, Gilbert W, Davis A, Temple S, Amo Fdel, Gendron-Maguire M, Swiatek P, Jenkins N, Copeland N, Gridley T, Dignam J, Doe C, Grove E, Williams B, Li D-Q, Hajihosseini M, Friedrich A, Price J, Guo M, Bier E, Jan L, Jan Y, Guo M, Jan L, Jan Y, Hartenstein V, Posakony J, Jan Y, Jan L, Kavanaugh W, Williams L, Knoblich J, Jan L, Jan Y, Kornack D, Rakic P, Langman J, Guerrant R, Freeman B, Luskin M, Pearlman A, Sanes J, Luskin M, Parnavelas J, Barfield J, Martin A, McConnell S, Minoe M, Danevic C, Boardman P, Harris B, Parnavelas J, Musacchio A, Wilmanns M, Saraste M, Parnavelas J, Barfield J, Franke E, Luskin M, Price J, Thurlow L, Rakic P, Rakic P, Reid C, Liang I, Walsh C, Rhyu M, Jan L, Jan Y, Smart IH, Smart IH, Smith D, Johnson K, Spana E, Kopczynski C, Goodman C, Doe C, Takahashi T, Nowakowski R, Caviness V, Towbin H, Staehelin T, Gordon J, Uemura T, Shepherd S, Ackerman L, Jan L, Jan Y, Vaessin H, Grell E, Wolff E, Bier E, Jan L, Jan Y, van der Geer P, Pawson T, Walsh C, Cepko C, Walsh C, Cepko C, Weinmaster G, Roberts V, Lemke G, Weinmaster G, Roberts V, Lemke G, Zamenhof S, Zhong W, Mirkovitch J, Darnell J, Zhou M-M, Ravichandran K, Olejniczak E, Petros A, Meadows R, Sattler M, Harlan J, Wade W, Burakoff S, Fesik S, Zipursky S, Venkatesh T, Teplow D, Benzer S. Asymmetric localization of a mammalian numb homolog during mouse cortical neurogenesis. Neuron. 1996;17:43–53. doi: 10.1016/s0896-6273(00)80279-2. [DOI] [PubMed] [Google Scholar]
- 8.Nowell PC. The clonal evolution of tumor cell populations. Science. 1976;194:23–8. doi: 10.1126/science.959840. [DOI] [PubMed] [Google Scholar]
- 9.Cleary AS, Leonard TL, Gestl SA, Gunther EJ. Tumour cell heterogeneity maintained by cooperating subclones in Wnt-driven mammary cancers. Nature. 2014;508:113–117. doi: 10.1038/nature13187. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Michor F, Polyak K. The origins and implications of intratumor heterogeneity. Cancer Prev Res. 2010;3:1361–1364. doi: 10.1158/1940-6207.CAPR-10-0234. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Yates LR, Campbell PJ. Evolution of the cancer genome. Nat Rev Genet. 2012;13:795–806. doi: 10.1038/nrg3317. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Junttila MR, de Sauvage FJ. Influence of tumour micro-environment heterogeneity on therapeutic response. Nature. 2013;501:346–54. doi: 10.1038/nature12626. [DOI] [PubMed] [Google Scholar]
- 13.Easwaran H, Tsai HC, Baylin SB. Cancer epigenetics: tumor heterogeneity, plasticity of stem-like states, and drug resistance. Mol Cell. 2014;54:716–27. doi: 10.1016/j.molcel.2014.05.015. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Wirtz-Peitz F, Nishimura T, Knoblich JA. Linking Cell Cycle to Asymmetric Division: Aurora-A Phosphorylates the Par Complex to Regulate Numb Localization. Cell. 2008;135:161–173. doi: 10.1016/j.cell.2008.07.049. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Atwood SX, Prehoda KE. aPKC Phosphorylates Miranda to Polarize Fate Determinants during Neuroblast Asymmetric Cell Division. Curr Biol. 2009;19:723–729. doi: 10.1016/j.cub.2009.03.056. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Shen Q, Zhong W, Jan YN, Temple S. Asymmetric Numb distribution is critical for asymmetric cell division of mouse cerebral cortical stem cells and neuroblasts. Development. 2002;129:4843–4853. doi: 10.1242/dev.129.20.4843. [DOI] [PubMed] [Google Scholar]
- 17.Cicalese A, Bonizzi G, Pasi CE, Faretta M, Ronzoni S, Giulini B, Brisken C, Minucci S, Di Fiore PP, Pelicci PG. The Tumor Suppressor p53 Regulates Polarity of Self-Renewing Divisions in Mammary Stem Cells. Cell. 2009;138:1083–1095. doi: 10.1016/j.cell.2009.06.048. [DOI] [PubMed] [Google Scholar]
- 18.Zimdahl B, Ito T, Blevins A, Bajaj J, Konuma T, Weeks J, Koechlein CS, Kwon HY, Arami O, Rizzieri D, Broome HE, Chuah C, Oehler VG, Sasik R, Hardiman G, Reya T. Lis1 regulates asymmetric division in hematopoietic stem cells and in leukemia. Nat Genet. 2014;46:245–52. doi: 10.1038/ng.2889. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Habib SJ, Chen BC, Tsai FC, Anastassiadis K, Meyer T, Betzig E, Nusse R. A localized Wnt signal orients asymmetric stem cell division in vitro. Science. 2013;339:1445–8. doi: 10.1126/science.1231077. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Macara IG. Parsing the polarity code. Nat Rev Mol Cell Biol. 2004;5:220–231. doi: 10.1038/nrm1332. [DOI] [PubMed] [Google Scholar]
- 21.Adams AE, Johnson DI, Longnecker RM, Sloat BF, Pringle JR. CDC42 and CDC43, two additional genes involved in budding and the establishment of cell polarity in the yeast Saccharomyces cerevisiae. J Cell Biol. 1990;111:131–42. doi: 10.1083/jcb.111.1.131. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Wodarz A, Ramrath A, Grimm A, Knust E. Drosophila atypical protein kinase C associates with Bazooka and controls polarity of epithelia and neuroblasts. J Cell Biol. 2000;150:1361–74. doi: 10.1083/jcb.150.6.1361. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Etienne-Manneville S. Cdc42–the centre of polarity. J Cell Sci. 2004;117:1291–1300. doi: 10.1242/jcs.01115. [DOI] [PubMed] [Google Scholar]
- 24.Conibear E, Davis NG. Palmitoylation and depalmitoylation dynamics at a glance. J Cell Sci. 2010;123:4007–4010. doi: 10.1242/jcs.059287. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Linder ME, Deschenes RJ. Palmitoylation: policing protein stability and traffic. Nat Rev Mol Cell Biol. 2007;8:74–84. doi: 10.1038/nrm2084. [DOI] [PubMed] [Google Scholar]
- 26.Wang W, Runkle KB, Terkowski SM, Ekaireb RI, Witze ES. Protein depalmitoylation is induced by Wnt5a and promotes polarized cell behavior. J Biol Chem. 2015;290:15707–15716. doi: 10.1074/jbc.M115.639609. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Resh MD. Covalent lipid modifications of proteins. Curr Biol. 2013;23:R431–5. doi: 10.1016/j.cub.2013.04.024. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Runkle KB, Kharbanda A, Stypulkowski E, Cao XJ, Wang W, Garcia BA, Witze ES. Inhibition of DHHC20-Mediated EGFR Palmitoylation Creates a Dependence on EGFR Signaling. Mol Cell. 2016;62:385–396. doi: 10.1016/j.molcel.2016.04.003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Laude AJ, I, Prior A. Palmitoylation and localisation of RAS isoforms are modulated by the hypervariable linker domain. J Cell Sci. 2008;121:421–427. doi: 10.1242/jcs.020107. [DOI] [PubMed] [Google Scholar]
- 30.Aicart-Ramos C, Valero RA, Rodriguez-Crespo I. Protein palmitoylation and subcellular trafficking. Biochim Biophys Acta - Biomembr. 2011;1808:2981–2994. doi: 10.1016/j.bbamem.2011.07.009. [DOI] [PubMed] [Google Scholar]
- 31.Levental I, Lingwood D, Grzybek M, Coskun Ü, Simons K. Palmitoylation regulates raft affinity for the majority of integral raft proteins. Proc Natl Acad Sci U S A. 2010;107:22050–22054. doi: 10.1073/pnas.1016184107. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Chen B, Zheng B, DeRan M, Jarugumilli GK, Fu J, Brooks YS, Wu X. ZDHHC7-mediated S-palmitoylation of Scribble regulates cell polarity. Nat Chem Biol. 2016;12:686–93. doi: 10.1038/nchembio.2119. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Fukata Y, Fukata M. Protein palmitoylation in neuronal development and synaptic plasticity. Nat Rev Neurosci. 2010;11:161–175. doi: 10.1038/nrn2788. [DOI] [PubMed] [Google Scholar]
- 34.Azzam DJ, Zhao D, Sun J, Minn AJ, Ranganathan P, Drews-Elger K, Han X, Picon-Ruiz M, Gilbert Ca, Wander Sa, Capobianco AJ, El-Ashry D, Slingerland JM. Triple negative breast cancer initiating cell subsets differ in functional and molecular characteristics and in γ-secretase inhibitor drug responses. EMBO Mol Med. 2013;5:1502–22. doi: 10.1002/emmm.201302558. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Moore N, Houghton J, Lyle S. Slow-cycling therapy-resistant cancer cells. Stem Cells Dev. 2012;21:1822–30. doi: 10.1089/scd.2011.0477. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Prat A, Parker JS, Karginova O, Fan C, Livasy C, Herschkowitz JI, He X, Perou CM. Phenotypic and molecular characterization of the claudin-low intrinsic subtype of breast cancer. Breast cancer Res. 2010;12:R68. doi: 10.1186/bcr2635. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Mohseny AB, Machado I, Cai Y, Schaefer K-L, Serra M, Hogendoorn PCW, Llombart-Bosch A, Cleton-Jansen A-M. Functional characterization of osteosarcoma cell lines provides representative models to study the human disease. Lab Invest. 2011;91:1195–1205. doi: 10.1038/labinvest.2011.72. [DOI] [PubMed] [Google Scholar]
- 38.Tirino V, Desiderio V, D’Aquino R, Francesco FDe, Pirozzi G, Galderisi U, Cavaliere C, Rosa ADe, Papaccio G. Detection and characterization of CD133+ cancer stem cells in human solid tumours. PLoS One. 2008;3 doi: 10.1371/journal.pone.0003469. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Duncan JA, Gilman AG. A cytoplasmic acyl-protein thioesterase that removes palmitate from G protein ?? subunits and p21(RAS) J Biol Chem. 1998;273:15830–15837. doi: 10.1074/jbc.273.25.15830. [DOI] [PubMed] [Google Scholar]
- 40.Kong E, Peng S, Chandra G, Sarkar C, Zhang Z, Bagh MB, Mukherjee AB. Dynamic palmitoylation links cytosol-membrane shuttling of acyl-protein thioesterase-1 and acyl-protein thioesterase-2 with that of proto-oncogene H-Ras product and growth-associated protein-43. J Biol Chem. 2013;288:9112–9125. doi: 10.1074/jbc.M112.421073. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.Vartak N, Papke B, Grecco HE, Rossmannek L, Waldmann H, Hedberg C, Bastiaens PIH. The autodepalmitoylating activity of APT maintains the spatial organization of palmitoylated membrane proteins. Biophys J. 2014;106:93–105. doi: 10.1016/j.bpj.2013.11.024. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42.Piperno G, LeDizet M, Chang XJ. Microtubules containing acetylated alpha-tubulin in mammalian cells in culture. J Cell Biol. 1987;104:289–302. doi: 10.1083/jcb.104.2.289. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43.Biel M, Deck P, Giannis A, Waldmann H. Synthesis and evaluation of acyl protein thioesterase 1 (APT1) inhibitors. Chem - A Eur J. 2006;12:4121–4143. doi: 10.1002/chem.200501128. [DOI] [PubMed] [Google Scholar]
- 44.Ohno Y, Kihara A, Sano T, Igarashi Y. Intracellular localization and tissue-specific distribution of human and yeast DHHC cysteine-rich domain-containing proteins. Biochim Biophys Acta - Mol Cell Biol Lipids. 2006;1761:474–483. doi: 10.1016/j.bbalip.2006.03.010. [DOI] [PubMed] [Google Scholar]
- 45.Wan J, Roth AF, Bailey AO, Davis NG. Palmitoylated proteins: purification and identification. Nat Protoc. 2007;2:1573–84. doi: 10.1038/nprot.2007.225. [DOI] [PubMed] [Google Scholar]
- 46.Babina IS, McSherry EA, Donatello S, Hill AD, Hopkins AM. A novel mechanism of regulating breast cancer cell migration via palmitoylation-dependent alterations in the lipid raft affiliation of CD44. Breast Cancer Res. 2014;16:R19. doi: 10.1186/bcr3614. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47.Adamson P, Marshall CJ, Hall A, Tilbrook PA. Post-translational modifications of p21rho proteins. J Biol Chem. 1992;267:20033–20038. [PubMed] [Google Scholar]
- 48.Thankamony SP, Knudson W. Acylation of CD44 and its association with lipid rafts are required for receptor and hyaluronan endocytosis. J Biol Chem. 2006;281:34601–34609. doi: 10.1074/jbc.M601530200. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49.Wang DA, Sebti SM. Palmitoylated cysteine 192 is required for RhoB tumor-suppressive and apoptotic activities. J Biol Chem. 2005;280:19243–19249. doi: 10.1074/jbc.M411472200. [DOI] [PubMed] [Google Scholar]
- 50.Huber AH, Nelson WJ, Weis WI. Three-Dimensional Structure of the Armadillo Repeat Region of β-Catenin. Cell. 1997;90:871–882. doi: 10.1016/s0092-8674(00)80352-9. [DOI] [PubMed] [Google Scholar]
- 51.Daugherty RL, Gottardi CJ, Luan L, Ding T, Stinnett A, Reese J, Paria BC, Lin G, Aranda V, Muthuswamy SK, Tonks NK, Burkhalter RJ, Symowicz J, Hudson LG, Stack MS, Luckert K, Götschel F, Sorger PK, Hecht A, Joos TO, Pötz O. Phospho-regulation of Beta-catenin adhesion and signaling functions. Physiology (Bethesda) 2007;22:303–9. doi: 10.1152/physiol.00020.2007. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 52.Knoblich JA, Jan LY, Jan YN. The N terminus of the Drosophila Numb protein directs membrane association and actin-dependent asymmetric localization. Proc Natl Acad Sci. 1997;94:13005–13010. doi: 10.1073/pnas.94.24.13005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 53.Zhou F, Xue Y, Yao X, Xu Y. CSS-Palm: Palmitoylation site prediction with a clustering and scoring strategy (CSS) Bioinformatics. 2006;22:894–896. doi: 10.1093/bioinformatics/btl013. [DOI] [PubMed] [Google Scholar]
- 54.Zwahlen C, Li SC, Kay LE, Pawson T, Forman-Kay JD. Multiple modes of peptide recognition by the PTB domain of the cell fate determinant Numb. EMBO J. 2000;19:1505–1515. doi: 10.1093/emboj/19.7.1505. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 55.Dietzen DJ, Hastings WR, Lublin DM. Caveolin is palmitoylated on multiple cysteine residues. Palmitoylation is not necessary for localization of caveolin to caveolae. J Biol Chem. 1995;270:6838–42. doi: 10.1074/jbc.270.12.6838. [DOI] [PubMed] [Google Scholar]
- 56.Atwood SX, Chabu C, Penkert RR, Doe CQ, Prehoda KE. Cdc42 acts downstream of Bazooka to regulate neuroblast polarity through Par-6 aPKC. J Cell Sci. 2007;120:3200–3206. doi: 10.1242/jcs.014902. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 57.Vanni C, Ottaviano C, Guo F, Puppo M, Varesio L, Zheng Y, Eva A. Constitutively active Cdc42 mutant confers growth disadvantage in cell transformation. Cell Cycle. 2005;4:1675–1682. doi: 10.4161/cc.4.11.2170. [DOI] [PubMed] [Google Scholar]
- 58.Court H, Sudbery P. Regulation of Cdc42 GTPase Activity in the Formation of Hyphae in Candida albicans. Mol Biol Cell. 2006;18:265–281. doi: 10.1091/mbc.E06-05-0411. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 59.Freisinger T, Klünder B, Johnson J, Müller N, Pichler G, Beck G, Costanzo M, Boone C, Cerione RA, Frey E, Wedlich-Söldner R. Establishment of a robust single axis of cell polarity by coupling multiple positive feedback loops. Nat Commun. 2013;4:1807. doi: 10.1038/ncomms2795. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 60.Gladfelter AS, Bose I, Zyla TR, Bardes ESG, Lew DJ. Septin ring assembly involves cycles of GTP loading and hydrolysis by Cdc42p. J Cell Biol. 2002;156:315–326. doi: 10.1083/jcb.200109062. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 61.Nishimura A, Linder ME. Identification of a novel prenyl and palmitoyl modification at the CaaX motif of Cdc42 that regulates RhoGDI binding. Mol Cell Biol. 2013;33:1417–1429. doi: 10.1128/MCB.01398-12. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 62.Lasagni L, Ballerini L, Angelotti ML, Parente E, Sagrinati C, Mazzinghi B, Peired A, Ronconi E, Becherucci F, Bani D, Gacci M, Carini M, Lazzeri E, Romagnani P. Notch activation differentially regulates renal progenitors proliferation and differentiation toward the podocyte lineage in glomerular disorders. Stem Cells. 2010;28:1673–1685. doi: 10.1002/stem.492. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 63.Brimer N, Lyons C, Wallberg AE, Vande Pol SB. Cutaneous papillomavirus E6 oncoproteins associate with MAML1 to repress transactivation and NOTCH signaling. Oncogene. 2012;31:4639–46. doi: 10.1038/onc.2011.589. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 64.Morrisey EE, Tang Z, Sigrist K, Lu MM, Jiang F, Ip HS, Parmacek MS. GATA6 regulates HNF4 and is required for differentiation of visceral endoderm in the mouse embryo. Genes Dev. 1998;12:3579–3590. doi: 10.1101/gad.12.22.3579. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 65.Jiang J, Chan YS, Loh YH, Cai J, Tong GQ, Lim CA, Robson P, Zhong S, Ng HH. A core Klf circuitry regulates self-renewal of embryonic stem cells. Nat Cell Biol. 2008;10:353–60. doi: 10.1038/ncb1698. [DOI] [PubMed] [Google Scholar]
- 66.Kong XB, Zhang C. Dickkopf (Dkk) 1 promotes the differentiation of mouse embryonic stem cells toward neuroectoderm. Vitr Cell Dev Biol - Anim. 2009;45:185–193. doi: 10.1007/s11626-008-9157-2. [DOI] [PubMed] [Google Scholar]
- 67.Wang Y, Dong J, Li D, Lai L, Siwko S, Li Y, Liu M. Lgr4 regulates mammary gland development and stem cell activity through the pluripotency transcription factor Sox2. Stem Cells. 2013;31:1921–1931. doi: 10.1002/stem.1438. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 68.Franken NAP, Rodermond HM, Stap J, Haveman J, van Bree C. Clonogenic assay of cells in vitro. Nat Protoc. 2006;1:2315–9. doi: 10.1038/nprot.2006.339. [DOI] [PubMed] [Google Scholar]
- 69.Rangel MC, Bertolette D, Castro NP, Klauzinska M, Cuttitta F, Salomon DS. Developmental signaling pathways regulating mammary stem cells and contributing to the etiology of triple-negative breast cancer. Breast Cancer Res Treat. 2016;1–16 doi: 10.1007/s10549-016-3746-7. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 70.Xu J, Prosperi JR, Choudhury N, Olopade OI, Goss KH. β-catenin is required for the tumorigenic behavior of triple-negative breast cancer cells. PLoS One. 2015;10 doi: 10.1371/journal.pone.0117097. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 71.Jung K, Gupta N, Wang P, Lewis JT, Gopal K, Wu F, Ye X, Alshareef A, Abdulkarim BS, Douglas DN, Kneteman NM, Lai R. Triple negative breast cancers comprise a highly tumorigenic cell subpopulation detectable by its high responsiveness to a Sox2 regulatory region 2 (SRR2) reporter. Oncotarget. 2015;6:10366–73. doi: 10.18632/oncotarget.3590. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 72.Azzam DJ, Zhao D, Sun J, Minn AJ, Ranganathan P, Drews-Elger K, Han X, Picon-Ruiz M, Gilbert CA, Wander SA, Capobianco AJ, El-Ashry D, Slingerland JM. Triple negative breast cancer initiating cell subsets differ in functional and molecular characteristics and in γ-secretase inhibitor drug responses. EMBO Mol Med. 2013;5:1502–1522. doi: 10.1002/emmm.201302558. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 73.Fuentealba LC, Eivers E, Geissert D, Taelman V, De Robertis EM. Asymmetric mitosis: Unequal segregation of proteins destined for degradation. Proc Natl Acad Sci U S A. 2008;105:7732–7737. doi: 10.1073/pnas.0803027105. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 74.Neumüller RA, Betschinger J, Fischer A, Bushati N, Poernbacher I, Mechtler K, Cohen SM, Knoblich JA. Mei-P26 regulates microRNAs and cell growth in the Drosophila ovarian stem cell lineage. Nature. 2008;454:241–5. doi: 10.1038/nature07014. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 75.Fichelson P, Moch C, Ivanovitch K, Martin C, Sidor CM, Lepesant JA, Bellaiche Y, Huynh JR. Live-imaging of single stem cells within their niche reveals that a U3snoRNP component segregates asymmetrically and is required for self-renewal in Drosophila. Nat Cell Biol. 2009;11:685–693. doi: 10.1038/ncb1874. [DOI] [PubMed] [Google Scholar]
- 76.Cau J, Hall A. Cdc42 controls the polarity of the actin and microtubule cytoskeletons through two distinct signal transduction pathways. J Cell Sci. 2005;118:2579–2587. doi: 10.1242/jcs.02385. [DOI] [PubMed] [Google Scholar]
- 77.Stamnes M. Regulating the actin cytoskeleton during vesicular transport. Curr Opin Cell Biol. 2002;14:428–433. doi: 10.1016/s0955-0674(02)00349-6. [DOI] [PubMed] [Google Scholar]
- 78.Nishimura T. Role of Numb in Dendritic Spine Development with a Cdc42 GEF Intersectin and EphB2. Mol Biol Cell. 2005;17:1273–1285. doi: 10.1091/mbc.E05-07-0700. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 79.Altschuler SJ, Angenent SB, Wang Y, Wu LF. On the spontaneous emergence of cell polarity. Nature. 2008;454:886–889. doi: 10.1038/nature07119. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 80.Johnson JM, Jin M, Lew DJ. Symmetry breaking and the establishment of cell polarity in budding yeast. Curr Opin Genet Dev. 2011;21:740–746. doi: 10.1016/j.gde.2011.09.007. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 81.Gupta A. STOCHASTIC MODEL FOR CELL POLARITY. Ann Appl Probab. 2012;22:827–859. [Google Scholar]
- 82.Sudhaharan T, Goh WI, Sem KP, Lim KB, Bu W, Ahmed S. Rho GTPase Cdc42 is a direct interacting partner of adenomatous polyposis coli protein and can alter its cellular localization. PLoS One. 2011;6 doi: 10.1371/journal.pone.0016603. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 83.Watanabe T, Wang S, Noritake J, Sato K, Fukata M, Takefuji M, Nakagawa M, Izumi N, Akiyama T, Kaibuchi K. Interaction with IQGAP1 links APC to Rac1, Cdc42, and actin filaments during cell polarization and migration. Dev Cell. 2004;7:871–883. doi: 10.1016/j.devcel.2004.10.017. [DOI] [PubMed] [Google Scholar]
- 84.Jeselsohn R, Brown NE, Arendt L, Klebba I, Hu MG, Kuperwasser C, Hinds PW. Cyclin D1 Kinase Activity Is Required for the Self-Renewal of Mammary Stem and Progenitor Cells that Are Targets of MMTV-ErbB2 Tumorigenesis. Cancer Cell. 2010;17:65–76. doi: 10.1016/j.ccr.2009.11.024. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 85.Moumen M, Chiche A, Decraene C, Petit V, Gandarillas A, Deugnier M-A, Glukhova Ma, Faraldo MM. Myc is required for β-catenin-mediated mammary stem cell amplification and tumorigenesis. Mol Cancer. 2013;12:132. doi: 10.1186/1476-4598-12-132. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 86.Bouras T, Pal B, Vaillant F, Harburg G, Asselin-Labat ML, Oakes SR, Lindeman GJ, Visvader JE. Notch Signaling Regulates Mammary Stem Cell Function and Luminal Cell-Fate Commitment. Cell Stem Cell. 2008;3:429–441. doi: 10.1016/j.stem.2008.08.001. [DOI] [PubMed] [Google Scholar]
- 87.Stoelzle T, Schwarb P, Trumpp A, Hynes NE. c-Myc affects mRNA translation, cell proliferation and progenitor cell function in the mammary gland. BMC Biol. 2009;7:63. doi: 10.1186/1741-7007-7-63. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 88.Schmidt EV. The role of c-myc in regulation of translation initiation. Oncogene. 2004;23:3217–3221. doi: 10.1038/sj.onc.1207548. [DOI] [PubMed] [Google Scholar]
- 89.Cerami E, Gao J, Dogrusoz U, Gross BE, Sumer SO, Aksoy BA, Jacobsen A, Byrne CJ, Heuer ML, Larsson E, Antipin Y, Reva B, Goldberg AP, Sander C, Schultz N. The cBio Cancer Genomics Portal: An Open Platform for Exploring Multidimensional Cancer Genomics Data. Cancer Discov. 2012;2 doi: 10.1158/2159-8290.CD-12-0095. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 90.Gao J, Aksoy BA, Dogrusoz U, Dresdner G, Gross B, Sumer SO, Sun Y, Jacobsen A, Sinha R, Larsson E, Cerami E, Sander C, Schultz N. Integrative analysis of complex cancer genomics and clinical profiles using the cBioPortal. Sci Signal. 2013;6:pl1. doi: 10.1126/scisignal.2004088. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Movie S1. Single channel of a U2 OS cell ectopically expressing APT1WT-CFP (blue).
Movie S10. Merge of Movie S1 of a U2 OS cell ectopically expressing APT1WT-CFP (blue), YFP-CDC42 Palm (yellow), and mCherry-Histone-H2B (red).
Movie S2. Single channel of a U2 OS cell from Movie S1 ectopically expressing NumbWT-YFP (yellow).
Movie S3. Merge of Movie S1 of a U2 OS cell ectopically expressing APT1WT-CFP (blue), NumbWT-YFP (yellow), and mCherry-Histone-H2B (red).
Movie S4. Single channel of a U2 OS cell ectopically expressing APT1WT-CFP (blue).
Movie S5. Merge of Movie S4 of a U2 OS cell ectopically expressing APT1WT-CFP (blue) and mCherry-Histone-H2B (red).
Movie S6. Single channel of a U2 OS cell ectopically expressing APT1S119A-CFP (blue).
Movie S7. Merge of Movie S6 of a U2 OS cell ectopically expressing APT1S119A-CFP (blue) and mCherry-Histone-H2B (red).
Movie S8. Single channel of a U2 OS cell ectopically expressing APT1WT-CFP (blue).
Movie S9. Single channel of a U2 OS cell ectopically expressing YFP-CDC42 Palm (yellow).
Fig. S1. Scoring method for determining asymmetric divisions.
Fig. S2. DHHC20 and APT1 localization in MDA-MB-231 cells.
Fig. S3. Effect of CDC42 and PARD3 knockdown on asymmetric Numb and β-catenin localization.
Fig. S4. CDC42 activity and lipidation promote asymmetric APT1, Numb, and β-catenin localization.
Fig. S5. Validation of RNA-seq and additional GSEA analyses relating to Figure 6.
Fig. S6. Staining of asymmetric APT1 in mouse embryonic stem cell.
Fig. S7. Colony counts, growth curves, and reporter expression relating to Figure 7.
Fig. S8. Gating scheme for ALDH+ cells on dissociated colonies or adherent cells.
Table S1. Excel spreadsheet of RNA-seq annotated genes.