

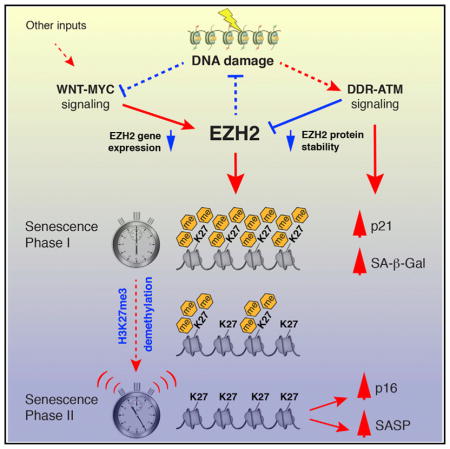
SUMMARY
Polycomb group (PcG) factors maintain facultative heterochromatin and mediate many important developmental and differentiation processes. EZH2, a PcG histone H3 lysine-27 methyltransferase, is repressed in senescent cells. We show here that downregulation of EZH2 promotes senescence through two distinct mechanisms. First, depletion of EZH2 in proliferating cells rapidly initiates a DNA damage response prior to a reduction in the levels of H3K27me3 marks. Second, the eventual loss of H3K27me3 induces p16 (CDKN2A) gene expression independent of DNA damage and potently activates genes of the senescence-associated secretory phenotype (SASP). The progressive depletion of H3K27me3 marks can be viewed as a molecular “timer” to provide a window during which cells can repair DNA damage. EZH2 is regulated transcriptionally by WNT and MYC signaling and posttranslationally by DNA damage-triggered protein turnover. These mechanisms provide insights into the processes that generate senescent cells during aging.
In Brief
Ito et al. show that downregulation of EZH2 rapidly elicits DNA damage and triggers the onset of senescence without loss of H3K27me3 marks. With slower kinetics, the depletion of H2K27me3 results in the upregulation of p16 and proinflammatory factors. WNT-MYC and ATM signaling are identified as upstream regulators of EZH2.
INTRODUCTION
Cellular senescence is the irreversible loss of replicative capacity triggered by genotoxic stresses such as telomere erosion (Sulli et al., 2012). It is an important component of normal physiological processes such as development, wound healing, and aging (Campisi, 2013). Expression of activated oncogenes (such as the G12V variant of HRAS) in normal cells leads to what is known as oncogene-induced senescence (OIS) (Serrano et al., 1997). Senescent cells accumulate to low levels in normal tissues with age, and their clearance in mice delays age-associated phenotypes (Baker et al., 2016; Herbig et al., 2006).
Senescent cells show characteristic changes in gene expression, including upregulation and secretion of proinflammatory cytokines, chemokines, and extracellular matrix-remodeling enzymes, referred to as the senescence-associated secretory phenotype (SASP) (He and Sharpless, 2017). Formation of localized regions of heterochromatin known as senescence-associated heterochromatic foci (SAHF) can be observed in some cell types (Narita et al., 2003). The cell cycle arrest is mediated by upregulation of the cyclin-dependent kinase inhibitors p21 (CDKN1A) and p16 (CDKN2A). p21 is upregulated by the tumor suppressor p53 (TP53), an important sensor of stresses, including DNA damage. p16 is regulated in a more complex manner that includes the Polycomb group (PcG) of chromatin regulators (Gil and Peters, 2006).
The PcG proteins are epigenetic regulators that maintain gene expression patterns established during development or differentiation (epigenetic is defined as heritable effects without changes in DNA sequence). They are found in two families of complexes, known as Polycomb repressive complex 1 and 2 (PRC1 and PRC2). PRC2 contains EZH2, the histone lysine methyltransferase that catalyzes the trimethylation of lysine 27 on histone H3 (H3K27me3) through its conserved Su(var)3–9, Enhancer-of-zeste and Trithorax (SET) domain. PRC1 and PRC2 act in concert to promote an inactive chromatin environment that represses transcription. During senescence, some PcG proteins are down-regulated, leading to loss of H3K27me3 at the CDKN2A locus, which results in the upregulation of p16 (Bracken et al., 2007). However, PcG proteins likely have additional roles beyond suppression of p16 during senescence (Shah et al., 2013).
Growing evidence suggests that EZH2 plays vital functions independent of its role to catalyze trimethylation of H3K27. EZH2 has been reported to act as a coactivator of the androgen receptor (Xu et al., 2012). EZH2 can also methylate non-histone substrates (Kim et al., 2013; Lee et al., 2012). Additionally, certain cancer cells appear to depend on both methyltransferase-dependent and -independent functions of EZH2 (Kim et al., 2015). However, it remains unclear whether loss of EZH2 affects senescence through catalytic or non-catalytic mechanisms.
Few studies have addressed the regulation of PcG during the onset of senescence. One possible upstream effector is wingless-type mouse mammary tumor virus integration site (WNT) signaling, whose downregulation has been found to induce senescence (Ye et al., 2007). In the canonical WNT pathway, extracellular WNT proteins ultimately upregulate a variety of target genes, including MYC. Further evidence for a possible regulatory link is that the BMI1 gene, a PRC1 component, is a direct MYC target and that downregulation of MYC can promote senescence (Guney et al., 2006).
Here we report that downregulation of EZH2 induces senescence by an H3K27me3-independent mechanism by activating a DNA damage response (DDR) during DNA replication. However, with delayed kinetics, loss of the H3K27me3 modification at the CDKN2A locus induces senescence by upregulating p16 in a DNA damage-independent manner. The loss of H3K27me3 also promotes the transcription of SASP genes. Upstream regulators of EZH2 include posttranslational modification by the ataxia telangiectasia mutated (ATM) kinase and transcriptional regulation by the WNT-MYC pathway.
RESULTS
Downregulation of EZH2 Leads to Cellular Senescence with Features of SASP
Expression profiling of proliferating and replicatively senescent human diploid fibroblasts (HDFs) showed that EZH2 is among the genes that are reproducibly downregulated in senescent cells (Lackner et al., 2014). Real-time qPCR) confirmed that EZH2 transcripts were decreased in senescent cells of three commonly used strains of normal HDFs (LF1, WI-38, and IMR90) (Figure 1A). In addition, cells made senescent by expression of HRAS(G12V) or exposure to DNA-damaging agents also showed reduced expression of EZH2 (Figures 1B and 1C). The levels of EZH2 protein decreased gradually as cells approached replicative senescence (Figures 1D and S1A). EZH2 protein became undetectable after 2 weeks of senescence; however, depletion of the H3K27me3 mark was not apparent until much later. Although we observed some loss of core histone H3 during senescence (Ivanov et al., 2013), this did not account for the reduction of H3K27me3 marks. Similar to LF1 cells, WI-38 and IMR90 cells showed loss of EZH2 at (or shortly after) the onset of senescence, whereas the disappearance of H3K37me3 lagged (Figure S1B). Although EZH2 levels are reduced in quiescence (~2-fold), senescent cells show an essentially complete depletion (Figures S1C and S2D). Synchronous induction of senescence by DNA damage (etoposide) rapidly reduced EZH2 levels without accompanying changes in global H3K27me3 marks (Figures S1E and S1F).
Figure 1. EZH2 Expression Decreases during Senescence, and Its Acute Downregulation Elicits Premature Senescence.

(A) EZH2 mRNA levels were determined by real-time qPCR in early-passage proliferating and replicatively senescent cells of strains LF1, WI-38, and IMR90 (**p < 0.01, n = 3).
(B) Early passage LF1 cells were infected with a lentivirus vector expressing HRAS(G12V) cDNA (or empty vector control), and expression of EZH2 mRNA was determined 6 days after infection. (**p < 0.01, n = 3).
(C) Cells were irradiated with ionizing radiation (10 Gy) or treated with 200 ng/mL neocarzinostatin (NCS), and EZH2 mRNA levels were determined after 6 days (**p < 0.01, n = 3).
(D) EZH2 protein levels and the presence of K27me3 marks on H3 were examined in LF1 cells by immunoblotting, from 18 population doublings before senescence until 8 weeks after the onset of senescence. EZH2 and H3K27me3 levels were normalized to GAPDH (green, red); H3K27me3 is also shown normalized to total H3 (blue).
(E) Cells were infected with a lentivirus vector expressing shRNA 3 against EZH2 (Figure S1H). shRNA to GFP was the control. The levels of EZH2 and H3K27me3 were examined by immunoblotting.
(F) EZH2 was knocked down as in (E), and proliferation was assessed by counting cell numbers (**p < 0.01, n = 3).
(G) The presence of SAHF, SA-β-Gal activity, and EdU incorporation were determined 2 days after shEZH2 infection. SA-β-Gal or SAHF-positive cells were scored as percent of total cells in randomly selected fields (*p < 0.05, **p < 0.01, n = 3).
(H) EZH2 was knocked down as in (E), and unsupervised hierarchical clustering of SASP gene Z scores from RNA-seq data is shown as heatmaps. Error bars represent SD. See also Figures S1 and S2 and Tables S1, S4, and S5.
To confirm that downregulation of EZH2 is sufficient to induce senescence, we introduced short hairpin RNAs (shRNAs) targeting its transcript into early-passage LF1 cells (Figure S1G). Three distinct shRNAs were tested using lentivirus vectors and resulted in a 63%–97% decrease of EZH2 mRNA (Figure S1H). Knockdown of EZH2 resulted in rapid depletion of EZH2 protein 2 days after infection, whereas significant depletion of H3K27me3 was not evident until 8 days (Figure 1E). Downregulation of EZH2 induced premature senescence by several criteria. First, cell proliferation was arrested within 2 days (Figure 1F). Second, the cells expressed senescence-associated β-galactosidase (SA-β-Gal), displayed SAHF, and did not incorporate 5-ethynyl-2′ deoxyuridine (EdU) (Figures 1G and S1I). In accordance with the presence of SAHF, the levels of the histone variant macroH2A increased (Figures S1J and S1K; Ye et al., 2007). Third, RNA sequencing (RNA-seq) analysis showed expression of SASP and inflammatory genes after EZH2 knockdown (Figures 1H and S2A and S2B). Interestingly, this phenotype became prominent only at late times after infection (6–8 days), whereas growth arrest, SA-β-Gal, and SAHF were induced much more rapidly (2–4 days). Collectively, downregulation EZH2 is sufficient to rapidly induce senescence accompanied by a robust SASP response.
Downregulation of EZH2 Results in DNA Damage and Induces p21
Because global levels of H3K27me3 were not affected at the onset of replicative senescence or senescence induced by knockdown of EZH2, we examined the presence of H3K27me3 marks at the CDKN2A locus by performing chromatin immunoprecipitation (ChIP) coupled with qPCR (ChIP-qPCR). No significant changes in H3K27me3 enrichment were observed 4 days after infection with shEZH2, with a decrease becoming apparent at 8 days (Figures 2A and 2B). These data reflect closely the global levels of H3K27me3 and suggest that loss of EZH2 can induce senescence independent of epigenetic changes at the CDKN2A locus.
Figure 2. Downregulation of EZH2 Induces DNA Damage and Senescence in a DNA Replication-Dependent Manner.

(A) H3K27me3 enrichment at the CDKN2A locus was determined 4 and 8 days after shEZH2 infection using ChIP. For locations of primer pairs, see (B). Normal rabbit immunoglobulin G (IgG) was used as the immunoprecipitation (IP) control (**p < 0.01, n = 3).
(B) Schematic of the CDKN2A locus.
(C) The frequency of p21-expressing cells was scored by IF after knockdown of EZH2 (percent of total cells, random fields, more than 400 cells per condition, **p < 0.01).
(D) The frequency of p16-expressing cells was determined as in (C) (*p < 0.05).
(E) Early passage LF1 cells were grown in the presence of DZNep (5 μM), and the levels of p16, p21, and EZH2 proteins and H3K27me3 marks were examined by immunoblotting after 9 and 18 days of treatment.
(F) p21 shRNAs or the empty vector were combined with an EZH2 shRNA (+) or a shGFP control (−), and SA-β-Gal-positive cells were scored 4 days after infection (*p < 0.05, **p < 0.01, n = 3).
(G) 53BP1 foci were visualized by IF 4 and 8 days after EZH2 knockdown. The stacked bars depict the fraction of cells with 1 focus (blue) or 2 (red), 3 (green), and more than 4 (black) foci per nucleus (**p < 0.01, n = 4).
(H) shATM and shEZH2 knockdowns were combined, and SA-β-Gal-positive cells were scored as in (F) (**p < 0.01, n = 3).
(I) At the time of shEZH2 infection (time [t] = 0; Figure S1G), cells were reseeded into either normal medium (Pro, 15% FBS), quiescence-inducing medium (Qui, 0.25% FBS), or medium supplemented with thymidine (Thy). 53BP1 foci were scored after 4 days (**p < 0.01, n = 3).
(J) EZH2 was knocked down as in (A), and cells were pulse-labeled with EdU for 1 hr 2 days after infection. A representative image showing colocalization of EdU (red) and 53BP1 (green) signals is shown. Right: magnified views of the indicated areas. Scale bar, 3 μm.
Error bars represent SD. See also Figures S3 and S4 and Tables S1 and S2.
Four days after knockdown of EZH2, RNA-seq analysis of differential gene expression implicated p53 and p21 activation as the top upstream regulators (Figure S2C). To examine the activation of senescence pathways elicited by the downregulation of EZH2 in more detail, we monitored the time course of p21 and p16 expression changes. p21 mRNA was induced 4 days after EZH2 knockdown (Figure S3A), whereas p16 mRNA was unchanged at this time, becoming upregulated only later (8 days; Figure S3B). The earliest time after infection we could assess was 2 days, at which point SA-β-Gal staining showed that the majority of cells (93%) were already senescent (Figure 1G). Likewise, examining single cells by immunofluorescence, we found that, at the 2-day time point, 83% of cells were positive for p21 protein (Figure 2C). The frequency of p21-positive cells declined over time, with only 10% of cells being positive at 8 days.
A similar analysis of p16 showed no expression at early time points, with the appearance of p16-positive cells becoming significant at 6 and 8 days (Figure 2D). In agreement, treatment with the drug 3-deazaneplanocin A (DZNep), which is known to deplete cellular EZH2 protein levels (Tan et al., 2007), induced senescence characterized by early activation of p21 followed by upregulation of SASP genes and p16 and loss of H3K27me3 marks (Figures 2E and S3C and S3D). To examine whether upregulation of p21 is required for the induction of senescence by EZH2 depletion, we performed double knockdowns and found that p21 knockdown opposed the appearance of senescent cells (Figures 2F and S3E–S3G). Collectively, these results indicate that p21 is an important and early effector engaged by EZH2 downregulation, and that p16 upregulation occurs later and coincides with the loss of H3K27me3 at the CDKN2A locus.
Because the primary regulator of p21 in senescence is p53, which, in turn, is activated by DNA damage, we examined components of the DDR after knockdown of EZH2. Knockdown of EZH2 led to a strong and rapid induction of 53BP1 DNA damage foci. Quantification of individual cells showed a significant increase in the frequency of both focus-containing cells as well as the number of foci per nucleus (Figure 2G).
To determine whether activation of ATM is required for the induction of senescence by EZH2 downregulation, we performed double knockdowns of ATM and EZH2. We found that ATM knockdown significantly alleviated the onset of senescence (Figures 2H and S3H and S3I). To determine whether the DDR elicited by EZH2 downregulation is localized to telomeres, we performed an assay that colocalizes DNA damage foci with telomeres (Herbig et al., 2006). We did not find significant changes in DNA damage occurring specifically at telomeres upon knockdown of EZH2 (Figures S3J and S3K). Similarly, knockdown of EZH2 in LF1 cells immortalized by expression of telomerase robustly induced senescence, as indicated by SA-β-Gal staining (Figure S3L), suggesting that telomere maintenance is not sufficient to prevent senescence because of EZH2 depletion. Altogether, these data indicate that senescence elicited by EZH2 downregulation is triggered initially by non-telomeric double-strand breaks (DSBs) and mediated by the ATM-p53-p21 DDR pathway.
Induction of DNA Damage by EZH2 Depletion Is Dependent on DNA Replication
Because EZH2 and other PRC2 components localize to sites of DNA replication (Hansen et al., 2008; Leung et al., 2013), and RNA-seq analysis showed the top downregulated ontologies upon knockdown of EZH2 to be associated with DNA strand elongation, DNA replication, and G1-S phase transition (Figure S2A), we examined the effect of cell cycle progression on the ability of EZH2 knockdown to elicit a DDR. We found that arresting the cell cycle, either in G0 by serum withdrawal or in late G1 by a thymidine block, strongly protected against DDR induction, upregulation of p21, and onset of senescence (Figures 2I and S3M and S3N). To determine whether DNA damage localizes to sites of DNA replication, we knocked down EZH2 under proliferative conditions and pulse-labeled with EdU before the cultures became fully senescent. We found that, in many of the cells still capable of EdU incorporation, 53BP1 foci frequently colocalized with sites of EdU labeling (Figure 2J). This is in agreement with recent studies showing that loss of PcG proteins affects the symmetry and progression of DNA replication forks and promotes their collapse (Piunti et al., 2014). Altogether, these data suggest that the DDR and ensuing senescence triggered by EZH2 depletion are dependent on DNA replication.
To further examine the mechanism by which EZH2 depletion induces replication fork stress, we ablated the recognition of the H3K27me3 mark by the PRC2 component EED. EED-H3K27me3 interaction was perturbed in two ways: with a small-molecule inhibitor (EED226) that blocks the H3K27me3-binding pocket of EED (Qi et al., 2017) and by expressing EED mutants unable to bind H3K27me3 marks (Margueron et al., 2009). Treatment with EED226 strongly induced the presence of SA-β-Gal-positive cells, upregulation of both p21 and p16, the appearance of 53BP1 DNA damage foci, and activation of a SASP response (Figures S4A–S4F). Similarly, ectopic expression of a mutant EED (F97A, W364A, or Y365A) unable to bind H3K27me3 elicited a DDR and upregulated SASP genes (Figures S4G and S4H), albeit to a lesser extent than treatment with EED226. In aggregate, these data suggest that the mislocalization or depletion of the PRC2 complex away from the replication fork induces a DDR through replication fork stress.
Loss of H3K27me3 Marks Induces Senescence by Upregulating p16 and SASP in a DNA Damage-Independent Manner
The temporal correlations between declining H3K27me3 levels, p16 upregulation, and activation of SASP genes suggest a possible contribution of H3K27me3 loss to the phenotypes of senescent cells. We therefore examined the effects of GSK126, a highly selective S-adenosyl-methionine (SAM)-competitive inhibitor of EZH2, on early-passage HDFs (McCabe et al., 2012). GSK126 inhibited EZH2 enzymatic activity within 4 days of treatment, as indicated by the loss of H3K27me3, without altering EZH2 protein levels (Figure 3A). Treatment with GSK126 also elicited senescence in a dose-dependent manner, as evidenced by the inhibition of proliferation and increase in SA-β-Gal staining. These effects were first evident at 8–10 days and continued to increase over a period of 30 days (Figures 3B and 3C and S5A).
Figure 3. Inhibition of EZH2 Activity Induces Senescence in the Absence of DNA Damage by Depleting H3K27me3 Marks and Activating p16 and SASP Genes.

(A) Early passage LF1 cells were grown continuously in the presence of GSK126, and levels of EZH2 protein and H3K27me3 marks were examined by immunoblotting.
(B and C) Cells were treated with GSK126 as in (A), and proliferation (B) or SA-β-Gal staining (C) were determined (**p < 0.01, n = 3).
(D and E) Cells were treated as in (A), and the frequency of p21-positive (D) and p16-positive (E) cells was scored by IF (percent of total cells, random fields, more than 400 cells per condition, *p < 0.05, **p < 0.01).
(F) Cells were treated with GSK126 for 8 days and examined for 53BP1 foci as in Figure 2G (n = 3).
(G) Cells were infected with lentivirus vectors expressing EZH2 cDNA, EZH2 with an in-frame deletion of the SET domain (EZH2ΔSET), or empty vector control, and expression of the indicated genes was determined by real-time qPCR 9 days after infection. Data are represented as heatmaps relative to control cells (n = 3).
(H) Cells were treated with GSK126 (5 μM), JQ1 (100 nM), or both, and expression of the indicated genes was determined by real-time qPCR after 10 days. Data are represented as heatmaps relative to DMSO-treated cells (n = 3).
(I) Cytokine array analysis of conditioned medium from cells treated with GSK126 (5 μM) or GSK126 plus JQ1 (100 nM) for 17 days. For comparison, cells were infected with shRNAs against EZH2 (shEZH2) or GFP (control [CTR]) for 8 days. IL1A and IL1B were secreted at undetectable levels, as reported previously (Orjalo et al., 2009), because they remain cell surface-associated. Data are represented as heatmaps relative to CTR (n = 2). See Table S3 for the measured cytokine concentrations.
(J) Cells were treated with GSK126 (5 μM) or DMSO for 10 days, and H3K27me3 and H3K27ac enrichment at the IL1A-IL1B-IL37 loci was determined by ChIP-qPCR. Primer pairs were chosen in regions of putative enhancers, and their locations are shown in the schematic as red bars. H3K27ac and H3K4me1 tracks were obtained from ENCODE (GEO: GSM469966 and GSM521895). Normal rabbit IgG was used as the IP control (*p < 0.05, **p < 0.01, n = 3).
Error bars represent SD. See also Figure S5 and Tables S1, S2, and S3.
To gain further insights into how loss of the H3K27me3 modification induces senescence, we investigated the expression of p21 and p16 proteins using single-cell immunofluorescence (IF) assays. Treatment of cells with GSK126 significantly increased the frequency of p16-positive cells but had no effect on p21 expression (Figures 3D and 3E). Furthermore, in contrast to knockdown of EZH2, inhibition of its activity with GSK126 did not elicit a DDR (Figure 3F), which agrees with the absence of p21 induction. To explore another method of inhibiting EZH2 activity, we used a catalytically inactive EZH2 mutant that lacks the conserved SET domain (EZH2ΔSET). Similar to what was observed after treatment with GSK126, infection with an EZH2ΔSET-expressing lentivirus vector induced senescence, accompanied by upregulation of p16 without triggering a DDR or upregulating p21 (Figures S5B–S5E).
To investigate the effects of EZH2 inhibition in non-replicating cells, quiescent LF1 cells were treated with GSK126 and monitored for changes in H3K27me3 and p16 levels. Although GSK126 treatment effectively depleted H3K27me3 marks within 4 days in proliferating cells (Figure 3A), it took at least 21 days to observe appreciable changes in quiescent cells (Figures S5F and S5G). Hence, loss of EZH2 methyltransferase activity can deplete H3K27me3 marks and induce p16 independent of cell cycle progression, albeit with much reduced kinetics.
H3K27me3 marks have been reported to decrease at enhancer regions of many SASP genes during senescence (Shah et al., 2013). In contrast, senescence leads to an increase in histone H3 lysine 27 acetyl (H3K27ac) at enhancers adjacent to SASP genes (Tasdemir et al., 2016). SASP factors were broadly upregulated and secreted following treatment with GSK126 or EZH2ΔSET-expressing lentivirus (Figures 3G–S3I). We therefore examined whether loss of H3K27me3 is associated with enrichment of H3K27ac at enhancers of SASP genes. Indeed, we found, using ChIP-qPCR, that treatment of proliferating cells with GSK126 resulted in a decrease of H3K27me3 and increase of H3K27ac marks at annotated enhancers adjacent to IL1A and IL1B (Figure 3J) and MMP3 (Figure S5H) genes.
Members of the bromodomain and extraterminal domain (BET) family of proteins are the “readers” that recognize acetylated lysine residues and facilitate transcriptional activation by recruitment of co-regulatory complexes. Accordingly, treatment with the small molecule BET inhibitor JQ1 antagonized the expression and secretion of GSK126-induced SASP factors (Figures 3H and 3I). However, JQ1 did not affect the GSK126-elicited upregulation of p16 or rescue senescence (Figures S5I and S5J). These data suggest that the H3K27 methylation-to-acetylation switch at enhancers of SASP genes mediates their activation but does not have broader effects on senescence.
SASP gene expression in senescent cells is also regulated, in part, by the mammalian target of rapamycin (mTOR) (Laberge et al., 2015). mTOR activity promotes the translation of the cytokine IL1A, which is then processed at the membrane and binds the IL1R receptor, which, in turn, activates the degradation of IκB, an inhibitor of nuclear factor κB (NF-κB) transcription factors. We found that, in GSK126-treated cells, the mTOR inhibitor rapamycin reduced the transcript levels of some SASP genes upregulated by GSK126 (Figure S5K). Treatment with GSK126 also decreased the levels of IκBα (Figure S5L), and this was rescued by treatment with rapamycin, Hence, NF-κB and mTOR contribute to the regulation of SASP genes during GSK126-induced senescence in a manner that is shared with other forms of senescence. In aggregate, these experiments indicate that inhibition of EZH2 methyltransferase activity is sufficient to induce senescence in the absence of DNA damage and that this response is characterized by the upregulation of p16 and accompanied by SASP.
EZH2 Suppresses Entry into Oncogene-Induced Senescence
We next examined whether preventing the downregulation of EZH2 that normally occurs during senescence could antagonize its establishment. We induced OIS using a 4-hydroxytamoxifen (4-OHT)-inducible estrogen receptor (ER):RAS(G12V) fusion protein and combined this with ectopic expression of EZH2 cDNA delivered with a lentivirus vector (Figure 4A). Expression of EZH2 strongly alleviated entry into OIS, as indicated by cell proliferation, frequency of SA-β-Gal positive cells, and p16 expression (Figures 4B–4D). Additionally, EZH2 also broadly alleviated the induction of SASP genes (Figure 4E). These data indicate that maintenance of EZH2 expression and, hence, H3K27me3 marks suppresses entry into senescence in response to stress.
Figure 4. Expression of EZH2 Opposes Entry into OIS and Induction of SASP.

(A) LF1 cells were engineered using lentivirus vectors to stably express EZH2 cDNA and a 4-OHT-inducible ER:RAS(G12V) protein. Control cell lines were constructed using empty vectors. Cells were treated with 4-OHT (200 nM) or vehicle (ethanol) for the indicated times, and the levels of EZH2 and ER:RAS proteins were determined by immunoblotting.
(B) Proliferation in the experiment shown in (A) was assessed by counting cell numbers (*p < 0.05, **p < 0.01, n = 3).
(C) SA-β-Gal-positive cells were scored after 12 days of 4-OHT treatment in the experiment shown in (A) (**p < 0.01, n = 3).
(D) p16 mRNA expression was determined by real-time qPCR as in (C) (**p < 0.01, n = 3).
(E) Expression of the indicated SASP genes was determined by real-time qPCR as in (C) (*p < 0.05, **p < 0.01, n = 3).
Error bars represent SD. See also Table S1.
Regulation of EZH2 Expression in Cellular Senescence
ATM kinase-mediated phosphorylation of EZH2 has been reported to decrease its stability (Li et al., 2013). In genotoxic stress-induced senescence, we found that cells with high numbers of phosphorylated ATM foci contained low levels of EZH2 (Figures 5A and 5B). In a time course experiment, the ATM inhibitor KU-55933 significantly delayed the disappearance of EZH2 protein (Figures 5C and S6A), and the MG132 proteasome inhibitor delayed the loss of EZH2 (Figure S6B). The phosphorylation of EZH2 on serine residues elicited by etoposide was inhibited by shRNA knockdown of ATM (Figure 5D). EZH2 is phosphorylated by ATM at Ser652 and Ser734 during neurode-generation (Li et al., 2013). EZH2 serine phosphorylation and turnover in response to etoposide were both alleviated in cells ectopically expressing an S652A/S743A double mutant of EZH2 (Figures 5E and 5F). Collectively, these data indicate that EZH2 protein, in a process initiated by ATM, is rapidly degraded during the DDR at the onset of senescence.
Figure 5. DNA Damage-Mediated Degradation of EZH2.

(A) LF1 cells were treated with etoposide (40 μM) for 8 hr and immunostained with antibodies to phospho-ATM (S1981, green) or EZH2 (red). Nuclei were counterstained with DAPI (blue). The same image is shown in the three channels. Scale bar, 10 μm.
(B) Cells were treated with etoposide for 20 hr, stained as in (A), and scored for EZH2 intensity and the number of phospho-ATM (S1981) foci per nucleus (random fields, more than 200 cells per condition).
(C) Cells were treated with etoposide (Eto) plus the ATM inhibitor KU-55933 (10 μM) or carrier (DMSO), and the levels of EZH2 protein were determined by immunoblotting.
(D) Cells infected with lentivirus vectors expressing shRNA against ATM (or empty vector as control) were treated for 8 hr with etoposide (+) or DMSO vehicle (−). Cell lysates were immunoprecipitated with anti-EZH2 antibody, followed by immunoblotting analysis with anti-phospho-serine or anti-EZH2 antibodies. Aliquots of whole cell lysates (WCLs) were immunoblotted for GAPDH to verify equivalent total protein input.
(E) Cells engineered using lentivirus vectors to stably express EZH2 or EZH2(S652A/S734A) mutant cDNAs were processed and analyzed as in (D).
(F) EZH2 or EZH2(S652A/S734A) mutant cDNA-expressing cells were treated with etoposide and cycloheximide (CHX, 50 μg/mL), and the levels of EZH2 protein were determined by immunoblotting.
(G) Cells were treated with etoposide in a time course, and the relative levels of EZH2 mRNA and EZH2 protein were determined by real-time qPCR and immunoblotting, respectively. The p values shown are pairwise comparisons with the zero time point (*p < 0.05, **p < 0.01, n = 3).
Interestingly, EZH2 mRNA expression was also downregulated during the onset of senescence, but with slower kinetics than that of EZH2 protein (Figure 5G). This suggests that DNA damage also affects EZH2 expression transcriptionally. Expression of the WNT ligand WNT2 has been reported to decrease in both replicative senescence and OIS (Ye et al., 2007), and MYC is a well-known downstream effector of WNT signaling (Herbst et al., 2014). We found that WNT2 and MYC are both downregulated, that the activity of β-catenin declines in cells undergoing replicative senescence (Figure 6A and S6C), and that knockdown of either WNT2 or MYC is sufficient to induce senescence and proliferation arrest (Figures 6B and S6D–S6F).
Figure 6. EZH2 Is Required for Senescence Induced by Downregulation of WNT or MYC Signaling.

(A) WNT2 and MYC mRNA levels were determined by real-time qPCR in early-passage proliferating and replicatively senescent (2 weeks, Figure 1D) LF1 fibroblasts (**p < 0.01, n = 3).
(B) LF1 cells were infected with lentivirus vectors expressing shRNAs against WNT2 or MYC, and SA-β-Gal-positive cells were scored 4 days after infection. shGFP was used as the control (**p < 0.01, n = 3).
(C) MYC enrichment at the promoter region of the EZH2 gene was determined by ChIP after knockdown of WNT2 or MYC as in (B). Locations of primer pairs are shown on the left. Normal rabbit IgG was used as the IP control (*p < 0.05, **p < 0.01, n = 3).
(D) Cells were infected with shRNA (#1) targeting MYC as in (B), and the levels of MYC, EZH2, SUZ12, and EED proteins were determined by immunoblotting.
(E) Cells were infected with shRNA (#1) against WNT2 as in (B), and the levels of active β-catenin (ABC), EZH2, SUZ12, and EED proteins were examined by immunoblotting.
(F) Cells were infected with a lentivirus vector expressing MYC cDNA, and the levels of EZH2 and MYC proteins were determined by immunoblotting. Empty pLX vector (–) was used as the control.
(G) Cells were treated for 8 days with the indicated concentrations of the GSK3 inhibitor CHIR99021, and the levels of EZH2 and ABC proteins were determined by immunoblotting.
(H) Ectopic expression of EZH2 cDNA was combined with a shRNA knockdown of WNT2. Controls were empty pLX vector for EZH2 (–) and shGFP for WNT2. The levels of p16, p21, and EZH2 proteins were determined by immunoblotting. Note that, although endogenous EZH2 expression is effectively downregulated by shWNT2 (lane 3; see also E), ectopic expression is maintained by the pLX-EZH2 vector (lane 4).
(I) p21 mRNA expression was determined by real-time qPCR in the experiment shown in (H) (*p < 0.05, n = 3).
(J) The frequency of SA-β-Gal-positive cells was scored in the experiment shown in (H) (*p < 0.05, **p < 0.01, n = 3).
(K) The double intervention shown in (H) was performed with MYC shRNA (instead of WNT2 shRNA), and the levels of p16, p21, and EZH2 proteins were examined. Note again that, although endogenous EZH2 expression is effectively downregulated by shMYC (lane 3; see also D), ectopic expression is maintained by the pLX-EZH2 vector (lane 4).
(L) p21 mRNA expression was determined in the experiment shown in (K) (**p < 0.01, n = 3).
(M) SA-β-Gal-positive cells were quantified in the experiment shown in (K).
Error bars represent SD. See also Figure S6 and Tables S1 and S2.
MYC is a direct regulator of BMI1, a PRC1 component (Guney et al., 2006). We thus examined whether WNT and MYC signaling also regulate EZH2 during senescence. Examination of Encyclopdia of DNA elements (ENCODE) datasets revealed a broad region of MYC binding centered at 150 bp upstream of the transcriptional start site (TSS) of the EZH2 gene. We therefore performed quantitative ChIP analysis with several primer pairs and found that MYC is indeed recruited to this region in proliferating cells (Figure 6C). Binding was reduced in cells after knockdown of WNT2 and eliminated after knockdown of MYC. These results show that EZH2 is a direct transcriptional target of MYC.
Knockdown of MYC strongly downregulated EZH2, with weaker effects observed on other PRC2 proteins (Figure 6D). Similar results were found after knockdown of WNT2 (Figure 6E). Conversely, ectopic expression of MYC cDNA increased EZH2 protein levels (Figure 6F). Finally, activation of WNT signaling with the GSK-3 inhibitor CHIR99021 increased the levels of both active β-catenin and EZH2 proteins in a concentration-dependent manner (Figure 6G). These data indicate that WNT signaling and MYC act upstream of EZH2 to regulate its expression.
To test whether EZH2 is a necessary component of this pathway, we knocked down WNT2 or MYC in cells that were previously engineered to ectopically express EZH2 cDNA. Ectopic expression of EZH2 suppressed the activation of p21 and partially rescued the onset of senescence after WNT2 knockdown (Figures 6H–6J and S6G). Similarly, expression of EZH2 alleviated the activation of p21 and onset of senescence after knockdown of MYC (Figures 6K–6M and S6G). This analysis thus places EZH2 downstream of both WNT2 and MYC. To confirm that MYC is acting downstream of WNT2, we knocked down WNT2 in cells ectopically expressing MYC cDNA and found that activation of p21 and entry into senescence were alleviated (Figures S6H–S6J). These data further confirm the existence of a WNT-MYC-EZH2 pathway for the regulation of senescence.
DISCUSSION
In this study, we have explored how PcG regulates cellular senescence. The EZH2 protein is the catalytic core of PcG complexes, depositing the H3K27me3 mark that is the characteristic feature of facultative heterochromatin. We found that downregulation of EZH2 with shRNA in normal, proliferating cells rapidly induced senescence that was initiated by widespread DNA damage, leading to a DDR mediated by the ATM-p53-p21 pathway. The ensuing cell cycle arrest clearly preceded the depletion of H3K27me3 marks and is therefore likely to be independent of expression changes at PcG-regulated loci. PcG complexes are known to be localized at replication forks during S phase (Hansen et al., 2008; Piunti et al., 2014), and their depletion can cause DNA damage through replication fork collapse. The DDR caused by loss of EZH2 likely proceeds through this mechanism because it was dependent on cell cycle progression, and DNA damage (53BP1) foci frequently colocalized with sites of DNA replication. Additionally, prevention of PRC2 localization to the replication fork by antagonizing the binding of EED to H3K27me3 marks phenocopied the effect of EZH2 depletion. Replication fork collapse because of over-replication elicited by oncogenes such as H-RAS has been implicated as the triggering mechanism of OIS (Di Micco et al., 2006).
The CDK inhibitor p16 (CDKN2A) is an important effector of senescence and was rapidly upregulated upon EZH2 knockdown. However, the upregulation of p16 was a late event after EZH2 depletion, clearly separated from the appearance of DNA damage, induction of SA-β-Gal activity, and cell cycle arrest. The induction of senescence by PcG downregulation thus consists of two distinct phases: an early phase characterized by DNA damage and a late phase caused by depletion of H3K27me3 marks. That these processes can be independent was evidenced by the activation of senescence by the simple inhibition of EZH2 activity, either with the drug GSK126 or expression of a catalytically inactive EZH2 protein. These senescence responses were late in onset and characterized by the upregulation of p16 but devoid of DNA damage and activation of p21.
Although irreversible cell cycle arrest is the classical feature of senescence, senescent cells display many gene expression changes that are involved in unrelated processes. Among these, the SASP is of considerable interest because it is likely to be responsible for many of the deleterious effects of senescent cells. p16 is an important regulator of senescence, but its upregulation alone, although sufficient to cause cell cycle arrest, does not induce the SASP (Coppé et al., 2011). We found that GSK126-mediated loss of H3K27me3 marks resulted not only in the upregulation of p16 but also a robust activation of SASP genes. This was associated with a replacement of H3K27me3 by H3K27ac at enhancers of SASP genes, whose activation was opposed by BET inhibitors, suggesting that these epigenetic changes have functional significance. The observation that loss of EZH2 activity can upregulate SASP genes in the absence of a DDR underscores the importance of PcG-mediated epigenetic regulation.
How is EZH2 regulated during senescence? At the transcriptional level, we document here the existence of a WNT2-MYC-EZH2 regulatory cascade. Downregulation of MYC closely phenocopied the senescence response induced by depletion of EZH2, including DNA damage, biphasic kinetics, and activation of SASP. We also found that EZH2 is a direct transcriptional target of MYC. MYC has been reported to downregulate miR-26a/b, a negative effector of EZH2 (Sander et al., 2008), and, through its effects on the AKT pathway, to indirectly activate and stabilize the EZH2 protein (Kaur and Cole, 2013). These mechanisms likely reinforce the direct regulation of the EZH2 gene by MYC we document here and underscore the importance of MYC as a regulator of EZH2 and, by extension, the PcG pathway.
Because canonical WNT signaling is known to activate MYC expression, we examined the regulatory relationships between WNT2, MYC, and EZH2 during the onset of senescence, a situation of reduced WNT and MYC signaling. We did this by an epistasis analysis, performing a series of double interventions. In particular, ectopic expression of MYC antagonized the senescence-inducing effects of WNT2 depletion, and, likewise, expression of EZH2 partially reversed the effects of both MYC and WNT2 downregulation. These compensatory effects were significant but not complete, most likely because other PcG genes in addition to EZH2 are involved, and overexpression of MYC in normal cells can trigger either apoptosis or senescence. Nevertheless, the genetic relationships are clearly indicative of a WNT2-MYC-EZH2 hierarchy.
EZH2 is also regulated posttranslationally by ATM-triggered protein degradation (Li et al., 2013). We found that genotoxic stress-induced senescence was accompanied by the rapid disappearance of EZH2 protein that was faster than its transcriptional repression and was antagonized by the ATM inhibitor KU-55933 and the proteasome inhibitor MG132. EZH2 is rapidly recruited to sites of DNA damage after laser micro-irradiation (Chou et al., 2010). These findings suggest that PcG plays a role in the response to DNA damage and is then processed for degradation. We propose that the rapid disappearance of both EZH2 mRNA and protein we have documented at the onset of cellular senescence can act as a “clock” that determines the length of the first, DDR-associated phase of senescence. The slow, DNA replication-independent turnover of H3K27me3 marks would be the timing mechanism, whose depletion eventually triggers the upregulation of p16 and SASP genes and, thus, ushers in the second (probably irreversible) phase of senescence. In agreement, ectopic expression of EZH2 significantly delayed the upregulation of p16 as well as many SASP genes.
The SASP is activated by DNA damage through an ATM-mediated DDR (Rodier et al., 2009). IL1A is an important SASP regulator (Laberge et al., 2015) that, in concert with mTOR, activates the NF-κB transcription factors. Our data indicate that EZH2 is an important factor in the initial upregulation of the IL1A gene. First, GSK126 was an effective inducer of SASP in the absence of a DDR, thus effectively placing EZH2 between the DNA damage signal and IL1A upregulation. Second, mTOR and NF-κB influenced SASP gene expression in GSK126-induced senescence, again placing EZH2 in an upstream position. Finally ectopic expression of EZH2 delayed SASP during OIS. NF-κB activity is also promoted by GATA4 during senescence (Kang et al., 2015), and GATA4 has been reported to be repressed by EZH2 methylation (He et al., 2012). This mechanism, which acts upstream of the IL1A-mTOR-NF-κB pathway, would thus also be enforced by loss of EZH2 activity.
Our model, as shown in the Graphical Abstract, places DNA damage both upstream and downstream of EZH2. Following acute DNA damage, such as telomere uncapping or irradiation, the ATM-mediated DDR is likely sufficient to upregulate p21 and initiate the first phase of senescence. However, during OIS, which is ultimately caused by replication fork collapse, the DNA damage elicited by the depletion of EZH2 at these sites may be an important reinforcing mechanism. Furthermore, during senescence mediated by the WNT-MYC pathway in the absence of upstream DNA damage, DNA damage because of EZH2 downregulation provides the signal to establish cell cycle arrest and, hence, is an important component of the “timer” that provides an opportunity for repair.
Our results suggest that inhibiting components of PcG complexes is likely to induce senescence phenotypes through a variety of mechanisms. Understanding these will be critical for teasing out the anti-cancer effects of PcG inhibitors from possible deleterious consequences. For example, compromise of facultative heterochromatin maintenance might lead to upregulation of the SASP and proinflammatory effects in normal resting cells. Given that PcG complexes are localized at replication forks for the transmission of H3K27me3 marks to daughter cells, targeting this process may be an effective way to provoke DNA damage that is largely confined to cycling cells.
EXPERIMENTAL PROCEDURES
Culture Conditions
All HDF cultures were passaged (1:4) and incubated at 37°C in an atmosphere of 2.5% O2, 92.5% N2, and 5% CO2. 293T cells were cultured in air supplemented with 5% CO2. LF1 cells were cultured in Ham’s F10 nutrient mixture (Invitrogen) supplemented with 15% fetal bovine serum (FBS, HyClone). Quiescence was induced by reducing FBS to 0.25%. IMR90, WI-38, and 293T cells were cultured in DMEM supplemented with 10% FBS. All media were supplemented with 2 mM L-glutamine and penicillin/streptomycin (Sigma-Aldrich).
Lentiviral Infections
293T cells were co-transfected with plasmid DNAs of a lentiviral vector and the helper vectors pVSV-G and pCMV Δ8.9 using FuGENE HD (Promega). Medium was collected 24, 36, and 48 hr after transfection and used to infect HDF cells. Infected cells were selected with 2 μg/mL puromycin for 2 days, 500 μg/mL G418 for 3 days, or 5 μg/mL blasticidin for 5 days (see Figure S1G for a schematic of the time line and Supplemental Experimental Procedures for details of individual vectors).
Real-Time qPCR
Real-time qPCR was performed using the SYBR Green system (Applied Bio-systems) on the ABI 7900 Fast Sequence Detection instrument as specified by the manufacturer. Total RNA was harvested from cells using Trizol reagent (Invitrogen) and further purified using RNeasy columns (QIAGEN). 1 μg of total RNA was transcribed into cDNA in 50-μL reactions using the TaqMan kit (Applied Biosystems). 1 μL of this reaction was used in subsequent real-time qPCR reactions. GAPDH was used as the normalization control. All reactions were performed in triplicates. The primers used for real-time qPCR are provided in Table S1.
ChIP
The procedure was based on the Magna ChIP protocol (Millipore, 17-10085) and Chromatrap Premium ChIP qPCR (Chromatrap, 500115). Cells (2 × 106) were cross-linked in 1% formaldehyde for 10 min at room temperature, harvested by scraping, centrifuged, and resuspended in SDS lysis buffer. After 10 min on ice, lysed cells were diluted 2-fold in ChIP dilution buffer and disrupted with the S220 Focused Ultrasonicator (Covaris) to yield genomic DNA fragments of 300 bp. The samples were immunoprecipitated overnight at 4°C with the indicated antibodies. Immunocomplexes were pulled down with 20 μL of magnetic protein A beads as described in the Magna ChIP protocol. The DNA was recovered by 2 hr digestion with proteinase K at 65°C, followed by a 10-min incubation at 95°C. The DNA was cleaned up by phenol/chloroform extraction and ethanol precipitation and finally resuspended in 30 μL of Tris-EDTA (TE).
The ChIP and input were then used for qPCR with the primers described in Table S2.
Immunofluorescence and Immuno-FISH
Cells were fixed for 20 min with freshly prepared 4% paraformaldehyde in PBS. Permeabilization was performed with 0.2% Triton X-100 in PBS for 20 min at room temperature. After blocking nonspecific binding, antibodies were diluted in the blocking solution and incubated with the specimens overnight at 4°C in a humidified chamber. Samples were washed three times with PBS and incubated for 1 hr at room temperature with secondary antibodies diluted in blocking solution. Nuclei were counterstained with 2 μg/mL DAPI in PBS containing 0.2% Triton X-100 for 15 min. The telomere dysfunction-induced focus (TIF) assay, an immunofluorescence combined with fluores-cence in situ hybridization (Immuno-FISH) procedure (Herbig et al., 2006) is described in detail in the Supplemental Experimental Procedures, as is a listing of the antibodies used. For EdU pulse labeling, cells were incubated with 10 μM of EdU stock solution (Click-iT, Invitrogen) for 1 hr prior to fixation.
Statistical Analysis
Data are shown as means with SD. Statistical significance was determined using the two-tailed Student’s test (t test). p < 0.05 was considered significant.
DATA AND SOFTWARE AVAILABILITY
The accession number for the RNA-seq data reported in this paper is GEO: GSE109064.
Supplementary Material
Highlights.
Depletion of EZH2 elicits DNA damage in a replication-dependent manner
EZH2 is regulated by the WNT-MYC pathway and DDR-mediated proteolysis
Removal of H3K27me3 induces senescence in the absence of DNA damage
H3K27me3 marks act as a timer that links DNA damage with the induction of SASP
Acknowledgments
This work was supported by NIH/NIA grants R37 AG016694 and P01 AG051449 (to J.M.S.). T.I. was supported in part by NIH grants T32 GM007601 and F31 AG043189. The Brown Genomics Core was supported in part by the COBRE award from NIH/NIGMS P30 GM0103410.
Footnotes
AUTHOR CONTRIBUTIONS
Conceptualization and Methodology, T.I. and J.M.S.; Investigation, T.I., S.A.E., and Y.V.T.; Writing – Original Draft and Review and Editing, T.I. and J.M.S.; Supervision, N.N. and J.M.S.
DECLARATION OF INTERESTS
The authors declare no competing interests.
Supplemental Information includes Supplemental Experimental Procedures, six figures, and five tables and can be found with this article online at https://doi.org/10.1016/j.celrep.2018.03.002.
References
- Baker DJ, Childs BG, Durik M, Wijers ME, Sieben CJ, Zhong J, Saltness RA, Jeganathan KB, Verzosa GC, Pezeshki A, et al. Naturally occurring p16(Ink4a)-positive cells shorten healthy lifespan. Nature. 2016;530:184–189. doi: 10.1038/nature16932. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bracken AP, Kleine-Kohlbrecher D, Dietrich N, Pasini D, Gargiulo G, Beekman C, Theilgaard-Mönch K, Minucci S, Porse BT, Marine JC, et al. The Polycomb group proteins bind throughout the INK4A-ARF locus and are disassociated in senescent cells. Genes Dev. 2007;21:525–530. doi: 10.1101/gad.415507. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Campisi J. Aging, cellular senescence, and cancer. Annu Rev Physiol. 2013;75:685–705. doi: 10.1146/annurev-physiol-030212-183653. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Chou DM, Adamson B, Dephoure NE, Tan X, Nottke AC, Hurov KE, Gygi SP, Colaiácovo MP, Elledge SJ. A chromatin localization screen reveals poly (ADP ribose)-regulated recruitment of the repressive poly-comb and NuRD complexes to sites of DNA damage. Proc Natl Acad Sci USA. 2010;107:18475–18480. doi: 10.1073/pnas.1012946107. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Coppé JP, Rodier F, Patil CK, Freund A, Desprez PY, Campisi J. Tumor suppressor and aging biomarker p16(INK4a) induces cellular senescence without the associated inflammatory secretory phenotype. J Biol Chem. 2011;286:36396–36403. doi: 10.1074/jbc.M111.257071. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Di Micco R, Fumagalli M, Cicalese A, Piccinin S, Gasparini P, Luise C, Schurra C, Garre’ M, Nuciforo PG, Bensimon A, et al. Oncogene-induced senescence is a DNA damage response triggered by DNA hyper-replication. Nature. 2006;444:638–642. doi: 10.1038/nature05327. [DOI] [PubMed] [Google Scholar]
- Gil J, Peters G. Regulation of the INK4b-ARF-INK4a tumour suppressor locus: all for one or one for all. Nat Rev Mol Cell Biol. 2006;7:667–677. doi: 10.1038/nrm1987. [DOI] [PubMed] [Google Scholar]
- Guney I, Wu S, Sedivy JM. Reduced c-Myc signaling triggers telomere-independent senescence by regulating Bmi-1 and p16(INK4a) Proc Natl Acad Sci USA. 2006;103:3645–3650. doi: 10.1073/pnas.0600069103. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hansen KH, Bracken AP, Pasini D, Dietrich N, Gehani SS, Monrad A, Rappsilber J, Lerdrup M, Helin K. A model for transmission of the H3K27me3 epigenetic mark. Nat Cell Biol. 2008;10:1291–1300. doi: 10.1038/ncb1787. [DOI] [PubMed] [Google Scholar]
- He S, Sharpless NE. Senescence in Health and Disease. Cell. 2017;169:1000–1011. doi: 10.1016/j.cell.2017.05.015. [DOI] [PMC free article] [PubMed] [Google Scholar]
- He A, Shen X, Ma Q, Cao J, von Gise A, Zhou P, Wang G, Marquez VE, Orkin SH, Pu WT. PRC2 directly methylates GATA4 and represses its transcriptional activity. Genes Dev. 2012;26:37–42. doi: 10.1101/gad.173930.111. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Herbig U, Ferreira M, Condel L, Carey D, Sedivy JM. Cellular senescence in aging primates. Science. 2006;311:1257. doi: 10.1126/science.1122446. [DOI] [PubMed] [Google Scholar]
- Herbst A, Jurinovic V, Krebs S, Thieme SE, Blum H, Göke B, Kolligs FT. Comprehensive analysis of β-catenin target genes in colorectal carcinoma cell lines with deregulated Wnt/β-catenin signaling. BMC Genomics. 2014;15:74. doi: 10.1186/1471-2164-15-74. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ivanov A, Pawlikowski J, Manoharan I, van Tuyn J, Nelson DM, Rai TS, Shah PP, Hewitt G, Korolchuk VI, Passos JF, et al. Lyso-some-mediated processing of chromatin in senescence. J Cell Biol. 2013;202:129–143. doi: 10.1083/jcb.201212110. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kang C, Xu Q, Martin TD, Li MZ, Demaria M, Aron L, Lu T, Yankner BA, Campisi J, Elledge SJ. The DNA damage response induces inflammation and senescence by inhibiting autophagy of GATA4. Science. 2015;349:aaa5612. doi: 10.1126/science.aaa5612. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kaur M, Cole MD. MYC acts via the PTEN tumor suppressor to elicit autoregulation and genome-wide gene repression by activation of the Ezh2 methyltransferase. Cancer Res. 2013;73:695–705. doi: 10.1158/0008-5472.CAN-12-2522. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kim E, Kim M, Woo DH, Shin Y, Shin J, Chang N, Oh YT, Kim H, Rheey J, Nakano I, et al. Phosphorylation of EZH2 activates STAT3 signaling via STAT3 methylation and promotes tumorigenicity of glioblastoma stem-like cells. Cancer Cell. 2013;23:839–852. doi: 10.1016/j.ccr.2013.04.008. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kim KH, Kim W, Howard TP, Vazquez F, Tsherniak A, Wu JN, Wang W, Haswell JR, Walensky LD, Hahn WC, et al. SWI/SNF-mutant cancers depend on catalytic and non-catalytic activity of EZH2. Nat Med. 2015;21:1491–1496. doi: 10.1038/nm.3968. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Laberge RM, Sun Y, Orjalo AV, Patil CK, Freund A, Zhou L, Curran SC, Davalos AR, Wilson-Edell KA, Liu S, et al. MTOR regulates the pro-tumorigenic senescence-associated secretory phenotype by promoting IL1A translation. Nat Cell Biol. 2015;17:1049–1061. doi: 10.1038/ncb3195. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lackner DH, Hayashi MT, Cesare AJ, Karlseder J. A genomics approach identifies senescence-specific gene expression regulation. Aging Cell. 2014;13:946–950. doi: 10.1111/acel.12234. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lee JM, Lee JS, Kim H, Kim K, Park H, Kim JY, Lee SH, Kim IS, Kim J, Lee M, et al. EZH2 generates a methyl degron that is recognized by the DCAF1/DDB1/CUL4 E3 ubiquitin ligase complex. Mol Cell. 2012;48:572–586. doi: 10.1016/j.molcel.2012.09.004. [DOI] [PubMed] [Google Scholar]
- Leung KH, Abou El Hassan M, Bremner R. A rapid and efficient method to purify proteins at replication forks under native conditions. Bio-techniques. 2013;55:204–206. doi: 10.2144/000114089. [DOI] [PubMed] [Google Scholar]
- Li J, Hart RP, Mallimo EM, Swerdel MR, Kusnecov AW, Herrup K. EZH2-mediated H3K27 trimethylation mediates neurodegeneration in ataxia-telangiectasia. Nat Neurosci. 2013;16:1745–1753. doi: 10.1038/nn.3564. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Margueron R, Justin N, Ohno K, Sharpe ML, Son J, Drury WJ, 3rd, Voigt P, Martin SR, Taylor WR, De Marco V, et al. Role of the polycomb protein EED in the propagation of repressive histone marks. Nature. 2009;461:762–767. doi: 10.1038/nature08398. [DOI] [PMC free article] [PubMed] [Google Scholar]
- McCabe MT, Ott HM, Ganji G, Korenchuk S, Thompson C, Van Aller GS, Liu Y, Graves AP, Della Pietra A, 3rd, Diaz E, et al. EZH2 inhibition as a therapeutic strategy for lymphoma with EZH2-activating mutations. Nature. 2012;492:108–112. doi: 10.1038/nature11606. [DOI] [PubMed] [Google Scholar]
- Narita M, Nũnez S, Heard E, Narita M, Lin AW, Hearn SA, Spector DL, Hannon GJ, Lowe SW. Rb-mediated heterochromatin formation and silencing of E2F target genes during cellular senescence. Cell. 2003;113:703–716. doi: 10.1016/s0092-8674(03)00401-x. [DOI] [PubMed] [Google Scholar]
- Orjalo AV, Bhaumik D, Gengler BK, Scott GK, Campisi J. Cell surface-bound IL-1alpha is an upstream regulator of the senescence-associated IL-6/IL-8 cytokine network. Proc Natl Acad Sci USA. 2009;106:17031–17036. doi: 10.1073/pnas.0905299106. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Piunti A, Rossi A, Cerutti A, Albert M, Jammula S, Scelfo A, Cedrone L, Fragola G, Olsson L, Koseki H, et al. Polycomb proteins control proliferation and transformation independently of cell cycle checkpoints by regulating DNA replication. Nat Commun. 2014;5:3649. doi: 10.1038/ncomms4649. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Qi W, Zhao K, Gu J, Huang Y, Wang Y, Zhang H, Zhang M, Zhang J, Yu Z, Li L, et al. An allosteric PRC2 inhibitor targeting the H3K27me3 binding pocket of EED. Nat Chem Biol. 2017;13:381–388. doi: 10.1038/nchembio.2304. [DOI] [PubMed] [Google Scholar]
- Rodier F, Coppé JP, Patil CK, Hoeijmakers WA, Muñoz DP, Raza SR, Freund A, Campeau E, Davalos AR, Campisi J. Persistent DNA damage signalling triggers senescence-associated inflammatory cytokine secretion. Nat Cell Biol. 2009;11:973–979. doi: 10.1038/ncb1909. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sander S, Bullinger L, Klapproth K, Fiedler K, Kestler HA, Barth TF, Möller P, Stilgenbauer S, Pollack JR, Wirth T. MYC stimulates EZH2 expression by repression of its negative regulator miR-26a. Blood. 2008;112:4202–4212. doi: 10.1182/blood-2008-03-147645. [DOI] [PubMed] [Google Scholar]
- Serrano M, Lin AW, McCurrach ME, Beach D, Lowe SW. Oncogenic ras provokes premature cell senescence associated with accumulation of p53 and p16INK4a. Cell. 1997;88:593–602. doi: 10.1016/s0092-8674(00)81902-9. [DOI] [PubMed] [Google Scholar]
- Shah PP, Donahue G, Otte GL, Capell BC, Nelson DM, Cao K, Aggarwala V, Cruickshanks HA, Rai TS, McBryan T, et al. Lamin B1 depletion in senescent cells triggers large-scale changes in gene expression and the chromatin landscape. Genes Dev. 2013;27:1787–1799. doi: 10.1101/gad.223834.113. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sulli G, Di Micco R, d’Adda di Fagagna F. Crosstalk between chromatin state and DNA damage response in cellular senescence and cancer. Nat Rev Cancer. 2012;12:709–720. doi: 10.1038/nrc3344. [DOI] [PubMed] [Google Scholar]
- Tan J, Yang X, Zhuang L, Jiang X, Chen W, Lee PL, Karuturi RK, Tan PB, Liu ET, Yu Q. Pharmacologic disruption of Polycomb-repressive complex 2-mediated gene repression selectively induces apoptosis in cancer cells. Genes Dev. 2007;21:1050–1063. doi: 10.1101/gad.1524107. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Tasdemir N, Banito A, Roe JS, Alonso-Curbelo D, Camiolo M, Tscha-harganeh DF, Huang CH, Aksoy O, Bolden JE, Chen CC, et al. BRD4 Connects Enhancer Remodeling to Senescence Immune Surveillance. Cancer Discov. 2016;6:612–629. doi: 10.1158/2159-8290.CD-16-0217. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Xu K, Wu ZJ, Groner AC, He HH, Cai C, Lis RT, Wu X, Stack EC, Loda M, Liu T, et al. EZH2 oncogenic activity in castration-resistant prostate cancer cells is Polycomb-independent. Science. 2012;338:1465–1469. doi: 10.1126/science.1227604. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ye X, Zerlanko B, Kennedy A, Banumathy G, Zhang R, Adams PD. Downregulation of Wnt signaling is a trigger for formation of facultative heterochromatin and onset of cell senescence in primary human cells. Mol Cell. 2007;27:183–196. doi: 10.1016/j.molcel.2007.05.034. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.