

ABSTRACT
The mechanisms underlying the systemic effects mediated by gut microbiota are under active investigation. In addition to local, direct effects of gut microbiota on the host, metabolic products from microbiota may act peripherally, reaching distal organs through the circulation. In our recent publication we demonstrated that gut microbiota influence bone remodeling distally, promoting both bone resorption and formation. We proposed that these effects are mediated, at least in part, by the induction of insulin like growth factor (IGF-1) by the microbiota metabolite short chain fatty acids (SCFA). Here we explore additional mechanisms by which microbial metabolites could directly or indirectly alter host bone remodeling. We discuss whether SCFA directly modulate bone resorption by their actions on osteoclasts, and test the possibility that serotonin is another gut microbiota derived long-distance mediator of effects on bone remodeling. A detailed understanding of the mechanisms of microbiota effect on bone remodeling could help establish potential therapeutic strategies to promote bone health.
KEYWORDS: gut microbiota, bone, SCFA, GPR109, serotonin
Introduction
Gut microbiota exerts systemic influence on diverse aspects of host physiology. Locally, gut microbiota affects nutrient and energy extraction and gut barrier function. Systemically, gut microbiota shape immune homeostasis and have been implicated in obesity, glucose intolerance, hypertension, and endocrine function in the host.1-4 A role for gut microbiota in brain development, behavior, and appetite has also been invoked,5 and additional aspects of host physiology regulated by gut microbiota are likely to emerge. For example, the effect of gut microbiota on bone remodeling and biomechanical properties has only been explored relatively recently.6-10 While early reports suggested that gut microbiota stimulates bone loss by activating bone resorbing osteoclasts,7 we recently found that gut microbiota also regulate bone formation by osteoblasts, thus providing an anabolic stimulus to the skeleton.6
The mechanism by which gut microbiota affect host organs distant from the gut is an area of active investigation. The distal effects of gut microbiota are thought to be mediated by circulating molecules including metabolites, hormones and neurotransmitters produced or induced by microbiota.4,11 The major fermentative products of the gut microbiota are SCFA, predominantly acetate, propionate and butyrate.12 Other microbial products with the potential to impact host physiology include secondary bile acids, serotonin, dopamine and norepinephrine. SCFA can directly affect host cell function (for example, T cell differentiation) and are also thought to regulate a number of host endocrine molecules including peptide YY (PYY), leptin and serotonin.4
We and others have demonstrated that colonization of germ-free (GF) mice with gut microbiota increases SCFA concentrations in the cecum.6,13 SCFAs have multiple cellular and physiological functions. SCFA can activate G-protein-coupled-receptors (GPCRs), inhibit histone deacetylases, and serve as energy substrates for intestinal cells. As butyrate is a preferred energy source for colonocytes, it may have diverse indirect effects on host physiology through effects on nutritional status. In contrast, SCFA have direct actions on cells by activating GPCRs, specifically GPR41, the receptor for propionate and butyrate, GPR43, the receptor for acetate and propionate, and GPR109, the receptor for butyrate and niacin.12 Although induced development of peripheral regulatory T cells by SCFA depends on the expression of GPR43,13 whether SCFA receptors are required for microbiota to mediate bone remodeling is unknown. Furthermore, the SCFA propionate and butyrate may have GPCR-independent effects through their activity as histone deacetylase (HDAC) inhibitors.14
Previous work by ourselves and others has demonstrated that colonization with gut microbiota increases bone resorption by increasing osteoclast activity, possibly mediated through immune stimulation and increase in pro-osteoclastogenic cytokines.6,7,9,10 Recently, we and others found that gut microbiota not only stimulate bone resorption but unexpectedly also stimulates bone formation, likely mediated by increases in IGF-1.6,8 We demonstrated that levels of IGF-1, a growth factor known to regulate skeletal formation, are dynamically modulated by changes in gut microbiota, thus modulating the anabolic stimulus to the skeleton. Colonization of GF mice increased serum IGF-1 levels, while antibiotic treatment (either broad-spectrum antibiotics or vancomycin alone) decreased serum IGF-1.6 Consistent with IGF-1 being a critical mediator between gut microbiota and bone formation, IGF-1 levels and skeletal growth in neonatal mice are impaired in GF compared to colonized neonatal mice (8), and chemical blockade of the IGF-1R decreased the growth advantage of colonized mice (8). As serum IGF-1 and cecal SCFA concentration were correlated, we tested if SCFA linked colonization with gut microbiota and modulation of host IGF-1 levels. Indeed, SCFA supplementation was sufficient to increase liver and adipose IGF-1, resulting in increased serum IGF-1 levels in antibiotic treated mice.6 This suggests that one mechanism by which gut microbiota impact bone formation is via generation of SCFA from non-digestible fiber, which then promotes host IGF-1 production. How SCFA modulate host IGF-1 production is not known but is an area of active investigation.
Although our previous work suggested that SCFA indirectly regulate bone remodeling through modulating circulating IGF-1, additional roles for SCFA in modulating bone physiology are possible. For example, SCFA have been reported to increase levels of peripheral serotonin, a neurotransmitter for which there is some evidence for a role in bone turnover.18-21 Furthermore, a direct role of SCFA in regulating the differentiation or function of bone cells is also possible. The repertoire of SCFA receptor expression on various bone cell types has not been reported, but sodium butyrate and gut microbiota have been shown to affect osteoblast precursors.2,3 One month after colonization, osteoclastic bone resorptive activity, as measured by C-terminal telopeptide of type I collagen (CTX-I), is increased.6 Sodium butyrate and the HDAC inhibitor trichostatin A are both reported to inhibit osteoclast differentiation in vitro.1,15 Thus, direct effects of SCFA to inhibit osteoclast formation or bone resorption either via activation of GPCR or through HDAC inhibition may contribute to the transient nature of increased osteoclastic bone resorption. Indeed, GPR41 and GPR109 are expressed by monocyte/macrophage cells after stimulation with LPS or IFN-γ,16,17 suggesting that monocyte-derived osteoclasts may express receptors for SCFA.
Osteoclast precursors express GPCR receptors for SCFA
The expression pattern of GPR41, 43 and 109 during osteoclast differentiation is not known. We examined the expression of SCFA receptor on bone marrow osteoclast precursors and differentiating osteoclasts by quantitative real-time PCR with normalization to the housekeeping gene Hprt. Bone marrow cells were expanded for 3 days in the presence of M-CSF (macrophage colony stimulating factor) to generate myeloid osteoclast precursors, followed by differentiation in the presence of RANKL (receptor activator of NF-κB ligand) and M-CSF. Osteoclast precursors express both Gpr41 and Gpr109, but expression is significantly and rapidly downregulated by 24h after exposure to RANKL (Fig. 1A). In contrast, Gpr43 mRNA expression was difficult to detect and is unchanged with exposure to RANKL. Future studies by western blot or flow cytometry are needed to confirm the protein levels of SCFA receptors on osteoclasts.
Figure 1.
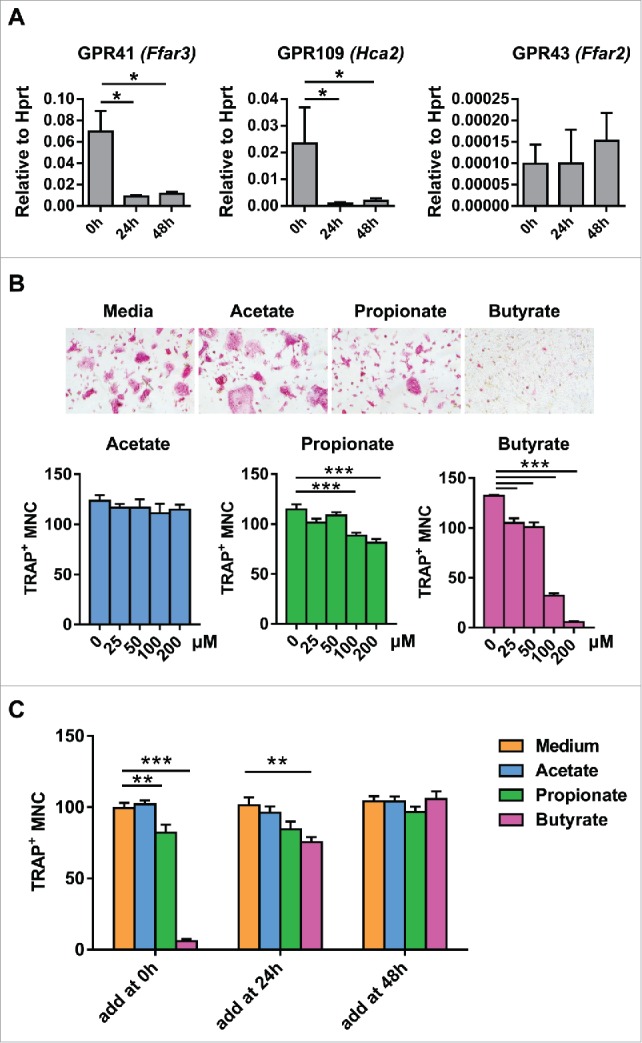
Butyrate and proprionate inhibit osteoclast differentiation in vitro. (A) Osteoclasts were differentiated from equivalent numbers of bone marrow derived macrophages (BMDM) in the presence of M-CSF and RANKL. GPR41, GPR109, and GPR43 expression was assessed by real-time PCR in osteoclast cultures before, 24 h, or 48 h after addition of RANKL. Gene expression was normalized to Hprt. (B) Equal numbers of BMDM were treated with acetate, proprionate, and butyrate at the indicated concentrations in the presence of RANKL and M-CSF and TRAP positive multinuclear cell (MNC) were counted. Representative TRAP stained are shown. (C) 200 μM of individual SCFA were added before, 24 h, or 48 h after addition of RANKL and TRAP positive MNC were counted. Data are shown as mean ± SEM. Difference between groups are determined by one-way ANOVA. * P < 0.05; ** P < 0.01; *** P < 0.001.
Propionate and butyrate inhibit OC differentiation in a dose and time dependent manner in vitro
As SCFA receptors are expressed on osteoclast precursors, we tested whether individual SCFA directly affect osteoclast differentiation. Acetate alone does not have a significant effect on osteoclast differentiation (Fig. 1B), even at a dose as high as 1 mM (data not shown). In contrast, butyrate and, to a lesser degree, propionate inhibit osteoclast differentiation in a dose and time dependent manner. Addition of butyrate at a concentration as low as 25 μM or proprionate at a concentration of 100 μM or more significantly inhibited differentiation from osteoclast precursors (Fig. 1B), as assessed by counting of multinucleated TRAP+ osteoclasts after 3 days of culture with RANKL. Consistent with the dramatic downregulation of Gpr41 and Gpr109 mRNA within 24 h of exposure to RANKL, addition of butyrate or proprionate at 24 h or 48 h after differentiation does not decrease osteoclast formation (Fig. 1C). Together, these data suggest that osteoclasts precursors express the receptors for SCFA and butyrate and proprionate are capable of directly inhibiting osteoclast differentiation. However, whether systemic concentrations of butyrate and proprionate are high enough to inhibit osteoclast formation and whether this inhibition occurs through activation of the cognate GPCR is unknown.
Inhibition of osteoclastogenesis by butyrate is independent of GPR109
Because osteoclast precursors express the receptors for proprionate and butyrate, and the window for inhibition of differentiation with these agents parallels receptor expression, we hypothesized that they inhibit osteoclast differentiation through GPCR. Thus, we examined osteoclast formation in mice lacking the receptor for butyrate, GPR109. We obtained GPR109 wild type (WT) and knockout (KO) littermates on the C57BL/6 background by mating heterozygous mice. 3-month-old female mice were used throughout the study. Consistent with the inhibitory effect of butyrate on early osteoclast differentiation, we found that RANKL induced osteoclast formation from total bone marrow cells is increased in GPR109 KO compared to the WT mice (Fig. 2A). However, butyrate at a concentration of 200 μM was able to suppress osteoclast formation even in the absence of GPR109 (Fig. 2B). The second GPR109 ligand, niacin, did not inhibit osteoclast differentiation in either WT or KO cells at a concentration of 200 μM (data not shown). It is possible that the inhibitory effect of butyrate occurs because of its role as an HDAC inhibitor15, or through other mechanisms and/or receptors. Although this data suggests that GPR109 has an inhibitory role on osteoclast differentiation under in vitro culture conditions, this could potentially reflect activation by ligands present in media as well; many GPCRs have a tonic level of activation in the absence of ligand22 and GPR109 may constitutively inhibit precursors from differentiating into osteoclasts.
Figure 2.
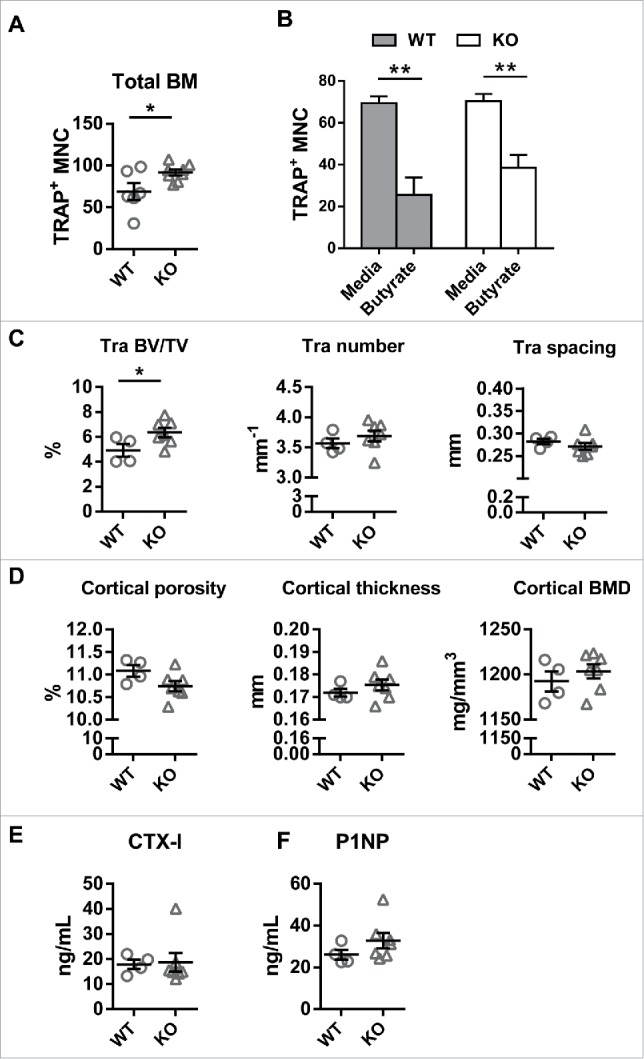
GPR109 is not required for butyrate inhibition of osteoclast differentiation and deficiency increases trabecular bone mass. (A) An equal number of total bone marrow cells isolated from WT and GPR109 KO mice was differentiated in the presence of M-CSF and RANKL and osteoclast formation was quantified based on number of TRAP positive MNC formed. (B) BMDM from WT and GPR109 KO mice were treated with 100 µM butyrate and RANKL was added simultaneously to induce osteoclast differentiation. (C) Trabecular and (D) cortical bone parameters determined by microCT. The region of interest was thresholded using a global threshold that set the bone/marrow cutoff at 352.3 mg HA/cm3 for trabecular bone and 589.4 mg HA/cm3 for cortical bone. (E) CTX-I and (F) P1NP levels in the serum from WT and GPR109 KO mice. Data are shown as mean ± SEM. Difference between groups are compared by Student's t test in panel A, C, D, E and F; and one-way ANOVA in panel B. * P < 0.05; ** P < 0.01.
GPR109 knockout mice have increased trabecular bone mass
If GPR109 constitutively inhibits osteoclast differentiation, mice lacking GPR109 would be expected to have low bone mass. Thus, we evaluated the bone phenotype of GPR109 KO mice. Micro-computed tomography (microCT) of femurs from 3 month old female littermates was used to quantitate bone mass and structural parameters. In contrast to our expectation, we found that trabecular bone volume/tissue volume (BV/TV), a measure of trabecular bone mass is modestly but significantly higher in GPR109-deficient mice (Fig. 2C), without any alteration in cortical bone parameters (Fig. 2D). This increased bone mass phenotype was confirmed in 8 month-old mice (data not shown). The observed increase in trabecular bone mass phenotype in KO mice cannot be explained by the effects of GPR109 deficiency on osteoclast formation, which leaves open the question of whether GPR109 plays a role in osteoclast activity, osteoblast/osteocyte function, or alternatively that the alteration in bone mass is secondary to effects of GPR109 loss on other physiologic processes.
To better understand whether the increased bone mass in GPR109 KO mice reflected a change in osteoclast or osteoblast activity, we compared bone turnover markers in the serum from GPR109 WT and KO mice. The level of the bone resorption marker CTX-I is similar between WT and KO mice (Fig. 2E), suggesting the osteoclast activity is similar between WT and KO mice. However, there is a trend toward higher levels of the bone formation marker P1NP in GPR109 KO mice (Fig. 2F), indicating that increased bone formation may explain the increased bone mass in the KO mice. We next examined serum IGF-1 levels and found that GPR109 deficiency does not alter IGF-1 levels (data not shown), suggesting that mechanisms other than increased IGF-1 are responsible for the slightly increased bone mass in GPR109 KO mice.
Independent of GPR109, butyrate and proprionate may be important in limiting osteoclast formation in the setting of colonization of GF mice as they have an inhibitory effect on osteoclast differentiation in vitro. However, SCFA are mainly produced in the gut and systemic levels are low.12 Acetate is the most abundant SCFA in the periphery, with a concentration of about 70 μM systemically and 250 μM in the portal vein. In contrast, butyrate is consumed locally by enterocytes, such that its peripheral concentration is only approximately 4 μM, well below the minimum concentration needed to inhibit osteoclast differentiation in vitro. Propionate is metabolized in the liver and is also present at low concentrations systemically. If butyrate and proprionate act directly inhibit osteoclasts in vivo, we would expect the concentrations of these SCFA in the bone marrow to be in the inhibitory range. Thus, we attempted to measure SCFA concentrations in both bone marrow and serum by HPLC and found concentrations lower than the limit of detection (0.2 μM for acetate, 0.4 μM for proprionate, and 2.5 μM for butyrate). Therefore, it appears unlikely that the SCFA concentration in the bone marrow is high enough to directly inhibit osteoclast differentiation in vivo.
Although direct effects of SCFA on osteoblasts remain possible, it seems likely that SCFA primarily regulate bone remodeling through indirect mechanisms. We have previously reported one such mechanism, in which SCFA modulate host production of circulating IGF-1, but regulation of other hormones or host factors may also contribute to the effects of SCFA and gut microbiota on bone.
Colonization, but not SCFA supplementation, increases peripheral serotonin levels
Serotonin is a potential candidate molecule to mediate signaling between microbiota and bone. Gut is the major site of peripheral serotonin synthesis, where it is produced by enterochromaffin cells (ECs), mucosal mast cells and myenteric neuron cells.23 Interestingly, metabolites produced by gut microbiota, including propionate, butyrate, and the secondary bile acid deoxycolate directly induced expression of tryptophan hydroxylase 1 (Tph1), the rate-limiting enzyme for serotonin synthesis, on a chromaffin cell line in vitro and colonization significantly increased peripheral serotonin levels in GF mice.23 Additionally, some species of bacteria also synthesize serotonin in the gut.24 Serotonin has been proposed to promote osteoclastogenesis, as Tph1−/− mice have decreased osteoclastogenesis.25 The role of peripheral serotonin in bone physiology is controversial. Studies using Tph1−/− mice came to opposing conclusions regarding the role of gut serotonin production on bone remodeling. Yadav et al. reported that Tph1−/− mice exhibited higher bone mass compared to WT controls,18 while a study by Cui et al. found no differences in bone mass between Tph1−/− and WT mice.19 Similarly, treatment with Tph1 inhibitors was reported to either prevent or have no effect on development of osteoporosis in ovariectomized rodent models.18,19 The effect of Tph1−/− on bone may depend on age, as others demonstrated increased BV/TV in Tph1−/− mice at 6 weeks but not at 16 weeks of age (20), 21 or 83 weeks of age (21). Thus, the role of gut derived peripheral serotonin in bone physiology and may depend on age, strain background or environment, such as the composition of the gut microbial community. Whether SCFA directly induce serotonin in vivo in not known, and we hypothesized that SCFA-mediated increases in serotonin could promote the transient increase in bone resorption observed after colonization of GF mice.
To test the effect of microbiota on peripheral serotonin production, we tested whether serotonin levels changed after colonization of GF mice with gut microbiota from specific pathogen free (SPF) mice. We confirmed a previous report that colonization significantly increased peripheral serotonin levels in GF mice (Fig. 3A). We next tested whether SCFA can directly modulate serotonin production by supplementing the drinking water of mice treated with either of two broad-spectrum antibiotic cocktails with a mix of proprionate, butyrate and acetate. Peripheral serotonin was measured in serum using a commercial ELISA kit (Eagle Biosciences, NH). Details of colonization, antibiotic and SCFA treatment protocols are reported in ref. 6. We found a trend towards reduced serum serotonin levels after antibiotic treatment. However, in contrast to IGF-1, supplementation with SCFA had no effect on serum serotonin levels with either antibiotic cocktail (Fig. 3B). Moreover, we found that while antibiotic treatment lead to decreased expression of Tph1 and Chga and increased Slc6a4 and Maoa expression in the colon, a gene signature consistent with decreased serotonin production in the gut, SCFA supplementation had no effect on the expression of these genes (Fig. 3C). Together, our data do not suggest a correlation between the microbiota metabolite SCFA and peripheral serotonin production in vivo.
Figure 3.
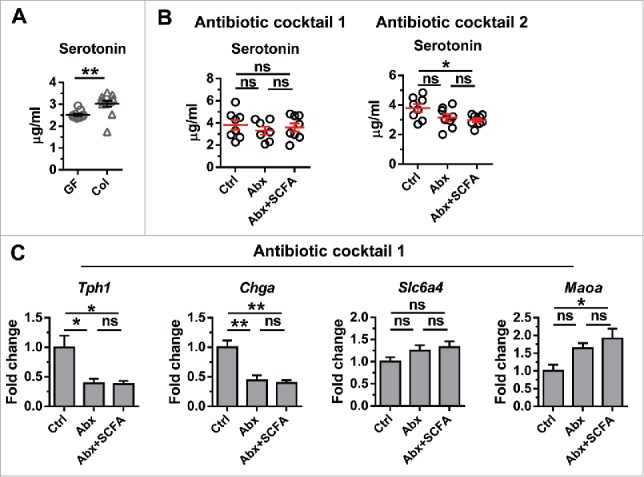
Regulation of serotonin by microbiota and its metabolites. (A) Peripheral Serotonin levels in GF mice and littermates colonized for 1 month starting at 8 weeks of age. Between group differences were compared by Mann-Whitney test; *** p < 0.001. (B) Peripheral Serotonin levels in SPF mice and SPF mice treated with two different antibiotic cocktails with or without SCFA supplementation. Antibiotic cocktail 1 contains 1 mg/mL each of ampicillin, metronidazole and neomycin and 0.5 mg/mL vancomycin. Antibiotic cocktail 2 contains 0.2 mg/mL gentamicin, 0.15 mg/mL ciprofloxacin, 2 mg/mL streptomycin, and 1 mg/mL bacitracin. (C) Expression of Tph1, Chga, Slc6a4 and Maoa in the colon from SPF mice treated with antibiotic cocktail 1 with or without SCFA supplementation. Data are shown as mean ± SEM. Differences between multiple groups were compared by one-way ANOVA followed with Kruskal-Wallis post-hoc test. * p < 0.05; ** p < 0.01; ns, not statistically significant.
Conclusions
As knowledge of microbiota-host interactions grows, manipulation of microbiota has been proposed as a promising strategy to promote host health. Thus, increased understanding of the broad effects of microbiota on host physiology and the mechanisms underlying these systemic effects is needed. Here we discuss our recent report demonstrating that gut microbiota affects bone remodeling distally by promoting both bone formation and bone resorption. Our previous work suggested that one mechanism responsible for this is the regulation of circulating IGF-1 levels by microbiota, potentially through the microbial metabolite SCFA. Here we expand on potential mechanisms connecting microbiota and bone by investigating direct effects of SCFA on osteoclasts and exploring whether SCFA also increase serotonin, another circulating molecule with signaling functions that is induced by microbiota. Although butyrate, and to a lesser extent, proprionate inhibit osteoclast formation in a dose and time dependent manner in vitro, a direct effect of these molecules on osteoclasts in vivo is unlikely as the circulating concentration of these molecules is lower than the concentration needed to affect osteoclast differentiation. Interestingly, GPR109 KO mice have a slight increase in bone mass despite increased osteoclast differentiation, suggestive of non-osteoclast functions of butyrate and GPR109 on bone. Indeed, the increase in trabecular bone mass in GPR109 deficient mice is on the order of the increased bone mass seen in GF mice, which have lower levels of butyrate and thus decreased GPR109-mediated signaling. While it is possible that SCFA directly affect the function of other bone cell types, an indirect mechanism is more likely given the low circulating concentrations of SCFA. A good candidate for mediating the effects of SCFA on bone is peripheral serotonin, which, like IGF-1, is increased after colonization. However, while SCFA supplementation was capable of recapitulating many aspects of microbiota effects on bone,6 peripheral serotonin levels were unchanged by SCFA supplementation. Thus, it does not appear that serotonin can be invoked as a mechanism by which microbiota-derived SCFA affect bone. Given the rapid expansion in our understanding of host-microbiota interactions and metabolomics, it is likely that other candidate pathways to explain microbiota effects on bone will be discovered in the near future. Dissecting the mechanisms underlying microbiota-mediated systemic effects on bone will likely provide insight into new therapeutic strategies to improve bone health as well as potential unintended effects of manipulating the microbiota for treatment of C. difficile infection, metabolic disorders and autoimmune disease.
Disclosure of potential conflicts of interest
No potential conflicts of interest were disclosed.
Acknowledgments
We thank Jeremy W. Herzog, Maureen Ann Bower, Ryan Balfour Sartor from National Gnotobiotic Rodent Resource Center, University of North Carolina at Chapel Hill for helpful advice and performing gnotobiotic studies.
Funding
This work was supported by NIH grants AG046257 from the NIA, AR062590 from NIAMS, a Faculty Career Development Award from the Brigham and Women's Hospital and the Bettina Looram Fund. Gnotobiotic experiments were performed at the National Gnotobiotic Rodent Resource Center at University of North Carolina at Chapel Hill, which is supported by funding from NIH grants 5-P30-DK034987 and 5-P40-OD010995 and the Crohn's and Colitis Foundation of America.
References
- [1].Iwami K, Moriyama T. Effects of short chain fatty acid, sodium butyrate, on osteoblastic cells and osteoclastic cells. Int J Biochemis. 1993;25:1631-1635. doi: 10.1016/0020-711X(93)90522-G. PMID:8288032 [DOI] [PubMed] [Google Scholar]
- [2].Katono T, Kawato T, Tanabe N, Suzuki N, Iida T, Morozumi A, Ochiai K, Maeno M. Sodium butyrate stimulates mineralized nodule formation and osteoprotegerin expression by human osteoblasts. Arch Oral Bio. 2008;53:903-909. doi: 10.1016/j.archoralbio.2008.02.016. PMID:18406397 [DOI] [PubMed] [Google Scholar]
- [3].Luo Y, Chen GL, Hannemann N, Ipseiz N, Kronke G, Bauerle T, Munos L, Wirtz S, Schett G, Bozec A. Microbiota from Obese Mice Regulate Hematopoietic Stem Cell Differentiation by Altering the Bone Niche. Cell Metab. 2015;22:886-894. doi: 10.1016/j.cmet.2015.08.020. PMID:26387866 [DOI] [PubMed] [Google Scholar]
- [4].Evans JM, Morris LS, Marchesi JR. The gut microbiome: the role of a virtual organ in the endocrinology of the host. J Endocrinol. 2013;218:R37-47. doi: 10.1530/JOE-13-0131. PMID:23833275 [DOI] [PubMed] [Google Scholar]
- [5].Fung TC, Olson CA, Hsiao EY. Interactions between the microbiota, immune and nervous systems in health and disease. Nat Neurosci. 2017;20:145-155. doi: 10.1038/nn.4476. PMID:28092661 [DOI] [PMC free article] [PubMed] [Google Scholar]
- [6].Yan J, Herzog JW, Tsang K, Brennan CA, Bower MA, Garrett WS, Sartor BR, Aliprantis AO, Charles JF. Gut microbiota induce IGF-1 and promote bone formation and growth. Proc Natl Acad Sci U S A. 2016;113:E7554-E7563. doi: 10.1073/pnas.1607235113. PMID:27821775 [DOI] [PMC free article] [PubMed] [Google Scholar]
- [7].Sjogren K, Engdahl C, Henning P, Lerner UH, Tremaroli V, Lagerquist MK, Backhed F, Ohlsson C. The gut microbiota regulates bone mass in mice. J Bone Miner Res: the official journal of the American Society for Bone and Mineral Research. 2012;27:1357-1367. doi: 10.1002/jbmr.1588. PMID:22407806 [DOI] [PMC free article] [PubMed] [Google Scholar]
- [8].Schwarzer M, Makki K, Storelli G, Machuca-Gayet I, Srutkova D, Hermanova P, Martino ME, Balmand S, Hudcovic T, Heddi A, et al.. Lactobacillus plantarum strain maintains growth of infant mice during chronic undernutrition. Science. 2016;351:854-857. doi: 10.1126/science.aad8588. PMID:26912894 [DOI] [PubMed] [Google Scholar]
- [9].Li JY, Chassaing B, Tyagi AM, Vaccaro C, Luo T, Adams J, Darby TM, Weitzmann MN, Mulle JG, Gewirtz AT, et al.. Sex steroid deficiency-associated bone loss is microbiota dependent and prevented by probiotics. J Clin Invest. 2016;126:2049-2063. doi: 10.1172/JCI86062. PMID:27111232 [DOI] [PMC free article] [PubMed] [Google Scholar]
- [10].Guss JD, Horsfield MW, Fontenele FF, Sandoval TN, Luna M, Apoorva F, Lima SF, Bicalho RC, Singh A, Ley RE, et al.. Alterations to the Gut Microbiome Impair Bone Strength and Tissue Material Properties. J Bone Miner Res: the official journal of the American Society for Bone and Mineral Research. 2017;32:1343-1353. doi: 10.1002/jbmr.3114. PMID:28244143 [DOI] [PMC free article] [PubMed] [Google Scholar]
- [11].Husted AS, Trauelsen M, Rudenko O, Hjorth SA, Schwartz TW. GPCR-Mediated Signaling of Metabolites. Cell Metab. 2017;25:777-796. doi: 10.1016/j.cmet.2017.03.008. PMID:28380372 [DOI] [PubMed] [Google Scholar]
- [12].Koh A, De Vadder F, Kovatcheva-Datchary P, Backhed F. From Dietary Fiber to Host Physiology: Short-Chain Fatty Acids as Key Bacterial Metabolites. Cell. 2016;165:1332-1345. doi: 10.1016/j.cell.2016.05.041. PMID:27259147 [DOI] [PubMed] [Google Scholar]
- [13].Smith PM, Howitt MR, Panikov N, Michaud M, Gallini CA, Bohlooly YM, Glickman JN, Garrett WS. The microbial metabolites, short-chain fatty acids, regulate colonic Treg cell homeostasis. Science. 2013;341:569-573. doi: 10.1126/science.1241165. PMID:23828891 [DOI] [PMC free article] [PubMed] [Google Scholar]
- [14].Candido EP, Reeves R, Davie JR. Sodium butyrate inhibits histone deacetylation in cultured cells. Cell. 1978;14:105-113. doi: 10.1016/0092-8674(78)90305-7. PMID:667927 [DOI] [PubMed] [Google Scholar]
- [15].Rahman MM, Kukita A, Kukita T, Shobuike T, Nakamura T, Kohashi O. Two histone deacetylase inhibitors, trichostatin A and sodium butyrate, suppress differentiation into osteoclasts but not into macrophages. Blood. 2003;101:3451-3459. doi: 10.1182/blood-2002-08-2622. PMID:12511413 [DOI] [PubMed] [Google Scholar]
- [16].Zandi-Nejad K, Takakura A, Jurewicz M, Chandraker AK, Offermanns S, Mount D, Abdi R. The role of HCA2 (GPR109A) in regulating macrophage function. FASEB journal: official publication of the Federation of American Societies for Experimental Biology. 2013;27:4366-4374. doi: 10.1096/fj.12-223933. PMID:23882124 [DOI] [PMC free article] [PubMed] [Google Scholar]
- [17].Chang PV, Hao L, Offermanns S, Medzhitov R. The microbial metabolite butyrate regulates intestinal macrophage function via histone deacetylase inhibition. Proceedings of the National Academy of Sciences of the United States of America. 2014;111:2247-2252. doi: 10.1073/pnas.1322269111. PMID:24390544 [DOI] [PMC free article] [PubMed] [Google Scholar]
- [18].Yadav VK, Balaji S, Suresh PS, Liu XS, Lu X, Li Z, Guo XE, Mann JJ, Balapure AK, Gershon MD, et al.. Pharmacological inhibition of gut-derived serotonin synthesis is a potential bone anabolic treatment for osteoporosis. Nat Med. 2010;16:308-312. doi: 10.1038/nm.2098. PMID:20139991 [DOI] [PMC free article] [PubMed] [Google Scholar]
- [19].Martinez RC, Wachsman M, Torres NI, LeBlanc JG, Todorov SD, Franco BD. Biochemical, antimicrobial and molecular characterization of a noncytotoxic bacteriocin produced by Lactobacillus plantarum ST71KS. Food Microbiol. 2013;34:376-381. doi: 10.1016/j.fm.2013.01.011. PMID:23541205 [DOI] [PubMed] [Google Scholar]
- [20].Bakhtiar SM, LeBlanc JG, Salvucci E, Ali A, Martin R, Langella P, Chatel JM, Miyoshi A, Bermudez-Humaran LG, Azevedo V. Implications of the human microbiome in inflammatory bowel diseases. FEMS Microbiol Lett. 2013;342:10-17. doi: 10.1111/1574-6968.12111. PMID:23431991 [DOI] [PubMed] [Google Scholar]
- [21].LeBlanc JG, Milani C, de Giori GS, Sesma F, van Sinderen D, Ventura M. Bacteria as vitamin suppliers to their host: a gut microbiota perspective. Curr Opin Biotechnol. 2013;24:160-168. doi: 10.1016/j.copbio.2012.08.005. PMID:22940212 [DOI] [PubMed] [Google Scholar]
- [22].Teitler M, Herrick-Davis K, Purohit A. Constitutive activity of G-protein coupled receptors: emphasis on serotonin receptors. Curr Top Med Chem. 2002;2:529-538. doi: 10.2174/1568026023393859. PMID:12052192 [DOI] [PubMed] [Google Scholar]
- [23].Yano JM, Yu K, Donaldson GP, Shastri GG, Ann P, Ma L, Nagler CR, Ismagilov RF, Mazmanian SK, Hsiao EY. Indigenous bacteria from the gut microbiota regulate host serotonin biosynthesis. Cell. 2015;161:264-276. doi: 10.1016/j.cell.2015.02.047. PMID:25860609 [DOI] [PMC free article] [PubMed] [Google Scholar]
- [24].Tsavkelova EA, Klimova S, Cherdyntseva TA, Netrusov AI. [Hormones and hormone-like substances of microorganisms: a review]. Prikl Biokhim Mikrobiol. 2006;42:261-268. PMID:16878539 [PubMed] [Google Scholar]
- [25].Chabbi-Achengli Y, Coudert AE, Callebert J, Geoffroy V, Cote F, Collet C, de Vernejoul MC. Decreased osteoclastogenesis in serotonin-deficient mice. Proceedings of the National Academy of Sciences of the United States of America. 2012;109:2567-2572. doi: 10.1073/pnas.1117792109. PMID:22308416 [DOI] [PMC free article] [PubMed] [Google Scholar]