

Regulated cell death plays an important role in a myriad of processes including development, host immunity and tissue homeostasis. The best understood type of regulated cell death is apoptosis. Often apoptosis is initiated by mitochondrial outer membrane permeabilisation or MOMP. Widespread MOMP effectively acts as cellular death sentence; usually it leads to caspase activation and apoptosis however even without caspases, cells die post-MOMP through a process called caspase-independent cell death or CICD.
The success of directly targeting apoptosis to treat cancer is perhaps best attested by the recent clinical approval of venetoclax, a BCL-2 targeting drug, to treat drug-refractory chronic lymphocytic leukemia (CLL). Nevertheless, engaging apoptosis as a means to treat cancer can be sub-optimal since therapies often fail to completely eradicate tumour cells. Moreover some studies suggest that apoptosis may have oncogenic effects [1]. These include, apoptosis induced compensatory proliferation, mitogen-induced cancer stem cell proliferation as well as establishment of an immunosuppressive microenvironment. Furthermore, sub-lethal engagement of caspase activity in cells can lead to genomic instability, potentially boosting tumour evolution [2]. Because caspases promote these effects, coupled to them not being required for MOMP-dependent killing, prompted us to ask whether CICD might be a better way to kill cancer cells.
To answer this question, we initially focused on understanding mechanisms underlying CICD. We found that inhibition of caspases downstream of MOMP can trigger necroptosis as a form of caspase-independent cell death (CICD), through the production and autocrine effects of pro-inflammatory TNF [3]. Recent studies underline anti-tumorigenic effects of inducing necroptosis, however RIPK3 – a protein typically essential for necroptosis execution – is silenced in the majority of cancers [4]. As such, the clinical utility of engaging necroptosis in cancer treatment remains unclear.
Nevertheless, we noticed that cells undergoing CICD, independent of necroptosis, produce several inflammatory cytokines that are important for recruitment and activation of both innate and adaptive immune responses [3]. In a tumourigenesis experiment setup to mimic partial therapeutic response, engagement of CICD but not apoptosis led to tumour infiltration of inflammatory polarised macrophages and activated T-cells (both cytotoxic and helper) [3]. Whereas engagement of apoptosis had no beneficial effect, triggering CICD in vivo was sufficient to completely eradicate tumours in 50% of the cases in a manner, dependent on intact immunity. This suggests that triggering CICD in tumours rather than apoptosis may be a better strategy in cancer treatment.
Given that mitochondrial apoptosis is typically considered immunologically silent how can CICD be pro-inflammatory? Addressing this, we found that the anti-tumourigenic and pro-inflammatory nature of MOMP is dependent on NF-κB – a master transcriptional regulator of pro-inflammatory cytokines [3]. Under caspase deficient conditions, we found that mitochondrial permeabilisation activates NF-κB in a manner dependent on IAP down-regulation and NIK activation [3]. Interestingly, previous studies have shown that caspase deficiency also allows MOMP to trigger type I IFN response via the cGAS/STING pathway, activated by mitochondrial DNA [5,6]. We found that although NF-κB and STING can be activated independently during CICD, both are required for optimal cytokine production [3]. How do caspases silence inflammation? One possibility might be cleavage dependent inactivation of molecules important for engaging NF-κB and STING signalling. Alternatively, it may simply be a difference of kinetics; apoptosis occurs very rapidly post-MOMP, within minutes, whereas CICD can take many hours to days, affording the cell time to produce cytokines before death.
In our view, engaging CICD as an anti-cancer treatment offers two potential benefits compared with apoptosis: 1) it prevents caspase dependent oncogenic effects of apoptosis 2) it can activate anti-tumour immunity (Figure 1). The second point is intriguing, since immunogenicity of death is largely considered a key feature of uncontrolled necrosis which allows the passive release of intracellular molecules named DAMPs (damage-associated molecular patterns) capable of alerting an immune response. Our work supports recent findings from others whereby dying cells initiate anti-tumour immunity in a regulated manner dependent on de novo synthesis of inflammatory signals [3,7]. Therefore, in order to find a more effective way to kill cancer cells, we might turn our focus on the signals accompany a cell during its death rather than simply the execution of cell death per se. Going forward, several key questions remain. How exactly does CICD induce an immune response? Are there additional mitochondrial pathways that signal immunity and how do caspases silence these effects? Most importantly, can we clinically translate these findings, engaging CICD to improve cancer treatment?
Figure 1.
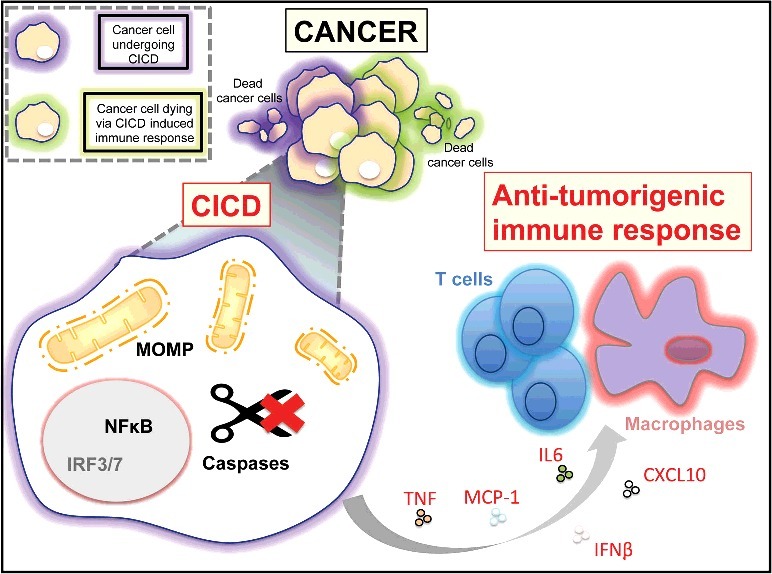
Under conditions of partial therapeutic response, tumour CICD can have two anti-cancer effects. Engaging MOMP-under caspase inhibited conditions triggers cell death and anti-tumour immunity dependent upon NFκB in the dying cell. Anti-tumour immunity can kill remaining tumour cells.
Funding Statement
Cancer Research UK [ID C40872/A20145].
Disclosure of Potential Conflicts of Interest
No potential conflicts of interest were disclosed.
References
- [1].Ichim G, Tait SW. A fate worse than death: apoptosis as an oncogenic process. Nat Rev Cancer. 2016;16(8):539–548. doi: 10.1038/nrc.2016.58. PMID:27364482 [DOI] [PubMed] [Google Scholar]
- [2].Ichim G, Lopez J, Ahmed SU, et al. Limited mitochondrial permeabilization causes DNA damage and genomic instability in the absence of cell death. Mol Cell. 2015;57(5):860–872. doi: 10.1016/j.molcel.2015.01.018. PMID:25702873 [DOI] [PMC free article] [PubMed] [Google Scholar]
- [3].Giampazolias E, Zunino B, Dhayade S, et al. Mitochondrial permeabilization engages NF-kappaB-dependent anti-tumour activity under caspase deficiency. Nat Cell Biol. 2017;19(9):1116–1129. doi: 10.1038/ncb3596. PMID:28846096 [DOI] [PMC free article] [PubMed] [Google Scholar]
- [4].Koo GB, Morgan MJ, Lee DG, et al. Methylation-dependent loss of RIP3 expression in cancer represses programmed necrosis in response to chemotherapeutics. Cell Res. 2015;25(6):707–725. doi: 10.1038/cr.2015.56. PMID:25952668 [DOI] [PMC free article] [PubMed] [Google Scholar]
- [5].White MJ, McArthur K, Metcalf D, et al. Apoptotic caspases suppress mtDNA-induced STING-mediated type I IFN production. Cell. 2014;159(7):1549–1562. doi: 10.1016/j.cell.2014.11.036. PMID:25525874 [DOI] [PMC free article] [PubMed] [Google Scholar]
- [6].Rongvaux A, Jackson R, Harman CC, et al. Apoptotic caspases prevent the induction of type I interferons by mitochondrial DNA. Cell. 2014;159(7):1563–1577. doi: 10.1016/j.cell.2014.11.037. PMID:25525875 [DOI] [PMC free article] [PubMed] [Google Scholar]
- [7].Yatim N, Jusforgues-Saklani H, Orozco S, et al. RIPK1 and NF-kappaB signaling in dying cells determines cross-priming of CD8(+) T cells. Science. 2015;350(6258):328–334. doi: 10.1126/science.aad0395. PMID:26405229 [DOI] [PMC free article] [PubMed] [Google Scholar]