

Abstract
A mechanism that enables direct aldehyde C–H functionalization has been achieved via the synergistic merger of photoredox, nickel, and hydrogen atom transfer (HAT) catalysis. This mild, operationally simple protocol transforms a wide variety of commercially available aldehydes, along with aryl or alkyl bromides, into the corresponding ketones in excellent yield. This C–H abstraction coupling technology has been successfully applied to the expedient synthesis of the medicinal agent haloperidol.
Graphical Abstract
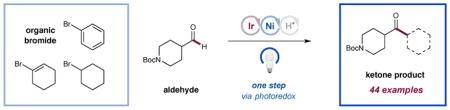
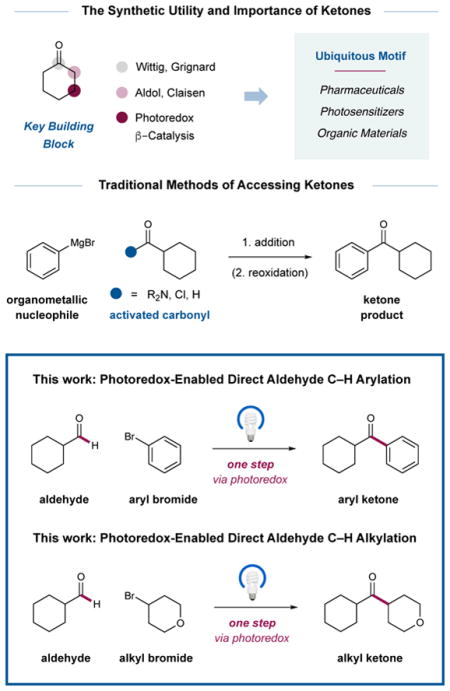
Ketones are important and ubiquitous structural motifs found in natural products, pharmaceuticals, photosensitizers, flavors, fragrances, and organic materials.1 Moreover, within the field of synthetic organic chemistry, they represent a versatile building block that can be utilized in a diverse array of powerful bond-forming reactions. Conventional methods for the synthesis of ketones include (i) the addition of organometallic species to carbonyl moities that include amides, anhydrides, and acid chlorides, (ii) aromatic substitution protocols such as Friedel–Crafts acylations,2 and (iii) transition metal-catalyzed couplings of acid chlorides with aryl and alkyl stannanes and boronic esters. 3,4 Recently, an alternative yet equally attractive approach to ketone formation has emerged that involves metal-catalyzed formyl C–H functionalization, wherein aryl iodides and aldehydes were successfully combined as coupling partners. 5 While these transformations require the use of stoichiometric oxidants, reductants, or directing groups, it is important to recognize that these seminal studies were the first to employ widely available aldehydes (enolizable and non-enolizable) in a C–H arylation cross-coupling mechanism. Recently, we questioned if this overall strategy might be accomplished in a general sense using widely available aryl bromides (and for the first time alkyl halides) via the combination of three interconnected catalytic cycles, namely photoredox, nickel, and HAT catalysis. Given the intrinsic utility of ketones along with the widespread availability of both coupling partners, we envisioned that this new C–H abstraction–arylation/alkylation redox mechanism would be of conceptual and practical interest to chemists in both academic and industrial settings.
The merger of photoredox and transition metal catalysis (now termed metallaphotoredox) has enabled the invention of a large and varied range of cross-coupling reactions, 6 many of which are now being adopted by the pharmaceutical industry.7 In this context, our laboratory has previously demonstrated a direct sp3 C–H bond functionalization method wherein the synergistic combination of photoredox, nickel, and hydrogen atom transfer catalysis in a triple catalytic activation mechanism allows amines and ethers to undergo direct α-C–H arylation and alkylation. 8 As a critical design element, the exploitation of a polarity-matched hydrogen abstraction step using an electrophilic quinuclidinium radical cation enables a high degree of kinetic selectivity, in that bond dissociation enthalpy (BDE) considerations are less important than C–H bond polarity.9 As a result, electron-rich sp3 C–H bonds undergo selective and efficient H-abstraction/nickel-catalyzed arylation in the presence of weak, acidic or weak, neutral C–H systems found in methyls, methylenes, and methines. With this selectivity paradigm in mind, we recently questioned whether aldehydic sp2 H–C(O) bonds might be activated toward arylation and alkylation using this strategy, thereby providing a new and generic pathway to ketone construction. Herein, we describe the successful implementation of these ideals and present a broadly applicable protocol for metallaphotoredox-mediated aldehyde C–H arylation, vinylation, or alkylation.
The mechanistic details of our proposed transformation are outlined in Scheme 1. Photoexcitation with visible light of photocatalyst Ir[dF(CF3)ppy]2(dtbbpy)PF6 (1) is known to produce the strongly oxidizing complex *Ir[dF(CF3)ppy]2 (dtbbpy)+ (2) (E1/2red [*IrIII/IrII] = +1.21 V vs. the saturated calomel electrode (SCE) in CH3CN).10 The *Ir(III) excited state 2 can effect the oxidation of quinuclidine (3) (E1/2ox = +1.1 V vs. SCE in CH3CN) 11 to form cationic radical 4 and the reduced Ir(II) complex 5. At this stage, we proposed that quinuclidinium radical cation (4) should engage in a HAT event with any given aldehyde (N-Boc-4-piperidinecarboxaldehyde (6) is shown) to generate the corresponding acyl radical 7.12 With respect to regiocontrol, we hypothesized that the hydrogen abstraction event should be selective for the formyl C–H bond given that it is both hydridic and relatively weak in comparison to most other C–H moieties. For example, the difference in the bond dissociation enthalpies of aldehyde C–H bonds vs. α-amino C–H bonds (both of which exhibit hydridic bond polarization) might be sufficient to ensure exclusive formation of the acyl radical species 7. At this stage, concurrent oxidative addition of aryl bromide 10 to LnNi0 species 9 should deliver the aryl–NiII species 11, which we hoped would be rapidly intercepted by the radical 7 to form the acyl–NiIII complex 12.13 Thereafter, reductive elimination would afford the desired ketone product 14 and NiI species 13. As a critical step, both the nickel and photoredox catalytic cycles would simultaneously turn over via single electron transfer from the reduced IrII species 5 (E1/2red [IrIII/IrII] = −1.37 V vs. SCE in CH3CN)10 to the NiI complex 13 (E1/2red[NiII/Ni0] = −1.2 V vs. SCE in DMF).14 Finally, the quinuclidine catalyst would be regenerated via deprotonation of the quinuclidinium ion 8 with inorganic base.
Scheme 1.

Proposed Mechanism for Aldehyde C–H Arylation and Alkylation via a Triple Catalysis Mechanism.
Our investigation into this new HAT-metallaphotoredox-mediated aldehyde C–H arylation began with exposure of N-Boc-4-piperidinecarboxaldehyde and 5-bromo-2-(trifluoromethyl)pyridine to visible light in the presence of Ir[dF(CF3)ppy]2(dtbbpy)PF6 (1), NiBr2·dtbbpy, quinuclidine and K2CO3. Given the requirement of high dielectric solvents in our previous C–H abstraction/arylation studies, we anticipated that the desired transformation would be possible using DMSO or CH3CN; however, the efficiency of ketone product formation was poor (Table 1, entries 1–2, 2–8% yield). To our surprise, we observed that a competing α-amino C–H arylation mechanism was operating in both media. Remarkably, by switching to 1,4-dioxane, we observed exclusive formation of the desired ketone product (entry 3, 87% isolated yield) without any observable α-amino C–H arylation. We surmised that solvent dielectric constant might play an important role in stabilizing the ionic nature of the quinuclidinium radical cation species and likely impacts the relative roles of BDEs vs. degree of bond polarization. It is worth noting that, under the optimized conditions, the nickel catalyst loading could be reduced without an impact on the efficiency of this reaction (entry 4, 80% yield). Control experiments (entries 5–7) revealed that the photocatalyst, nickel catalyst, quinuclidine, base, and visible light were all essential for the success of this new aldehyde C–H arylation.
Table 1.
Optimization of the Aldehyde C–H Arylation.a
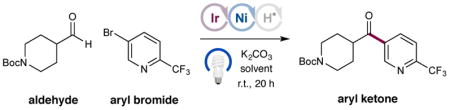
| |||
|---|---|---|---|
| entry | conditions | solvent | yieldb |
| 1 | as shown | DMSO | 2% |
| 2 | as shown | CH3CN | 8% |
| 3 | as shown | dioxane | 92% (87%) |
| 4 | 2 mol% Ni catalyst | dioxane | 80% |
| 5 | no photocatalyst | dioxane | 0% |
| 6 | no Ni catalyst | dioxane | 0% |
| 7 | no light | dioxane | 0% |
Photocat 1 (1 mol%), NiBr2·dtbbpy (10 mol%), quinuclidine (10 mol%), aryl halide (1.0 equiv), aldehyde (2.0 equiv), and K2CO3 (1.5 equiv). Yield by 1H NMR analysis.
Isolated yields in parentheses.
With the optimized conditions in hand, we next sought to determine the generality of the aryl halide component in this new ketone-forming reaction. As shown in Table 2, electron-rich arenes containing alkyl and methoxy groups performed well (15 and 16, 82% and 81% yield, respectively). Moreover, a diverse array of electron-deficient bromoarenes that incorporate a variety of substituents (ketone, ester, nitrile, trifluoromethyl, sulfone, and trifluoromethoxy groups) were found to be competent substrates (17–22, 82–92% yield).15 Bicyclic aromatics such as phthalides and phthalimides were also coupled with high levels of efficiency (23 and 24, 93% and 81% yield, respectively). Notably, the efficiency of the reaction was not impeded by ortho substituents on the aromatic ring (25–26, 88–90% yield). With respect to hetero-aromatic coupling partners, we have found that a range of substituted pyridyl bromides are also effective electrophiles (29–32, 50–90% yield). Perhaps most importantly, this transformation is not limited to electron-deficient pyridines. For example, quinoline, isoquinoline, and pyrimidine can be readily employed in this HAT-metallaphotoredox-mediated aldehyde C–H arylation (33–35, 79–87% yield).
Table 2.
From Aldehydes to Ketone Adducts: Scope of Triple Catalytic Cross-Coupling with Aryl, Alkyl Bromides and Aldehydes.a

|
Isolated Yields. Performed with photocat 1 (1 mol%), NiBr2·dtbbpy (10 mol%), quinuclidine (10 mol%), aryl/alkyl bromide, aldehyde, and K2CO3. See Supporting Information.
K2CO3 (2.0 equiv).
Aldehyde (3.0 equiv).
Aldehyde (10.0 equiv).
Aldehyde (6.0 equiv).
Having demonstrated the capacity of aryl bromides to participate in this new ketone-forming reaction, we were delighted to find that vinyl electrophiles can also be incorporated (36–38, 56–74% yield). Perhaps more important was the finding that alkyl halides can also be employed to generate non-conjugated ketones. Indeed, we have found that this transformation can accommodate cyclic and acyclic aliphatic bromides with useful levels of efficiency (39–41, ≥55% yield). To our knowledge this is the first time that aldehydes have been merged with aliphatic bromides to generate saturated ketones in one chemical step.
We next turned our attention to the scope of the formyl component. As shown in Table 2, an assortment of readily available aldehydes are viable. For example, primary aldehydes are effective coupling partners, including substrates that incorporate carbamate, phenyl, unprotected alcohol, and tert-butyl groups (42–48, 70–92% yield). Notably, acetaldehyde, which is extremely volatile, can be readily employed (44, 70% yield). Moreover, α-branched alkanals were found to readily undergo this C–H arylation (49 and 50, both 91% yield). Ring-bearing formyl systems were also successful, including cyclohexyl, cyclopentyl, cyclopropyl and tetrahydropyranyl carboxaldehyde (51–54, 81–90% yield). Lastly, aromatic aldehydes were found to couple with aryl halides proficiently despite the diminished hydridic nature of these formyl C–H bonds (55–57, 70–73% yield).
To highlight the synthetic utility of this triple catalytic mechanism and its potential application to drug-like molecules, we have accomplished a two-step synthesis of haloperidol, a well-established antipsychotic medication.16 As shown in Figure 1, 4-chlorobutanal 58 and 1-bromo-4-fluorobenzene 59 were successfully combined using our aldehyde coupling protocol to forge ketone 60 in good yield (77%). Exposure of this γ-chloroarylketone to the piperidine nucleophile 61 subsequently delivered haloperidol in relatively short order.
Figure 1.
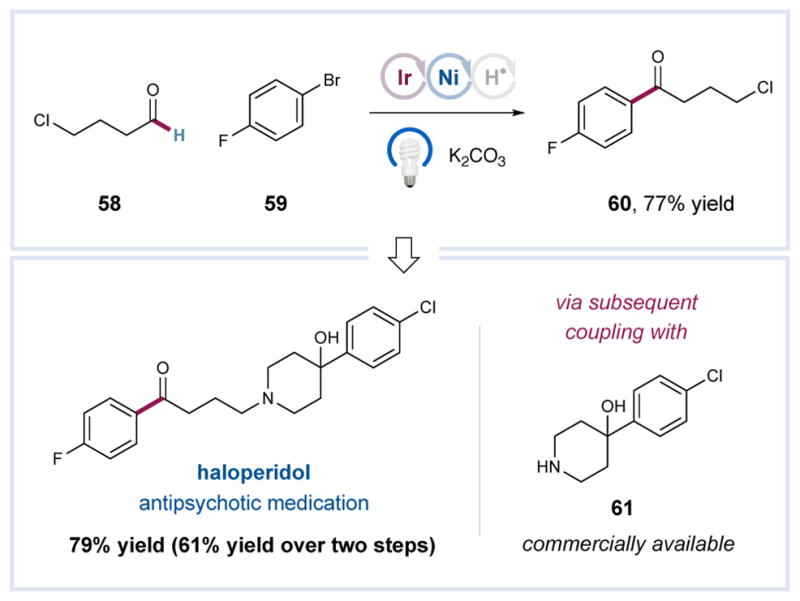
Two-Step Synthesis of Haloperidol.
Supplementary Material
Acknowledgments
Financial support provided by the NIHGMS (R01 GM103558-05) and kind gifts from Merck, BMS, Janssen, and Eli Lilly. X. Z. is grateful for a postdoctoral fellowship from the Shanghai Institute of Organic Chemistry.
Footnotes
The Supporting Information is available free of charge on the ACS Publications website at DOI: (link to DOI)
Experimental procedures and compound characterization data (PDF)
References
- 1.(a) McDaniel R, Thamchaipenet A, Gustafsson C, Fu H, Betlach M, Betlach M, Ashley G. Proc Natl Acad Sci USA. 1999;96:1846. doi: 10.1073/pnas.96.5.1846. [DOI] [PMC free article] [PubMed] [Google Scholar]; (b) Cuquerella MC, Lhiaubet-Vallet V, Cadet J, Miranda MA. Acc Chem Res. 2012;45:1558. doi: 10.1021/ar300054e. [DOI] [PubMed] [Google Scholar]; (c) Kamat PV. Chem Rev. 1993;93:267. [Google Scholar]
- 2.Nahm S, Weinreb SM. Tetrahedron Lett. 1981;22:3818. [Google Scholar]; (b) Sartori G, Maggi R. Chem Rev. 2006;106:1077. doi: 10.1021/cr040695c. [DOI] [PubMed] [Google Scholar]; (c) Bechara WS, Pelletier G, Charette AB. Nat Chem. 2012;4:228. doi: 10.1038/nchem.1268. [DOI] [PubMed] [Google Scholar]
- 3.For selected reviews, see: Miyaura N, Suzuki A. Chem Rev. 1995;95:2457.Willis MC. Chem Rev. 2010;110:725. doi: 10.1021/cr900096x.Wu XF, Neumann H, Beller M. Chem Soc Rev. 2011;40:4986. doi: 10.1039/c1cs15109f.Moragas T, Correa A, Martin R. Chem Eur J. 2014;20:8242. doi: 10.1002/chem.201402509.
- 4.For selected examples, see: Takemiya A, Hartwig JF. J Am Chem Soc. 2006;128:14800. doi: 10.1021/ja064782t.Gooßen LJ, Rudolphi F, Oppel C, Rodríguez N. Angew Chem, Int Ed. 2008;47:3043. doi: 10.1002/anie.200705127.Weires NA, Baker EL, Garg NK. Nat Chem. 2016;8:75. doi: 10.1038/nchem.2388.Amani J, Molander GA. J Org Chem. 2017;82:1856. doi: 10.1021/acs.joc.6b02897.
- 5.(a) Huang YC, Majumdar KK, Cheng CH. J Org Chem. 2002;67:1682. doi: 10.1021/jo010289i. [DOI] [PubMed] [Google Scholar]; (b) Pucheault M, Darses S, Genet JP. J Am Chem Soc. 2004;126:15356. doi: 10.1021/ja044749b. [DOI] [PubMed] [Google Scholar]; (c) Ko S, Kang B, Chang S. Angew Chem Int Ed. 2005;44:455. doi: 10.1002/anie.200462006. [DOI] [PubMed] [Google Scholar]; (d) Ruan J, Saidi O, Iggo JA, Xiao J. J Am Chem Soc. 2008;130:10510. doi: 10.1021/ja804351z. [DOI] [PubMed] [Google Scholar]; (e) Suchand B, Satyanarayana G. J Org Chem. 2016;81:6409. doi: 10.1021/acs.joc.6b01064. [DOI] [PubMed] [Google Scholar]
- 6.(a) Kalyani D, McMurtrey KB, Neufeldt SR, Sanford MS. J Am Chem Soc. 2011;133:18566. doi: 10.1021/ja208068w. [DOI] [PMC free article] [PubMed] [Google Scholar]; (b) Sahoo B, Hopkinson MN, Glorius F. J Am Chem Soc. 2013;135:5505. doi: 10.1021/ja400311h. [DOI] [PubMed] [Google Scholar]; (c) Shu X-z, Zhang M, He Y, Frei H, Toste FD. J Am Chem Soc. 2014;136:5844. doi: 10.1021/ja500716j. [DOI] [PMC free article] [PubMed] [Google Scholar]; (d) Zuo Z, Ahneman DT, Chu L, Terrett JA, Doyle AG, MacMillan DWC. Science. 2014;345:437. doi: 10.1126/science.1255525. [DOI] [PMC free article] [PubMed] [Google Scholar]; (e) Tellis JC, Primer DN, Molander GA. Science. 2014;345:433. doi: 10.1126/science.1253647. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.(a) Corcoran EB, Pirnot MT, Lin S, Dreher SD, DiRocco DA, Davies IW, Buchwald SL, MacMillan DWC. Science. 2016;353:279. doi: 10.1126/science.aag0209. [DOI] [PMC free article] [PubMed] [Google Scholar]; (b) Oderinde MS, Frenette M, Robbins DW, Aquila B, Johannes JW. J Am Chem Soc. 2016;138:1760. doi: 10.1021/jacs.5b11244. [DOI] [PubMed] [Google Scholar]; (c) Oderinde MS, Jones NH, Juneau A, Frenette M, Aquila B, Tentarelli S, Robbins DW, Johannes JW. Angew Chem Int Ed. 2016;55:13219. doi: 10.1002/anie.201604429. [DOI] [PubMed] [Google Scholar]
- 8.(a) Shaw MH, Shurtleff VW, Terrett JA, Cuthbertson JD, MacMillan DWC. Science. 2016;352:1304. doi: 10.1126/science.aaf6635. [DOI] [PMC free article] [PubMed] [Google Scholar]; (b) Le C, Liang Y, Evans RW, Li X, MacMillan DWC. Nature. 2017;547:79. doi: 10.1038/nature22813. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Roberts BP. Chem Soc Rev. 1999;28:25. [Google Scholar]
- 10.Lowry MS, Goldsmith JI, Slinker JD, Rohl R, Pascal RA, Jr, Malliaras GG, Bernhard S. Chem Mater. 2005;17:5712. [Google Scholar]
- 11.Jeffrey JL, Terrett JA, MacMillan DWC. Science. 2015;349:1532. doi: 10.1126/science.aac8555. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.(a) Fontana F, Minisci F, Barbosa MCN, Vismara E. J Org Chem. 1991;56:2866. [Google Scholar]; (b) Chatgilialoglu C, Crich D, Komatsu M, Ryu I. Chem Rev. 1999;99:1991. doi: 10.1021/cr9601425. [DOI] [PubMed] [Google Scholar]
- 13.Chu L, Lipshultz JM, MacMillan DWC. Angew Chem Int Ed. 2015;54:7929. doi: 10.1002/anie.201501908. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Durandetti M, Devaud M, Périchon J. New J Chem. 1996;20:659. [Google Scholar]
- 15.Electron-deficient aryl chlorides are also competent substrates for this arylation protocol, albeit with reduced levels of efficiency.
- 16.Kook CS, Reed MF, Digenis GA. J Med Chem. 1975;18:533. doi: 10.1021/jm00239a023. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.