

Summary
Autosomal dominant mutations in the sodium-gated potassium channel subunit gene KCNT1 have been associated with two distinct seizure syndromes, nocturnal frontal lobe epilepsy (NFLE) and malignant migrating focal seizures of infancy (MMFSI). To further explore the phenotypic spectrum associated with KCNT1, we examined individuals affected with focal epilepsy or an epileptic encephalopathy for mutations in the gene. We identified KCNT1 mutations in 12 previously unreported patients with focal epilepsy, multifocal epilepsy, cardiac arrhythmia, and in a family with sudden unexpected death in epilepsy (SUDEP), in addition to patients with NFLE and MMFSI. In contrast to the 100% penetrance so far reported for KCNT1 mutations, we observed incomplete penetrance. It is notable that we report that the one KCNT1 mutation, p.Arg398Gln, can lead to either of the two distinct phenotypes, ADNFLE or MMFSI, even within the same family. This indicates that genotype–phenotype relationships for KCNT1 mutations are not straightforward. We demonstrate that KCNT1 mutations are highly pleiotropic and are associated with phenotypes other than ADNFLE and MMFSI. KCNT1 mutations are now associated with Ohtahara syndrome, MMFSI, and nocturnal focal epilepsy. They may also be associated with multifocal epilepsy and cardiac disturbances.
Keywords: KCNT1, Autosomal dominant nocturnal frontal lobe epilepsy, Epileptic encephalopathy, Cardiac arrhythmia, Sudden unexpected death in epilepsy
KCNT1 encodes a sodium-gated potassium channel subunit that plays an important role in regulating excitability in neurons.1 We recently reported heterozygous KCNT1 mutations in a severe form of autosomal dominant nocturnal frontal lobe epilepsy (ADNFLE)2 in which a significant proportion of individuals have comorbidities of intellectual disability (ID), psychiatric features, and refractory seizures.2 Heterozygous KCNT1 mutations were also described in patients with malignant migrating focal seizures of infancy (MMFSI).3 A heterozygous KCNT1 mutation has also been described in a patient with leukoencephalopathy and severe epilepsy4 and a homozygous mutation was reported in a patient with Ohtahara syndrome.5
The involvement of KCNT1 in the phenotypically distinct disorders ADNFLE and MMFSI suggests that KCNT1 mutations may be associated with a spectrum of phenotypes encompassing focal epilepsy, psychiatric disorders, and epileptic encephalopathies. To investigate this, we studied patients and families with focal epilepsies, including 22 with psychiatric comorbidities, as well as patients with epileptic encephalopathies, including MMFSI, for mutations in KCNT1. We also sought to further explore the mutational spectrum, that is, the positions and classes of KCNT1 mutations, in epileptic disorders as these may provide insights into the resulting pathogenic mechanisms.
Methods
The study was approved by the ethics committees of Western Sealand, the University of South Australia, and Austin Health, Melbourne, Australia. Genomic DNA from study participants was extracted from peripheral blood leukocytes. One hundred forty-three patients, including 92 with focal epilepsies (22 with psychiatric comorbidities), who were negative for CHRNA2, CHRNA4, and CHRNB2 mutations, and 51 unrelated patients with epileptic encephalopathies including 19 with MMFSI, were tested for mutations in KCNT1 using high-resolution melting-curve (HRM) analysis as described previously.2 Additional Danish, Dutch, and Italian samples underwent targeted resequencing for selected epilepsy genes including KCNT1 using gene panels as described in Data S1 and Table S1. Variants were validated and familial segregation was analyzed by Sanger sequencing. Whole exome sequencing (WES) was performed as described in Data S1.
Results
We identified a total of 12 new unrelated patients with missense mutations in KCNT1. The mutations are listed in Table 1 along with the clinical features found in each individual. The positions of these mutations within the KCNT1 protein are shown in Figure 1B.
Table 1.
Clinical and molecular features of patients with KCNT1 mutations identified during this study
| Family | KCNT1 mutation | Inheritance | Individual | Phenotype | Age of seizure onset |
Seizure type | Other features | Origin |
|---|---|---|---|---|---|---|---|---|
| 1 | c.2782C>T; p.Arg928Cys | Familial | III.3 | Unaffected | – | – | – | Danish |
| III.6 | ADNFLE | 1 year | Nocturnal frontal seizures, GTCS | Learning impairment, psychiatric features | ||||
| IV.2 | ADNFLE | 12 years | Nocturnal frontal seizures | Learning impairment, psychiatric features | ||||
| IV.3 | ADNFLE | 15 years | Nocturnal frontal seizure (only one) | – | ||||
| IV.4 | ADNFLE | 7 years | Nocturnal frontal seizures | Learning impairment | ||||
| IV.5 | ADNFLE | 6 years | Nocturnal frontal seizures evolving to GTCS, FDS | Learning impairment, psychiatric features, cardiac arrhythmia | ||||
| 2 | c.1193G>A; p.Arg398Gln | Familial | II.3 | Likely ADNFLE | Unknown | Unconfirmed nocturnal events | – | Australian |
| III.2 | ADNFLE | 8–14 months | Nocturnal frontal seizures evolving to GTCS | |||||
| 18 years | NFLE | |||||||
| III.3 | MMFSI | 3 months | Asymmetric tonic, FDS, status epilepticus | Profound intellectual disability, spastic quadriparesis, progressive microcephaly scoliosis, sleep apnea, gastroesophageal reflux | ||||
| III.4 | Focal epilepsy | 6 months | Nocturnal focal epilepsy (mild) | Learning impairment | ||||
| III.5 | MMFSI | 5 months | Asymmetric tonic, FDS | Profound intellectual disability, microcephaly, scoliosis, precocious puberty | ||||
| 3 | c.769C>G; p.His257Asp | De novo | MMFSI | 2 weeks | Asymmetric tonic, TCS, status epilepticus, FDS | Intellectual disability, spastic quadriparesis, precocious puberty, cortical visual impairment | Australian | |
| 4 | c.785G>A; p.Arg262Gln | De novo | MMFSI | 8 weeks | Hemiclonic, GTCS, hemitonic, myoclonic | Developmental delay, hypotonia, hyperreflexia, progressive microcephaly, visual impairment | Australian, European origin | |
| 5 | c.862G>A; p.Gly288Ser | De novo | MMFSI | 2 months | Hemiclonic, NCSE TCS | Developmental delay, hypotonia | Dutch | |
| 6 | c.862G>A; p.Gly288Ser | Unknown | MMFSI | 3 months | Hemiclonic, spasms, FDS, asymmetric tonic, status epilepticus | Profound intellectual disability, acquired microcephaly, choreiform movements | American | |
| 7 | c.862G>A; p.Gly288Ser | Unknown | MMFSI | 5 weeks | Focal clonic, eye deviation, oculoclonic, TCS, focal | Global developmental delay | Australian Italian/Egyptian Origin | |
| 8 | c.1018G>A; p.Val340Met | Unknown | Multifocal epilepsy | 3 years | Atypical absences, atonic seizures, focal atonic seizures, TCS | Limbic encephalitis at seizure onset (encephalopathy, psychiatric features, Anti-GAD65 antibodies). Psychiatric features resolved, behavioral problems, learning impairment and epilepsy persisted | Dutch | |
| 9 | c.1283G>A; p.Arg428Gln | De novo | MMFSI | 3 weeks | Spasms, tonic, FDS | Profound early developmental arrest, visual impairment, acquired microcephaly, craniosynostosis | Canadian | |
| 10 | c.2782C>T; p.Arg928Cys | Unknown | NFLE | 5 years | Tonic seizures, TCS | Learning impairment | Dutch | |
| 11 | c.2800G>A; p.Ala934Thr | Maternal mosaicism | MMFSI | 8 weeks | FDS, asymmetric tonic, aura, myoclonic, NCSE | Profound developmental delay, generalized spasticity, hyperreflexia, precocious puberty | Australian | |
| 12 | c.2849G>A; p. Arg950Gln | De novo | MMFSI | 5 months | Focal, tonic–clonic, | Developmental delay, hypotonia, hyperreflexia, progressive microcephaly, autistic tracts | Italian |
FDS, focal dyscognitive seizure; TCS, tonic–clonic seizures; NCSE, nonconvulsive status epilepticus; MMFSI, malignant migrating focal seizures of infancy; ADNFLE, autosomal dominant nocturnal frontal lobe epilepsy; NFLE, nocturnal frontal lobe epilepsy.
Figure 1.
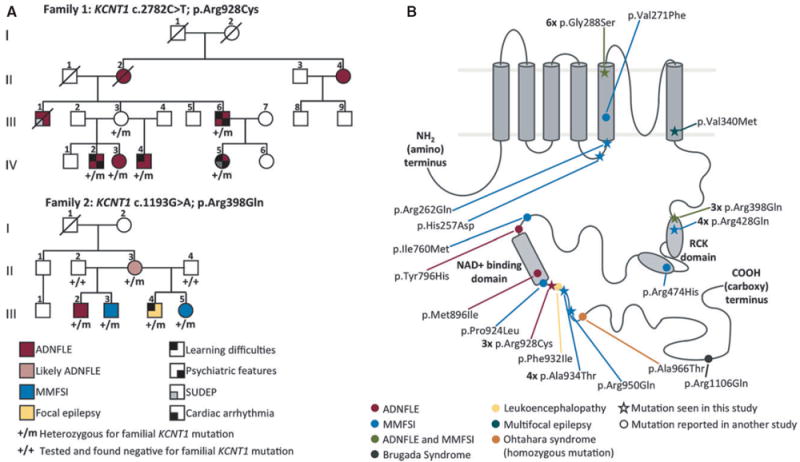
(A) Pedigrees of families 1 and 2 with KCNT1 mutations. Diagonal lines indicate deceased individuals. (B) Diagram of the KCNT1 protein showing the protein structure and the locations of the mutations identified in this and previous studies.2-9,13 The KCNT1 protein consists of a small amino-terminal domain, a transmembrane domain containing six transmembrane segments with the pore-loop between segments 5 and 6 and a large intracellular carboxy-terminal domain containing tandem RCK domains and an NAD+ binding domain. Mutations seen in this study are indicated by stars and those seen in other studies are indicated by dots. ADNFLE mutations are marked in red, MMFSI mutations in blue, the leukoencephalopathy mutation in yellow, the mutation seen in the multifocal epilepsy patient in blue-green, and mutations seen in both ADNFLE and MMFSI patients in green. The homozygous mutation seen in Ohtahara syndrome is marked in orange and the mutation seen in Brugada syndrome is marked in dark gray. Mutations observed in multiple families/patients are indicated by the numbers in bold type.
Epilepsia © ILAE
A heterozygous missense mutation KCNT1 c.2782C>T; p.Arg928Cys was identified in family 1 of Danish origin. The mutation cosegregates with the nocturnal frontal lobe epilepsy (NFLE) phenotype (Fig. 1A) and affected individuals showed a mean age of onset of nocturnal seizures of 9 years (range 1–15 years). Learning impairment, memory deficit, and psychiatric problems, including depression, suicide attempt, anxiety, and attention-deficit/hyperactivity disorder (ADHD) occurred in four of five affected family members, consistent with our earlier report of KCNT1 causing ADNFLE with intellectual disability (ID) and psychiatric features.2 Interictal EEG data available in two individuals (III.6 and IV.5) showed epileptiform abnormalities in the frontotemporal regions (Fig. S1). Individual III.1 (Fig. 1A) had frequent nocturnal seizures and died suddenly and unexpectedly during the night at 23 years of age. His death was classified as sudden unexplained death in epilepsy (SUDEP). Although the KCNT1 mutation segregated in his family, we were unable to test this deceased individual for the KCNT1 mutation. Video-polygraphic study of individual IV-5 with the KCNT1 mutation and ADNFLE showed irregularity in the heart rhythm during wakefulness, increasing significantly during sleep especially during the rapid eye movement (REM) periods. These abnormalities included an ST elevation in the J point plus several supraventricular extra systoles, suggesting Brugada syndrome. Whole exome sequencing of individual IV-5 revealed no mutations in any gene listed in Online Mendelian Inheritance in Man (OMIM) as associated with cardiac arrhythmia.
In the analysis of 92 unrelated families with focal epilepsy, a heterozygous KCNT1 mutation c.1193G>A; p.Arg398Gln co-segregating with affected status was identified in family 2 (Fig. 1A). The family comprised five affected individuals, one with ADNFLE, one with likely ADNFLE, one with focal epilepsy, and two with MMFSI. Notably, in this family the same KCNT1 mutation, p.Arg398Gln can lead to either ADNFLE or MMFSI.
Patient 10 with sporadic NFLE had the KCNT1 mutation c.2782C>T; p.Arg928Cys. Prior to seizure onset the patient had normal intelligence, with subsequent regression (TIQ 78 at 8 years of age), and did not exhibit any psychiatric features. Patient 8, who had multifocal epilepsy with onset associated with limbic encephalitis due to anti-GAD65 antibodies, had the KCNT1 mutation c.1018G>A; p.Val340- Met.
Eight unrelated patients with MMFSI (patients 3–7, 9, 11, and 12) were found to have KCNT1 mutations. These were c.769C>G; p.His257Asp, c.785G>A; Arg262Gln, c.862G>A; p.Gly288Ser (seen in three patients), c.1283G>A; p.Arg428Gln, c.2800G>A; p.Ala934Thr and c.2849G>A; p.Arg950Gln. None of the mutations were present in publicly available variant datasets (dbSNP, NHLBI, Exome Variant Server) and each was predicted to be possibly damaging by both the PolyPhen-2 and SIFT in silico prediction tools, consistent with being pathogenic. Mutations were shown to have occurred de novo in five of the eight patients, consistent with previous reports of de novo KCNT1 mutations in MMFSI.3,6,7 One patient (patient 11) inherited a mutation (p.Ala934Thr) from an unaffected parent who was shown to be mosaic for the KCNT1 mutation by restriction fragment length assays (HhaI and BstUI digests). Quantitation of bands from the BstUI digest indicated that the mutation was present in approximately 60% of peripheral blood derived cells (data not shown).
Discussion
We have identified a total of 12 new unrelated cases (2 families and 10 sporadic) with mutations in KCNT1. All KCNT1 mutations identified both previously2-9 and here are missense mutations, with no nonsense or other truncating mutations reported. This suggests that perturbation of normal KCNT1 protein function, rather than loss of function, underlies the pathogenicity associated with KCNT1 mutations. The mutations initially reported in ADNFLE and MMFSI were clustered around the RCK and NAD+ binding domains of the protein.2-4 More recently, mutations have been reported within the S5 transmembrane segment of the protein,6,7,9 indicating that alteration of other regions of KCNT1 is also pathogenic. Here we report three novel mutations in patients with multifocal epilepsy and MMFSI in or near the S5 and S6 region of the KCNT1 channel subunit. These findings provide further evidence for the contribution of mutations in this region of KCNT1 to the pathogenesis of MMFSI.
Although ID and psychiatric disorders have previously been reported in ADNFLE, these phenotypic features are infrequent in ADNFLE families with mutations in the other ADNFLE-associated genes CHRNA4, CHRNB2, and CHRNA2.2 Our finding of ADNFLE, intellectual disability, and psychiatric features associated with mutation of KCNT1 in family 1 provides further evidence that KCNT1 mutations cause a distinct form of ADNFLE that is more severe and includes comorbidities.2,10 Patients presenting with these features should be tested for KCNT1.
In addition to the neurologic phenotypes observed in the families in this study, one individual in family 1 had cardiac arrhythmia and another had SUDEP, which is thought to have cardiac involvement.11,12 This suggests the involvement of KCNT1 mutations in cardiac disorders. Indeed, a KCNT1 mutation has recently been reported in a patient with epilepsy and Brugada syndrome.13 The coexistence of epilepsy and Brugada syndrome has been reported previously in a family with a sodium channel mutation (SCN5A).14 Further clinical examination of patients with KCNT1 mutations to look for cardiac features and functional studies of the KCNT1 protein are required to explore the possible relationship between KCNT1 mutations and cardiac arrhythmias.
ADNFLE families with KCNT1 mutations have previously shown 100% penetrance of the mutation.2 In contrast, in the current study one individual (family 1, III.3) with a KCNT1 mutation did not have seizures. The nonpenetrance in this individual is unlikely to be due to somatic mosaicism, as she is inferred to have inherited a KCNT1 mutation from her affected mother (II.2, deceased and unavailable for mutation analysis). Thus her unaffected status is most likely due to incomplete penetrance, which has not been previously described for KCNT1 mutations.
In this study, we have identified recurrent mutations in KCNT1, with p.Gly288Ser found three times and p.Arg928Cys seen twice, suggesting that there are mutational “hot spots” in KCNT1. Consistent with this, some mutations identified in the 12 patients described here have been seen previously in other patients, with 5 of the 16 different KCNT1 mutations described to date being observed multiple times (Fig. 1B). This is likely due to the occurrence of these mutations in CpG di-nucleotides, which are particularly susceptible to mutation.15 Therefore, it is emerging that there are recurrent pathogenic mutations in KCNT1. This is important when making diagnoses for patients.
Somatic mosaicism of the KCNT1 mutation p.Ala934Thr was observed in the unaffected parent of one patient (patient 11), with the mutation estimated to be present in 60% of peripheral blood cells. This indicates that significant levels of somatic mosaicism of KCNT1 mutations can exist without causing an overt phenotype. Parental mosaicism of a KCNT1 mutation poses the risk of recurrence of affected offspring; thus this finding has important implications for the genetic counseling of families with KCNT1 mutations.
Although seizures are the predominant feature so far associated with KCNT1 mutations, this study highlights the pleiotropic effects of mutations in this gene. The severity of the observed epilepsy phenotypes ranges from mild (a single seizure) to severe (epileptic encephalopathy). Moreover, we report here that the same KCNT1 mutation, p.Arg398Gln can lead to either ADNFLE or MMFSI within the same family, indicating that genotype–phenotype correlations are not straightforward. The association of both phenotypes with the p.Arg398Gln mutation was unexpected, as previous data indicated that mutations associated with MMFSI cause a significantly larger increase in current amplitude than those associated with ADNFLE in mutant KCNT1 channels expressed in vitro in Xenopus oocytes, providing an explanation for how different mutations cause different phenotypes. 8 However, a more recent study9 shows that the p.Gly288Ser mutation can also cause both phenotypes. Furthermore, that study9 did not show the same pattern of differences in the current increases as the original study,8 casting doubt on the conclusion that differences in the increase in current amplitude caused by the different mutations explain the different phenotypes associated with KCNT1 mutations. The association of the different phenotypes with the same mutation may instead be the result of either genetic modifiers or environmental factors.
In summary, our study confirms the role of KCNT1 in severe ADNFLE with intellectual and psychiatric comorbidities and in MMFSI. We show that KCNT1 also plays a role in focal and possibly in multifocal epilepsy and raise the possibility of an additional role in cardiac disorders and SUDEP. As with a number of genes containing mutations of major effect in neurologic disorders, deciphering genotype– phenotype correlations for KCNT1 mutations is emerging as challenging. However, the identification of a KCNT1 mutation in individuals presenting with a seizure phenotype will provide patients with a molecular diagnosis for their previously unsolved disorder. As more is learned about the effects of KCNT1 mutations, we anticipate there will be improvements in the therapeutic treatment of patients.
Supplementary Material
Acknowledgments
We thank the patients and their families for their participation in this study. We thank Bev Johns and Robert Schultz for technical assistance. This work was supported by grants from the National Health and Medical Research Council of Australia (Early Career Fellowship 1016715 to SEH, Career Development Fellowship 1032603 to LMD, and Program Grant 628952 to LMD and IES).
Biographies

Rikke Steensbjerre Møller is an assistant professor at the Danish Epilepsy Centre and University of Southern Denmark.

Sarah Heron is a research fellow in the Epilepsy Research Group, University of South Australia.
Footnotes
Supporting Information
Additional Supporting Information may be found in the online version of this article:
Figure S1. Interictal EEG features of patients from family 1, III.6 (to the left) and IV.5 (to the right), respectively, during drowsiness and during stage 2 non–rapid eye movement (NREM) sleep.
Table S1. List of other genes analyzed for patients described in this study.
Data S1. Methods.
Disclosure
None of the authors has any conflicts of interest to disclose. We confirm that we have read the Journal’s position on issues involved in ethical publication and affirm that this report is consistent with those guidelines.
References
- 1.Battacharjee A, Kaczmarek LK. For K+ channel, Na+ is the new Ca2+ Trends Neurosci. 2005;28:422–428. doi: 10.1016/j.tins.2005.06.003. [DOI] [PubMed] [Google Scholar]
- 2.Heron SE, Smith KR, Bahlo M, et al. Missense mutations in the sodium-gated potassium channel gene KCNT1 cause severe autosomal dominant nocturnal frontal lobe epilepsy. Nat Genet. 2012;44:1188–1190. doi: 10.1038/ng.2440. [DOI] [PubMed] [Google Scholar]
- 3.Barcia G, Fleming MR, Deligniere A, et al. De novo gain-of-function KCNT1 channel mutations cause malignant migrating partial seizures of infancy. Nat Genet. 2012;44:1255–1259. doi: 10.1038/ng.2441. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Vanderver A, Simons C, Schmidt JL, et al. Identification of a novel de novo p.Phe932Ile KCNT1 mutation in a patient with leukoencephalopathy and severe epilepsy. Pediatr Neurol. 2014;50:112–114. doi: 10.1016/j.pediatrneurol.2013.06.024. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Martin HC, Kim GE, Pagnamenta AT, et al. Clinical whole-genome sequencing in severe early-onset epilepsy reveals new genes and improves molecular diagnosis. Hum Mol Genet. 2014;23:3200–3211. doi: 10.1093/hmg/ddu030. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Ishii A, Shioda M, Okumura A, et al. A recurrent KCNT1 mutation in two sporadic cases with malignant migrating partial seizures in infancy. Gene. 2013;531:467–471. doi: 10.1016/j.gene.2013.08.096. [DOI] [PubMed] [Google Scholar]
- 7.McTague A, Appleton R, Avula S, et al. Migrating partial seizures of infancy: expansion of the electroclinical, radiological and pathological disease spectrum. Brain. 2013;136:1578–1591. doi: 10.1093/brain/awt073. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Milligan CJ, Li M, Gazina EV, et al. KCNT1 gain of function in 2 epilepsy phenotypes is reversed by quinidine. Ann Neurol. 2014;75:581–590. doi: 10.1002/ana.24128. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Kim GE, Kronengold J, Barcia G, et al. Human Slack potassium channel mutations increase positive cooperativity between individual channels. Cell Rep. 2014;9:1661–1672. doi: 10.1016/j.celrep.2014.11.015. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Derry CP, Heron SE, Phillips F, et al. Severe autosomal dominant nocturnal frontal lobe epilepsy associated with psychiatric disorders and intellectual disability. Epilepsia. 2008;49:2125–2129. doi: 10.1111/j.1528-1167.2008.01652.x. [DOI] [PubMed] [Google Scholar]
- 11.Aurlien D, Leren TP, Taubøll E, et al. New SCN5A mutation in a SUDEP victim with idiopathic epilepsy. Seizure. 2008;18:158–160. doi: 10.1016/j.seizure.2008.07.008. [DOI] [PubMed] [Google Scholar]
- 12.Hindocha N, Nashef L, Elmslie F, et al. Two cases of sudden unexpected death in epilepsy in a GEFS+ family with an SCN1A mutation. Epilepsia. 2008;49:360–365. doi: 10.1111/j.1528-1167.2007.01439_2.x. [DOI] [PubMed] [Google Scholar]
- 13.Juang JMJ, Lu TP, Lai LC, et al. Disease-targeted sequencing of ion channel genes identifies de novo mutations in patients with non-familial Brugada syndrome. Sci Rep. 2014;4:6733. doi: 10.1038/srep06733. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Parisi P, Oliva A, Coll Vidal M, et al. Coexistence of epilepsy and Brugada syndrome in a family with SCN5A mutation. Epilepsy Res. 2013;105:415–418. doi: 10.1016/j.eplepsyres.2013.02.024. [DOI] [PubMed] [Google Scholar]
- 15.Holliday R, Grigg GW. DNA methylation and mutation. Mutat Res. 1993;285:61–67. doi: 10.1016/0027-5107(93)90052-h. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.