

Abstract
Mannitol, a sugar alcohol, is a major primary photosynthetic product in celery (Apium graveolens L. cv Giant Pascal). We report here on purification, characterization, and cDNA cloning of cytosolic non-reversible glyceraldehyde-3-P dehydrogenase (nr-G3PDH, EC 1.2.1.9), the apparent key contributor of the NADPH required for mannitol biosynthesis in celery leaves. As determined by sodium dodecyl sulfate-polyacrylamide gel electrophoresis, purified nr-G3PDH showed a molecular mass of 53 kD. A 1,734-bp full-length cDNA clone (accession no. AF196292) encoding nr-G3PDH was identified using polymerase chain reaction and rapid amplification of cDNA ends techniques. The cDNA clone has an open reading frame of 1,491 bp encoding 496 amino acid residues with a calculated molecular weight of 53,172. Km values for the celery nr-G3PDH were low (6.8 μm for NADP+ and 29 μm for d-glyceraldehyde-3-P). NADPH, 3-phosphoglycerate, and ATP were competitive inhibitors, and cytosolic levels of these three metabolites (as determined by nonaqueous fractionation) were all above the concentrations necessary to inhibit activity in vitro, suggesting that nr-G3PDH may be regulated through feedback inhibition by one or more metabolites. We also determined a tight association between activities of nr-G3PDH and mannose-6-P reductase and mRNA expression levels in response to both leaf development and salt treatment. Collectively, our data clearly show metabolic, developmental, and environmental regulation of nr-G3PDH, and also suggest that the supply of NADPH necessary for mannitol biosynthesis is under tight metabolic control.
The NADP-dependent, non-reversible glyceraldehyde-3-P dehydrogenase (nr-G3PDH, EC 1.2.1.9) catalyzes the oxidation of d-glyceraldehyde-3-P (D-G3P) to 3-phosphoglycerate (3-PGA) with generation of NADPH.
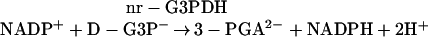 |
The enzyme has been localized in the cytosol of leaf mesophyll cells based on cell fractionation studies of various plants including celery (Apium graveolens L. cv Giant Pascal) (Kelly and Gibbs, 1973; Rumpho et al., 1983). It has been proposed that in green leaf tissues nr-G3PDH is a component of a photosynthetic shuttle transferring reducing equivalents from the chloroplasts to the cytosol, and that the reductant generated in the cytosol may be used to meet several biosynthetic requirements (Kelly and Gibbs, 1973; Scagliarini et al., 1990; Trost and Pupillo, 1993), including mannitol biosynthesis (Rumpho et al., 1983). In this role, the nr-G3PDH shuttle should have an advantage as it transfers reducing equivalents with no net gain or loss of carbon or phosphate between the plastidic and cytosolic compartments. In a survey of microalgae and higher plants, Mateos and Serrano (1992) found that the occurrence of nr-G3PDH seems to be a specific feature of those organisms with chloroplasts or cyanelles, which is consistent with the proposed function of the enzyme in photosynthesis.
There have been several reports on the (partial) purification and characterization of nr-G3PDH from different plants (Kelly and Gibbs, 1973; Iglesias and Losada, 1988; Scagliarini et al., 1990; Trost and Pupillo, 1993; Habenicht et al., 1994). These studies have shown that nr-G3PDH Km values for both NADP+ and D-G3P are low and suggest that activity in vivo may be highly regulated. Inhibition of the enzyme activity by its products and several other compounds was found under in vitro conditions whereas the use of NADPH was suggested to be important to relieve inhibition of the enzyme's activity in vivo (Scagliarini et al., 1990; Trost and Pupillo, 1993). Scagliarini et al. (1990) also speculated that nr-G3PDH activity may only be important under periods of high photo-assimilation. Recently, the study of nr-G3PDH has attracted further attention and genes encoding nr-G3PDH have been cloned and sequenced from pea (Pisum sativum), maize (Zea mays; Habenicht et al., 1994), tobacco (Nicotiana plumbaginifolia; GenBank no. U87848), and Arabidopsis (GenBank no. AC005967). Sequence comparisons indicate that nr-G3PDH is a member of the aldehyde dehydrogenase superfamily with no relationship to the phosphorylating G3PDH found in the chloroplast and cytosol (Habenicht et al., 1994; Michels et al., 1994).
In spinach (a non-sugar alcohol synthesizing species), it was proposed that the main function of nr-G3PDH was to supply NADPH for photorespiration (Scagliarini et al., 1990). These authors have also suggested (a) that nr-G3PDH may compete with other cytosolic enzymes including Fru bisphosphate aldolase and NAD-triose-P dehydrogenase and (b) that competition with aldolase may not be compatible with subsequent synthesis of translocatable carbohydrates like Suc. However, in extracts of celery leaves, where mannitol is a major translocated photosynthetic product, nr-G3PDH activities are as much as five times higher than those from leaves of plants that do not synthesize sugar alcohols (Rumpho et al., 1983). This and other studies of celery leaf tissues suggest that a major function for nr-G3PDH is to generate NADPH for the reduction of Man-6-P via an NADPH-dependent Man-6-P reductase (M6PR), which is located in the cytosol of mesophyll cells (for reviews, see Loescher and Everard, 1996, 2000). It is not known, however, how nr-G3PDH protein from celery might compare with those found in non-sugar alcohol synthesizing species.
It is clear that mannitol biosynthesis via M6PR in celery is subject to both developmental and environmental controls, e.g. increasing with photosynthetic capacity and salt stress (Loescher and Everard, 1999). Consistent with the requirements for mannitol biosynthesis, a substantial quantity of NADPH is needed in the cytosol of leaf mesophyll cells. This suggests a tight regulation of nr-G3PDH activity, perhaps in a fashion similar to M6PR regulation. However, nr-G3PDH has yet to be purified or characterized from celery (or from any other sugar-alcohol synthesizing species), and its biochemical, developmental, and environmental regulation, especially as related to mannitol synthesis, has yet to be defined. Such information may also be important to understanding the requirements for mannitol biosynthesis in transgenic plants and how such plants may resist environmental stresses (Tarczynski et al., 1993; Shen et al., 1997).
The goal of the present study was to characterize nr-G3PDH enzyme from celery leaves and to clarify the role of the enzyme in supplying the NADPH necessary for mannitol biosynthesis. We also determined how nr-G3PDH could be regulated at the biochemical and molecular levels relative to mannitol synthesis, leaf development, and salt stress.
RESULTS
nr-G3PDH Activity in Developing and Salt-Treated Leaves
Activity of nr-G3PDH increased as leaves developed, peaking in fully expanded, mature leaves and decreasing in older, senescing leaves (Fig. 1). When similar celery plants were treated with 50 or 150 mm NaCl in the irrigation nutrient solutions for 15 d, there was a significant increase in nr-G3PDH activity with salt levels in premature and mature leaves (around leaf no. 5), but not in older leaves (Fig. 1). Plant growth was strongly inhibited by 300 mm NaCl, and nr-G3PDH activity was decreased to the level of that in controls except in the youngest leaves (Fig. 1).
Figure 1.

nr-G3PDH activity in developing celery leaves and the effect of salt on the activities. Celery plants were grown with 0 (control), 50, 150, and 300 mm NaCl in the irrigation solutions for 15 d prior to sampling.
Purification and Characterization of nr-G3PDH
The nr-G3PDH was purified from mature celery leaves in several steps: (NH4)2SO4 and acetone fractionation, and DEAE-Sepharose, Sephadex-200, and Affi-Blue Sepharose chromatography, with 30% overall recovery and specific activity of 30.7 μmol min−1 mg−1 protein (Table I). The enzyme from the final purification step was apparently homogeneous electrophoretically with a single band at approximately 53 kD in SDS-PAGE (Fig. 2).
Table I.
Purification scheme for nr-G3PDH from 250 g of mature celery leaves
| Step | Activity | Protein | Specific Activity | Purification | Yield |
|---|---|---|---|---|---|
| μmol min−1 | mg | μmol min−1 mg−1 | -fold | % | |
| Homogenate | 229.8 | 5,530 | 0.042 | NAa | NA |
| (NH4)2SO4 | 200.4 | 914 | 0.22 | 5 | 87 |
| Acetone | 164.8 | 352 | 0.47 | 11 | 72 |
| DEAE-Sepharose | 144.7 | 29 | 4.93 | 119 | 63 |
| Sephadex-200 | 101.3 | 8.2 | 12.4 | 299 | 44 |
| Affi-Blue | 68.9 | 2.25 | 30.7 | 738 | 30 |
NA, Not applicable.
Figure 2.

An SDS-PAGE analysis showing the various stages of purification of nr-G3PDH from celery leaves. Lane M, Molecular mass standards (Bio-Rad Laboratories, broad range); lane 1, after acetone precipitation (20 μg); lane 2, pooled active fractions from a DEAE-Sepharose column (20 μg); lane 3, pooled active fractions from a Sephadex-200 column (20 μg); lane 4, pooled active fractions from an Affi-Blue Sepharose column (20 μg). Proteins were stained with Coomassie Blue. Molecular mass of nr-G3PDH is indicated on the right side of the gel as 53 kD.
The kinetics of nr-G3PDH were investigated with the purified enzyme. Km values for the celery leaf nr-G3PDH were low for both NADP+ and D-G3P, with calculated Kms of 6.8 and 29 μm, respectively. High D-G3P concentrations, above 250 μm, however, inhibited activity (data not shown). Varying NADP concentrations at fixed D-G3P concentrations (i.e. 12.5, 25, 50, or 100 μm) resulted in linear double reciprocal plots except at 12.5 μm D-G3P where the plot was hyperbolic (data not shown). No reversible activity was detected with NADPH or NADH and 3-PGA as substrates. NAD did not substitute for NADP (data not shown).
Product inhibition patterns indicated mixed inhibition by NADPH, i.e. competitive at high concentrations (> 10 μm) and uncompetitive at low concentrations (<10 μm), with a calculated Ki for NADPH of 7.3 μm. Inhibition by 3-PGA was competitive with a Ki for 3-PGA of 0.75 mm (Fig. 3, A and B). The inhibition by 3-PGA with varying NADP was non-competitive (Fig. 3C). In our standard assay for nr-G3PDH, addition of 0.1 mm NADPH or 10 mm 3-PGA to the reaction buffer caused a 50% decrease in activity. By lowering NADPH to 0.01 mm or 3-PGA to 1 mm, the inhibition was about 20%. Relative to other compounds that may modulate nr-G3PDH activity, we determined the effects of several sugars and sugar phosphates, as well as Pi, PPi, ATP, ADT, and phosphoenolpyruvate (PEP) (Table II). ATP was a competitive inhibitor with respect to both NADP and D-G3P (Fig. 4, A and B). ADP and PEP were also inhibitory, but less so than ATP (Table II).
Figure 3.

Inhibition of nr-G3PDH activity by NADPH and 3-PGA. Assays were performed by varying NADP+ concentrations at a fixed D-G3P (100 μm) with addition of 0, 10, 20, or 40 μm NADPH (A), by varying D-G3P concentrations at a fixed NADP+ (100 μm) with addition of 0, 1.25, 2.5, or 5 mm 3-PGA (B), and by varying NADP+ concentrations at a fixed D-G3P (100 μm) with addition of 0, 1.25, or 2.5 mm 3-PGA (C).
Table II.
Effect of metabolites on nr-G3PDH activity
| Metabolite | Concentration | Activity |
|---|---|---|
| mm | % | |
| ATP | 5 | 38 |
| ATP | 2.5 | 56 |
| ATP | 1 | 68 |
| ATP | 0.5 | 80 |
| ADP | 5 | 71 |
| ADP | 2.5 | 79 |
| ADP | 0.5 | 100 |
| PEP | 5 | 64 |
| PEP | 0.5 | 94 |
| Fru-1,6-bisP | 2.5 | 87 |
Enzyme activity was measured in the presence of metabolite at the concentration indicated. The results are means of duplicate assays and activity is expressed as percentage of control. Other metabolites tested, but without significant effects on activity, were Man-6-P (5 mm), Fru-6-P (5 mm), Glc-6-P (5 mm), UDP-Glc (5 mm), Glc-1-P (5 mm), PPi (1 mm), Pi (2.5 mm), Fru-2,6-bisP (2.5 mm), and 50 mm mannitol, Suc, Fru, or Glc.
Figure 4.

Competitive inhibition by ATP against NADP+ (A) and D-G3P (B). Assays were performed by varying NADP+ or D-G3P with a fixed concentration (100 μm) of the other substrate. ATP concentrations (mm) in the assays are indicated. The apparent competitive inhibition shifted toward a mixed-type inhibition at ATP concentrations >2.5 mm, but these are probably not physiologically important and thus are not shown.
Estimation of Subcellular Metabolite Content
Using nonaqueous density gradient centrifugation, chloroplasts, cytosol, and vacuoles were partially separated into six fractions. As determined by the marker enzymes, chloroplasts were mainly in the top fractions, cytosolic components predominantly in the middle fractions, with vacuolar constituents in the bottom fractions (Fig. 5). The metabolites were then measured, and their concentrations calculated in the different compartments as shown in Table III.
Figure 5.

Distribution of marker enzymes from fractions derived from a nonaqueous density gradient of freeze-dried, sonicated celery leaves. Typically, distribution of α-mannosidase (EC 3.2.1.24) is in the vacuole, NADP-dependent reversible glyceraldehyde-3-P dehydrogenase (GAP-DH, EC 1.2.1.13) is in the chloroplast, and PEP carboxylase (PEPcase, EC 4.1.1.31) is in the cytosol. Recoveries of the marker enzymes were in the range of 93% to 106% based on total activities in the unfractionated materials.
Table III.
Estimated subcellular metabolite concentrations in mature celery leaves
| Metabolite | Chloroplast | Cytosol |
|---|---|---|
| mm | ||
| 3-PGA | 4.80 | 3.40 |
| ATP | 0.2 | 0.4 |
| Glc-6-P | 0.9 | 16.5 |
| Fru-6-P | 3 | 10.1 |
| Glc-1-P | 0.4 | 2.3 |
| NADP | 0.13 | 0.25 |
| NADPH | 0.09 | 0.24 |
Vacuole data are not shown since none of the metabolites were found in the vacuole at more than 2% of the totals. Although repeated several times, these estimates are the results from a single separation. The total metabolic pools were (in μmol g−1 dry wt) 3-PGA (4.2), ATP (0.15), Glc-6-P (3.2), Fru-6-P (2.6), Glc-1-P (0.6), and NADP(H) (0.29).
Molecular Cloning of nr-G3PDH cDNA
PCR amplification of the first strand cDNA from celery leaves with two degenerate primers resulted in a major band of 900-bp DNA (data not shown). With a BLASTN search of the GenBank Database, the 900-bp clone showed high homology (more than 80%) to maize, pea, and tobacco nr-G3PDH cDNAs (data not shown). To obtain the complete sequence of celery nr-G3PDH cDNA, nested-, gene-specific primers were designed from the sequence of the 900-bp fragment. 5′-RACE and 3′-RACE were performed with gene-specific and anchor primers as described in the kit. The 5′- and 3′-end sequences were then assembled with the 900-bp fragment, resulting in a full length cDNA of 1,734 bp. The full cDNA clone included a 1,491-bp open reading frame (stop codon included), encoding a 496-amino acid polypeptide with a predicted Mr of 53,172 (Fig. 6). This corresponded well with the size of nr-G3PDH polypeptide estimated by SDS-PAGE (Fig. 2). The nucleotide sequence for the nr-G3PDH gene from celery leaves has been deposited in the GenBank Database (accession no. AF196292).
Figure 6.

Sequence alignment of celery nr-G3PDH with other sequences. The deduced amino acid sequence of celery nr-G3PDH (a) aligned with the nr-G3PDH gene from tobacco (b; accession no. U87848), the nr-G3PDH gene from pea (c; Habenicht et al., 1994), a putative nr-G3PDH peptide from Arabidopsis (d; accession no. AC005967), and the nr-G3PDH gene from maize (e; Habenicht et al., 1994). The amino acids used for the degenerate primers to obtain the 900-bp fragment are underlined. The three most conserved regions present in all non-phosphorylating ALDHs are marked with asterisks. Cys-298, indicated by the arrow, is postulated to be involved in the formation of the thioacylenzyme intermediate for all ALDH enzymes (Lindahl, 1992; Habenicht et al., 1994).
Northern-Blot Analysis
To check whether the expression of nr-G3PDH gene in celery leaves is influenced during leaf development and salt treatment, total RNA was isolated and subjected to northern analysis. This hybridization analysis revealed that nr-G3PDH mRNA was low in young leaves, increased following leaf expansion to mature leaves, and was hardly detectable in older, yellowing, beginning-to-senesce leaves (Fig. 7 A). Salt treatments substantially increased nr-G3PDH gene expression. Compared with the controls, nr-G3PDH mRNA in mature leaves was much higher (at least 4-fold in 100 and 150 mm, and even higher in 300 mm NaCl treatments), and it was detectably higher in 50 mm NaCl treatments (Fig. 7 B).
Figure 7.

A, Northern analysis of nr-G3PDH mRNA from total RNA (10 μg per lane) extracted from young leaves (Y), immature leaves (IM), mature leaves (M), and senescent yellowish leaves (S) of celery plants. B, Northern analysis of nr-G3PDH mRNA from total RNA (10 μg per lane) extracted from mature leaves of celery plants treated with 0, 50, 100, 150, and 300 mm NaCl. The blots were probed with a 900-bp PCR fragment of celery nr-P3PDH as described in “Materials and Methods.” The loading of each lane was verified by reprobing the blot with an 18S rRNA probe from Arabidopsis.
DISCUSSION
In this paper we describe enzyme purification and characterization and cDNA cloning of the nr-G3PDH from celery leaves. For characterization, nr-G3PDH was purified to apparent electrophoretic homogeneity (Fig. 2). The apparent role for nr-G3PDH is to supply NADPH in the cytosol from triose-P that is produced and exported from the chloroplasts during photosynthesis. In mature celery leaves, which may partition as much as 50% of the newly assimilated carbon into mannitol in the cytosol, a large quantity of cytosolic NADPH is clearly necessary as a substrate for the reduction of Man-6-P to mannitol-1-P. Subsequently, the mannitol-1-P can be dephosphorylated to form mannitol (Rumpho et al., 1983; Loescher et al., 1992; Loescher and Everard, 1996).
Determining a role or roles for nr-G3PDH in green leaves has been complicated by the presence of other cytosolic enzymes involved in the production of NADPH. Glc-6-P dehydrogenase uses Glc-6-P in the oxidative pentose-P pathway to generate NADPH, but nr-G3PDH produces NADPH without requiring any cytosolic ATP, ADP, inorganic phosphate (Pi) or sugars, so there is an obvious advantage compared with cytosolic Glc-6-P dehydrogenase. The data here also show several properties of nr-G3PDH that strongly support this enzyme's proposed role as the main source of NADPH for mannitol synthesis. There is a tight association between nr-G3PDH activity (Fig. 1) and mRNA (Fig. 7, A and B), and that of M6PR as related to mannitol synthesis, which is both developmentally and environmentally controlled (Everard and Loescher, 2000). The activity pattern of nr-G3PDH in developing leaves and the increased activities under saline conditions (Fig. 1) were similar to those found for the key enzyme M6PR in mannitol biosynthesis in celery (Everard et al., 1994). The close association between supply (nr-G3PDH) and use (M6PR) of NADPH in the mannitol biosynthetic pathway suggests a tight control of carbon flux to mannitol. This association was also apparent in transcriptional regulation as both M6PR and nr-G3PDH mRNA show a similar pattern of responses to leaf development and salt treatments (Fig. 7, A and B; Everard et al., 1997; J.D. Everard and W.H. Loescher, unpublished data).
The nr-G3PDH activity in vivo also appears to be tightly regulated, primarily by feedback inhibition from products NADPH and 3-PGA. The low Ki of NADPH (7.3 μm, Fig. 3A) as compared with a relative high Ki of 3-PGA (0.75 mm, Fig. 3B) suggests that turnover (use) of NADPH may be the control step to relieve such inhibition. However, our nonaqueous gradient fractionation data (Table III) indicate that cytosolic NADPH and 3-PGA were above the concentrations necessary to inhibit nr-G3PDH activity in vitro. Collectively, these results indicate that nr-G3PDH activity may only proceed by relieving product inhibition, e.g. by the use of NADPH in mannitol and other biosynthetic processes. It is interesting to note that in celery the Ki for 3-PGA (0.75 mm) is much lower than that reported in spinach (10 mm) by Scagliarini et al. (1990). This may facilitate maintaining appropriate triose-P concentrations in both the cytosol and chloroplast of sugar alcohol synthesizers. It is also possible, given our results, that physiological concentrations of 3-PGA in celery leaves (i.e. 3.4 mm, Table III) may inhibit nr-G3PDH activity. The level of 3-PGA in the cytoplasm is in turn mainly regulated by the Pi translocator, which catalyzes the obligatory counter exchange of triose-P for Pi, 3-PGA, or D-G3P (Flugge and Heldt, 1991). Further, in plant leaves, the ability of the triose/Pi translocator to transport 3-PGA is mainly dependent on the cytosolic pH (Stitt, 1990). Lowering the cytosolic pH increases the concentration of 3-PGA in its transportable form, 3-PGA2-, which may reduce the cytosolic level of 3-PGA. However, we do not know how much cytosolic pH may change during celery leaf photosynthesis.
d-Erythrose-4-P, an analog of D-G3P, and the l-isomer of G3P are powerful inhibitors of nr-G3PDH from pea and spinach leaves (Kelly and Gibbs, 1973). However, the physiological significance of these two compounds and their effects on nr-G3PDH activity may be negligible during leaf photosynthesis (Scagliarini et al., 1990). Of the other metabolites we tested (Table II), ATP, ADP, and PEP are all potentially nr-G3PDH inhibitors. The free ATP pool in the cytoplasm was estimated here to be as high as 0.4 mm (Table III), which is close to the Ki of ATP (Fig. 4). The competitive inhibition by ATP for both NADP+ and D-G3P (Fig. 4) was similar to the results reported for spinach nr-G3PDH (Trost and Pupillo, 1993), whereas the Ki of ATP for celery nr-G3PDH was much lower. As shown in Table II, physiologically relevant concentrations of 0.5 and 1 mm ATP with NADP+ (100 μm) and D-G3P (100 μm) caused 20% and 30% inhibition of nr-G3PDH activity, respectively. Possible inhibitory mechanisms of these adenylate compounds on nr-G3PDH have been suggested and discussed (Trost and Pupillo, 1993). However, in situ ATP levels are quite difficult to estimate, and thus it is not clear from either their report or from our data whether ATP plays a role in regulating nr-G3PDH activity in vivo.
Nonetheless, despite these analytical uncertainties, specific metabolites and the turnover rate of NADPH would appear to control the partitioning of triose-P through nr-G3PDH and Suc/mannitol synthesis pathway in celery leaves. Under conditions favoring mannitol synthesis, e.g. salinity stress (Everard et al., 1994; Loescher and Everard, 1996), the increased activity of M6PR is certain to accelerate turnover of NADPH in the cytosol as Man-6-P is reduced to mannitol-1-P (Loescher, 1987). These conditions would relieve inhibition of nr-G3PDH activity and thus be tightly linked to mannitol synthesis. This also provides further support for the hypothesis that nr-G3PDH is an important source of cytosolic NADPH for mannitol-synthesis in celery leaves. In addition, previous studies in our laboratory (Everard et al., 1994) and by Stoop and Pharr (1994) have both found that extractable activity of Suc-P synthase was unaffected by salt even though the labeling data showed that partitioning to Suc was decreased. A tight association between nr-G3PDH and M6PR may also explain why salinity stress reduces Suc synthesis in celery leaves, as both nr-G3PDH and M6PR are competing for the substrates for Suc-P synthase at the level of aldolase and Fru-6-P, respectively. Thus, any metabolic processes that relieve inhibition of nr-G3PDH activity may also reduce carbon partitioning in the direction of Suc in celery plants.
From BLASTN and BLASTP database searches, the high degree of nr-G3PDH sequence homology between the cDNA from celery leaves and those reported from other plants, indicates that the enzyme is highly conserved (Fig. 6). The celery nr-G3PDH sequence closely resembles nr-G3PDH from pea (accession no. X75327; 90% identity), tobacco (accession no. U87848; 90% identity), maize (Zea mays; accession no. X75326; 86% identity), and Arabidopsis (accession no. 4115387; 87% identity). Although the nr-G3PDH enzymes from these different sources have quite similar biochemical characteristics, e.g. low Km values for substrates and inhibition by products (Kelly and Gibbs, 1973; Scagliarini et al., 1990), there are differences in Km and Ki. Other database comparisons of the deduced amino acid sequence identified nr-G3PDH to be, as expected, a member of the aldehyde dehydrogenase superfamily (ALDHs). Enzymes of this superfamily catalyze the irreversible oxidation of a wide variety of aldehydes to their corresponding acids via the formation of a thioacyl intermediate (Lindahl, 1992). The celery nr-G3PDH protein sequence shares three highly conserved regions with all known non-phosphorylating ALDHs (Fig. 6). More detailed comparisons of other nr-G3PDH and ALDHs are available (Habenicht et al., 1994; Michels et al., 1994). Further studies on the molecular aspects of celery nr-G3PDH are otherwise currently under way in our laboratory where we are focusing on developing more information on binding sites and on molecular, developmental, and environmental regulation.
MATERIALS AND METHODS
Plant Material
Celery (Apium graveolens L. cv Giant Pascal) was grown under greenhouse conditions in East Lansing, Michigan, during September to December of 1998, March to June of 1999, and August to October of 1999, essentially as described by Davis et al. (1988). The average daily temperature was maintained around 20°C to 25°C. Metal halide lamps were used as supplemental lighting to provide a minimum photosynthetic photon flux density of 750 μmol m−2 s−1 for 14 h during the winter months. Plants were watered and fertilized as previously described (Everard et al., 1994). Salt treatments were stepped up in 25 mm NaCl d−1 increments and were maintained for 15 d once the final concentrations of 0, 50, 150, and 300 mm were achieved. The plants in this study were approximately 4 months old with typically 10 to 14 leaves. Senescent leaves from seedling stages were routinely removed. Mature, just fully expanded leaves or leaves at various developmental stages were harvested at noon and frozen in liquid N2 prior to storage at −80°C. Leaves were sequentially numbered by position relative to the center of the plant with number 1 being the youngest visible light green leaf, and numbers 12 to 14 the oldest (typically thick and leathery). The shoot meristem was not numbered and not used in this study. Chemicals and enzymes were purchased from Sigma (St. Louis) or Roche (Indianapolis), or as otherwise specified.
Assay for nr-G3PDH
nr-G3PDH was assayed as described by Kelly and Gibbs (1973) with some modifications. The standard reaction mixture contained 50 mm Tris (tris[hydroxymethyl]aminomethane) buffer, pH 7.7, 3 mm reduced glutathione, 5 units of triose-P-isomerase, 2 mm dihydroxyacetone-P, and 0.1 mm NADP+. Assays were monitored with an spectrophotometer (model U-3100, Hitachi, Tokyo) at 340 nm and 30°C. The dihydroxyacetone-P was obtained from dihydroxyacetone-P dimethyl ketal according to instructions by Sigma, and was prepared as a 20 mm stock solution. D-G3P, the substrate for nr-G3PDH, was generated from dihydroxyacetone-P by triose-P-isomerase (2 mm dihydroxyacetone-P generated 0.1 mm D-G3P under the above conditions; Kelly and Gibbs, 1973).
Activities of nr-G3PDH in Developing and Salt-Affected Leaves
The activities of nr-G3PDH in developing leaves were estimated in clarified homogenates with the standard assay as described above. Leaves of various ages, either from control plants or from plants treated with 50, 150, or 300 mm NaCl, were homogenized in a chilled mortar with 4 volumes of chilled extraction buffer. The buffer contained 50 mm Tris, pH 7.8, 5 mm dithiothreitol (DTT), 1 mm EDTA, 2 mm MgCl2, 0.1 mm phenylmethylsulfonyl fluoride, and 2% (w/v) soluble polyvinylpyrrolidone. After centrifugation at 18,000g for 20 min, the clear supernatant was desalted by passing through a 5-mL Sephadex G-25 column and used as the crude enzyme extract. Desalting the extracts resulted in no detectable loss of nr-G3PDH activity in the crude extract, and this step was beneficial in reducing the background in standard nr-G3PDH assays (data not shown).
Purification of nr-G3PDH
nr-G3PDH was purified using procedures modified from those previously described by Scagliarini et al. (1990) and Michels et al. (1994). Mature celery leaves (250 g) were homogenized on ice with a Polytron PT-3000 (Kinematica AG, Littau, Switzerland) in 2 volumes of chilled buffer (buffer A) containing 50 mm HEPES (4-[2-hydroxyethyl]-1-piperazineethanesulfonic acid), pH 7.8, 5 mm DTT, 1 mm EDTA, 2 mm MgCl2, 0.1 mm phenylmethylsulfonyl fluoride, and 2% (w/v) soluble polyvinylpyrrolidone. The homogenate was filtered through a layer of Polycloth (150 mesh, Setar America Inc., Depew, NY) and centrifuged at 25,000g for 20 min. The supernatant was fractionated with ammonium sulfate and precipitated proteins were collected from the 30% to 60% saturation fraction. After dissolving the precipitate in 25 mL of buffer (25 mm HEPES, pH 7.8, and 0.5 mm DTT [buffer B]), an equal volume of acetone at −18°C was added in drops. The precipitated proteins from the acetone fraction were collected after centrifuging at 18,000g for 10 min, resuspended in 50 mL of buffer B, and again centrifuged at 18,000g for 20 min. The supernatant was dialyzed in buffer B overnight and then applied to an ion-exchange column (1.2 × 20 cm, DEAE-Sepharose CL-6B, Pharmacia Biotech, Piscataway, NJ) previously equilibrated with buffer B. Unbound proteins were eluted with buffer B and bound proteins eluted at a flow rate of 1 mL min−1 with a linear gradient of 0 to 0.4 m NaCl in buffer B. Fractions (3 mL each) were collected and assayed for nr-G3PDH activity. Active fractions were pooled and concentrated by dialysis against solid Suc. The concentrated fractions were chromatographed on a previously equilibrated-gel filtration column (2.5 × 90 cm, Sephadex-200, Pharmacia Biotech) using buffer B, containing 150 mm NaCl, and at a flow rate of 0.5 mL min−1. Active fractions (3 mL fraction−1) were pooled and dialyzed in buffer B for 8 h prior to loading onto an Affi-Gel Blue column (1.2 × 10 cm, equilibrated with buffer B, Bio-Rad Laboratories, Hercules, CA). Proteins were first eluted with buffer B (plus 2 mm NADP+) and then with a 60 mL of linear gradient of 0 to 0.4 m NaCl in buffer B. Active fractions from the salt gradient were pooled and concentrated by reverse dialysis as described above. The purified nr-G3PDH was used directly for assaying enzyme properties or stored at −20°C in the presence of 0.1 mm NADP+ and 10% (v/v) glycerol.
Enzyme Properties
Kinetic assays were performed with 0.2 μg of the protein in 0.5-mL reaction buffer containing 50 mm Tris, pH 7.7, 3 mm reduced glutathione, and 5 units of triose-P-isomerase. For example, for kinetic assays of NADP and D-G3P, respectively, NADP+ was varied from 2.5 to 100 μm with 100 μm D-G3P, or D-G3P was varied from 2.5 to 150 μm with 100 μm NADP+. Effects of metabolites on nr-G3PDH activity were measured in the presence of Man-6-P, Glc-6-P, Fru-6-P, Glc-1-P, UDP-Glc, ATP, ADP, 3-PGA, PEP, PPi, Pi, Fru-1, 6-bisP, Fru-2,6-bisP, NADPH, NAD(H), or 50 mm mannitol, Suc, Glc, or Fru. Activities were also measured with NADPH, NAD, or NADH as cofactor. Km values for NADP+ and D-G3P were determined by regression analysis of Woolf-Augustinsson-Hofstee plots. The type of inhibitions by NADPH, 3-PGA, or ATP were similarly determined by regression analysis of both Lineweaver-Burk and Woolf-Augustinsson-Hofstee plots. The Ki values for NADPH, 3-PGA, and ATP were calculated using the equations for different inhibition as described by Segel (1976).
SDS-PAGE
SDS-PAGE was carried out with a Hoefer Mighty Small II (Pharamacia Biotech) using a 1-mm-thick slab gel containing 12% (v/v) acrylamide according to the procedure of Laemmli (1970). Gels were stained with Coomassie Brilliant Blue R-250 and destained in a methanol-acetic acid-water solution. Molecular-mass standards were obtained from Bio-Rad Laboratories.
Nonaqueous Gradient Fractionation
Nonaqueous fractionation of celery leaf materials was carried out using methods similar to those of Stitt et al. (1989) and Sharkey and Vanderveer (1989). Frozen leaf samples (after removing large ribs) were ground in liquid N2 in a mortar, and the leaf powder was lyophilized in a Labconco freeze dryer at −50°C for 100 h. Approximately 300 mg of dry powder was transferred to 20 mL of heptane on dry ice and ultrasonicated with a sonicator (model XL 2020, Misonix, Farmingdale, NY) for a total 2 min. The suspension was concentrated by centrifugation for 2 min at 3,000g. The clear supernatant was discarded and the sediment was resuspended in 1.5 mL of tetrachloroethylene:heptane (2:1, v/v). Two 100-μL samples were withdrawn for determination of enzyme activities and metabolites in the unfractionated materials, and the remainder was layered onto a density gradient, prepared in 12-mL polyallomer tubes (103 × 16 mm) by sequentially layering 1.5-mL fractions of tetrachloroethylene-heptane mixtures of the following densities: 1.62, 1.58, 1.53, 1.48, 1.44, 1.39, and 1.34 g mL−1. Fractionation was accomplished by centrifugation at 25,000g for 4 h at −10°C in a swing-out rotor. The top 3 mL of clear yellow-colored fraction, which contained no enzyme activity or metabolites, was discarded, and the remaining volume was divided into six 1.5-mL fractions for marker enzyme and metabolite assays as described by Stitt et al. (1989).
We measured NADP-dependent reversible glyceraldehyde-3-P dehydrogenase (EC 1.2.1.13), PEP carboxylase (EC 4.1.1.31), and α-mannosidase (EC 3.2.1.24) activities as markers for chloroplasts, cytosol, and vacuoles, respectively (Stitt et al., 1989). The metabolites, 3-PGA, ATP, Glc-6-P, Fru-6-P, and Glc-1-P were assayed spectrophotometrically with appropriately coupled enzyme reactions (Stitt et al., 1989). NADP and NADPH were assayed by enzymatic cycling (Passonneau and Lowry, 1974) with an Aminco-Bowman spectrophotofluorometer (American Instrument Co., Rochester, NY). Appropriate recovery experiments were included in all measurements by analyzing marker enzyme activities and the total pool of metabolites in the unfractionated materials. To evaluate subcellular compartmentation of the metabolites, a two-compartment analysis was introduced as described by Stitt et al. (1989). The marker enzyme activities and metabolite contents were expressed in terms of the protein content of the fraction. The measured metabolite content in a given fraction was assumed to derive partly from the chloroplasts and partly from the cytosol, since the tonoplast is generally regarded as impermeable to these metabolites. In addition, the relative volumes of different cellular spaces were estimated from transmission electron microscopy micrographs (Everard et al., 1993). The chloroplasts were approximately 12%, cytosol 8%, and vacuoles 77% of a cell's volume. The reliability of the nonaqueous gradient fractionation techniques and the data calculations have been fully discussed elsewhere (Stitt et al., 1989).
PCR Cloning of nr-G3PDH cDNA
Total RNA was extracted from approximately 200 mg of mature celery leaves using TRIzol Reagent (Gibco-BRL, Cleveland) according to the manufacturer's instructions. First strand cDNA synthesis was accomplished using SUPERSCRIPT II RNase H-reverse transcriptase (Gibco-BRL). Two degenerate oligonucleotide primers were designed according to highly conserved regions in the nr-G3PDH sequences from pea (Pisum sativum; Habenicht et al., 1994) as follows: primer WHL11, “antisense”-primer based on region 462/467-KDSGIG in the sequence, 5′-CC(TGA) AT(CTAG) CC(GA) CT(GA) TC(CT) TT-3′; primer WHL13, “sense”-primer based on region 202/207-LHMVHC in the sequence, 5′-CT(TCAG) CA(TC) ATGGT(TCAG) CA(TC) TG-3′. PCR was performed using 2 μL of the 20-μL reverse transcriptase reaction, 20 pmol of each primer, and 2.5 units of Taq DNA polymerase (Gibco-BRL), according to the manufacturer's instructions. The PCR products, after purification with GENECLEAN I kit (BIO 101, La Jolla, CA), were cloned by the pGEM-T Easy Vector System (Promega, Madison, WI), and introduced into JM109 competent cells (Promega). 5′- and 3′-RACE were performed using the 5′/3′ RACE kit (Boehringer Mannheim) according to the manufacturer's instructions. All clones were sequenced on both strands with at least two replications. Sequence alignment and comparison with nr-G3PDH sequences from other organisms were performed by using DNASTAR software (1999 version) and a BLAST search of GenBank and EMBL DNA sequence databases.
Northern Analysis
Ten micrograms of total RNA, as quantified spectrophotometrically, was separated on 1.3% (w/v) agarose gels under denaturing (formaldehyde) conditions. RNA was capillary transferred (Sambrook et al., 1989) to nylon membranes (Hybond N+, Amersham) overnight in 10× SSC and fixed to the membrane at 80°C for 1.5 h. Random priming was used to 32P-label a 900-bp PCR fragment of the nr-G3PDH clone with NEBlot kit (New England Biolabs, Beverly, MA) according to the manufacturer's instructions. Membranes were prehybridized for 2 h at 65°C in phosphate hybridization buffer containing 1 mm EDTA, 250 mm Na2HPO4, pH 7.4, 1% (w/v) casein, and 7% (w/v) SDS. Hybridization was performed overnight in the same buffer under the same conditions as for prehybridization with addition of the 32P-labeled probes. Following hybridization, membranes were rinsed briefly (two times for 1 min) in low-salt washing buffer containing 40 mm NaHPO4, pH 6.8, 1% (w/v) SDS, and 1 mm EDTA. Three further 20-min washes in the low-salt washing buffer were performed at 65°C prior to image capture on a phosphor imaging screen (Molecular Dynamics, Sunnyvale, CA). The same blots were washed at 90°C for 30 min in a solution containing 2% (w/v) SDS, 0.5 m Tris, pH 7, and 0.1 mm EDTA to strip off the nr-G3PDH probe, and rehybridized with a 32P-labeled probe of 18S rRNA from Arabidopsis to standardize for RNA loading.
Protein Estimation
Protein content was determined by the method of Bradford (1976) using bovine serum albumin as a standard.
ACKNOWLEDGMENTS
We wish to acknowledge the staff of the Sequencing and Macromolecular Structure Facilities at Michigan State University, Dr. Abed Janoudi for suggestions, Dr. John D. Everard for providing many valuable suggestions and some RNA extracts, and Dr. Steve van Nocker for aid and assistance during northern blotting.
Footnotes
This work was supported by the U.S. Department of Agriculture National Research Initiative Competitive Grants Program (grant no. 93–37100–8907 to W.H.L.).
LITERATURE CITED
- Bradford MM. A rapid and sensitive method for the quantitation of microgram quantities of protein utilizing the principle of protein-dye binding. Anal Biochem. 1976;72:248–254. doi: 10.1016/0003-2697(76)90527-3. [DOI] [PubMed] [Google Scholar]
- Davis JM, Fellman JK, Loescher WH. Biosynthesis of sucrose and mannitol as a function of leaf age in celery (Apium graveolens L.) Plant Physiol. 1988;86:129–133. doi: 10.1104/pp.86.1.129. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Everard JD, Cantini C, Grumet R, Plummer J, Loescher WH. Molecular cloning of mannose-6-phosphate reductase and its developmental expression in celery. Plant Physiol. 1997;113:1427–1435. doi: 10.1104/pp.113.4.1427. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Everard JD, Franceschi VR, Loescher WH. Mannose-6-phosphate reductase, a key enzyme in photoassimilate partitioning, is abundant and located in the cytosol of photosynthetically active cells of celery (Apium graveolens L.) source leaves. Plant Physiol. 1993;102:345–356. doi: 10.1104/pp.102.2.345. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Everard JD, Gucci R, Kann SC, Flore JA, Loescher WH. Gas exchange and carbon partitioning in the leaves of celery (Apium graveolens L.) at various leaves of root zone salinity. Plant Physiol. 1994;106:281–292. doi: 10.1104/pp.106.1.281. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Flugge UI, Heldt HW. Metabolite translocators of the chloroplast envelope. Annu Rev Plant Physiol Plant Mol Biol. 1991;42:129–144. [Google Scholar]
- Habenicht A, Hellman U, Cerff R. Non-phosphorylating GAPDH of higher plants is a member of the aldehyde dehydrogenase superfamily with no sequence homology to phosphorylating GAPDH. J Mol Biol. 1994;237:165–171. doi: 10.1006/jmbi.1994.1217. [DOI] [PubMed] [Google Scholar]
- Iglesias AA, Losada M. Purification and kinetic and structural properties of spinach leaf NADP-dependent nonphorylating glyceraldehyde-3-phosphate dehydrogenase. Arch Biochem Biophys. 1988;260:830–840. doi: 10.1016/0003-9861(88)90514-0. [DOI] [PubMed] [Google Scholar]
- Kelly GJ, Gibbs M. Non-reversible d-glyceraldehyde 3-phosphate dehydrogenase of plant tissues. Plant Physiol. 1973;52:111–118. doi: 10.1104/pp.52.2.111. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Laemmli UK. Cleavage of structural protein during the assembly of the head of bacteriophage T4. Nature. 1970;227:680–685. doi: 10.1038/227680a0. [DOI] [PubMed] [Google Scholar]
- Lindahl R. Aldehyde dehydrogenases and their role in carcinogenesis. Crit Rev Biochem Mol Biol. 1992;27:283–335. doi: 10.3109/10409239209082565. [DOI] [PubMed] [Google Scholar]
- Loescher WH. Physiology and metabolism of sugar alcohols in higher plants. Physiol Plant. 1987;70:553–557. [Google Scholar]
- Loescher WH, Everard JD. Sugar alcohol metabolism in sinks and sources. In: Zamski E, Schaffer AA, editors. Photoassimilate Distribution in Plant and Crops: Source-Sink Relationships. New York: Marcel Dekker; 1996. pp. 185–207. [Google Scholar]
- Loescher WH, Everard JD. Regulation of sugar alcohol biosynthesis. In: Leegood RC, Sharkey TD, von Caemmerer S, editors. Photosynthesis: Physiology and Metabolism. Dordrecht, The Netherlands: Kluwer Academic Publisher; 2000. pp. 275–299. [Google Scholar]
- Loescher WH, Tyson RH, Everard JD, Redgwell RJ, Bieleski RL. Mannitol synthesis in higher plants: evidence for the role and characterization of a NADP-dependent mannose-6-phosphate reductase. Plant Physiol. 1992;98:1396–1402. doi: 10.1104/pp.98.4.1396. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Mateos MI, Serrano A. Occurrence of phosphorylating and non-phosphorylating NADP+-dependent glyceraldehyde-3-phosphate dehydrogenases in photosynthetic organisms. Plant Sci. 1992;84:163–170. [Google Scholar]
- Michels S, Scagliarini S, Seta FD, Carles C, Riva M, Trost P, Branlant G. Arguments against a close relationship between non-phosphorylating and phosphorylating glyceraldehyde-3-phosphate dehydrogenases. FEBS Lett. 1994;339:97–100. doi: 10.1016/0014-5793(94)80393-5. [DOI] [PubMed] [Google Scholar]
- Passonneau JV, Lowry OH. Measurement by enzymatic cycling. In: Bergmeyer HU, editor. Methods of Enzymatic Analysis. Vol. 4. Deerfield Beach, FL: Verlag Chemie International; 1974. pp. 2059–2072. [Google Scholar]
- Rumpho ME, Edwards GE, Loescher WH. A pathway for photosynthetic carbon flow to mannitol in celery leaves: activity and localization of key enzymes. Plant Physiol. 1983;73:869–873. doi: 10.1104/pp.73.4.869. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sambrook J, Fritsch EF, Maniatis T. Molecular Cloning: A Laboratory Manual. Cold Spring Harbor, NY: Cold Spring Harbor Laboratory Press; 1989. [Google Scholar]
- Scagliarini S, Trost P, Valenti V, Pupillo P. Glyceraldehyde 3-phosphate: NADP+ reductase of spinach leaves. Plant Physiol. 1990;94:1337–1344. doi: 10.1104/pp.94.3.1337. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Segel IH. Biochemical Calculations. 2nd Ed. New York: John Wiley & Sons; 1976. , 246–273. [Google Scholar]
- Sharkey TD, Vanderveer PJ. Stromal phosphate concentration is low during feedback limited photosynthesis. Plant Physiol. 1989;91:679–684. doi: 10.1104/pp.91.2.679. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Shen B, Jensen RG, Bohnert HJ. Increased resistance to oxidative stress in transgenic plants by targeting mannitol biosynthesis to chloroplasts. Plant Physiol. 1997;113:1177–1183. doi: 10.1104/pp.113.4.1177. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Stitt M. The flux of carbon between chloroplast and cytoplasm. In: Dennis DT, Turpin DH, editors. Plant Physiology, Biochemistry and Molecular Biology. New York: John Wiley & Sons; 1990. pp. 309–326. [Google Scholar]
- Stitt M, Lilley RM, Gerhardt R, Heldt H. Metabolite levels in specific cells and subcellular compartments of plant leaves. Methods Enzymol. 1989;174:518–552. [Google Scholar]
- Stoop JMH, Pharr DM. Mannitol metabolism in celery stressed by excess macronutrients. Plant Physiol. 1994;106:503–511. doi: 10.1104/pp.106.2.503. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Tarczynski MC, Jensen RG, Bohnert HJ. Stress protection of transgenic tobacco by production of the osmolyte mannitol. Science. 1993;259:508–510. doi: 10.1126/science.259.5094.508. [DOI] [PubMed] [Google Scholar]
- Trost P, Pupillo P. Inhibition of spinach d-glyceraldehyde 3-phosphate: NADP+ oxidoreductase (nonphosphorylating) by adenylate compounds: the effect of dead-end inhibitors on a steady state random reaction mechanism. Arch Biochem Biophys. 1993;306:76–82. doi: 10.1006/abbi.1993.1483. [DOI] [PubMed] [Google Scholar]