

Ionizing radiation (IR) remains one of the most frequently used treatments for cancer. Therapeutically, IR produces DNA damage, especially highly toxic DNA double strand breaks (DSB). Depending upon the cellular context and severity of damage, cells can opt to repair these DSBs or undergo apoptotic death. Master regulators of the DNA damage response include three related protein kinases DNA-PK, ATM, and ATR, all members of the PIKK (phosphatidylinositol 3' kinase-related kinases) family of proteins having both discreet and redundant targets and functions. The cellular choice between DNA repair and cell death can be dictated by a number of factors, notably the presence and fidelity of the DNA damage response pathway proteins (including the PIKKs), the cell cycle phase, and the activity of pro-apoptotic regulators, notably the tumor suppressor p53. How the cell responds to damage is important in causation of cancer, dictating tumor response to genotoxic therapies, and mitigating side effects. The cell type itself is also an important determinant of the response to IR and other genotoxic agents. Whole body radiation damages all cells, but the unique cell type dependent responses argue that cellular fate is not simply dependent upon the level of damage induced, but rather whether the cell is predisposed to undergo either DNA repair or apoptosis. Understanding the interacting roles of genetic and cellular context in the response to IR is critical for determining how in vitro cellular models, tumors, tissues, or even whole organisms will respond to therapy.
We examined the response of gastrointestinal progenitor cells compared to terminally differentiated cells following whole body irradiation in mouse models with genetic deficiencies in the PIKKs and Trp53 [1]. The response, be it repair or apoptosis, of “non-reproductive” terminally differentiated cells was less influential on organismal survival compared to the response of progenitor cells (stem cells and transit amplifying cells). Progenitor cells can repopulate lost terminally differentiated cells, but if irreparably damaged themselves, may cause failure of tissue regeneration and mortality. The relative roles of DNA repair versus apoptosis in IR response was studied by (a) mapping the spatiotemporal cellular response and (b) comparing the response of single DNA-PK, ATM and Trp53 mutant mice and compound mutant genotypes. The result was surprising, as loss of pro-apoptotic p53 was expected to increase the survival of radiosensitive DNA-PK mutant mice, but instead, toxicity and mortality were enhanced in mice lacking both DNA-PK and Trp53. The role of p53 in inducing cell cycle arrest as opposed to apoptosis was more impactful on cell survival after IR. Without p53 mediated G1/S arrest, intestinal progenitor cells with damaged DNA abnormally entered S phase. The absence of functional DNA-PK prevented not only DNA repair, but also activation of both intra-S and G2/M checkpoints, leading to progression into G2/M despite replete DNA damage [1,2]. This culminated in mitotic cell death, depletion of stem cells, and enhanced animal mortality (Figure 1).
Figure 1.
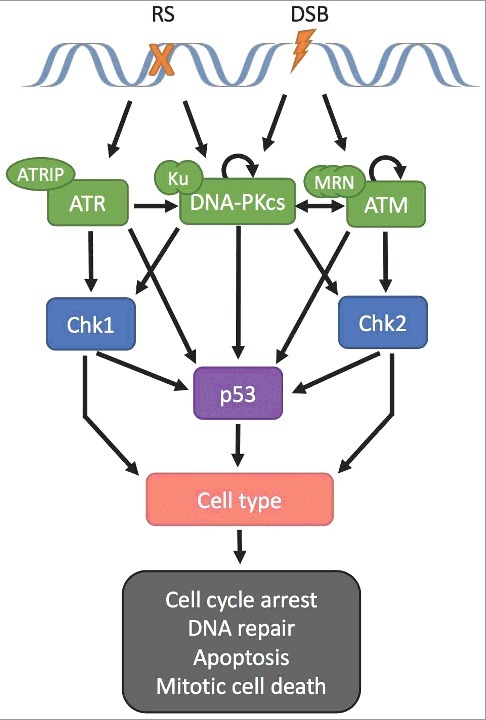
PIKK regulation of p53 and cell fate. Both genetic makeup and cell type dictate cellular fate. Replication stress (RS) or double strand breaks (DSB) activate PIKK proteins ATR, DNA-PKcs, and/or ATM, causing activation of target proteins p53, Chk1, Chk2 regulating the G1/S, intra-S, or G2/M checkpoints, respectively. Both PIKK signaling and cell type modulate the ultimate cellular outcome.
Notably, although DNA-PK and p53 are both well-studied proteins, the phenotype of the double mutants could not be predicted a priori, highlighting the importance of studying genetic interactions in dictating phenotype. Further, these responses were specific to crypt base columnar stem cells highlighting the importance of cell type specificity. Because stem cells divide infrequently, they are more dependent upon nonhomologous end-joining (NHEJ) mediated DNA DSB repair, compared to repair via homologous recombination (HR) which requires cell replication [3]. This makes this cell population more susceptible to loss of DNA-PK activity. Stem cell loss led to an inability to produce the transit amplifying cells that repopulate other terminally differentiated cells of the gastrointestinal tract, causing mortality. Other examples of genetic interactions between PIKKs and Trp53 include synthetic lethality between DNA-PK and ATM and between ATM and ATR as well as p53 independent apoptosis in mice lacking both DNA-PK and p53 [4,5]. Additional genetic interactions include enhanced lethality in mice lacking p53 with aberrant ATR and increased survival in mice with dual mutations in p53 and ATM [6].
Certain cancer types are critically reliant upon functional DNA damage response pathways to survive, the classic example being the dependence of homologous recombination-deficient (BRCA1 or BRCA2 mutants) cancers on PARP functionality for survival. Dysregulation of DSB repair pathways is now recognized as a targetable phenotype for many cancers, permitting inhibitor-mediated enhancement of cancer cell death either alone or in combination with genotoxic therapy. Inhibitors for DNA-PK, ATM, and ATR are currently in various phases of development, as are drugs targeting other DNA damage response genes including Chk1, Chk2, Wee1, Cdc7, and PARP. Further research will assist in elucidating how these “addictions” to DNA damage sensors and responders may be exploited therapeutically, but many inhibitors appear clinically promising. Due to the high frequency of p53 alterations in cancer and its central role in the DNA damage response, understanding the interplay between these targets and p53 will be critical for successful development of these targeted agents. Empirical testing of targeted agents and combinations in genetically complex patient derived tumor cells is a promising new approach to identify the optimal therapy for a given patient [7].
Funding Statement
NIH/NCI [grant numbers U01 CA176303, U01 CA217883, U54 CA132381, U54 132383]; NIH/NIGMS [IDeA grant number P20GM103451].
Disclosure of potential conflicts of interest
No potential conflicts of interest were disclosed.
References
- [1].Gurley KE, Ashley AK, Moser RD, et al. . Synergy between Prkdc and Trp53 regulates stem cell proliferation and GI-ARS after irradiation. Cell Death Differ. 2017;24(11):1853–1860. doi: 10.1038/cdd.2017.107. PMID:28686579 [DOI] [PMC free article] [PubMed] [Google Scholar]
- [2].Ashley AK, Shrivastav M, Nie J, et al. . DNA-PK phosphorylation of RPA32 Ser4/Ser8 regulates replication stress checkpoint activation, fork restart, homologous recombination and mitotic catastrophe. DNA Repair (Amst). 2014;21:131–139. doi: 10.1016/j.dnarep.2014.04.008. PMID:24819595 [DOI] [PMC free article] [PubMed] [Google Scholar]
- [3].Mandal PK, Blanpain C, Rossi DJ. DNA damage response in adult stem cells: pathways and consequences. Nat Rev Mol Cell Biol. 2011;12:198–202. doi: 10.1038/nrm3060. PMID:21304553 [DOI] [PubMed] [Google Scholar]
- [4].Gurley KE, Kemp CJ. Synthetic lethality between mutation in Atm and DNA-PK(cs) during murine embryogenesis. Current biology: CB. 2001;11:191–194. doi: 10.1016/S0960-9822(01)00048-3. PMID:11231155 [DOI] [PubMed] [Google Scholar]
- [5].Gurley KE, Moser R, Gu Y, et al. . DNA-PK suppresses a p53-independent apoptotic response to DNA damage. EMBO Reports. 2009;10:87–93. doi: 10.1038/embor.2008.214. PMID:19057578 [DOI] [PMC free article] [PubMed] [Google Scholar]
- [6].Blackford AN, Jackson SP. ATM, ATR, and DNA-PK: The Trinity at the Heart of the DNA Damage Response. Mol Cell. 2017;66:801–817. doi: 10.1016/j.molcel.2017.05.015. PMID:28622525 [DOI] [PubMed] [Google Scholar]
- [7].Pauli C, Hopkins BD, Prandi D, et al. . Personalized In Vitro and In Vivo Cancer Models to guide precision medicine. Cancer Discov. 2017;7:462–477. doi: 10.1158/2159-8290.CD-16-1154. PMID:28331002 [DOI] [PMC free article] [PubMed] [Google Scholar]