

ABSTRACT
Clostridium difficile infection (CDI) is a common complication in inflammatory bowel disease (IBD) and has been associated with poor IBD outcome. Intestinal microbiota composition in IBD patients with CDI has not been specifically evaluated to date. The fecal microbiota of 56 IBD patients, including 8 in flare with concomitant CDI, 24 in flare without CDI, and 24 in remission, as well as 24 healthy subjects, was studied using 16S sequencing. Analysis was performed using the Qiime pipeline. Compared to IBD patients without CDI, IBD patients with CDI had more pronounced dysbiosis with higher levels of Ruminococcus gnavus and Enterococcus operational taxonomic units (OTUs) and lower levels of Blautia and Dorea OTUs. Correlation network analysis suggested a disrupted ecosystem in IBD patients in flare, particularly in those with CDI. In patients with IBD, CDI is associated with a more pronounced intestinal dysbiosis with specific alterations in intestinal microorganisms.
KEYWORDS: IBD, Clostridium difficile
Introduction
Clostridium difficile infection (CDI) is a common complication of inflammatory bowel disease (IBD) and has been associated with a poor IBD outcome.1 CDI typically occurs after a transient microbiota perturbation in the gut. Thus, antibiotic intake is a major risk factor in the general population. In IBD, as the gut microbiota is basally perturbed (i.e., dysbiosis),2 CDI can occur even without an antibiotic trigger.
Microbiota composition in patients with CDI is imbalanced and characterized by decreased biodiversity,3,4 as well as notable increases and decreases in several taxa that are known to be increased and decreased in IBD,4 respectively. For example, Bifidobacterium and members of Clostridium clusters IV and XIVa (such as Faecalibacterium prausnitzii or Roseburia intestinalis) were decreased, while Enterobacteriaceae and Enterococcus were increased.5 However, intestinal microbiota composition in IBD patients with CDI has not been specifically evaluated to date.
Results
Population, microbiota composition and diversity
Our study population was composed of 56 IBD patients, including 8 in flare with concomitant CDI, 24 in flare without CDI, and 24 patients in remission, as well as 24 healthy subjects (HS) (Table 1). IBD patients in flare without CDI and patients in remission were matched to patients in flare with CDI (3:1) based on IBD subtype and topography. None of the study participants reported having taken antibiotics or using colon-cleansing products for at least 2 months before enrollment.
Table 1.
Characteristics of patients.
| IBD flare CDI (n = 8) | IBD flare (n = 24) | IBD remission (n = 24) | HS (n = 24) | ||||
|---|---|---|---|---|---|---|---|
| Age: Year (mean +/− SD) | 33.4 +/− 10.5 | 36.5 +/− 11.6 | 36.5 +/− 11.6 | 33.0 +/− 10.2 | |||
| Male: n (%) | 5 (62.6%) | 14 (58.3%) | 14 (58.3%) | 15 (62.5%) | |||
| Active smoking: n(%) | 2 (25.0%) | 9 (37.5%) | 9 (37.5%) | 1 (4.2%) | |||
| CD/UC: n(%) | CD | UC | CD | UC | CD | UC | NA |
| n (%) | 6 (75%) | 2 (25%) | 18 (75%) | 6 (25%) | 18 (75%) | 6 (25%) | NA |
| Montreal classification | |||||||
| A1/A2/A3 (n) | 6/0/0 | NA | 4/12/2 | NA | 3/15/0 | NA | NA |
| L1/L2/L3 (n) | 2/2/2 | NA | 6/6/6 | NA | 6/6/6 | NA | NA |
| B1/B2/B3 (n) | 4/0/2 | NA | 9/2/7 | NA | 6/2/10 | NA | NA |
| E1/E2/E3 (n) | NA | 0/1/1 | NA | 0/3/3 | NA | 0/3/3 | NA |
| Treatment: n(%) | NA | ||||||
| 5-ASA | 3 (37.5%) | 10 (41.7%) | 6 (25.9%) | 0 | |||
| Coticosteroids | 1 (12.5%) | 10 (41.7%) | 1 (4.2%) | 0 | |||
| Thiopurine or MTX | 1 (12.5%) | 10 (41.7%) | 10 (41.7%) | 0 | |||
| anti-TNFa | 2 (25.0%) | 13 (54.2%) | 19 (79.2%) | 0 | |||
| Antibiotics | 0 | 0 | 0 | 0 | |||
β diversity analysis showed a clustering of samples by disease phenotype and a progressive microbiota shift from HS to IBD in remission, to IBD in flare and finally to IBD in flare with CDI (Fig. 1A-B). Compared with the HS samples, the α diversity (assessed by 4 different indexes) was significantly decreased in IBD patients, particularly in samples from patients in flare (Fig. 1C, Fig. S1A-B). There was no significant difference between patients with and without CDI.
Figure 1.
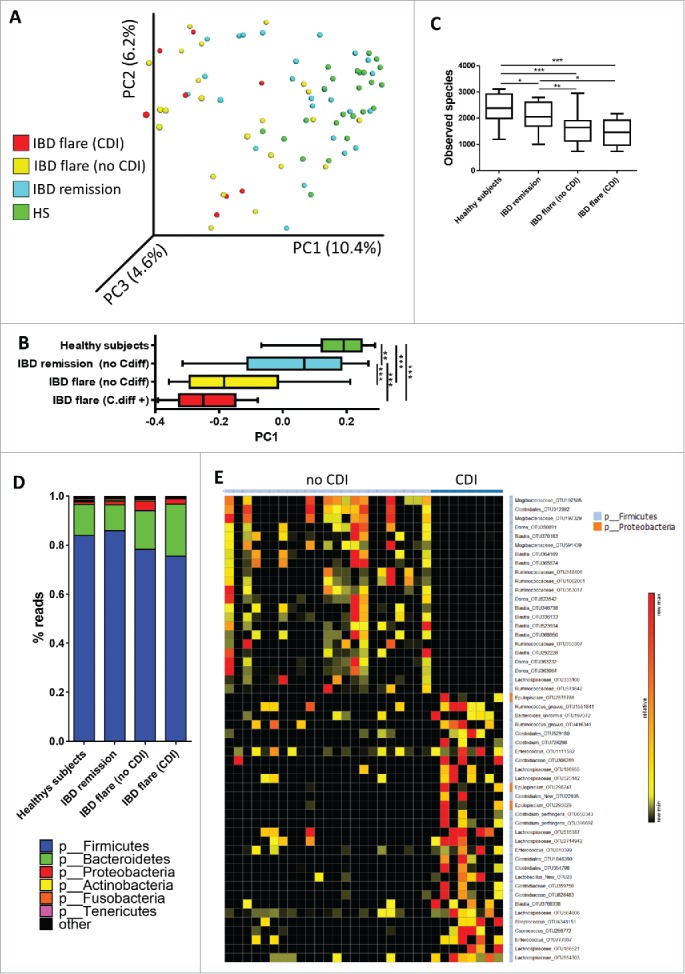
Bacterial microbiota biodiversity and composition. (A) β diversity. Principal coordinate analysis of the Bray Curtis distance with each sample colored by the disease phenotype. PC1, PC2, and PC3 represent the top 3 principal coordinates that captured the most diversity, with the fraction of diversity captured by that coordinate shown as a percentage. (B) PC1 value according disease phenotype (t test, ** = p < 0.01; *** = p < 0.001). (C) OTU number describing the diversity of the bacterial microbiota in the different groups studied (t test,* = p < 0.05; ** = p < 0.01; *** = p < 0.001). (D) Global composition of bacteria at the phyla level. HS and patient sub-groups are labeled on the X-axis and expressed as the relative OTU abundance per group. (E) Bacterial taxa differentially enriched in IBD patients in flare both with and without CDI (generated using LeFSE analysis, LDA score >2).
As expected, most of the bacteria from both the IBD and HS samples belonged to the phyla Firmicutes, Bacteroidetes, Proteobacteria and Actinobacteria (Fig. 1D), and the distribution of these phyla in the patients was in agreement with published data.6 Firmicutes and Bacteroidetes were the most abundant in HS. In IBD patients, particularly those in flare, there was a decrease in Firmicutes (mostly from the Ruminococcaceae family) associated with an increase of Proteobacteria (mostly from the Enterobacteriaceae family; Fig. 1D, Fig. S1C).
Differential bacterial composition and altered microbial network in CDI
To identify more precisely the impact of CDI on IBD microbiota, we directly compared the bacterial composition of IBD patients in flare, both with and without CDI, using LEfSe (Linear discriminant analysis Effect Size).7 We identified several OTU that differentiated patients with and without CDI (Fig. 1E, Table S1). Several Blautia and Dorea OTUs were decreased in patients with CDI, whereas several Clostridium, Ruminococcus gnavus and Enterococcus OTUs were increased in patients with CDI.
To evaluate globally the microbial gut ecosystem and its alterations in IBD and CDI, we built a microbial correlation network by phenotype (Fig. 2). In HS, we observed a highly connected network between many bacterial OTUs, suggesting the presence of a balanced and cooperative ecosystem. Although smaller than that of HS, the correlation network was also well-connected in IBD patients in remission. In contrast, the correlation network was disrupted in IBD patients in flare, with separated bacterial clusters being detected. For IBD patients in flare who also had CDI, the alterations were even more pronounced with a lower relative connectedness (2.1 vs 5.4 in IBD patients in flare without CDI, p < 10−8) and no interaction between several groups of bacteria, suggesting a loss of cooperation in the ecosystem.
Figure 2.
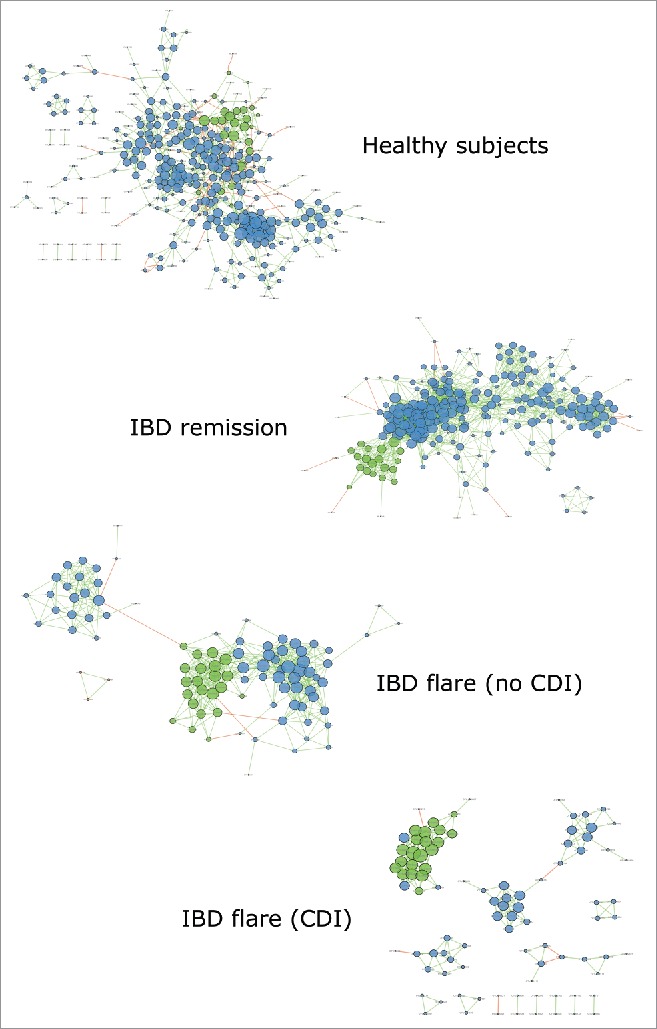
Bacterial microbiota correlation network. Correlation network for the HS, IBD in remission, and IBD in flare both without and with CDI was generated using Cytoscape. Each circle (node) represents an OTU, the color represents its bacterial phylum (blue: Firmicutes; green: Bacteroidetes; orange: Proteobacteria), and its size represents the number of direct edges that it has. The edge color indicates the magnitude of the distance correlation; green indicates a positive correlation and red indicates a negative correlation (determined using the Spearman test). Statistical significance was determined for all pairwise comparisons; only significant correlations (p value < 0.05 after FDR correction) are displayed.
Discussion
To the best of our knowledge, this report presents the first detailed analysis of the intestinal microbiota of IBD patients both with and without CDI. In accordance with previous studies, we confirmed that the intestinal microbiota of IBD patients is dysbiotic. The current study showed that CDI in patients with IBD flare is associated with a more severe dysbiosis than in an IBD flare without CDI. This infection is characterized by specific alterations in intestinal microorganisms with an impaired microbial ecosystem. Interestingly, among the decreased bacterial OTUs in patients with CDI, we identified many genera known to be deficient in IBD compared with HS, such as Blautia and Dorea.6 In addition, the Ruminococcus gnavus and Enterococcus OTUs that were increased in the patients with CDI have also been shown to be overrepresented in IBD patients compared with HS.8,9 This finding indicates a deeper dysbiosis in IBD patients with CDI. Moreover, several CDI features in non-IBD patients, such as increased numbers of Enterococcus,5 were also found in the IBD patients.
In conclusion, this study showed that in patients with IBD, CDI is associated with a more pronounced intestinal dysbiosis. Whether this feature is a cause or a consequence of CDI is not established. Therefore, the next step will be to investigate microbiota factors associated with the risk of developing CDI in IBD as a prerequisite for preventive intervention.
Materials and methods
Patients and sample collection
All patients were recruited at the Gastroenterology Department of the Saint Antoine Hospital (Paris, France) and provided informed consent. Approval was obtained from the local ethics committee (Comite de Protection des Personnes Ile-de-France IV, Suivitheque study). A diagnosis of IBD was defined by clinical, radiological, endoscopic and histological criteria. A flare was determined by the physician in charge of the patient based on clinical and biologic data. None of the study participants reported having taken antibiotics or using colon-cleansing products for at least 2 months before enrollment. Patient characteristics are presented in Table 1. Samples from healthy subjects (HS) and from IBD patients without a Clostridium difficile infection were described previously.2 Whole stools were collected in sterile boxes and immediately homogenized, and 0.2 g aliquots were frozen at −80°C for further analysis.
Fecal DNA extraction
Genomic DNA was extracted from 200 mg of feces as described previously.1 Following microbial lysis with both mechanical and chemical steps, nucleic acids were precipitated in isopropanol for 10 minutes at room temperature, incubated for 15 minutes on ice and centrifuged for 30 minutes at 15,000 g and 4°C. Pellets were suspended in 112 µL of phosphate buffer and 12 µL of potassium acetate. After RNase treatment and DNA precipitation, nucleic acids were recovered via centrifugation at 15,000 g and 4°C for 30 minutes. The DNA pellet was suspended in 100 µL of TE buffer.
16S rRNA gene sequencing
After extraction, total DNA concentration was measured using PicoGreen (Invitrogen), and global 16S gene DNA copy numbers were measured using a qPCR method adapted from Maeda et al.,10 enabling inhibition effect estimation and DNA concentration adjustment. The sequence region of the 16S rRNA gene spanning the variable region V3-V5 was amplified using the broad-range forward primer, For16S_519 (CAGCMGCCGCGGTAATAC), and reverse primer, Rev16S_926 (CCGTCAATTCMTTTGAGTTT). The amplification reaction (initial activation step at 94°C for 1 min, followed by 30 cycles of 94°C for 15 s, 43°C for 15 s, 68°C for 45 s and a final incubation step at 68°C for 1 min) was performed in a total volume of 100 µL containing 1X PCR buffer, 2 mM MgSO4, 1 U of DNA High Fidelity Taq Polymerase (Invitrogen, Carlsbad, CA), 625 nM of each barcoded primer (IDT), 250 µM of each dNTP (Invitrogen) and the concentration-adjusted DNA sample. A bidirectional library was prepared using the One Touch2 Template Kit and sequenced on PGM Ion Torrent using the Ion PGM Sequencing 400 Kit (Life Technologies, Carlsbad, CA).
16S rRNA gene sequence analysis
The sequences were demultiplexed and quality filtered using the Quantitative Insights Into Microbial Ecology (QIIME, version 1.8.0) software package.11 The sequences were trimmed for barcodes and PCR primers and were binned for a minimal sequence length of 200 bp. The sequences were then assigned to (OTUs) using the UCLUST algorithm12 with a 97% threshold pairwise identity and taxonomically classified using the Greengenes reference database.13 Rarefaction was performed (10,000 sequences per sample) and used to compare OTU abundances across samples. Principal component analyses (PCA) of the Bray Curtis distance, with each sample colored according to disease phenotype, were built and used to assess the variation between experimental groups. The number of observed species, as well as the Shannon, Simpson and Chao1 diversity indexes, were calculated using rarefied data (depth = 10,000 sequences/sample) and used to characterize species diversity in a community.
Clostridium difficile testing
Diagnosis of CDI was performed using a stool culture on selective medium (TCCA, taurocholate cycloserine cefoxitine medium) and a stool cytotoxicity assay on MRC-5 cells. In cases in which a positive culture and negative stool cytotoxicity assay were obtained, a toxigenic culture (determination of the isolate's ability to produce toxins in vitro) was performed.
Statistical analysis
GraphPad Prism version 6.0 (San Diego, CA) was used for all analyses and graph preparation. For all graphical data, the results are expressed as the mean ± SEM, and statistical analyses were performed using the 2-tailed nonparametric Mann–Whitney U-test or the Kruskal-Wallis test with Dunn's multiple comparison test. Statistical significance of the sample grouping for the β diversity analysis was performed using the Permanova method (9999 permutations). Differences with a P value less than 0.05 were considered to be significant.
Comparisons between IBD patients with and without CDI was performed using the linear discriminant analysis (LDA) effect size (LEfSe) pipeline.7
A correlation network was built using an OTU table and the Spearman correlation. Only OTUs present in >50% of the samples were used. The p values were corrected using the Benjamini and Hochberg procedure to control for the false discovery rate (FDR). Correlation network figures were built using Cytoscape 3.1.1. The relative connectedness of the networks was calculated by dividing the number of significant interactions (edges) by the number of taxa (nodes) in the network. Statistical significance was assessed by a chi-square test.
Supplementary Material
Abbreviations
- CD
Crohn's disease
- HS
healthy subjects
- IBD
inflammatory bowel diseases
- UC
ulcerative colitis
Disclosure of potential conflicts of interest
The authors disclose no conflicts of interest.
Acknowledgments
We thank URC-EST and CRB HUEP-UPMC for their help in collecting and storing the samples used in this study.
Funding
This study was supported by an unrestricted Biocodex grant.
Authors' contributions
HS designed and supervised the study. VL, SJ and CM provided technical help. HS, CL, JK, GL, AB, INL, JC, PS, LB, recruited the patients, HS performed the analysis and interpreted the data. HS wrote the manuscript.
The authors state that the manuscript, including related data, figures and tables, has not been previously published and that the manuscript is not under consideration elsewhere.
References
- [1].Ananthakrishnan AN, McGinley EL, Saeian K, Binion DG. Temporal trends in disease outcomes related to Clostridium difficile infection in patients with inflammatory bowel disease. Inflamm Bowel Dis. 2011. April;17(4):976-83. doi: 10.1002/ibd.21457 [DOI] [PubMed] [Google Scholar]
- [2].Sokol H, Leducq V, Aschard H, Pham HP, Jegou S, Landman C, Cohen D, Liguori G, Bourrier A, Nion-Larmurier I, et al.. Fungal microbiota dysbiosis in IBD. Gut. 2017. June;66(6):1039-1048. doi: 10.1136/gutjnl-2015-310746 [DOI] [PMC free article] [PubMed] [Google Scholar]
- [3].van Nood E, Vrieze A, Nieuwdorp M, Fuentes S, Zoetendal EG, de Vos WM, Visser CE, Kuijper EJ, Bartelsman JF, Tijssen JG, et al.. Duodenal infusion of donor feces for recurrent Clostridium difficile. N Engl JMed. 2013. January 31;368(5):407-15. doi: 10.1056/NEJMoa1205037 [DOI] [PubMed] [Google Scholar]
- [4].Milani C, Ticinesi A, Gerritsen J, Nouvenne A, Lugli GA, Mancabelli L, TurroniF DS, Mangifesta M, Viappiani A, et al.. Gut microbiota composition and Clostridium difficile infection in hospitalized elderly individuals: a metagenomic study. Sci Rep. 2016. May 11;6:25945. doi: 10.1038/srep25945 [DOI] [PMC free article] [PubMed] [Google Scholar]
- [5].Seekatz AM, Young VB. Clostridium difficile and the microbiota. J Clin Invest. 2014. October;124(10):4182-9. doi: 10.1172/JCI72336 [DOI] [PMC free article] [PubMed] [Google Scholar]
- [6].Morgan XC, Tickle TL, Sokol H, Gevers D, Devaney KL, Ward DV, Reyes JA, Shah SA, LeLeiko N, Snapper SB, et al.. Dysfunction of the intestinal microbiome in inflammatory bowel disease and treatment. Genome Biol. 2012. April 16;13(9):R79. doi: 10.1186/gb-2012-13-9-r79 [DOI] [PMC free article] [PubMed] [Google Scholar]
- [7].Segata N, Izard J, Waldron L, Gevers D, Miropolsky L, Garrett WS, Huttenhower C. Metagenomic biomarker discovery and explanation. Genome Biol. 2011. June 24;12(6):R60. doi: 10.1186/gb-2011-12-6-r60 [DOI] [PMC free article] [PubMed] [Google Scholar]
- [8].Hoffmann TW, Pham HP, Bridonneau C, Aubry C, Lamas B, Martin-Gallausiaux C, Moroldo M, Rainteau D, Lapaque N, Six A, et al.. Microorganisms linked to inflammatory bowel disease-associated dysbiosis differentially impact host physiology in gnotobiotic mice. ISME J. 2016. February;10(2):460-77. doi: 10.1038/ismej.2015.127 [DOI] [PMC free article] [PubMed] [Google Scholar]
- [9].Willing BP, Dicksved J, Halfvarson J, Andersson AF, Lucio M, Zheng Z, Järnerot G, Tysk C, Jansson JK, Engstrand L. A pyrosequencing study in twins shows that gastrointestinal microbial profiles vary with inflammatory bowel disease phenotypes. Gastroenterology. 2010. December;139(6):1844-1854.e1. doi: 10.1053/j.gastro.2010.08.049 [DOI] [PubMed] [Google Scholar]
- [10].Maeda H, Fujimoto C, Haruki Y, Maeda T, Kokeguchi S, Petelin M, Arai H, Tanimoto I, Nishimura F, Takashiba S. Quantitative real-time PCR using TaqMan and SYBR Green for Actinobacillus actinomycetemcomitans, Porphyromonas gingivalis, Prevotella intermedia, tetQ gene and total bacteria. FEMS Immunol Med Microbiol. 2003; 39:81-86. doi: 10.1016/S0928-8244(03)00224-4. PMID:14557000 [DOI] [PubMed] [Google Scholar]
- [11].Caporaso JG, Kuczynski J, Stombaugh J, Bittinger K, Bushman FD, Costello EK, Fierer N, Peña AG, Goodrich JK, Gordon JI, et al.. QIIME allows analysis of high-throughput community sequencing data. Nat Methods. 2010. May;7(5):335-6. doi: 10.1038/nmeth.f.303 [DOI] [PMC free article] [PubMed] [Google Scholar]
- [12].Edgar RC. Search and clustering orders of magnitude faster than BLAST. Bioinformatics. 2010; 26:2460-2461. doi: 10.1093/bioinformatics/btq461. PMID:20709691 [DOI] [PubMed] [Google Scholar]
- [13].McDonald D, Price MN, Goodrich J, Nawrocki EP, DeSantis TZ, Probst A, Andersen GL, Knight R, Hugenholtz P. An improved Greengenes taxonomy with explicit ranks for ecological and evolutionary analyses of bacteria and archaea. ISME J. 2012; 6:610-618. doi: 10.1038/ismej.2011.139. PMID:22134646 [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.