

ABSTRACT
Diarrheal diseases remain the second most common cause of mortality in young children in developing countries. Efforts have been made to explore the impact of diarrhea on bacterial communities in the human gut, but a thorough understanding has been impeded by inadequate resolution in bacterial identification and the examination of only few etiological agents. Here, by profiling an extended region of the 16S rRNA gene in the fecal microbiome, we aimed to elucidate the nature of gut microbiome perturbations during the early phase of infectious diarrhea caused by various etiological agents in Vietnamese children. Fecal samples from 145 diarrheal cases with a confirmed infectious etiology before antimicrobial therapy and 54 control subjects were analyzed. We found that the diarrheal fecal microbiota could be robustly categorized into 4 microbial configurations that either generally resembled or were highly divergent from a healthy state. Factors such as age, nutritional status, breastfeeding, and the etiology of the infection were significantly associated with these microbial community structures. We observed a consistent elevation of Fusobacterium mortiferum, Escherichia, and oral microorganisms in all diarrheal fecal microbiome configurations, proposing similar mechanistic interactions, even in the absence of global dysbiosis. We additionally found that Bifidobacterium pseudocatenulatum was significantly depleted during dysenteric diarrhea regardless of the etiological agent, suggesting that further investigations into the use of this species as a dysentery-orientated probiotic therapy are warranted. Our findings contribute to the understanding of the complex influence of infectious diarrhea on gut microbiome and identify new opportunities for therapeutic interventions.
KEYWORDS: diarrhea, microbiome, enterotype, developing country, Bifidobacterium, Fusobacterium, oral microbiome
Introduction
Diarrheal diseases result in approximately 1.7 billion new infections and 0.75 million deaths in children aged under 5 y annually, making it the second most common cause of mortality in young children in developing countries.1-3 Repeated diarrheal episodes cumulatively increase the risk of malnutrition and stunting, which are associated with cognitive impairment and development of cardiovascular diseases and glucose intolerance in adulthood.4-6 Such syndromes exhaust societal resources, especially in impoverished regions; therefore there is a substantial demand for efficacious treatments and prophylaxes.
The human gastrointestinal tract is populated with an immensely rich and diverse microbial communities, with the large intestine harboring the greatest density of bacteria.7 Studies regarding the gut microbiota have highlighted the impact that microbial communities exert on human health, specifically in relation to nutrition,8 metabolic diseases,9 and cancer.10 Various techniques, including metagenomics, have been used to investigate microbial disturbances in persistent Clostridium difficile infections11 and inflammatory bowel disease,12 but such dysbiosis remains insufficiently characterized for infectious diarrhea in high incidence settings. The highly dynamic succession of microbial colonization in young children confounds analysis in this target group.13,14 Nevertheless, several 16S rRNA gene profiling studies have shown consistent patterns in the initial gut microbiota response following acute diarrheal episodes, in which a transition toward Proteobacteria and Streptococcus is observed in an increasingly oxygenated environment.15-17 This shift is additionally coupled with a reduction in specific Firmicutes and Bacteroidetes colonizers; their relative abundance is restored during post-diarrheal recovery state.15,17-19 These previous studies have either focused on diarrhea associated with Vibrio cholerae,15,18 or did not offer sufficiently detailed granularity for understanding microbiome alternation.16,19 Here, we aimed to characterize modifications of the gut microbiota in the early phase of infectious diarrhea in Vietnamese children by examining their fecal bacterial 16S rRNA composition.
Results
16S rRNA gene sequencing of diarrheal fecal samples
We subjected a collection of 200 fecal samples (55 samples from non-diarrheal controls and 145 samples from diarrheal patients) from Vietnamese children, to DNA extraction and 16S rRNA gene sequencing (Table 1 and Table S1). 15/54 controls showed presence of at least one pathogen after detection, including Norovirus, Rotavirus, Salmonella, and Campylobacter. However, the isolation of these organisms in asymptomatic carriers is frequently observed in endemic setting.20 One hundred and ninety nine of the fecal samples produced 16S rRNA sequences (median library size = 2,880,968 paired-end reads). Subsampling and quality filtering generated sub-libraries with a median depth of 756,829 paired-end reads, which served as the inputs for EMIRGE assemblies of the V3-V6 region. A mean of 74% of the reads (range: 37% to 92.5%) were successfully mapped and retrieved. A total of 131,702 assembled and clustered sequences, representing a pool of individual sample OTUs (operational taxonomic units), were produced from all samples. Subsequent filtering, OTU reconstruction, and chimeric sequence removal resulted in 7,479 OTUs clustering at 97% similarity in the 199 fecal samples.
Table 1.
Summary of the etiologies associated with 142 diarrheal samples contributing to this study.
| Etiological agent | Number | Proportion (%) |
|---|---|---|
| Viral | 39 | 27.5 |
| Norovirus (NoV) | 19 | 13.4 |
| Rotavirus (RoV) | 20 | 14.1 |
| Bacterial | 80 | 56.3 |
| Campylobacter | 14 | 9.9 |
| Salmonella | 15 | 10.6 |
| Shigella | 49 | 34.5 |
| Plesiomonas | 2 | 1.4 |
| Mixed | 23 | 16.2 |
| Campylobacter + NoV/RoV | 7 | 4.9 |
| Plesiomonas + NoV/RoV | 1 | 0.7 |
| Salmonella + NoV/RoV | 4 | 2.8 |
| Shigella + NoV/RoV | 11 | 7.7 |
| Total | 142 | 100 |
Microbial structures within the fecal samples
We examined the microbial composition of the fecal specimens from controls and diarrheal cases using the relative abundance of 205 agglomerated unique genera and their phylogenetic relationships. The average relative abundance of the various OTUs in the fecal specimens, up to genus level, is shown in Fig. S1. De novo clustering of the weighted Unifrac dissimilarity matrix, representing the pairwise diversity between samples, segregated the samples into 4 community state types (CSTs). The optimal number of CST groups was determined by gap statistics (Fig. S2) and supported by a prediction strength of > 75%. A principal coordinate analysis (PCoA) using the weighted Unifrac matrix provided further support for the structuring of the 4 different microbial CSTs (Fig. S3). Constrained analysis on the principal coordinates of 195 samples with their associated metadata indicated that age, weight-for-age Z (WAZ) score, and disease status (diarrhea/asymptomatic) best explained the variation between CSTs (Fig. 1). A random forest algorithm was used to reclassify each sample to one of the 4 CSTs based on the relative abundance of 205 genera, yielding a predicted error rate of 11.06%. Twenty-eight genera with the highest calculated variable importance measurement (> 0.002) were selected as the most significant taxa segregating the 4 CSTs. Random forest classification was then repeated with these taxa only to re-assess the most predictive genera for CST clustering, yielding a similar misclassification error rate of 11.56% (Fig. S4A and B). The following taxa predominated in each of the CSTs (with species included based on their abundances): CST1, Bifidobacterium-dominant (B. longum, B. breve, B. pseudocatenulatum, B. bifidum); CST2, Bacteroides-dominant (B. fragilis, B. vulgatus, B. uniformis, B. caccae); CST3, Streptococcus-dominant (S. gallolyticus and S. salivarius); and CST4, Escherichia-dominant (Fig. 2). A multivariate homogeneity of group dispersions test showed that CST3 possessed the highest measurement. This high variability was explained by the fact that several samples from this CST were rich in Megamonas, as opposed to Streptococcus. For diarrheal cases, the median reported duration of diarrhea onset was 2 d (IQR: 1–3 days). Thus, we classified our findings as representing the early phase of infectious diarrhea since the gut microbiota composition differs as diarrhea progresses to later stages.17,18
Figure 1.

The impact of diarrhea and demographics on the gut bacterial configurations. Constrained analysis of principal coordinates (CAP) biplot displaying the relationship between bacterial compositions (colored circles and triangles; see legend) and selected metadata. CAP was performed on the genus-level agglomerated weighted Unifrac dissimilarity matrix of 195 fecal samples with complete metadata. Black arrows denote the magnitudes and directions of quantitative demographic variables (age and WAZ score), and black squares represent qualitative variables (centered around relative diarrhea status).
Figure 2.

The bacterial compositions of the 4 community state types (CSTs). Heatmap showing the fractional abundance of the 15 most important genera determining the clustering of 199 diarrheal and non-diarrheal children fecal samples, as defined by random forest classification. Clustering was performed on the genus level weighted Unifrac dissimilarity matrix (with 205 genera) using the partitioning around meloids algorithm and identified 4 CSTs. The nature of sample is indicated at the head of the diagram: non-diarrheal control (gray); viral infection (chartreuse); mixed bacterial and viral infection (brown); bacterial infection (salmon); missing data (white). The confirmed major etiologies of infection are indicated in the middle row: non-diarrheal control (gray); Rotavirus (green); Norovirus (blue); Campylobacter (orange); Salmonella (magenta); Shigella (red); Plesiomonas (yellow).
The majority of the asymptomatic samples were within CST1 (n = 27) and CST2 (n = 22), which were generally enriched for taxa known to correspond with a healthy gut microbiota in young children.13,21 CST2 harbored a greater degree of bacterial diversity than CST1 and encompassed various genera that delineate an increasingly mature gut microbiota, including Faecalibacterium, Prevotella, Clostridium, Lachnospiraceae, and Phascolarctobacterium.8,13 The fecal samples from the diarrheal cases were distributed across the 4 CSTs, with CST3 (n = 52), and CST4 (n = 29) comprised almost exclusively of diarrheal cases. This structure corresponded with the dominance of Streptococcus and Escherichia species in gut microbiota of diarrheal children, as reported previously.16,17 The estimated Shannon diversity index was elevated in both controls and the cases in CST2 in comparison to the other CSTs (p < 0.05, ANOVA-Tukey's test) (Fig. 3). We additionally observed that bacterial diversity was lower in the diarrheal cases relative to the controls within CST1 and CST2, however this difference between the groups was not statistically significant (p > 0.05, ANOVA-Tukey's test).
Figure 3.

Shannon diversity index for 195 fecal samples, classified by diarrheal status and CST membership. Boxplots showing the Shannon diversity index, the upper whisker extends from the 75th percentile to the highest value within the 1.5 * interquartile range (IQR) of the hinge, the lower whisker extends from the 25th percentile to the lowest value within 1.5 * IQR of the hinge. Data points beyond the end of the whiskers are outliers. The asterisk indicates statistical significant in the pairwise comparison between CST2 and the other CSTs (ANOVA-Tukey's test, p < 0.05).
Factors affecting microbial structure in the feces of children with diarrhea
The CST grouping of the samples from diarrheal children (n = 136), and their respective demographic and clinical metadata, were input into a multinomial logistic regression model to identify factors explaining the grouping of the 4 CSTs (Table 2). CST2 was selected as a reference as it represented a mature gut microbiota configuration. We found that age, WAZ score, infection type (bacterial, viral, or mixed infection (Table 1)), and feeding practices remained significantly associated with CST classification (p < 0.05; likelihood ratio test). Furthermore, younger age was significantly associated with CST1 (median age = 16 months; p = 0.015; 2-tailed Z test) and CST3 (median age = 15 months; p = 0.042; 2-tailed Z test) (Table S2), while the fecal samples in CST2 and CST4 were generally associated with older children (median age = 22 and 23 months, respectively). This age differential may be explained by the maturation of the gastrointestinal microbiota in children as the Bifidobacterium-rich stage (CST1) precedes the Bacteroides-Firmicutes stage (CST2).14 Additionally, we found that a lower WAZ score was associated with CST4 (median WAZ score = −1.1; p = 0.008; 2-tailed Z test), while CST3 contained a significantly greater proportion of fecal samples from children infected with bacterial diarrheal pathogens (73%; p = 0.001; 2-tailed Z test; Table S2). Lastly, children that were exclusively breastfed were significantly associated with CST1 (p = 0.015; 2-tailed Z test). However, there was no significant difference in feeding patterns according to age (p = 0.422, Kruskal-Wallis rank sum test), suggesting that the association of breastfeeding with CST1 was not a product of clustering with younger age group.
Table 2.
Demographic and clinical predictors used in multinomial logistic regression for community state types (CSTs).
| Odds ratio of associated factor (95% CI*); p value$ |
|||
|---|---|---|---|
| Patient characteristics | CST1 (Bifidobacterium#; N = 25) v CST2 | CST3 (Streptococcus; N = 49) v CST2 | CST 4 (Escherichia; N = 29) v CST2 |
| Age in months | 0.92 [0.86–0.98]; p = 0.015 | 0.95 [0.91–1.00]; p = 0.042 | 0.99 [0.94–1.04]; p = 0.574 |
| Male | 2.14 [0.62–7.41]; p = 0.230 | 2.81 [0.98–8.03]; p = 0.054 | 2.12 [0.67–6.65]; p = 0.200 |
| Weight-for-age Z score | 0.63 [0.35–1.12]; p = 0.113 | 0.69 [0.42–1.12]; p = 0.135 | 0.46 [0.26–0.81]; p = 0.008 |
| Breastfed and formula-milk fed (compared with breastfed-only) | 0.10 [0.01–0.64]; p = 0.015 | 0.81 [0.24–2.76]; p = 0.737 | 0.44 [0.11–1.71]; p = 0.234 |
| Formula-milk fed only (compared with breastfed-only) | 0.26 [0.06–1.21]; p = 0.087 | 1.38 [0.38–5.01]; p = 0.621 | 0.25 [0.05–1.36]; p = 0.108 |
| Urban residence (compared with rural) | 1.60 [0.29–8.95]; p = 0.593 | 2.17 [0.51–9.3]; p = 0.297 | 1.5 [0.33–6.8]; p = 0.600 |
| Monthly income 145–483 USD (compared with monthly income < 145 USD) | 1.35 [0.31–5.93]; p = 0.690 | 0.53 [0.16–1.79]; p = 0.305 | 0.48 [0.13–1.74]; p = 0.262 |
| Monthly income > 484 USD (compared with monthly income < 145 USD) | 3.75 [0.15–94.77]; p = 0.422 | 3.28 [0.26–41.97]; p = 0.360 | 2.42 [0.14–41.78]; p = 0.544 |
| Vomiting | 0.19 [0.05–0.77]; p = 0.020 | 0.68 [0.20–2.26]; p = 0.525 | 0.42 [0.11–1.61]; p = 0.203 |
| Dysentery | 0.63 [0.17–2.35]; p = 0.496 | 0.76 [0.26–2.24]; p = 0.624 | 0.56 [0.17–1.86]; p = 0.342 |
| Bacterial infection (compared with viral) | 2.12 [0.43–10.52]; p = 0.358 | 13.61 [3.01–61.52]; p = 0.001 | 4.02 [0.81–20.04]; p = 0.089 |
| Mixed infection (compared with viral) | 1.21 [0.19–7.78]; p = 0.844 | 5.07 [0.9–28.58]; p = 0.066 | 3.31 [0.59–18.74]; p = 0.175 |
Note. Figures in bold indicate p < 0.05.
CI: Confidence Interval.
p value calculated through 2 tailed Z test.
Bacterial taxon which is predominant in each CST
Changes in taxonomic abundance in the feces of children with diarrhea
Streptococcus and Escherichia species were highly abundant in the diarrheal gut communities associated with CST3 and CST4, respectively. However, 64/145 (44%) of the fecal bacterial community structures from diarrheal patients were more similar to those from the control groups (CSTs 1 and 2). We used DESeq2 to identify the OTUs that were differentially abundant in the diarrheal samples of all 4 CSTs, taking into account the overarching discrepancy in composition due to different CST membership, aiming to minimize effects associated with confounding factors such as age, WAZ scores, and infection type. Seventeen OTUs were found to be at least 4-fold higher in abundance in diarrheal samples than control samples (p < 0.05); the majority of these were Escherichia species or organisms associated with the oral cavity, including Fusobacterium, Gemella, and Actinomyces (Fig. 4A). The most significant difference in a bacterial species in diarrheal fecal samples was associated with Fusobacterium mortiferum, which was 210 fold more abundant in diarrheal fecal samples than control samples (p < 0.0001). Furthermore, Sutterella, Megamonas, and Enterococcus species were also in higher abundance in diarrheal fecal samples in comparison to control samples. This analysis also identified 19 OTUs that were significantly depleted in diarrheal samples (p < 0.05). The majority of these OTUs (13/19) were gut colonizers belonging to the orders Clostridiales and Erysipelotrichales, including Subdoligranulum, Roseburia, Eubacterium, Coprococcus, Catenibacterium, and Ruminococcaceae. Among these, the most depleted taxon was the short chain fatty acid (SCFA) producer Blautia hansenii, which was significantly reduced in samples from those with diarrhea, regardless of the overall community group of the sample (p < 0.0001) (Fig. S5). B. bifidum, B. uniformis, and L. reuteri were also significantly associated with a healthy microbial configuration, albeit with different proportions in the various CSTs, likely reflecting age-specific development of the gut microbiota (Fig. S5). In addition, we performed the same analysis on a subset of samples belonging to CSTs 1 and 2, and this produced comparable results to the aforementioned.
Figure 4.

Bacterial taxa showing significantly different abundance among the examined classes. OTUs were identified to be of significantly differential abundance between groups in examination, as detected and filtered by DESeq2. In short, only OTUs with adjusted p values < 0.05, estimated fold change >4 or <1/4, and estimated base mean >30 were considered significantly differentially abundant and included in the plot. (A) Ratio of the log2 fold change of OTUs that differ between diarrheal and control stools, accounting for the different general microbial configurations in 4 CSTs (N = 199). (B) Ratio of the log2 fold change of OTUs that differ between bacterial and viral diarrheal infections, accounting for the different general microbial configurations in 4 CSTs (N = 119). (C) Relative abundances of OTUs that differ between dysenteric and non-dysenteric stools, accounting for different types of infection (N = 142).
We next constructed a correlation network that encompassed representative fecal taxa from both control and diarrheal samples. The resulting correlation network formed a tightly connecting interaction between members of the Clostridiales, Erysipelotrichales, and Bacteroidales, which indicated a positive interaction between these colonizers of a healthy gut (Fig. 5). A further positive interacting cluster was composed of genera that are frequently associated with the human oral cavity, including Fusobacterium nucleatum, Parvimonas micra, Peptostreptococcus stomatis, Solobacterium moorei, Gemmella haemolysans, Actinomyces odontolyticus, Lachnoanaerobaculum orale, Abiotrophia defectiva, Atopobium parvulum, Rothia mucilaginosa, and Streptococcus salivarius (Fig. 5). Conversely, Rothia was negatively correlated with Bacteroides vulgatus and Faecalibacterium prausnitzii, which are both indicators of a healthy and mature gastrointestinal microbiota.8,13 We additionally found that Fusobacterium mortiferum, the significantly enriched taxon in fecal samples from diarrheal children, exhibited a negative association with a key component of the gut Clostridiales network (Clostridium innocuum). This suggests that the colonization of F. mortiferum may inhibit the proliferation of related dependent taxa such as Blautia, Ruminococcus, and several Bacteroides. The negative correlation observed between Bifidobacterium longum subsp. infantis and Bifidobacterium breve likely reflects a competitive ecology as the gut microbiota matures.22
Figure 5.
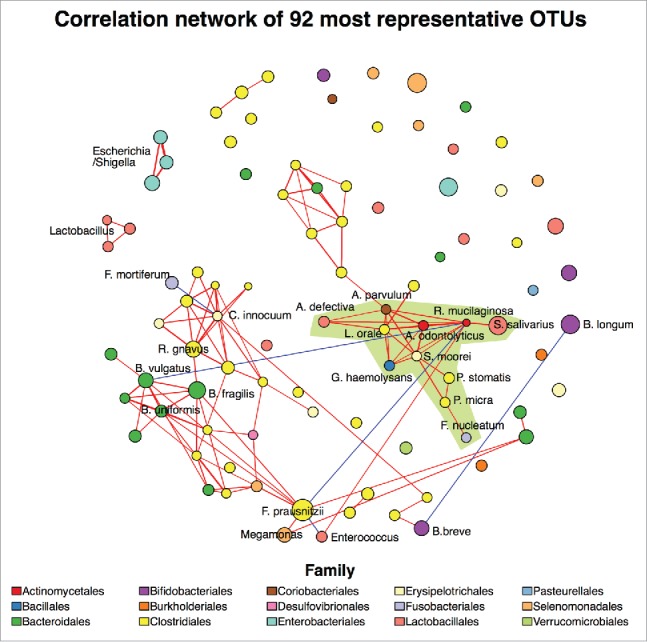
Correlation network of the healthy and diarrheal microbiome. Figure shows the correlation network of the 92 most represented OTUs sampled from 199 stool isolates, defined as OTUs with occurrence in at least 10 samples, constructed using the SparCC wrapper from package ‘SpiecEasi’. Only correlations with calculated p value ≤ 0.05 and absolute magnitude ≥ 0.25 were shown in the network. Positive and negative interactions were denoted as a red and blue solid line respectively, with line weight proportional to correlation strength. The OTUs (nodes) were colored based on taxonomic family (see legend), with sizes proportional to their relative abundances. The light green shaded area covers OTUs identified as members of normal human oral microbiota (through comparison with the Human Oral Microbiome Database).
Taxonomic changes associated with bacterial induced diarrhea and dysentery
To explore the differential composition of the gut microbiota in response to the different etiological agents (viral vs. bacterial), we examined a subset of 119 diarrheal fecal samples (viral infections = 39; bacterial infections = 80) to eliminate potential misclassification induced by 23 cases of mixed infections. Analysis using DESeq2 identified a group of 19 OTUs that were significantly more abundant in bacterial infections across the 4 CSTs. Among these, 8 taxa were more commonly associated with human oral microbiota (Fig. 4B); Actinomyces and Rothia were the most prevalent (assessed by the DESeq2 calculated base mean value). Other organisms with high relative proportion in the fecal samples from children with bacterial induced diarrhea included C. ramosum and B. vulgatus. Conversely, 4 OTUs were significantly elevated in the viral diarrheal infections; these included the Bifidobacterium species, B. breve and B. pseudocatenulatum (Fig. 4B).
We lastly categorized 142 diarrheal fecal samples based on the diagnosis of dysentery (the visible presence of mucoid and/or blood in the stool; n = 60), accounting for the differential microbiota changes due to types of infectious agents (bacterial, viral, or mixed infection). We identified 2 OTUs that were significantly associated with fecal samples from non-dysenteric diarrhea (DESeq2, p < 0.05) (Fig. 4C). B. pseudocatenulatum was almost 25 fold less abundant in dysenteric feces in comparison to non-dysenteric diarrheal feces; this was consistent across all of the corresponding etiological agents (Fig. 4C). For bacterial infections, L. reuteri was uniformly more depleted in the examined dysenteric fecal samples, but its variability was higher in dysenteric stools in those with a viral or mixed infection. When we accounted only for the stool with visible blood (n = 10), the analysis revealed that S. gallolyticus was the most depleted taxon in dysentery compared with watery diarrhea (DESeq2 Wald test without effect of infection types, p < 0.05). Other taxa of decreased abundance included Escherichia, B. pseudocatenulatum, Enterococcus and several oral microbiome species. However, these interpretations are to be confirmed in larger studies.
Discussion
Here we assessed the impact of diarrheal infection on the composition of the gut microbiota in Vietnamese children. Our results predict that the gut microbiota in young children exhibits differential responses during the early phase of diarrhea, which can be grouped into microbial community structures that either closely resemble or are highly divergent from those of healthy children. Approximately 44% of the diarrheal cases in this study retained a Bifidobacterium- or Bacteroides-rich structure. This association largely resembles the well-studied age-dependent normal gut microbiota in children,14 outlining a lack of global dysbiosis in these children. Among the various perturbed microbial states, we found that age, nutritional status, breastfeeding, and diarrheal etiology contributed to the composition of the bacterial communities during the early phase of diarrhea. However, since the WAZ score was collected after the onset of diarrhea, weight could be subjected to temporary fluctuation and its surrogate for nutritional status should be interpreted with caution. A recent study on diarrheal microbiome from U.S patients reported that diarrheal microbial structures could be grouped into 4 major clusters irrespective to the etiologies, of which 2 are almost exclusively restricted to diarrheal patients and associated with higher prevalence of Escherichia.23 In our study, Streptococcus was associated with younger age and bacterial infection, while Escherichia was associated with older children and a poor nutritional status. Indeed, analysis of the microbiota in diarrheal samples from young children (<2 y old) predominantly infected with pathogenic E. coli revealed that Streptococcus (S. gallolyticus and S. salivarius) was the most abundant organisms during the early phase of diarrhea.24 These 2 species were also dominant in our study. In contrast, Escherichia were overrepresented in the fecal samples from children (2–3 y old) infected with cholera in Bangladesh.15 Due to limitations of the study, we were unable to assess how these initial differential microbial communities affect diarrheal severity and recovery. Dysentery and antimicrobial treatment may further confound these associations. However, the ratio of Streptococcus/Bifidobacterium showed a significant positive correlation with diarrheal fecal output and duration of hospitalization, suggesting that the higher degree of divergence from the healthy microbiota may predict a more severe clinical presentation.24 Here, for bacterial induced diarrhea cases, we attempted to identify the pathogens' attributed sequenced reads in the fecal samples. However, only one Salmonella and one Campylobacter infections showed detectable 16S rRNA sequences classified to the corresponding pathogens. Shigella infections were not considered since its sequences are indistinguishable from Escherichia. This concurs with previous finding that by using 16S rRNA profiling on cholera fecal samples, V. cholerae is only detectable during the first day of symptom onset, and its presence is significantly reduced even on the second day of diarrhea.17
We additionally observed that obligate anaerobes belonging to the orders Clostridiales and Erysipelotrichales were consistently depleted in the diarrheal cases in comparison to the controls. This group included multiple SCFA producers including Blautia, Subdoligranulum, Roseburia, and Eubacterium. This finding may explain the transient depletion of SCFA following diarrhea, resulting in poor water and electrolyte absorption and a shortage of metabolic energy for enterocytes, which may lead to dehydration and further complications.25 These data are in accordance with previous studies, which have shown that Clostridiales become gradually enriched during the diarrheal recovery period.15,17,18 This loss of obligate anaerobes is coupled with the proliferation of facultative anaerobes, such as the Streptococcus and Escherichia species.16,17 Unpredictably, we observed that a substantial component of the microbiota from the human oral cavity was consistently represented in the diarrheal fecal samples. We propose that the transiently oxygenated environment and potential decrease in bacterial competition in the diarrheal gut enhances the colonization potential of these species.17 This may explain why S. salivarius, a dominant taxon of the oral cavity of young children, is capable of establishing an exceptionally high rate of colonization in suckling infants. Oral commensal organisms, predominantly S. salivarius and F. nucleatum, may serve as nucleators for the adherence of other bacteria, eventually forming polymicrobial biofilms that are capable of surviving mechanical and chemical stresses during their passage through the large intestine.26,27 Alternatively, the diarrhea-associated obligate anaerobe, F. mortiferum, which was found not to be associated with oral organisms, exhibited antagonism with the Clostridiales. This finding is in agreement with the spike of Fusobacterium observed in early phase diarrhea.18,19 Unlike the majority of Fusobacterium, F. mortiferum is extremely pleomorphic and bile resistant, with the ability to rapidly accumulate and metabolize a broad spectrum of sugars independent of amino acid fermentation.28-30 Furthermore, poultry isolated F. mortiferum has been shown to secrete a bacteriocin that can inhibit the growth of Gram-positive Bacillus species.31 Taken together, this evidence implies that F. mortiferum is more resilient to environmental stresses and able to occupy the anaerobic niche of the gut, possibly accelerating the depletion of Clostridiales via bacteriocin-mediated competition.
Dysentery denotes a more severe form of diarrhea, frequently requiring antimicrobial treatment and resulting in longer periods of hospitalization.20,32 To date, only Lactobacillus ruminis has been found to be depleted in dysenteric fecal samples.16 Here, we found that L. reuteri was significantly diminished in dysenteric diarrhea; we speculate that this discrepancy in species level identification may be induced by variation in environmental and genetic factors of the studied populations. In addition, we report here that a Bifidobacterium species, B. pseudocatenulatum, is substantially depleted in fecal specimens following bacterial infection; this effect is amplified during dysentery. Previous research examining the V1-V2 region of the 16S rRNA failed to efficiently identify Bifidobacterium, thus discounting its contribution.16 The oral administration of live B. pseudocatenulatum in induced cirrhotic and diabetic mice has been shown to restore the gut integrity and initiate an anti-inflammatory cytokine profile in the intestine, leading to a diminished systematic inflammatory response.33-35 Further, this potential probiotic also induces monocyte-derived macrophages to undergo transition into the anti-inflammatory M2 phase.36 It has been shown that the colonization of B. pseudocatenulatum or B. breve, but not other Bifidobacterium, protect mice from lethal Shiga toxin producing E. coli (STEC) infections.37 Treatment for dysentery is currently focused on antimicrobial therapy, and the usage of broad spectrum antimicrobials quickly depletes the balanced microbiota derived metabolites such as SCFAs, leading to disruption of the gut immune homeostasis.38 Therefore, we propose that B. pseudocatenulatum may be a potential dysentery-oriented probiotic candidate to reduce inflammation associated pathological conditions and accelerate the recovery of the microbiota to a healthy state.
Some limitations should be considered in the context of our investigation. First, the EMIRGE mediated read recovery rate was consistently lower in diarrheal samples. This means that a proportion of uncharacterized taxa were unavailable for examination, and it may impact on the true estimation of the diversity within these samples. Second, an exhaustive search for other diarrheagenic etiologies by molecular methods was not possible during this study; this may underestimate the true incidence of mixed infections. We propose that more comprehensive approaches, such as the Luminex xTAG multi-pathogen panel assay, could be incorporated in further studies to improve diagnostic accuracy.39 Third, the non-diarrhea controls included are not ideally representative of the healthy population, given that some of these children sought medical help for nutritional concerns. Notwithstanding these limitations, through sequencing an extended region of the 16S rRNA gene, it was possible to obtain high confidence taxonomic assignment up to the species level, portraying the highly complex gut microbiota in greater resolution. The inclusion of various diarrheal etiologies and the statistical methods used here further permitted us to reveal the general impact of infectious diarrhea on the gut microbiota. Diarrhea cases with unknown etiologies were excluded from this study, and this may underrepresent the true heterogeneity of diarrheal causes and their differing influences on the microbiota. Furthermore, we cannot dismiss that additional underlying mechanisms such as inflammation induced changes in immune response and oxygen gradient,40 aside from the physical manifestation of diarrhea, may contribute to observed changes in the microbiota. Regarding the correlation network analysis, it is apparent that SparCC detects fewer negative (competitive relationships) than positive correlations (mutualism and commensalism). Unlike positive correlations, which are more readily associated with the co-occurrence of OTUs, negative correlations are typically inferred by the mutual exclusions of OTUs. SparCC has been shown to possess increased precision in detecting negative correlations in comparison to other methods, but correlation detection is limited to those with greater power and low noise.41
In conclusion, by studying the microbiota of children with diarrhea associated with a diverse range of confirmed etiological agents, we have generated a comprehensive insight into the effects of infectious diarrhea on the gut microbiota. Therefore, our study provides a pivotal understanding of the impact of infectious diarrhea on the gut microbiota of children, particularly ones in low-middle income urban settings in Southeast Asia. Future work, through longitudinal study designs and the employment of shotgun metagenomics, should address how the initial differential responses in gut bacterial composition may impact disease progression, such as diarrhea clearance and recovery rate of the depleted microbiota.
Materials and methods
Study design, sample collection and microbiological procedures
Samples in this study originated from a previously described prospective observational study of pediatric diarrhea conducted at 3 major hospitals in Ho Chi Minh City (HCMC), Vietnam: Children's Hospital 1 (CH1), Children's Hospital 2 (CH2), and the Hospital for Tropical Diseases (HTD).20 The Oxford Tropical Research Ethics Committee (OxTREC) and the Scientific and Ethics Committed of CH1, CH2, and HTD provided ethical approval for the primary and this subsequent analysis. The general inclusion criteria for both diarrheal cases and non-diarrheal controls were children aged under 5 years, residing within HCMC and reporting no antimicrobial usage within 3 d before hospital admission. Children admitted to the study sites with diarrhea (defined as 3 or more loose stools or at least one bloody loose stool within a 24 hour period) were included as cases. Children who presented for health checks, nutritional, or gastrointestinal issues but reported no diarrheal or respiratory illnesses within 7 d of admission were recruited as controls. For each enrollee, clinical data regarding the symptoms and duration of diarrhea was obtained from a case report form completed by study clinicians, while demographic, feeding behavior and socioeconomic details were provided through a confidential questionnaire. Weight-for-age Z (WAZ) score was used to evaluate the nutritional status of all enrolled children based on WHO standards.42
A fecal sample was collected from each participant, both cases and controls, before any prescribed antimicrobial treatment. Both case and control fecal samples were subjected to standard microbiological culturing and biochemical testing to identify common diarrheal bacteria, including Shigella, Salmonella, Campylobacter, Plesiomonas, Yersinia, and Aeromonas, as described previously43 (https://wwwnc.cdc.gov/EID/article/19/6/11–1862-Techapp1.pdf). For the detection of Rotavirus and Norovirus, reverse transcriptase polymerase chain reaction (RT-PCR) was performed on total viral RNA extracted from all specimens.44 Further detection of Giardia lamblia, Entamoeba histolytica, and Cryptosporidium cysts were performed by microscopy on fresh fecal samples diluted in phosphate buffered saline. Stool samples were stored at −80°C until being subjected to DNA extraction up to 6 months after initial storage.
DNA extraction and sequencing
The prospective study from which the fecal specimens originated successfully recruited 1,419 cases and 609 controls. A subset of 200 fecal samples was selected for this investigation; these included 55 randomly selected controls and 145 cases from multiple confirmed etiologies. For diarrheal cases, random subsamples from each identified etiology were included to represent the epidemiology reported in our previous prospective study,20 with emphasis on Shigella as it was the major cause of dysentery (Table 1). Total DNA was extracted from these fecal samples using a phenol-chloroform extraction method. Briefly, 200 μl of the fecal sample was suspended in a solution containing 500 μl of DNA extraction buffer, 50 μl NaCl 200mM, 50 μl SDS 10%, 500 μl of phenol:chloroform:isoamyl (25:24:1), and 0.7 g of 0.5-mm-diameter zirconia/silica beads. Cells were lysed by mechanical disruption with a bead beater for one minute and subjected to 3 rounds of phenol:chloroform extraction. DNA was resuspended in TE buffer supplemented with RNase.
All DNA samples were shipped on dry ice to the Genome Institute of Singapore (GIS) for 16S rRNA sequencing using the GIS Efficient Rapid Microbial Sequencing (GERMS) platform. To obtain high resolution taxonomic identification up to the species level, all samples were PCR amplified using a previously optimized primer set (338F-1061R), which produces >700 bp amplicons covering the 4 variable regions (V3-V6) of the 16S rRNA region and can be assembled in silico to retrieve > 92% of the sequences in the Greengenes database.45 Long PCR fragments were cleaned using 1X AMPURE beads and randomly fragmented with Covaris (model LE220) shearing to ∼200bp. Library preparation was performed using the GeneRead DNA Library I Core Kit (Qiagen, Germany) according to the manufacturer's instructions, and the prepared DNA was subjected to sequencing on an Illumina HiSeq2500 platform to produce 75 bp paired-end reads.
EMIRGE assembly of 16S rRNA amplicons and OTU clustering
EMIRGE (Expectation Maximization Iterative Reconstruction of Genes from the Environment) is a database dependent assembler built on an iterative expectation-maximization (EM) algorithm, which is used to reconstruct full length 16S rRNA small subunit gene sequences (by simultaneously mapping and clustering) and estimate their relative abundance.46 The SILVA small subunit (SSU) rRNA database version 119 was filtered to remove potential large subunit (LSU) rRNA sequences, and closely related sequences were clustered at 97% identity by USEARCH; this was achieved by using PhyloFlash v2.0 (https://github.com/HRGV/phyloFlash). The resulting database was used as the reference template for EMIRGE. To limit the computational effort related to using the complete data set, one million paired-end reads were randomly subsampled without replacement from each sample library using seqtk (https://github.com/lh3/seqtk). Reads were trimmed using Sickle to remove those with quality <30 and length <60.47 We inputted the trimmed reads from each subsampled library into the amplicon-optimized version of EMIRGE, with 40 iterations and a 97% joining threshold. The EMIRGE output, a set of assembled and clustered sequences, for each sample indicates its representative OTUs with their estimated abundances. Assembled sequences with sample-wise normalized relative abundances less than 0.01% were removed from further analysis.
A pseudo-count for each sequence per subsampled library was calculated by scaling the number of successfully mapped reads to the EMIRGE estimated relative abundance of each sequence. All filtered sequences and a count table detailing their respective abundance were pooled and imported into the 16S rRNA processing platform mothur v.1.36.0.48 To minimize the length differences in assembled sequences and to facilitate more accurate OTU clustering, sequences were aligned to a trimmed version of the SILVA reference database to include only the amplified region (338F-1061R). Gap-only columns were filtered from this alignment, and sequences with a maximum of 7 (∼1%) ambiguous sites were retained for downstream analyses. Sequences were dereplicated, and ambiguous sites were replaced randomly with one of the 4 nucleotides (ATCG). These unique sequences and their respective counts served as input for the UPARSE clustering algorithm, which clustered all pooled sequences at a 97% similarity threshold.49 Chimeric sequences were stringently predicted and removed by UCHIME v6.0, set at both de novo mode and against the ChimeraSlayer informed reference database (e.g. ‘gold’ database). A total of 7,479 OTUs were reconstructed for 199 of the successfully sequenced samples. An alignment consisting of the most abundant representative sequences from each OTU was used to construct a phylogenetic tree using FastTree 2 under default parameters.50 Taxonomic assignments of OTU representatives up to the genus level were performed using the mothur implemented Ribosomal Database Project (RDP) classifier, with a minimum support threshold of 80%.51
Data analysis
All analyses were conducted in R52 using multiple packages, including ‘phyloseq’, ‘cluster’, ‘randomForestSRC’, ‘ggplot2’, ‘nnet’, ‘lmtest’, ‘vegan’, ‘DESeq2’, ‘SpiecEasi’ and other packages.53-61 An OTU count table, taxonomy classification table, related clinical and demographic data and the OTU phylogenetic tree were imported and analyzed as a ‘phyloseq’ object, allowing a unified and interactive analysis approach.53
Clustering into community state types (CSTs)
To gain overall insight into the microbial compositional structure of both controls and diarrheal cases, we applied a previously described clustering approach.62 To reduce sparsity as well as account for the divergence and the functional similarity shared between members of the same genus, which is frequently reported for human gut microbiota, the OTU count table was agglomerated at the unique genus level for clustering as recommended.63 Recent benchmark studies have proposed that the weighted Unifrac distance produces desirable accuracy and power in exploring β-diversity.64-66 A weighted Unifrac dissimilarity matrix between all samples was calculated using the collapsed genera's relative abundances and their phylogenetic relatedness.67 The partitioning around meloids (pam) algorithm was applied to this matrix to cluster all samples into distinct CSTs, with the optimal number of CSTs (k = 4) determined using gap statistics, a goodness of clustering measure.68 Further assessments such as prediction strength, average silhouette width (asw) were used to validate this optimal number of clusters, as recommended previously.63 Microbial CST grouping has been applied extensively to profile the human vaginal microbiome and has produced consistent findings.62,69,70 To assess the performance of this clustering approach and identify the most important genera contributing to the separation of these CSTs, the random forest classification algorithm was applied to the genera relative abundance table, using the samples' CST memberships as the response variable.71 A single tree assembly (5,000 trees) was used for all 199 samples of 4 CSTs, and other parameters were set as default according to the ‘rfsrc’ function in package ‘randomforestSRC’.55
Statistical analysis and regression modeling
To examine the relationship between the microbiome structures and associated explanatory variables for 195 samples with complete metadata, we applied constrained analysis of principal coordinates (CAP) on the calculated weighted Unifrac dissimilarity matrix and a set of demographic variables (age, sex, WAZ score, feeding pattern, income, rural residence), as well as diarrheal status. This was performed using the ‘capscale’ function in the ‘vegan’ package.59,72 Significant variables were identified and included in the final model based on Akaike information criterion (AIC) in a stepwise model selection approach. The α diversity of these 195 samples was estimated by Shannon diversity index. Analysis of variance (ANOVA) with post-hoc Tukey test and Bonferroni correction for multiple comparisons was used to compare control and diarrheal α-diversity among and within each CST. Multinomial logistic regression modeling was applied using the ‘multinom’ function in ‘nnet’ package to evaluate the association of the various aforementioned demographic factors and clinical features (vomiting, dysentery, and infection type) to the CST membership of diarrheal cases, with the Bacteroides rich CST (CST2) serving as the reference group.57 These predictors were included on the basis of limited missing data and capable of being assessed by clinicians upon patient's admission. Five samples with WAZ scores >3 or <-3, and one CST1 sample with age exceeding 2 standard deviations were considered as outliers and removed, resulting in 136 diarrheal samples with full metadata being subjected to regression modeling.
Evaluating differential abundance
DESeq2 was used to normalize the 7,479-OTUs table and to detect OTUs that show significantly differential relative abundance between 2 assessed groups per comparison (diarrhea against controls, bacterial infection against viral infection, and dysentery against non-dysentery), as recommended in recent benchmark studies.60,64,65 To identify consistent trends observed across all CSTs (diarrhea against controls, bacterial infection against viral infection) or infection types (dysentery against non-dysentery), we applied the likelihood ratio test (LRT) approach with the reduced models incorporating only either CST or infection type as coefficients. OTUs with adjusted p values < 0.05, estimated fold change >4 or <1/4, and estimated base mean >30 were considered significantly differentially abundant between 2 examined classes. Taxonomic identification to the species level was performed by manually comparing the reconstructed 16S rRNA sequences of OTUs to either the Human Oral Microbial Database (HOMD; http://www.homd.org/) or the Human Microbiome Project (HMP; http://www.hmpdacc.org/resources/blast.php) by BLAST. OTU was assigned a species nomenclature if its sequence is of at least 97% similarity to that in the database. Using these approaches, Shigella sequences are indistinguishable from Escherichia.
Interaction network construction
A correlation network was constructed for all 199 control and diarrheal samples to characterize the potential interactions between most representative 92 OTUs, defined as OTUs detected in at least 10 samples. This filtering step did not substantially affect the representativeness of the data set, with the median sample retainment rate of 93% (IQR: 87% - 97%). The network was constructed using the SparCC wrapper in the package ‘SpiecEasi’.61,73 The statistical significance for each interaction was assessed by 100 bootstrap iterations, with p values adjusted for multiple comparison correction. To avoid spurious correlations, only those with adjusted p values no greater than 0.05 and absolute magnitude equal to or above 0.25 were considered as significant correlations and represented in the final plots.
Supplementary Material
Abbreviations
- AIC
Akaike Information Criterion
- ANOVA
Analysis of Variance
- CST
Community State Type
- CAP
Constrained analysis of principal coordinates
- EMIRGE
Expectation Maximization Iterative Reconstruction of Genes from the Environment
- HCMC
Ho Chi Minh City
- IQR
Interquartile range
- LRT
Likelihood Ratio Test
- OTU
Operational Taxa Unit
- SCFA
Short Chain Fatty Acid
- WAZ
Weight for Age Z score
Ethical approval
The Oxford Tropical Research Ethics Committee (OxTREC) and the Scientific and Ethics Committee of Ho Chi Minh city's Children Hospital 1 (CH1), Children Hospital 2 (CH2), and Hospital for Tropical Diseases (HTD) provided ethical approval for the primary and this subsequent analysis (OxTREC No. 0109).
Reagents and catalog number
GeneRead DNA Library I Core Kit (Qiagen, catalog no. 180434)
Disclosure of potential conflicts of interest
The authors report no potential conflict of interest.
Acknowledgements
We gratefully acknowledge Andreas Sundquist and Matthew Davis, who wrote and modified the C++ script that generated the plot in Supplementary Figure 1. Computational resources were funded by the Li Ka Shing – University of Oxford Global Health Program (LG05, SM27). We express our gratitude to Paul J. McMurdie, Susan P. Holmes and her team for developing the package ‘phyloseq’ and conducting invaluable benchmark studies on analysis of microbiome data.
Funding
This work was funded by the Wellcome Trust and the Genome Institute of Singapore. SB is a Sir Henry Dale Fellow, jointly funded by the Wellcome Trust and the Royal Society (100087/Z/12/Z). DPT is supported by an OAK foundation fellowship.
References
- [1].Fischer Walker CL, Perin J, Aryee MJ, Boschi-Pinto C, Black RE. Diarrhea incidence in low- and middle-income countries in 1990 and 2010: a systematic review. BMC Public Health [Internet]. 2012. [cited 2014 Nov 18];12:220 Available from: http://www.pubmedcentral.nih.gov/articlerender.fcgi?artid=3323412&tool=pmcentrez&rendertype=abstract. doi: 10.1186/1471-2458-12-220 [DOI] [PMC free article] [PubMed] [Google Scholar]
- [2].Liu L, Johnson HL, Cousens S, Perin J, Scott S, Lawn JE, Rudan I, Campbell H, Cibulskis R, Li M, et al.. Global, regional, and national causes of child mortality: an updated systematic analysis for 2010 with time trends since 2000. Lancet [Internet]. 2012. [cited 2014 Jul 10];379:2151-61. Available from: http://www.ncbi.nlm.nih.gov/pubmed/22579125. doi: 10.1016/S0140-6736(12)60560-1 [DOI] [PubMed] [Google Scholar]
- [3].WHO. World Health Statistics 2014 Geneva, Switzerland; 2014. [Google Scholar]
- [4].Checkley W, Buckley G, Gilman RH, Assis AM, Guerrant RL, Morris SS, Molbak K, Valentiner-Branth P, Lanata CF, Black RE. Multi-country analysis of the effects of diarrhoea on childhood stunting. Int J Epidemiol. 2008;37:816-30. doi: 10.1093/ije/dyn099 PMID:18567626 [DOI] [PMC free article] [PubMed] [Google Scholar]
- [5].Guerrant RL, DeBoer MD, Moore SR, Scharf RJ, Lima AAM. The impoverished gut–a triple burden of diarrhoea, stunting and chronic disease. Nat Rev Gastroenterol Hepatol [Internet]. 2013;10:220-9. Available from: https://doi.org/10.1038/nrgastro.2012.239. doi: 10.1038/nrgastro.2012.239 [DOI] [PMC free article] [PubMed] [Google Scholar]
- [6].Deboer MD, Lima AAM, Oría RB, Scharf RJ, Moore SR, Luna MA, Guerrant RL. Early childhood growth failure and the developmental origins of adult disease: Do enteric infections and malnutrition increase risk for the metabolic syndrome? Nutr Rev. 2012;70:642-53. doi: 10.1111/j.1753-4887.2012.00543.x PMID:23110643 [DOI] [PMC free article] [PubMed] [Google Scholar]
- [7].The Human Microbiome Project Consortium Structure, function and diversity of the healthy human microbiome. Nature [Internet]. 2012. [cited 2013 Nov 6];486:207-14. Available from: http://www.pubmedcentral.nih.gov/articlerender.fcgi?artid=3564958&tool=pmcentrez&rendertype=abstract. doi: 10.1038/nature11234 [DOI] [PMC free article] [PubMed] [Google Scholar]
- [8].Subramanian S, Huq S, Yatsunenko T, Haque R, Mahfuz M, Alam MA, Benezra A, DeStefano J, Meier MF, Muegge BD, et al.. Persistent gut microbiota immaturity in malnourished Bangladeshi children. Nature [Internet]. 2014;510:417-21. Available from: http://www.nature.com/doifinder/10.1038/nature13421. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [9].Turnbaugh PJ, Ley RE, Mahowald MA, Magrini V, Mardis ER, Gordon JI. An obesity-associated gut microbiome with increased capacity for energy harvest. Nature [Internet]. 2006;444:1027-31. Available from: http://www.ncbi.nlm.nih.gov/pubmed/17183312. doi: 10.1038/nature05414 [DOI] [PubMed] [Google Scholar]
- [10].Kostic AD, Gevers D, Pedamallu CS, Kostic AD, Gevers D, Pedamallu CS, Michaud M, Duke F, Earl AM, Ojesina AI, et al.. Genomic analysis identifies association of Fusobacterium with colorectal carcinoma. Genome Res. 2012;22:292-8. doi: 10.1101/gr.126573.111 PMID:22009990 [DOI] [PMC free article] [PubMed] [Google Scholar]
- [11].Buffie CG, Bucci V, Stein RR, McKenney PT, Ling L, Gobourne A, No D, Liu H, Kinnebrew M, Viale A, et al.. Precision microbiome reconstitution restores bile acid mediated resistance to Clostridium difficile. Nature [Internet]. 2014;517:205-8. Available from: http://www.pubmedcentral.nih.gov/articlerender.fcgi?artid=4354891&tool=pmcentrez&rendertype=abstract. doi: 10.1038/nature13828 [DOI] [PMC free article] [PubMed] [Google Scholar]
- [12].Morgan XC, Tickle TL, Sokol H, Gevers D, Devaney KL, Ward D V, Reyes JA, Shah SA, LeLeiko N, Snapper SB, et al.. Dysfunction of the intestinal microbiome in inflammatory bowel disease and treatment. Genome Biol [Internet]. 2012;13:R79 Available from: http://www.pubmedcentral.nih.gov/articlerender.fcgi?artid=3506950&tool=pmcentrez&rendertype=abstract. doi: 10.1186/gb-2012-13-9-r79 [DOI] [PMC free article] [PubMed] [Google Scholar]
- [13].Bäckhed F, Roswall J, Peng Y, Feng Q, Jia H, Kovatcheva-Datchary P, Li Y, Xia Y, Xie H, Zhong H, et al.. Dynamics and Stabilization of the Human Gut Microbiome during the First Year of Life. Cell Host Microbe [Internet]. 2015;17:690-703. Available from: http://www.ncbi.nlm.nih.gov/pubmed/25974306. doi: 10.1016/j.chom.2015.04.004 [DOI] [PubMed] [Google Scholar]
- [14].Dogra S, Sakwinska O, Soh S, Ngom-bru C, Brück M, Berger B, Brüssow H, Karnani N, Lee YS, Yap F, et al.. Rate of establishing the gut microbiota in infancy has Consequences for Future Health. Gut Microbes. 2015;6:321-5. doi: 10.1080/19490976.2015.1078051 PMID:26516657 [DOI] [PMC free article] [PubMed] [Google Scholar]
- [15].Monira S, Nakamura S, Gotoh K, Izutsu K, Watanabe H, Alam NH, Nakaya T, Horii T, Ali SI, Iida T, et al.. Metagenomic profile of gut microbiota in children during cholera and recovery. Gut Pathog [Internet]. 2013;5:1 Available from: http://www.gutpathogens.com/content/5/1/1. doi: 10.1186/1757-4749-5-1 [DOI] [PMC free article] [PubMed] [Google Scholar]
- [16].Pop M, Walker AW, Paulson J, Lindsay B, Antonio M, Hossain M, Oundo J, Tamboura B, Mai V, Astrovskaya I, et al.. Diarrhea in young children from low-income countries leads to large-scale alterations in intestinal microbiota composition. Genome Biol [Internet]. 2014;15:R76 Available from: http://genomebiology.com/2014/15/6/R76. doi: 10.1186/gb-2014-15-6-r76 [DOI] [PMC free article] [PubMed] [Google Scholar]
- [17].David L, Weil A, Ryan ET, Calderwood SB, Harris JB, Chowdhury F, Begum Y, Qadri F, Larocque RC, Turnbaugh J. Gut Microbial Succession Follows Acute Secretory Diarrhea in Humans. MBio. 2015;6:1-14. doi: 10.1128/mBio.00381-15 [DOI] [PMC free article] [PubMed] [Google Scholar]
- [18].Hsiao A, Ahmed AMS, Subramanian S, Griffin NW, Drewry LL, Petri WA, Haque R, Ahmed T, Gordon JI. Members of the human gut microbiota involved in recovery from Vibrio cholerae infection. Nature [Internet]. 2014;515:423-6. Available from: http://www.nature.com/doifinder/10.1038/nature13738. doi: 10.1038/nature13738 [DOI] [PMC free article] [PubMed] [Google Scholar]
- [19].Becker-dreps S, Allali I, Monteagudo A, Vilchez S, Hudgens MG, Rogawski ET, Carroll IM, Zambrana LE, Espinoza F, Azcarate-peril MA. Gut Microbiome Composition in Young Nicaraguan Children during Diarrhea Episodes and Recovery. Am J Trop Med Hyg. 2015;93:1187-93. doi: 10.4269/ajtmh.15-0322 PMID:26350452 [DOI] [PMC free article] [PubMed] [Google Scholar]
- [20].Thompson CN, Phan MVT, Hoang NVM, Minh P V, Vinh NT, Thuy CT, Nga TTT, Rabaa M a., Duy PT, Dung TTN, et al.. A Prospective Multi-Center Observational Study of Children Hospitalized with Diarrhea in Ho Chi Minh City, Vietnam. Am J Trop Med Hyg [Internet]. 2015;92:1045-52. Available from: http://www.ajtmh.org/cgi/doi/10.4269/ajtmh.14-0655. doi: 10.4269/ajtmh.14-0655 [DOI] [PMC free article] [PubMed] [Google Scholar]
- [21].Koenig JE, Spor A, Scalfone N, Fricker AD, Stombaugh J, Knight R, Angenent LT, Ley RE. Succession of microbial consortia in the developing infant gut microbiome. Proc Natl Acad Sci. 2011;108:4578-85. doi: 10.1073/pnas.1000081107 PMID:20668239 [DOI] [PMC free article] [PubMed] [Google Scholar]
- [22].Avershina E, Storrø O, Øien T, Johnsen R, Wilson R, Egeland T, Rudia K. Bifidobacterial succession and correlation networks in a large unselected cohort of mothers and their children. Appl Environ Microbiol. 2013;79:497-507. doi: 10.1128/AEM.02359-12 PMID:23124244 [DOI] [PMC free article] [PubMed] [Google Scholar]
- [23].Singh P, Teal TK, Marsh TL, Tiedje JM, Mosci R, Jernigan K, Zell A, Newton DW, Salimnia H, Lephart P, et al.. Intestinal microbial communities associated with acute enteric infections and disease recovery. Microbiome [Internet]. 2015;3:45 Available from: http://www.microbiomejournal.com/content/3/1/45. doi: 10.1186/s40168-015-0109-2 [DOI] [PMC free article] [PubMed] [Google Scholar]
- [24].Sarker SA, Sultana S, Reuteler G, Moine D, Descombes P, Charton F, Bourdin G, McCallin S, Ngom-Bru C, Neville T, et al.. Oral Phage Therapy of Acute Bacterial Diarrhea With Two Coliphage Preparations: A Randomized Trial in Children From Bangladesh. EBioMedicine [Internet]. 2016;4:124-37. Available from: http://www.sciencedirect.com/science/article/pii/S2352396415302516. doi: 10.1016/j.ebiom.2015.12.023 [DOI] [PMC free article] [PubMed] [Google Scholar]
- [25].Monira S, Hoq MM, Chowdhury AKA, Suau A, Magne F, Hp Endtz, Alam M, Rahman M, Pochart P, Desjeux J-F, et al.. Short-chain fatty acids and commensal microbiota in the faeces of severely malnourished children with cholera rehydrated with three different carbohydrates. Eur J Clin Nutr [Internet]. 2010;64:1116-24. Available from: doi: 10.1038/ejcn.2010.123 [DOI] [PubMed] [Google Scholar]
- [26].Bradshaw DJ, Marsh PD, Keith Watson G, Allison C. Role of Fusobacterium nucleatum and coaggregation in anaerobe survival in planktonic and biofilm oral microbial communities during aeration. Infect Immun. 1998;66:4729-32. PMID:9746571 [DOI] [PMC free article] [PubMed] [Google Scholar]
- [27].Levesque C, Lamothe J, Frenette M. Coaggregation of Streptococcus salivarius with periodontopathogens: evidence for involvement of fimbriae in the interaction with Prevotella intermedia. Oral Microbiol Immunol [Internet]. 2003;18:333-7. Available from: http://www.ncbi.nlm.nih.gov/entrez/query.fcgi?cmd=Retrieve&db=PubMed&dopt=Citation&list_uids=12930529. doi: 10.1034/j.1399-302X.2003.00085.x [DOI] [PubMed] [Google Scholar]
- [28].Robrish SA, Oliver C, Thompson J. Sugar metabolism by fusobacteria: regulation of transport, phosphorylation, and polymer formation by Fusobacterium mortiferum ATCC 25557. Infect Immun [Internet]. 1991;59:4547-54. Available from: http://www.pubmedcentral.nih.gov/articlerender.fcgi?artid=259076&tool=pmcentrez&rendertype=abstract. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [29].Krieg NR, Staley JT, Brown DR, Hedlund BP, Paster BJ, Ward NL, Ludwig W, Whitman WB. Volume Four The Bacteroidetes, Spirochaetes, Tenericutes (Mollicutes), Acidobacteria, Fibrobacteres, Fusobacteria, Dictyoglomi, Gemmatimonadetes, Lentisphaerae, Verrucomicrobia, Chlamydiae, and Planctomycetes [Internet] In: Brown DR, Hedlund BP, Paster BJ, Ward NL, Ludwig W, Whitman WB, editors. Bergey's Manual of Systematic Bacteriology. New York: Springer; 2010. page 949.Available from: http://link.springer.com/10.1007/978-0-387-68572-4. [Google Scholar]
- [30].Gupta RS, Bhandari V. Phylogeny and molecular signatures for the phylum Fusobacteria and its distinct subclades. Anaerobe [Internet]. 2014;28:182-98. Available from: http://www.ncbi.nlm.nih.gov/pubmed/21503713. doi: 10.1016/j.anaerobe.2014.06.007 [DOI] [PubMed] [Google Scholar]
- [31].Portrait V, Cottenceau G, Pons AM. A Fusobacterium mortiferum strain produces a bacteriocin-like substance(s) inhibiting Salmonella enteritidis. Lett Appl Microbiol [Internet]. 2000;31:115-7. Available from: http://www.ncbi.nlm.nih.gov/pubmed/10972711. doi: 10.1046/j.1365-2672.2000.00774.x [DOI] [PubMed] [Google Scholar]
- [32].Traa BS, Fischer Walker CL, Munos M, Black RE. Antibiotics for the treatment of dysentery in children. Int J Epidemiol. 2010;39:70-4. doi: 10.1093/ije/dyq024 [DOI] [PMC free article] [PubMed] [Google Scholar]
- [33].Moratalla A, Gómez-Hurtado I, Santacruz A, Á Moya, Peiró G, Zapater P, González-Navajas JM, Giménez P, Such J, Sanz Y, et al.. Protective effect of Bifidobacterium pseudocatenulatum CECT7765 against induced bacterial antigen translocation in experimental cirrhosis. Liver Int. 2014;34:850-8. doi: 10.1111/liv.12380 PMID:24267920 [DOI] [PubMed] [Google Scholar]
- [34].Moratalla A, Gómez-Hurtado I, Á Moya-Pérez, Zapater P, Peiró G, González-Navajas JM, Gómez Del Pulgar EM, Such J, Sanz Y, Francés R. Bifidobacterium pseudocatenulatum CECT7765 promotes a TLR2-dependent anti-inflammatory response in intestinal lymphocytes from mice with cirrhosis. Eur J Nutr [Internet]. 2016;55:197-206. Available from: doi: 10.1007/s00394-015-0837-x [DOI] [PubMed] [Google Scholar]
- [35].Moya-Pérez A, Neef A, Sanz Y. Bifidobacterium pseudocatenulatum CECT 7765 Reduces Obesity-Associated Inflammation by Restoring the Lymphocyte-Macrophage Balance and Gut Microbiota Structure in High-Fat Diet-Fed Mice. PLoS One [Internet]. 2015;10:e0126976. Available from: doi: 10.1371/journal.pone.0126976 [DOI] [PMC free article] [PubMed] [Google Scholar]
- [36].Moratalla A, Caparrós E, Juanola O, Portune K, Puig-Kröger A, Estrada-Capetillo L, Bellot P, Gómez-Hurtado I, Piñero P, Zapater P, et al.. Bifidobacterium pseudocatenulatum CECT7765 induces an M2 anti-inflammatory transition in macrophages from patients with cirrhosis. J Hepatol [Internet]. 2015;64:135-45. Available from: http://linkinghub.elsevier.com/retrieve/pii/S0168827815005929. doi: 10.1016/j.jhep.2015.08.020 [DOI] [PubMed] [Google Scholar]
- [37].Asahara T, Shimizu K, Nomoto K, Hamabata T, Ozawa A, Takeda Y. Probiotic Bifidobacteria Protect Mice from Lethal Infection with Shiga Toxin-Producing Escherichia coli O157 : H7. Infect Immun [Internet]. 2004;72:2240-7. Available from: http://www.ncbi.nlm.nih.gov/pmc/articles/PMC375161/. doi: 10.1128/IAI.72.4.2240-2247.2004 [DOI] [PMC free article] [PubMed] [Google Scholar]
- [38].Lopez CA, Kingsbury DD, Velazquez EM, Baumler AJ. Collateral damage: Microbiota-derived metabolites and immune function in the antibiotic era. Cell Host Microbe. 2014;16:156-63. doi: 10.1016/j.chom.2014.07.009 PMID:25121745 [DOI] [PMC free article] [PubMed] [Google Scholar]
- [39].Duong VT, Phat VV, Tuyen HT, Dung TTN, Trung PD, Van Minh P, Phuong Tu LT, Campbell JI, Le Phuc H, Thanh Ha TT, et al.. Evaluation of luminex xTAG gastrointestinal pathogen panel assay for detection of multiple diarrheal pathogens in fecal samples in Vietnam. J Clin Microbiol. 2016;54:1094-100. doi: 10.1128/JCM.03321-15 PMID:26865681 [DOI] [PMC free article] [PubMed] [Google Scholar]
- [40].Velly H, Britton RA, Preidis GA. Mechanisms of cross-talk between the diet, the intestinal microbiome, and the undernourished host. Gut Microbes [Internet]. 2017;8:98-112. Available from: doi: 10.1080/19490976.2016.1267888 [DOI] [PMC free article] [PubMed] [Google Scholar]
- [41].Weiss S, Van Treuren W, Lozupone C, Faust K, Friedman J, Deng Y, Xia LC, Xu ZZ, Ursell L. Correlation detection strategies in microbial data sets vary widely in sensitivity and precision. ISME J [Internet]. 2016;10:1669-81. Available from: doi: 10.1038/ismej.2015.235 [DOI] [PMC free article] [PubMed] [Google Scholar]
- [42].World Health Organization WHO child growth standards: Methods and Development [Internet]. Geneva, Switzerland; 2006. Available from: http://hpps.kbsplit.hr/hpps-2008/pdf/dok03.pdf. [Google Scholar]
- [43].My PVT, Thompson CN, Le Phuc H, Tuyet PTN, Vinh H, Nguyen VMH, Van Minh P, Vinh NT, Thuy CT, Tran TTN, et al.. Endemic norovirus infections in children, Ho Chi Minh City, Vietnam, 2009–2010. Emerg Infect Dis [Internet]. 2013;19:29-32. Available from: http://www.pubmedcentral.nih.gov/articlerender.fcgi?artid=3713809&tool=pmcentrez&rendertype=abstract%5Cnhttp://europepmc.org/abstract/MED/23735160. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [44].My PVT, Rabaa MA, Vinh H, Holmes EC, Hoang NVM, Vinh NT, Phuong LT, Tham NT, Bay PVB, Campbell JI, et al.. The emergence of rotavirus G12 and the prevalence of enteric viruses in hospitalized pediatric diarrheal patients in Southern Vietnam. Am J Trop Med Hyg. 2011;85:768-75. doi: 10.4269/ajtmh.2011.11-0364 PMID:21976585 [DOI] [PMC free article] [PubMed] [Google Scholar]
- [45].Ong SH, Kukkillaya VU, Wilm A, Lay C, Ho EXP, Low L, Hibberd ML, Nagarajan N. Species Identification and Profiling of Complex Microbial Communities Using Shotgun Illumina Sequencing of 16S rRNA Amplicon Sequences. PLoS One 2013;8:1-8. doi: 10.1371/journal.pone.0060811 [DOI] [PMC free article] [PubMed] [Google Scholar]
- [46].Miller CS, Baker BJ, Thomas BC, Singer SW, Banfield JF. EMIRGE: reconstruction of full-length ribosomal genes from microbial community short read sequencing data. Genome Biol [Internet]. 2011;12:R44 Available from: http://genomebiology.com/2011/12/5/R44. doi: 10.1186/gb-2011-12-5-r44 [DOI] [PMC free article] [PubMed] [Google Scholar]
- [47].Joshi N, Fass J. Sickle: A sliding-window, adaptive, quality-based trimming tool for FastQ files (Version 1.33) [Software]. Available at https://github.com/najoshi/sickle. 2011.
- [48].Schloss PD, Westcott SL, Ryabin T, Hall JR, Hartmann M, Hollister EB, Lesniewski RA, Oakley BB, Parks DH, Robinson CJ, et al.. Introducing mothur: open-source, platform-independent, community-supported software for describing and comparing microbial communities. Appl Environ Microbiol [Internet]. 2009. [cited 2014 Jul 9];75:7537-41. Available from: http://www.pubmedcentral.nih.gov/articlerender.fcgi?artid=2786419&tool=pmcentrez&rendertype=abstract. doi: 10.1128/AEM.01541-09 [DOI] [PMC free article] [PubMed] [Google Scholar]
- [49].Edgar RC. UPARSE, highly accurate OTU sequences from microbial amplicon reads. Nat Methods [Internet] 2013. [cited 2014 Jul 10];10:996-8. Available from: http://www.ncbi.nlm.nih.gov/pubmed/23955772. doi: 10.1038/nmeth.2604 [DOI] [PubMed] [Google Scholar]
- [50].Price MN, Dehal PS, Arkin AP. FastTree 2 – Approximately Maximum-Likelihood Trees for Large Alignments. PLoS One [Internet]. 2010;5:e9490. Available from: doi: 10.1371/journal.pone.0009490 [DOI] [PMC free article] [PubMed] [Google Scholar]
- [51].Wang Q, Garrity GM, Tiedje JM, Cole JR. Naive Bayesian classifier for rapid assignment of rRNA sequences into the new bacterial taxonomy. Appl Environ Microbiol [Internet]. 2007. [cited 2014 Jul 9];73:5261-7. Available from: http://www.pubmedcentral.nih.gov/articlerender.fcgi?artid=1950982&tool=pmcentrez&rendertype=abstract. doi: 10.1128/AEM.00062-07 [DOI] [PMC free article] [PubMed] [Google Scholar]
- [52].R Core Team R: A language and environment for statistical computing. [Internet]. 2016; Available from: https://www.r-project.org/. [Google Scholar]
- [53].McMurdie PJ, Holmes S. phyloseq: an R package for reproducible interactive analysis and graphics of microbiome census data. PLoS One [Internet]. 2013. [cited 2014 Jul 14];8:e61217 Available from: http://www.pubmedcentral.nih.gov/articlerender.fcgi?artid=3632530&tool=pmcentrez&rendertype=abstract. doi: 10.1371/journal.pone.0061217 [DOI] [PMC free article] [PubMed] [Google Scholar]
- [54].Maechler M, Rousseeuw P, Struyf A, Hubert M, Hornik K. cluster: Cluster Analysis Basics and Extensions. R package version 2.0.4. 2016. [Google Scholar]
- [55].Ishwaran H, Kogalur UB. Random Forests for Survival, Regression and Classification (RF-SRC), R package version 2.2.0. 2016. [Google Scholar]
- [56].Wickham H. ggplot2 [Internet]. 2009. Available from: http://link.springer.com/10.1007/978-0-387-98141–3%5Cnpapers3://publication/doi/10.1007/978-0-387-98141-3.
- [57].Venables W, Ripley B. Modern Applied Statistics with S. Fourth Edi. New York: Springer; 2002. [Google Scholar]
- [58].Zeileis A, Hothorn T. Diagnostic Checking in Regression Relationships. R News 2002;2:7-10. [Google Scholar]
- [59].Oksanen J, Blanchet G, Kindt R, Legendre P, Minchin P, O'Hara R, Simpson G, Solymos P, Stevens M, Wagner H. vegan: Community Ecology Package. R package version 2.3-5. 2016. [Google Scholar]
- [60].Love MI, Huber W, Anders S. Moderated estimation of fold change and dispersion for RNA-seq data with DESeq2. Genome Biol [Internet]. 2014;15:550 Available from: http://genomebiology.com/2014/15/12/550. doi: 10.1186/s13059-014-0550-8 [DOI] [PMC free article] [PubMed] [Google Scholar]
- [61].Kurtz ZD, Müller CL, Miraldi ER, Littman DR, Blaser MJ, Bonneau RA. Sparse and Compositionally Robust Inference of Microbial Ecological Networks. PLOS Comput Biol [Internet]. 2015;11:e1004226. Available from: doi: 10.1371/journal.pcbi.1004226 [DOI] [PMC free article] [PubMed] [Google Scholar]
- [62].DiGiulio DB, Callahan BJ, McMurdie PJ, Costello EK, Lyell DJ, Robaczewska A, Sun CL, Goltsman DS a, Wong RJ, Shaw G, et al.. Temporal and spatial variation of the human microbiota during pregnancy. Proc Natl Acad Sci [Internet]. 2015;112:201502875 Available from: http://www.pubmedcentral.nih.gov/articlerender.fcgi?artid=4568272&tool=pmcentrez&rendertype=abstract. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [63].Koren O, Knights D, Gonzalez A, Waldron L, Segata N, Knight R, Huttenhower C, Ley RE. A Guide to Enterotypes across the Human Body: Meta-Analysis of Microbial Community Structures in Human Microbiome Datasets. PLoS Comput Biol [Internet]. 2013;9:e1002863. Available from: doi: 10.1371/journal.pcbi.1002863 [DOI] [PMC free article] [PubMed] [Google Scholar]
- [64].McMurdie PJ, Holmes S. Waste not, want not: why rarefying microbiome data is inadmissible. PLoS Comput Biol [Internet]. 2014. [cited 2014 Jul 15];10:e1003531 Available from: http://www.pubmedcentral.nih.gov/articlerender.fcgi?artid=3974642&tool=pmcentrez&rendertype=abstract. doi: 10.1371/journal.pcbi.1003531 [DOI] [PMC free article] [PubMed] [Google Scholar]
- [65].Thorsen J, Brejnrod A, Mortensen M, Rasmussen MA, Stokholm J, Al-Soud WA, Sørensen S, Bisgaard H, Waage J. Large-scale benchmarking reveals false discoveries and count transformation sensitivity in 16S rRNA gene amplicon data analysis methods used in microbiome studies. Microbiome [Internet]. 2016;4:62 Available from: http://microbiomejournal.biomedcentral.com/articles/10.1186/s40168-016-0208-8. doi: 10.1186/s40168-016-0208-8 [DOI] [PMC free article] [PubMed] [Google Scholar]
- [66].Weiss S, Xu ZZ, Peddada S, Amir A, Bittinger K, Gonzalez A, Lozupone C, Zaneveld JR, Vázquez-Baeza Y, Birmingham A, et al.. Normalization and microbial differential abundance strategies depend upon data characteristics. Microbiome [Internet]. 2017;5:27. Available from: doi: 10.1186/s40168-017-0237-y [DOI] [PMC free article] [PubMed] [Google Scholar]
- [67].Lozupone CA, Hamady M, Kelley ST, Knight R. Quantitative and qualitative beta-diversity measures lead to different insights into factors that structure microbial communities. Appl Environ Microbiol. 2007;73:1576-85. doi: 10.1128/AEM.01996-06 PMID:17220268 [DOI] [PMC free article] [PubMed] [Google Scholar]
- [68].Tibshirani R, Walther G, Hastie T. Estimating the number of clusters in a data set via the gap statistic. J R Stat Soc [Internet]. 2001;63:411-23. Available from: doi: 10.1111/1467-9868.00293 [DOI] [Google Scholar]
- [69].Ravel J, Gajer P, Abdo Z, Schneider GM, Koenig SSK, McCulle SL, Karlebach S, Gorle R, Russell J, Tacket CO, et al.. Vaginal microbiome of reproductive-age women. Proc Natl Acad Sci [Internet]. 2011;108:4680-7. Available from: doi: 10.1073/pnas.1002611107 [DOI] [PMC free article] [PubMed] [Google Scholar]
- [70].MacIntyre DA, Chandiramani M, Lee YS, Kindinger L, Smith A, Angelopoulos N, Lehne B, Arulkumaran S, Brown R, Teoh TG, et al.. The vaginal microbiome during pregnancy and the postpartum period in a European population. Sci Rep [Internet]. 2015;5:8988 Available from: http://www.nature.com/srep/2015/150311/srep08988/full/srep08988.html. doi: 10.1038/srep08988 [DOI] [PMC free article] [PubMed] [Google Scholar]
- [71].Breiman L. Random forests. Mach Learn [Internet] 2001; 45:5-32. Available from: http://link.springer.com/article/10.1023/A:1010933404324. doi: 10.1023/A:1010933404324 [DOI] [Google Scholar]
- [72].Egendre PIL, Anderson MJ. Distance-based redundancy analysis: Testing multispecies responses in multifactorial ecological experiments. Ecol Monogr. 1999;69:1-24. doi: 10.1890/0012-9615(1999)069%5b0001:DBRATM%5d2.0.CO;2 [DOI] [Google Scholar]
- [73].Friedman J, Alm EJ. Inferring correlation networks from genomic survey data. PLoS Comput Biol [Internet]. 2012;8:e1002687 Available from: http://journals.plos.org/ploscompbiol/article?id=10.1371/journal.pcbi.100268. doi: 10.1371/journal.pcbi.1002687 [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.