

Abstract
The bottromycins belong to the ribosomally synthesized and posttranslationally modified peptide (RiPP) family of natural products. Bottromycins exhibit unique structural features, including a hallmark macrolactamidine ring and thiazole heterocycle for which divergent members of the YcaO superfamily have been biosynthetically implicated. Here we report the in vitro reconstitution of two YcaO proteins, BmbD and BmbE, responsible for the ATP-dependent cyclodehydration reactions that yield thiazoline- and macrolactamidine-functionalized products, respectively. We also establish the substrate tolerance for BmbD and BmbE and systematically dissect the role of the follower peptide, which we show serves a purpose similar to canonical leader peptides in directing the biosynthetic enzymes to the substrate. Lastly, we leverage the expanded capabilities of YcaO proteins to conduct an extensive bioinformatic survey to classify known YcaO chemistry. This analysis predicts new functions remain to be uncovered within the superfamily.
Graphical Abstract
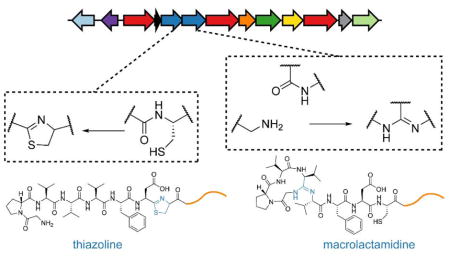
Ribosomally synthesized and posttranslationally modified peptides (RiPPs) are a growing natural product class with remarkable structural diversity.1 RiPP precursor peptides are direct gene products that undergo various posttranslational modifications by locally encoded biosynthetic enzymes.2 RiPP precursor peptides are functionally bipartite with the modifications typically being installed on the C-terminal “core peptide” region of the precursor. The N-terminal “leader peptide” region frequently contains recognition sequences that are bound by the biosynthetic enzymes. The bottromycin RiPP class possesses an unusually high density of posttranslational modifications on a small (8-mer) core peptide3–6 and inverts the RiPP precursor paradigm by displaying an N-terminal core peptide and C-terminal “follower peptide” (Figure 1). The bio-synthetic gene cluster for bottromycin was reported in 2012 by four independent groups, which demonstrated that the sequences of both the precursor peptide and the tailoring genes are strongly conserved in several Streptomyces species (Figure S1 and Table S1).7–10
Figure 1.

(A) Organization of the bottromycin biosynthetic gene cluster in Streptomyces bottropensis. (B) Precursor peptide sequence and reactions catalyzed by the two YcaO proteins (BmbD and BmbE). (C) Structure of bottromycin A2.
Despite considerable progress in characterizing several posttranslational modifications present in bottromycin,6,8–11 the formation of the macrolactamidine, which involves Gly1 and Val4, as well as a thiazoline derived from Cys8, have remained enigmatic (Figure 1).
Deletion of three genes encoding class B radical-S-adenosylmethionine (SAM) methytransferases produced bottromycin analogs lacking methyl groups on the β-carbons of Pro2, Val3/Val4 and Phe6.7–9 However, two proteins belonging to the YcaO superfamily (BmbD and BmbE, protein family PF02624)12 lacked clearly defined functions, but were hypothesized to be involved in modifications requiring activation of the peptide backbone.7,8 Work from our group has demonstrated that members of the YcaO superfamily utilize ATP to directly O-phosphorylate backbone amides resulting in the formation of azoline heterocycles using the nucleophilic side chains of Cys, Ser, and Thr residues.13,14 Individual deletion of bmbE and bmbH homologs in Streptomyces scabies were capable of producing a species consistent with a methylated, thiazoline-containing linear peptide, suggesting BmbE and BmbH cooperate to form the macrolactamidine. However, the precise roles of these proteins, as well as the origin of the thiazoline, has remained unresolved.11
All previous in vitro characterization of the YcaO superfamily has focused on biosynthetic gene clusters that encode a YcaO partner protein from either the E1 ubiquitin-activating enzyme15,16 or ocin-ThiF-like protein family (PF00899).14 These partner proteins are found either as N-terminal fusions to the YcaO or as discrete proteins. Both varieties contain a ~90-residue domain known as the RiPP precursor peptide recognition element (RRE) that physically engages the peptide substrate and delivers the core region to the biosynthetic enzymes.17 In contrast, bottromycin gene clusters contain RRE domains on the radical-SAM methyltransferases (Figure 1) and the BmbD and BmbE proteins bear minimal sequence similarity to canonical YcaOs outside of the conserved ATP-binding site (Figure S2).14 Intrigued by the putative involvement of RRE-independent YcaOs in macrolactamidine and thiazoline formation, we sought to characterize the catalytic activity of BmbD and BmbE in vitro.
The bmbD and bmbE genes were cloned into an expression vector that affords an N-terminal fusion to maltose-binding protein (Tables S2–S3). The corresponding proteins were overproduced in Escherichia coli and purified by amylose affinity chromatography (Figure S3). The precursor peptide, BmbC, was overproduced in a form that lacks the N-terminal Met residue. Treatment of BmbC with BmbD resulted in the ATP-dependent loss of 18 Da, consistent with dehydration as assessed by matrix-assisted laser desorption ionization time-of-flight mass spectrometry (MALDI-TOF-MS, Figure 2). An identical result was obtained when treating BmbC with BmbE. These findings are in accord with the known chemistry catalyzed by YcaO cyclode-hydratases.2 Furthermore, the Glu-rich, ATP-binding motifs conserved amongst YcaOs were essential for BmbD and BmbE processing, as substitution with Ala abolished activity (Figures S2, S4). Unlike canonical E1/ocin-ThiF-dependent, azoline-forming YcaOs,13 BmbD and BmbE do not contain Pro-rich C-termini; instead, their C-termini are relatively basic. Modest truncations (Δ20) to the C-termini did not abolish activity for either enzyme, which stands in contrast to YcaOs with Pro-rich C-termini (Figure S5).
Figure 2.

MALDI-TOF-MS analysis of (A) precursor peptide BmbC calc. m/z 4675 (B) Reaction with BmbD yields ions with an m/z value consistent with dehydration. (C) BmbD reaction omitting ATP. (D) Same as panel B but with BmbE. (E) BmbE reaction omitting ATP.
To determine which dehydration reaction was catalyzed by which protein, we exploited the fact that the thiazoline product contains a primary amino group (Gly1), whereas the macrolactamidine product contains a thiol group (Cys8). Thus, the products of the BmbD and BmbE reactions were subjected to an amine-specific reductive amination with formaldehyde and borane and thiol-specific alkylation with 2-bromoethylamine and subsequently analyzed by MALDI-TOF-MS. As expected, the thiazoline prod-uct underwent dimethylation on the Gly1 N-terminus while the macrolactamidine product was alkylated on the Cys8 side chain with no cross-products observed. Moreover, a BmbC-C8A variant was processed by BmbE, but not BmbD, which together implicated BmbD in thiazoline formation and BmbE in macrolactamidine formation (Figure S6, Table S4). High-resolution electrospray ionization tandem MS (HR-ESI-MS/MS) confirmed the locations of these posttranslational modifications (Figure S7, Table S5).
NMR spectroscopic analysis of a trypsin-treated BmbC-N15K variant was performed to further characterize the BmbC starting material and the BmbD/BmbE reaction products. 1H chemical shifts and the absence of characteristic nuclear Overhauser effect correlations in DMSO suggested the absence of secondary structure in unmodified BmbC. Comparison of the spectra for BmbC before and after BmbD processing showed that the only significant differences were in the region around Cys8, including: the amide protons for Asp7, Met9, Thr10, and Ala11; the Cα protons for Asp7 and Met9; the Cβ protons of Met9 (Figure S8). Moreover, the signals for the Cα and Cβ protons of Cys8 were shifted to values typically observed for thiazoline-containing peptides (Table S6).18 The same analysis was performed for BmbE-treated BmbC, where the Cβ and Cδ protons for Val3–5, as well as the Cα, Cβ, and Cγ protons of Pro2 were strongly shifted in comparison with unmodified BmbC (Figure S8). More modest changes were detected for the Phe6 and Asp7 protons. Collectively, the chemical labeling, processing of BmbC-C8A, HR-ESI-MS/MS, and NMR data allow the unambiguous assignment of thiazoline formation to BmbD and macrolactamidine formation to BmbE.
Typical YcaO cyclodehydratases bind their substrates via an RRE-containing partner protein;14 however, BmbD and BmbE were catalytically competent in vitro in the absence of any partner protein. For BmbE, this finding is in contrast to a previous in vivo metabolomics study that implicated BmbH in macrolactamidine formation.11 To assess if BmbH or BmbG (another functionally unassigned protein, Figure 1) enhanced BmbE activity in vitro, the proteins were individually supplied to the YcaO reaction mixture. Neither addition of BmbH or BmbG resulted in more product formation. Identical results were obtained when BmbD reactions were supplemented with BmbG/H (Figure S9).
A previous heterologous expression study10 showed that the bottromycin biosynthetic pathway was largely intolerant towards accepting BmbC variants. To determine if the same was true in vitro, we evaluated 21 BmbC variants as BmbD/BmbE substrates using a MALDI-TOF-MS endpoint assay (Figures 3, S7). As observed for other RiPPs,19 the variants were more widely tolerated as substrates in vitro as opposed to in vivo (Table S4). However, BmbD was still only able to process Cys8 and substitution of distal residues could abolish thiazoline formation. Similarly, BmbE exclusively formed a macrolactamidine with the fourth residue of BmbC and attempts to alter macrocycle size were unsuccessful (Table S2).
Figure 3.

Substrate tolerance of BmbD and BmbE (A) Core peptide variants processed by each YcaO. Δ indicates amino acid deletion at this position. (B) Activity of BmbC variants with Ala substitution in the follower peptide. The Phe13-Asp17 and Glu22-Glu32 regions were targeted based on their similarity to the lanthipeptide and cyanobactin recognition sequences, respectively. Circles indicate sites of Ala substitution. Filled circles indicate variants that inhibited BmbD or BmbE processing.
We next investigated if the BmbC follower peptide played a role in substrate recognition. Stop codons (denoted as asterisks) were introduced to produce C-terminal truncations of BmbC at intervals of five to identify the minimal sequence compatible with BmbD/BmbE processing. Consistent with follower peptide sequence conservation (Figure S1), BmbC-G35* was processed while E30* was not (Figure S10). Analysis of additional truncations confirmed that residues 1–34 of BmbC represents the minimum substrate for BmbD and BmbE.
The follower peptide of BmbC contains regions that are reminiscent of lanthipeptide and cyanobactin recognition sequences (Figure 3).20,21 To determine the relevance of these regions, 15 residues were individually replaced with Ala and assessed as BmbD/BmbE substrates. Despite their sequence divergence and the lack of a recognizable RRE, many of the same BmbC residues were important for both BmbD and BmbE processing (Figures 3, S11). To directly measure the binding affinity towards BmbD/BmbE, we prepared double variant BmbC-C8S/S43C to allow for the selective installation of fluorescein near the C-terminus of the peptide (Figure S12). Using a fluorescence polarization assay, the dissociation constants (KD) for BmbD and BmbE were estimated to be greater than 50 μM (Figure S13). As mentioned above, the bottromycin biosynthetic gene cluster does indeed encode proteins with RREs, but these domains are fused to the C-termini of the rSAM methyltransferases. To evaluate their role in BmbC binding, we excised the RRE domains from BmbB and BmbF and subjected the proteins to the same fluorescence polarization assay. The binding affinities for BmbC were significantly tighter relative to the YcaOs with KD values of 876 ± 67 nM (BmbB) and 597 ± 25 nM (BmbF). Further studies are warranted to reconcile how these binding and activity data fit into the yet-unresolved order of events during bottromycin biosynthesis.
Having confirmed a new activity for an RRE-independent YcaO in vitro, we wondered how often YcaOs are encoded in genomic contexts consistent with macrolactamidine and azoline formation. For this, we employed RODEO (Rapid ORF Description and Evaluation Online)22 to catalog the co-occurrence frequency of protein families for the ~13,000 YcaO proteins deposited in GenBank (Figure S14). Less than one-third of all YcaOs co-occur with a E1/ocin-ThiF-like member, which is the arrangement found in several RiPP classes, including linear azol(in)e-containing peptides, thiopeptides, and cyanobactins. A separate subset of YcaO proteins (~8% of the total) co-occur with “TfuA”, a protein of unknown function. Recent work from our group has shown that the TfuA-YcaO pair found in methanogenic archaea is responsible for installing a thioglycine moiety in methyl-coenzyme M reductase.23 This result suggests that the TfuA-YcaO present in the thioviridamide bio-synthetic gene cluster would similarly be responsible for thioamide formation.24
To shed light on the potential function of the remaining 60+% of YcaOs, we analyzed the local genomic region for Pfam co-occurrence (Table S7) as well as generated sequence similarity networks to assess phylogenetic and relatedness trends (Figure S15). We then created custom profile hidden Markov models25 based on known BmbD and BmbE homologs to discriminate between azoline- and amidine-forming YcaOs and reanalyzed the networks based on the resulting expectation values (Figures S16–17). While very few of the uncharacterized YcaOs were similar to BmbD (azoline-forming YcaO), one-third of the sequences showed significant similarity to BmbE, suggesting that amidine-forming YcaOs may be more prevalent than currently appreciated.26 The data further predict that RRE-independent azoline-forming YcaOs may be relatively rare (Figure S18).
In summary, our work has delineated the roles of BmbD, BmbE, and the follower peptide in bottromycin biosynthesis. BmbE has been confirmed to carry out a new enzymatic reaction for the YcaO superfamily. Our bioinformatic analysis not only implicates YcaO involvement in a diversity of new natural product biosynthetic gene clusters, but also a significant potential for new chemistry that would expand upon the established roles in azoline, thioamide, and amidine formation.
Supplementary Material
Acknowledgments
This work was supported by the National Institutes of Health (GM097142 to D.A.M.), the Chemistry–Biology Interface Training Program (GM070421 to C.J.S.), and the Seemon Pines Fellowship from the Department of Chemistry at the University of Illinois at Urbana-Champaign (to G.A.H.), Wolfson Research Merit Award (WM130033) from the Royal Society (to G.L.C.).
Footnotes
Notes
The authors declare no competing financial interests.
The Supporting Information, including experimental methods and supporting figures, is available free of charge on the ACS Publications website.
References
- 1.Arnison PG, Bibb MJ, Bierbaum G, Bowers AA, Bugni TS, Bulaj G, Camarero JA, Campopiano DJ, Challis GL, Clardy J, et al. Nat Prod Rep. 2013;30:108. doi: 10.1039/c2np20085f. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Burkhart BJ, Schwalen CJ, Mann G, Naismith JH, Mitchell DA. Chem Rev. 2017;117:5389. doi: 10.1021/acs.chemrev.6b00623. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Schipper D. J Antibiot. 1983;36:1076. doi: 10.7164/antibiotics.36.1076. [DOI] [PubMed] [Google Scholar]
- 4.Kaneda M. J Antibiot. 2002;55:924. doi: 10.7164/antibiotics.55.924. [DOI] [PubMed] [Google Scholar]
- 5.Shimamura H, Gouda H, Nagai K, Hirose T, Ichioka M, Furuya Y, Kobayashi Y, Hirono S, Sunazuka T, Omura S. Angew Chem Int Ed Engl. 2009;48:914. doi: 10.1002/anie.200804138. [DOI] [PubMed] [Google Scholar]
- 6.Mann G, Huo L, Adam S, Nardone B, Vendome J, Westwood NJ, Muller R, Koehnke J. Chembiochem. 2016;17:2286. doi: 10.1002/cbic.201600406. [DOI] [PubMed] [Google Scholar]
- 7.Crone WJK, Leeper FJ, Truman AW. Chem Sci. 2012;3:3516. [Google Scholar]
- 8.Gomez-Escribano JP, Song L, Bibb MJ, Challis GL. Chem Sci. 2012;3:3522. [Google Scholar]
- 9.Huo L, Rachid S, Stadler M, Wenzel Silke C, Müller R. Chem Biol. 2012;19:1278. doi: 10.1016/j.chembiol.2012.08.013. [DOI] [PubMed] [Google Scholar]
- 10.Hou Y, Tianero MD, Kwan JC, Wyche TP, Michel CR, Ellis GA, Vazquez-Rivera E, Braun DR, Rose WE, Schmidt EW, Bugni TS. Org Lett. 2012;14:5050. doi: 10.1021/ol3022758. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Crone WJ, Vior NM, Santos-Aberturas J, Schmitz LG, Leeper FJ, Truman AW. Angew Chem Int Ed Engl. 2016;55:9639. doi: 10.1002/anie.201604304. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Finn RD, Bateman A, Clements J, Coggill P, Eberhardt RY, Eddy SR, Heger A, Hetherington K, Holm L, Mistry J, Sonnhammer EL, Tate J, Punta M. Nucleic Acids Res. 2014;42:D222. doi: 10.1093/nar/gkt1223. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Dunbar KL, Chekan JR, Cox CL, Burkhart BJ, Nair SK, Mitchell DA. Nat Chem Biol. 2014;10:823. doi: 10.1038/nchembio.1608. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Dunbar KL, Tietz JI, Cox CL, Burkhart BJ, Mitchell DA. J Am Chem Soc. 2015;137:7672. doi: 10.1021/jacs.5b04682. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Dunbar KL, Melby JO, Mitchell DA. Nat Chem Biol. 2012;8:569. doi: 10.1038/nchembio.944. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Cox CL, Doroghazi JR, Mitchell DA. BMC Genomics. 2015;16:1. doi: 10.1186/s12864-015-2008-0. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Burkhart BJ, Hudson GA, Dunbar KL, Mitchell DA. Nat Chem Biol. 2015;11:564. doi: 10.1038/nchembio.1856. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Mocek U, Beale JM, Floss HG. J Antibiot. 1989;42:1649. doi: 10.7164/antibiotics.42.1649. [DOI] [PubMed] [Google Scholar]
- 19.Zhang Z, Hudson GA, Mahanta N, Tietz JI, van der Donk WA, Mitchell DA. J Am Chem Soc. 2016;138:15511. doi: 10.1021/jacs.6b08987. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Ortega MA, Velasquez JE, Garg N, Zhang Q, Joyce RE, Nair SK, van der Donk WA. ACS Chem Biol. 2014;9:1718. doi: 10.1021/cb5002526. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Sardar D, Pierce E, McIntosh JA, Schmidt EW. ACS Synth Biol. 2015;4:167. doi: 10.1021/sb500019b. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Tietz JI, Schwalen CJ, Patel PS, Maxson T, Blair PM, Tai HC, Zakai UI, Mitchell DA. Nat Chem Biol. 2017;13:470. doi: 10.1038/nchembio.2319. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Nayak DD, Mahanta N, Mitchell DA, Metcalf WW. eLife. 2017;6:e29218. doi: 10.7554/eLife.29218. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Izawa M, Kawasaki T, Hayakawa Y. Appl Environ Microbiol. 2013;79:7110. doi: 10.1128/AEM.01978-13. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Finn RD, Clements J, Arndt W, Miller BL, Wheeler TJ, Schreiber F, Bateman A, Eddy SR. Nucleic Acids Res. 2015;43:W30. doi: 10.1093/nar/gkv397. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Metelev M, Osterman IA, Ghilarov D, Khabibullina NF, Yakimov A, Shabalin K, Utkina I, Travin DY, Komarova ES, Serebryakova M, Artamonova T, Khodorkovskii M, Konevega AL, Sergiev PV, Severinov K, Polikanov YS. Nat Chem Biol. 2017;13:1129. doi: 10.1038/nchembio.2462. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.