

Abstract
Cell invasion through basement membrane (BM) barriers is crucial during development, leukocyte trafficking, and for the spread of cancer. Despite its importance in normal and diseased states, the mechanisms that direct invasion are poorly understood, in large part because of the inability to visualize dynamic cell-basement membrane interactions in vivo. This protocol describes multi-channel time-lapse confocal imaging of anchor cell invasion in live C. elegans. Methods presented include outline slide preparation and worm growth synchronization (15 min), mounting (20 min), image acquisition (20-180 min), image processing (20 min), and quantitative analysis (variable timing). Images acquired enable direct measurement of invasive dynamics including invadopodia formation, cell membrane protrusions, and BM removal. This protocol can be combined with genetic analysis, molecular activity probes, and optogenetic approaches to uncover molecular mechanisms underlying cell invasion. These methods can also be readily adapted for real-time analysis of cell migration, basement membrane turnover, and cell membrane dynamics by any worm laboratory.
INTRODUCTION
Basement membrane (BM) is a thin, dense, and highly cross-linked form of extracellular matrix (ECM) that underlies epithelia and endothelia, and surrounds muscle, nerve, and fat cells1,2. This sheet-like network functions, in part, as a barrier to the movement of cells across tissue boundaries. During normal development and immune surveillance, however, specialized cells acquire the ability to transmigrate through BM as they disperse to form organs or respond to injury or infection3–5. Cancer cells are thought to highjack normal programs for BM transmigration as they spread from a primary tumor to colonize distant sites3,4,6. Despite the central role in cancer lethality, cell invasion through BM remains one of the least understood steps in cancer progression7. Most BM transmigrations are difficult to visualize as they are dynamic, unpredictable, and often occur deep within tissues6,8. Because of this, research on cell invasion has almost exclusively used in vitro and ex vivo tissue culture models. While these systems have identified potential molecular mechanisms that govern cell invasion9,10, the complex milieu of extracellular signals, and biochemical and biophysical cell-cell and cell-matrix interactions that occur in vivo, cannot be recapitulated outside the normal physiological environment4. Thus, our understanding of BM transmigration remains incomplete.
Caenorhabditis elegans is a well-established model organism with a compact genome that is well suited for high-throughput genetic and functional genomic studies11. In addition, the worm’s transparency facilitates real-time visualization of many biological processes that are otherwise experimentally inaccessible in complex vertebrate tissues6,12. The anchor cell (AC) is a specialized uterine cell that invades through BM to connect the uterine and vulval tissues during hermaphrodite development13,14. These two tissues begin development juxtaposed but separated by the BM that lines each tissue. AC invasion initiates the connection of the uterine and vulval tissues, which is required for mating and egg-laying in the adult animal. The highly invariant nature of AC invasion, the ability to visualize the AC-BM interface, the ease of identifying genetic mutants and conducting RNA interference (RNAi) screens, and the robust transgenic approaches available for C. elegans, makes AC BM transmigration a uniquely powerful model to study cell invasion in vivo. Importantly, we have determined that AC invasion shares many of the same molecular and cellular mechanisms identified that regulate cancer cell invasion making it a relevant model to uncover conserved invasion programs affecting human health15–20.
Comparison with other models
In vivo, cell invasion through BM occurs in complex and dynamic cellular microenvironments where the invasive process is influenced by the mechanical properties and chemical cues of surrounding tissues. In addition, BM is comprised of more than 100 different proteins, glycoproteins and proteoglycans making it difficult to fully recapitulate BM invasion using 2D cell culture and 3D matrix models1,21–23. Since BM cannot be synthesized in vitro, many techniques for examining cell invasion rely on assays where cultured cells are placed on, or within a simplified matrix (e.g., gelatin or collagen), reconstituted BM (Matrigel), denuded BM explants or chick chorioallantoic membranes (CAM)10,24–26. New microfluidic platforms allow for real-time imaging of human cancer cell extravasation at single cell resolution within in vitro microvascular networks27. This assay highly recapitulates an important step in cancer metastasis, but basement membranes crossed during extravasation can only visualized statically by immunofluorescence approaches using antibodies after tissue fixation. These genetically and visually accessible models have been effective in identifying key pathways and cellular processes governing invasion. However, because these in vitro invasion assays either fail to recapitulate endogenous BM matrices or the native tissue environment, the physiological relevance of findings from these systems remain uncertain. Further, these models fail to reveal mechanisms that only manifest in complex in vivo environments.
A handful of in vivo models, have been developed in a variety of model organisms to study BM invasion in varying contexts4. Anterior-posterior axis polarization during mouse embryogenesis28, and the process of epithelial to mesenchymal transition during chick gastrulation29,30 are both similar to AC invasion in C. elegans in that they capture BM breaching during a predictable time point in animal development. However, BM visualization in both these models requires immunohistochemical analysis of fixed tissues using collagen IV or laminin antibodies since fluorescently tagged BM markers have not been developed. Drosophila imaginal disc eversion, is a well-studied developmental event that requires BM degradation at a precise stage in larval animals31,32. Importantly, as in C. elegans, BM proteins including collagen IV have been successfully tagged with fluorescent proteins in Drosophila33,34, but live-cell imaging of cell-BM interactions during disc eversion have not yet been successfully captured31. Drosophila tissue is highly pigmented making live-cell imaging deep within tissue more complex than imaging within transparent C. elegans35. Zebrafish xenographs36,37 and intravital imaging in mice38,39 examine the movements of transformed cells within and through blood vessels, essential steps in cancer cell metastasis. Through recent advances including use of an imaging window38 in mice and pigmentation mutants in zebrafish40, these models have overcome the problem of visual inaccessibility of imaging live events deep within tissues. However, BM cannot be directly visualized in either of these contexts and fluorescent proteins marking the vasculature are used to infer BM crossing in real-time. Direct visualization of BM in these systems also requires tissue fixation for antibody-based detection. Taken together, these models share some strengths and similarities to AC invasion in C. elegans. However, they have not yet allowed live-cell imaging approaches required to capture the dynamic events at the cell-BM interface during invasion.
Microfluidic technology is an alternative approach that could be used for imaging AC invasion in its entirety61,62. In particular, a recent device where worms are gently immobilized by flow and a gradual increase in pressure63, allows for high temporal resolution of imaging for extended durations. The downsides to microfluidic approaches are the complexity and cost of the systems. Further, these microfluidic techniques do not allow easy manipulation of the orientation of animals, which we have found helpful in visualizing AC invasion both laterally and ventrally. Finally, AC invasion is completed over a 2 hour window, where viability is not affected by anesthetic immobilization64, and thus microfluidic approaches are not necessary for optimal immobilization for imaging.
Development of the protocol
We present here a detailed protocol for using time-lapse microscopy and quantitative 4D image analysis to study AC invasion in C. elegans. Through our studies using this protocol, we have found that AC invasion can be broken down into distinct cellular processes19,20,41–43 (Supplementary Fig 1.; Supplementary Video 1). Beginning approximately six hours prior to invasion, a number of pro-invasive genes that regulate actin dynamics and BM composition are expressed and the encoded proteins polarize to the AC’s invasive cell membrane, the cell membrane at the AC-BM interface15,18,44. Shortly after this time (~1 hour), dynamic F-actin based AC-invadopodia form, depress the BM and then break down44. Each has a lifetime of approximately 1 minute. Triggered by a precisely timed secreted signal from the vulval cells, one or two invadopodia breach the BM during a narrow 30 minute window in the mid L3 larval stage15. The invadopodium that breaches the BM transforms into a large cellular protrusion that advances through a single hole in the underlying BM. As the protrusion enlarges, the BM is partially degraded and pushed aside as the protrusion extends between the underlying centrally located 10 fated vulval precursor cells (10 VPCs)19,45 (Supplementary Video 2). Taking advantage of the stereotyped nature and visual accessibility of AC invasion, we developed a time-lapse technique that allows convenient imaging of the entire AC invasion process, which is normally completed within 2 hours44. This model offers a unique opportunity to simultaneously examine dynamic events within the invading cell and in the BM being traversed.
Applications of the protocol
The imaging protocol described here can be used to study the entire process of cell invasion through BM—from initially polarizing the invasive cellular machinery towards the BM, to the formation of F-actin-based invadopodia, to the initial breach, to the formation of the invasive protrusion, and the modification, degradation and physical displacement of the BM. Using genetically encoded fluorescent proteins fused to your protein(s) of interest46–48, dual-color or multicolor imaging can be performed. This allows for simultaneous monitoring of two or more transgenes to uncover the dynamic interplay between the invading cell and the BM in real time. Alternatively, multiple proteins within the AC (often driven by AC-specific promoters “>” such as cdh-3>49, zmp-1>50, inft-1>51,52, lin-29>53,54) or the BM can be viewed simultaneously to uncover genetic interactions, sites of action, or possible colocalization between fluorophores15,43–45. Photodynamic imaging techniques, such as fluorescence recovery after photobleaching (FRAP)55 and fluorescence loss in photobleaching (FLIP)56 can also be implemented to assess cellular or BM dynamics during AC invasion16,18,45,53. Fluorescent activity probes for molecules such as the Rho GTPase can also be used15. Besides common fluorescent proteins, those with unique properties, such as photoconvertible fluorescent proteins, are particularly useful for tracking molecules of interest over time18,42,44. For instance, Dendra is a photoconvertible fluorescent protein that fluoresces green like GFP but fluoresces red after brief exposure to 405-nm light57. By fusing Dendra to laminin, a major component of BM, we tracked BM fate and found that physical displacement of the BM contributes to its removal during AC invasion44. Summaries of imaging techniques that have been implemented by our laboratory by adapting this protocol are summarized in Table 1.
TABLE 1.
Examples of 4D live cell imaging used to quantify invasion dynamics
| Description of experiment | Timing | Marker(s) used | Mutant/RNAi/Tx* | Ref |
|---|---|---|---|---|
| BM-Associated Proteins | ||||
| BM hole expansion | 60-90 min TL, 2 min Int | Laminin (LAM-1∷GFP) | WT, unc-40 (DCC) and him-4 (hemicentin) mutants | 44,45 |
| Hemicentin dynamics prior to and during AC invasion | 1 hr TL, 60 sec Int | Hemicentin (GFP∷HIM-4); Actin (mCh∷moeABP) | WT | 45 |
| FRAP analysis: | ||||
| Dystroglycan and Integrin membrane recovery rates | 9 min TL, 30 sec Int | Dystroglycan (DGN-1∷GFP); Integrin (PAT-3∷GFP) | WT and lin-29 (EGFR) mutants | 53 |
| Measuring BM collagen turnover | SS at 0 and 3 hr | Collagen (EMB-9∷mCh) | ost-1 (SPARC) overexpression | 16 |
| Landmark Photobleaching: | ||||
| Tissue shifting performed after photobleaching to measure BM linkage | SS before and after shifting | Laminin (LAM-1∷GFP) | WT and him-4 (hemicentin) mutants | 39 |
| Photoconversion: | ||||
| BM Sliding measured after optical highlighting | SS at 0 and 3-4 hrs | Photoconvertable laminin (LAM-1∷Dendra); type-IV collagen (EMB-9∷Dendra) | WT worms at differing larval stages | 42 |
| BM photoconverted before and after invasion to determine basement membrane physical displacement | SS at 0 and 2-3 hrs | Photoconvertable laminin (LAM-1∷Dendra); type-IV collagen (EMB-9∷Dendra) | WT and unc-40 (DCC) mutants | 44 |
| Anchor cell proteins | ||||
| Invadopodia size, lifetime, number and rate of formation | 24-60 min TL, 15-60 sec Int | F-actin (mCh∷moeABD) | WT, cdc-42 (Cdc42), lin-3 (EGF), and gdi-1 (GDI) RNAi | 44,18,15 |
| Imaging the transition from invadopodium to invasive protrusion | 1 hr TL, 60 sec Int | PI(4,5)P2 (mCh∷PLCδPH); Laminin (LAM-1∷GFP) | WT | 44 |
| AC invadopodial membrane trafficking and recycling from the endolysosome to the plasma membrane | 40 min TL,60 sec Int | PI(4,5)P2 (mCh∷PLCδPH); Laminin (LAM-1∷GFP) | WT, unc-60 (ADF/cofilin), cdc-42 (Cdc42), lin-3 (EGF), and gdi-1 (GDI) RNAi | 18,15 |
| Time-lapse showing cell division activity during AC invasion | 1 hr TL, 30 sec Int | F-actin (mCh∷moeABD); histone H2B (H2B∷GFP) | nhr-67/tlx (NR2E) RNAi | 51 |
| Oscillating F-actin clustering behavior | 1 hr TL, 60 sec Int | F-actin (mCh∷moeABD) | WT, unc-6 (netrin), unc-40 (DCC) and madd-2 (TRIM) mutants | 43 |
| Co-localization experiments with F-actin and DCC (netrin receptor) | 1 hr TL, 60 sec Int | DCC (UNC-40∷GFP); F-actin (mCh∷moeABD) | WT | 44,43 |
| Protein enrichment at BM breach sites | 60 min TL, 15-120 sec Int | F-actin (mCh∷moeABD); laminin (LAM-1∷GFP); DCC (UNC-40∷GFP) | WT, unc-40 (DCC) mutants | 20,44 |
| Photoconversion: | ||||
| Optical highlighting to measure actin disassembly at invadopodia | SS at 0, post-conversion and 30 min | Photoconvertable actin (Dendra2∷ACT-1) | unc-60 (ADF/cofilin) RNAi | 18 |
| Optical highlighting to lineage AC fate | SS at 0 and 6-8 hrs | Histone (H2B∷Dendra) | nhr-67/tlx (NR2E) RNAi | 51 |
| FLIP: | ||||
| Photobleaching experiments showing recycling through the endolysosome at the invadopodial membrane | 10 min TL, 30 sec Int | LAMP-1 (LMP-1∷GFP) | unc-60 (ADF/cofilin) RNAi | 18 |
| AC interaction with neighboring cells | ||||
| Ectopic membrane-tethered netrin attraction assay | 1 hr TL, 1 min Int | Netrin (UNC-6∷NLG-1 TM∷GFP); F-actin (mCh∷moeABD) | unc-40 (DCC) mutants | 43 |
Tx = treatment; WT = wild type; Int = intervals; TL = time-lapse; SS = snapshots.
The vertebrate orthologue encoded protein is listed in parentheses.
Variations on this protocol can also be used to investigate the interactions between the AC and neighboring cells that constitute the invasive microenvironment, such as the neighboring ventral uterine cells (to which the AC remains adherent during invasion) and the vulval cells into which the AC invades15,53. Lastly, the applications of this protocol are not limited to the study of AC invasion and could be applied, for example, to study membrane trafficking during neurite outgrowth58, or for real-time analysis of the turnover, repair, and aging of extracellular matrix16,42,53,59,60.
Recent upgrades to our imaging system has allowed for 4-D time-lapse acquisition of several animals simultaneously. We use micoromanager software to run a MS-2000 XYZ automated stage (Applied Scientific Instrumentation) through the “multiple position (xy)” option in the “Multi-dimension acquisition” control dialog. Using the automated stage, we have imaged up to six worms in parallel, which significantly reduces the overall imaging time needed to collect time-lapses from multiple animals.
Limitations of the protocol
There are technical and temporal limitations that preclude continuous imaging for some experiments. For example, we have used Dendra fused to laminin to determine BM removal over the course of AC invasion (described above). Converted (red) Dendra photobleaches rapidly, making continuous imaging prohibitive. In addition, the 2-3 hour time window necessary to photoconvert and then follow BM displacement during AC invasion exceeds the duration we can successfully image healthy animals that are anesthetized on coverslips, as they can enter a starvation arrest state59. To circumvent these limitations, we take initial (t=0) snapshots directly after photoconversion and then carefully recover worms from coverslips and place them back on agar plates with food. Then, worms are mounted on slides again for post-invasion imaging 2-3 hours later. We have successfully used this approach for several experiments involving optical highlighting or photobleaching (see Table 1).
Procedure overview and experimental design
In this protocol we outline methods for 4D imaging and analyzing AC invasion in live worms. Different parts of the procedure explain the synchronization of larval growth (Steps 1-4), mounting worms for time-lapse imaging (Steps 5-10), microscope parameters used for optimal resolution (Steps 11-16), processing of image files necessary for vibrant movies (Steps 17-24), and examination of the resulting data to obtain quantitative measurements of several aspects of cell invasion (Steps 25-27) (Fig. 1). The transgenic strains that are commonly used for studies of AC invasion are listed in Supplementary Table 2. Although the following methods are for imaging AC invasion during the L3 larval stage, we anticipate this protocol will be useful to C. elegans researchers who wish to employ live cell imaging in various tissues during any larval or adult stages. As these methods can be adapted for use with a number of different imaging systems and software, we have tried to use general descriptions throughout the protocol.
Figure 1.
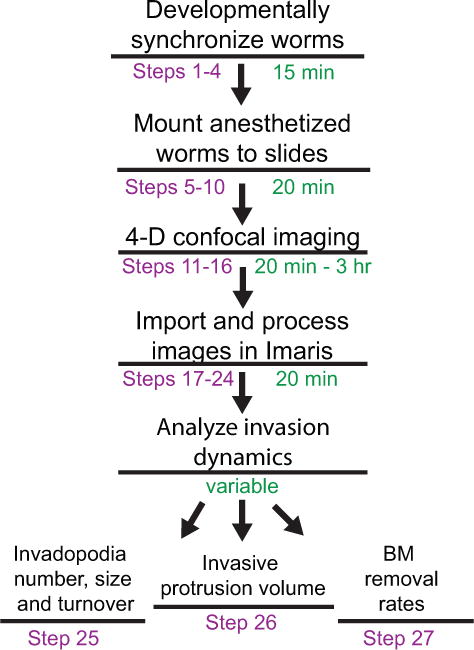
Overview of the protocol. Steps are shown in purple and the anticipated times to complete each step are shown in green.
Worm preparation prior to time-lapse imaging (Steps 1-10)
See Figure 2 for an overview of materials and workspace setup. The 0.5-2 hour continuous imaging needed to capture aspects of AC invasion requires the worms to be immobilized. We have adapted our immobilization technique from methods developed by Knobel et al.65 (see Steps 5-7). We use 0.2% tricaine combined with 0.02% levamisole in M9 buffer to anesthetize worms at the early-to-mid-L3 larval stage (see, Supplementary Table 1). A 12-minute incubation of the worms in this anesthetic solution in a 3-well glass slide (Fig. 3a, c) is sufficient to immobilize worms for 2 hours (if they are mounted/maintained in the solution). It is important to maintain the worms on the slide in M9 liquid, but also to allow for gas exchange during imaging. To accomplish this, we use a glass slide with an agarose pad on which anesthetized worms are dispersed (Fig. 3d, e). A coverslip is then placed on top of the worms followed by immediate application of VALAP66 (see MATERIALS for details) to seal the edge of the coverslip and underlying agarose pad (see Step 8). The VALAP helps prevent evaporation. To allow for gas exchange, two small gaps are left along the sides of the coverslip (Fig. 3f). This approach consistently keeps worms alive for 2 hours without negatively impacting worm development. Approximately 89% (17/19) of worms recovered from a 2-hour time-lapse imaging session survived and continued to grow into adulthood and produce healthy progeny. Furthermore, invasion occurs at the same rate in mounted and anesthetized worms as those not mounted and anesthetized (n=>40 animals observed). As an alternative to anesthetics, worms can be immobilized with polystyrene beads placed on 10% agar pads67. However, we have compared beads with the 5% agarose to the tricaine and levamisole mixture and found the tricaine and levamisole mixture to be superior for immobilization. This may be due to the fact that worms move more during L3 than during adult or other larval stages68.
Figure 2.

Tools and reagents for preparing larval stage C. elegans for time-lapse microscopy. Image shows basic tools and reagents used for standard worm maintenance and some of the additional equipment needed to prepare larval stage worms for time-lapse microscopy.
Figure 3.
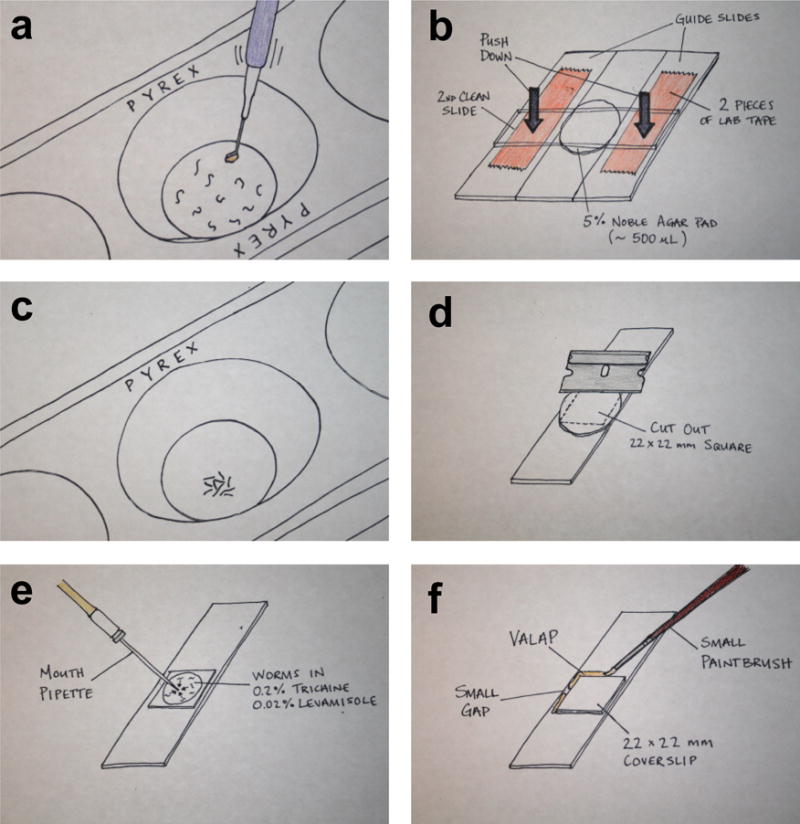
Anesthetizing and mounting worms for time-lapse microscopy. (a) Collect worms of interest in one round of picking then gently swirl the pick in the 50 μl of the anesthetic. Moving the pick in and out of the anesthetic will help to pull the worms off the pick. (b) Image shows an agar pad pouring assembly, which consists of two guide slides (each have two pieces of lab tape on them) and two clean microscope slides. (c) After 12-15 min. the worms should have minimal movements. By gently tapping on the glass spot plate, the worms will coalesce at the bottom of the well, allowing them to be collected by mouth pipette in a minimum volume of liquid. (d) Next, separate the agar pad pouring assembly and use a razor blade to cut the agar pad into a 22×22 mm square. (e) Dispel the collected worms onto the trimmed agar pad (they will remain in the anesthetic) and remove excess liquid anesthetic. (f) Place a 22×22 mm glass coverslip over the worms and seal with VALAP. Leave two small gaps on opposing sides for gas exchange.
Given the short (~2 hour) time window of AC invasion, we often synchronize worm growth to obtain a sufficient number of animals at the proper developmental stage (mid-L3 larval stage)69. To synchronize worms, 4-6 plates of gravid hermaphrodites are bleached and their embryos are collected and washed. These embryos are allowed to hatch in M9 buffer without food (Escherichia coli, OP50) and starved worms will arrest in the L1 larval stage. Worms and can survive in this condition for ~10 days70. Once the arrested L1 worms are given food, they begin to recover from arrest within an hour71, initiate growth in synchrony, and will be ready for imaging at the mid L3 stage approximately 30 hours later (at 20°C; 40 hours later at 15°C). The detailed synchronization protocol can be found in the PROCEDURE Steps 1-4.
Confocal imaging of AC invasion (Steps 11-16)
For image acquisition, a commercially available or customized spinning disk confocal microscope can be used (Fig. 4a shows our microscope set-ups). A scanning unit, high-power excitation lasers, and a 100× objective with high numerical aperture (NA) are essential for performing time-lapse of AC invasion. We have used a laser scanning confocal for imaging AC invasion to precisely photobleach or photoconvert fluorophores. However, we have only been successful at imaging for short intervals of time even when capturing a single plane due to rapid photobleaching. The high laser power of point illumination bleaches live samples more rapidly and faster than the area illumination of spinning disk confocals. In addition, the time required for image acquisition is longer (~10 fold) on a scanning confocal because of the time to complete the line scan for each imaging plane, which limits the number z-slices and the time intervals for acquisition (several minutes are often required versus less than 10 seconds using a spinning disk confocal). Since, we see measurable changes in actin dynamics at invadopodia using 15-second timed intervals, we have found the spinning disc to be optimal for this protocol.
Figure 4.
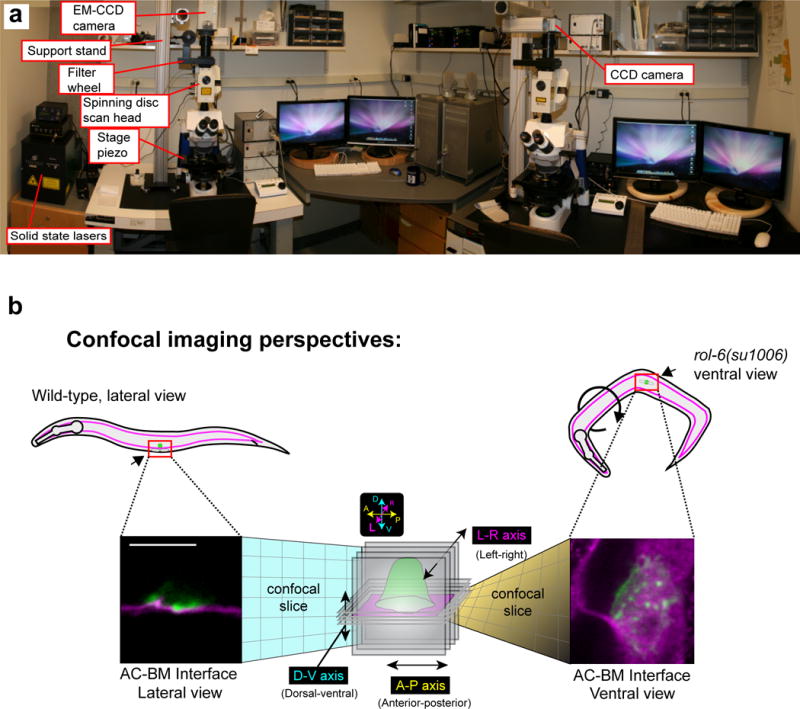
Hardware systems and imaging perspectives for time-lapse analysis of AC invasion. (a) Image shows two spinning disk confocals equipped for time-lapse microscopy. Both setups have solid state lasers and a motorized stage piezo. (b) Schematic diagram depicts the two imaging perspectives for time-lapse analysis of AC invasion. Worms normally lay on their sides (left), which allow AC invasion to be imaged laterally (lateral view). In contrast, in the rol-6(su1006)mutant background (right), approximately 20% of mounted worms orient with their ventral surface facing the objective. This orientation allows confocal sections to be taken in the plane of the AC-BM interface. Scale bar represents 5 μm (left and right images).
To observe the entire process of the AC invasion, worms at the early-to-mid L3 larval stage should be identified for imaging13. Worm staging can be done by examining the number of divisions of the primary vulval cells, the degree of gonad arm reflection and the number of ventral uterine cell descendants using differentiation interference contrast (DIC) imaging (Supplementary Table 1.)
Imaging parameters will need to be optimized for your particular imaging experiment to maintain a balance between sufficient image quality and the overall health of the animal, which can be negatively affected by phototoxicity and photobleaching. These adverse photo-effects are positively correlated with total exposure time, which in turn depends upon laser power, bin settings, gain, the objective used, the number of sections per z-stack, the frequency of time intervals, and the number of fluorescence channels acquired. The settings we have optimized for the imaging outlined in this protocol are listed in Step 11. Using these parameters, we do not observe severe phototoxicity and photobleaching when performing single or dual color imaging. We utilize the EM (electronic multiplication) function of an electron-multiplying charge-coupled device (EM-CCD) camera, allowing the exposure time for fluorescent proteins to be dramatically decreased further reducing phototoxicity and photobleaching. When analyzing data where voxels are measured and quantified, it is important to use the same imaging settings for the same transgene across all genetic backgrounds examined to ensure comparability. Please note that camera technology is developing rapidly. We anticipate that advanced imaging platforms will be available in the near future allowing for increased speed and resolution.
Mounting and orienting worms for imaging (Steps 5-10)
When placed on the agarose pad, the worms will naturally orient on their sides, which conveniently allows AC invasion to be imaged from the lateral perspective (Fig. 4b, left). However, this imaging perspective is not optimal for capturing the entire range of invasion events that occur at the cell-BM interface. To observe the complete cell-BM interface, we have found it optimal to image worms in the ventral plane (Fig. 4b, right). To perform ventral imaging, roller mutants72, such as rol-6(su1006) can be used by crossing the rol-6 mutation into the genetic backgrounds of the strains of interest. In our hands, 20% of rol-6(su1006)worms mounted on coverslips naturally lay with their ventral surface turned towards the objective. Ventral imaging is particularly useful when examining invadopodia formation and turnover (Step 25) and BM hole opening (Step 27).
Data analysis (Steps 17-27)
Currently, there are a variety of programs available for acquiring time-lapse imaging data, many of which come standard with commercially available confocal systems, such as iQ3 (Andor), Metamorph (Olympus), Zen (Zeiss), Elements (Nikon) and iVision (BioVision), as well as open source platforms, such as Micromanager. For 4D data analysis following data acquisition, we use Imaris (Bitplane), commercially available software that is compatible with a wide range of raw data formats. Details regarding how to use Imaris to correct image drift caused by movement of the animal during imaging and how to perform 4D image analysis of membrane protrusions and cytoskeletal structures can be found in Supplementary Videos 5–8. Imaris is specialized software designed for seamless import, visualization, manipulation and analysis of large 3D and 4D datasets. Importantly, separate ImageJ plugins are available for 3D and 4D analysis, including drift correction, free rotation, spot tracking, and volume measurement if access to Imaris is unavailable. We find that ImageJ (NIH) is sufficient and optimal for handing 2D datasets such as BM hole opening rates. There are several steps during image analysis that require the user to set intensity thresholds for fluorescent signals (Step 26 and 27) and manually edit computer-generated tracks when analyzing invadopodia lifetimes (Step 25). To minimize potential bias, we blind datasets, using the freely available ImageJ macro named “Randomizer.” We recommend having two researchers independently analyze datasets using these approaches to confirm results obtained. Using these approaches, we have achieved repeatable and robust results measuring invadopodia dynamics15,18,44,51 and BM clearance rates44,45.
MATERIALS
REAGENTS
Maintenance of C.elegans
-
●
Transgenic animals expressing fluorescent protein fusions, See Table 1 and Supplementary Table 2. As an example, in this protocol we use GFP-tagged laminin driven by its endogenous promoter (lam-1 > LAM-1∷GFP; henceforth written as LAM-1∷GFP), and the AC specific markers for actin (cdh-3 > mCherry∷moeABD; written as mCh∷MoeABD) and plasma membrane phospholipids (PI(4,5)P2) (cdh-3 > mCherry∷PLCδPH; written as mCh∷PLCδPH). Vital dyes can also be used. ! CAUTION Any experiments involving live C. elegans must conform to relevant institutional and national regulations. Many strains are available through the Caenorhabditis Genetic Center (CGC; https://cgc.umn.edu/). Published strains not found in the CGC can be requested by contacting the corresponding author listed on the manuscript.
-
●
Nematode growth medium (NGM) plates. Detailed protocols for culturing of C. elegans can be found here73: http://www.wormbook.org/chapters/www_strainmaintain/strainmaintain.html
-
●
Escherichia coli strain OP50 ordered from the Caenorhabditis Genetic Center (https://cgc.umn.edu/).
Anesthesia
-
●
Levamisole (Sigma, cat. no. 31742) ! CAUTION Acutely toxic orally. Wear respiratory protection. Avoid dust formation. Avoid breathing vapors, mist or gas. Ensure adequate ventilation. Evacuate personnel to safe areas. Avoid breathing dust.
-
●
Tricaine (Sigma, cat. no. 886-86-2) ! CAUTION Causes skin, eye and respiratory irritation. Harmful to aquatic life with long lasting effects. Avoid breathing dust/fume/ gas/mist/vapours/spray. Wash skin thoroughly after handling. Use only in a well-ventilated area and avoid release to the environment.
-
●
M9 buffer (see Reagent Setup).
Mounting worms
-
●
Agarose pad; see Reagent Setup (Agar Noble, BD, cat. no. 214230).
-
●
VALAP; see Reagent Setup.
Microscope
-
●
Immersion oil for microscope objective lens (type-F).
EQUIPMENT
Mounting worms
-
●
Dissecting stereoscope (Zeiss STEMI 2000).
-
●
3-well clear glass Pyrex micro spot plate [Sigma CLS722085].
-
●
Heat block set to ~70°C.
-
●
Standard microscope slides and coverslips (22mm × 22mm #1.5).
-
●
Flame (Bunsen burner or ethanol flame).
-
●
P200 pipetman and tips.
-
●
Worm pick (platinum wire melted into the end of cut glass Pasteur pipette).
-
●
Aspirator tube assembly (mouth pipette) [VWR 53507-278] ! CAUTION and pulled capillary glass pipettes [Kimble Kimax 34500 99].
-
●
Paint brush for applying VALAP sealant (smaller brush will be easier to control).
-
●
Kimwipes
-
●
Razor blades
Microscopy
-
●
Dissecting scope with oblique trans-illumination for mounting worms.
-
●
Spinning-disk confocal microscope. We prefer an upright set-up but an inverted scope can also be used. Many variations of hardware and acquisition software exist and different combinations can be assembled to work well. We use a Yokogawa CSU-10 (Yokogawa Electric Corporation; modified by Spectral Imaging) spinning disc head mounted on an upright Zeiss AxioImager.A1.
-
●
Objectives: 10× EC Plan-Neofluar 10×/0.3 M27 for locating worms and 100× Plan-Apochromat 100×/1.40 Oil DIC for imaging (Zeiss).
-
●
Solid state lasers: 488 Cyan (CDRH) from Spectral Physics; 561 Cobalt Jive (CDRH) DPSS laser.
-
●
Motorized Stage for z-stack acquisition: Piezo Z-axis Stage (PZM-2000; Applied Scientific Instrumentation, Inc.).
-
●
Camera: Hamamatsu Orca-R2 (C10600) or a Hamamatsu EM-CCD (C10600-10B) camera (Hamamatsu Photonics).
-
●
Filterwheel: FW-1000 (Applied Scientific Instrumentation, Inc.).
-
●
Filters: ZET448/561m, ZET405/448/561m, and ZET450/50m (Chroma)
Image processing software.
-
●
ImageJ 1.40g (NIH)
-
●
iVision (Image acquisition software). http://www.biovis.com/ivision.htm
-
●
Photoshop (Adobe)
-
●
IMARIS (Bitplane, Inc.): Used for confocal z-stack 4D reconstruction. Version number 7.4 is shown in the video tutorials (Supplementary videos 5–8), but steps in this protocol are consistent with the newer versions of the software (8.4 at the time of publication).
REAGENT SETUP
M9 buffer
6 g Na2HPO4, 3 g KH2PO4, 5 g NaCl, 1 ml of 1 M MgSO4, H2O to 1 liter. Sterilize by autoclaving and aliquot into 100ml bottles. Unopened stocks can be store at room temperature (~20-25°C.) for 3 months.
Levamisole solution
Dissolve levamisole powder in water to prepare 10 mM solution as a 10× stock, which can be stored at −20°C for several years. Dilute the 10× stock with M9 buffer immediately before use to make a 1× working solution.
Anesthesia buffer
Dissolve the levamisole and tricaine powder in water to prepare 0.2% (wt/vol) levamisole and 2% (wt/vol) tricaine as the 10× stock. These stocks remain stable for several years at −20°C. The 10× stock can be diluted with M9 immediately before use to make a 1× working solution that can be stored at RT for 1 week.
VALAP
Combine equal amount of vaseline, lanolin and paraffin wax by weight. Heat over a hot plate and mix together. Aliquot the stock at a volume of 1.5 ml per tube and store it at room temperature (stocks will last interminably). Melt it prior to use by placing it in a heat block at ~70°C.
Agarose
Prepare 5% (wt/vol) noble agar65 using a microwave and then aliquot the solution in disposable glass culture tubes (VWR, CAT# 47729-572) at a volume of 1.5 ml per tube and store at room temperature. We use plastic caps (Port City Diagnostics T3600CAP) to seal tubes and allowing them to be stored for at least 2 months without drying out. Melt the agar aliquots over a Bunsen burner and then place in a ~70°C heat block to keep molten. CRITICAL The high percentage (5%) of agar reduces Z-drift (the worms sinking into the agar over time).
EQUIPMENT SETUP
Confocal imaging system
Turn on the hardware and software components and set the proper parameters for time-lapse imaging, including exposure time, laser intensity, EM Gain, binning, z-stack range and step size, time-lapse duration, and time interval.
PROCEDURE
Synchronize worm cultures for imaging (optional) ● TIMING 15 min
CRITICAL Performing synchronized L1 larval plating facilitates worm-picking efficacy at the developmental stage optimal for time-lapse imaging (e.g., when imaging AC invasion you will want worms at the mid-to-late L3 larval stage).
-
1
Wash 2-3 plates containing the large number of gravid hermaphrodites with 4 ml water and transfer washed worms to a 15-ml conical tube.
-
2
Centrifuge at 400g for 1 min in an benchtop centrifuge. Aspirate the supernatant and resuspend the settled worms in 4 ml of M9 buffer. Add 5 ml alkaline bleach solution (made by mixing 2.3 ml of household bleach, 0.4 ml of 5M KOH, and 2.3 ml of autoclaved water). Shake the tube for 4-6 min in a rocker until worms are fully dissolved. Immediately centrifuge the tube at 400g for 1 min in the benchtop centrifuge.
-
3
Aspirate the supernatant and resuspend the pellet in autoclaved water by shaking the tube vigorously by hand 4-6 times, followed by centrifugation at 400g for 1 min in the benchtop centrifuge. Repeat this step twice.
-
4
Aspirate the supernatant and resuspend the pellet in M9 buffer by shaking the tube vigorously by hand 4-6 times. Centrifuge at 400g for 1 min Aspirate the supernatant and add 0.5-1 ml of M9 buffer to the tube. Place the tube on a rocker at 20°C overnight allowing embryos to hatch.
Mount worms on an agarose pad ● TIMING 20 min
-
5
Add 50 μl anesthesia solution into the small well of the 3-well glass slide (see Supplementary Video 3 for a detailed example of steps 5-9).
-
6
Flame your worm pick until it glows orange and allow it to cool for two seconds. Then, using a glob of OP50 E. coli from a fresh plate, pick up worms of interest. Gently swirl the pick in the 50 μl anesthetic in the glass spot plate (Fig. 3a; Supplementary Video 3). The surface tension of the liquid anesthetic will pull the glob of bacteria and worms off of the pick. Transfer 20-30 worms in total. If possible, try to collect all desired worms in one round of picking. Doing so will ensure all of the worms will be in the anesthetic for an equal amount of time. ▲CRITICAL STEP Minimize the amount of OP50 bacteria that are transferred along with worms into the anesthesia solution. We find that large amounts of bacteria in the anesthesia solution reduces worm viability during time-lapse imaging.
-
7
Wait for 12 min to allow worms to be anesthetized. Preform Step 8 during the incubation period.
-
8
Prepare an agar pad by placing a clean microscope slide between the two guide slides, each which have two pieces of lab tape on them. Next pour a drop (~500 μl) of molten 5% noble agar in the middle of the clean slide. Quickly, place a second clean slide on top of the drop of agar then press firmly down on the ends that overlap the taped guide slides (see Fig. 3b). Let this setup sit until the end of the incubation in Step 7.
▲CRITICAL STEP Agar pads must be made fresh to minimize drying out during imaging. It is better to pour an agar pad that is too large, which can then be trimmed down to the size of the 22 × 22 mm coverslip (see Fig. 3d). Maximizing the pad size allows for placement of more worms (n >20) on each slide. Even with synchronization, it can be difficult to find worms at the exact developmental stage you would like to image. An agar pad that is too small will leave large spaces of air after VALAP is applied, which can cause the pad to dry out more quickly.
-
9
Using a mouth pipette, transfer worms (in the liquid anesthetic) onto the trimmed agar pad (see Fig. 3e). With the same mouth pipette used to transfer the worms, remove excess liquid. Cover the worms with a standard glass coverslip, starting with one side and gently dropping it across the agar pad to avoid air bubbles. ! CAUTION Always use caution when using a mouth pipette to not ingest any fluid.
? TROUBLESHOOTING
-
10
Seal the coverslip with VALAP to minimize liquid evaporation (see Fig. 3f).
▲CRITICAL STEP We use a small paintbrush to apply VALAP to four sides of the coverslip and leave a small gap for gas exchange at two sides. Remove the residual VALAP that is high above the coverslip upper surface with a razor blade to minimize the risk of getting VALAP on the microscope objective.
? TROUBLESHOOTING
Time lapse imaging ● TIMING 20 -180 min
-
11
Turn on and configure the confocal imaging system.
▲CRITICAL STEP Before you begin you will need to determine the appropriate settings for some key image acquisition parameters. These will depend on your individual specimen and the dynamics of what you are trying to capture (see Supplementary Video 4). Below are two examples of specific image acquisitions and the appropriate settings on our systems:- Capturing invadopodia dynamics at the AC-BM interface (ventral view).
- Markers: AC-specific mCh∷moeABD (marking F-actin), LAM-1∷GFP (labeling laminin in the BM) and a dominant rol-6 mutation (urIs[rol-6(su1006); lam-1>lam-1∷GFP]) which causes worms to roll and results in ~20% of mounted worms being oriented with their ventral sides towards the microscope objective (Fig. 4b).
- Microscope: Spinning disk confocal (Fig. 4a)
- Camera: Hamamatsu EM-CCD
- Laser power: 100% for 25 mW 488 nm and 25 mW 561 nm laser lines
- Exposure: 50 ms (488 nm, LAM-1∷GFP), 100 ms (561nm, mCh∷moeABD)
- EM Gain: 220 (max is 255)
- Gain: 0
- Binning: 1 × 1
- Z-step size: 0.5 μm
- Total # of steps: 8 (covering 3.5 μm at 0.5 μm z-steps)
- Time intervals: 15 sec
- Time-lapse duration: 30 min
- Imaging invasive protrusion dynamics during BM transmigration (lateral view).
- Markers: AC-specific mCh∷PLCδPH (PI(4,5)P2 probe in the AC) and LAM-1∷GFP (labeling laminin in the BM).
- Microscope: Spinning disk confocal (Fig. 4a)
- Camera: Hamamatsu R2
- Laser power: 85% for 20 mW 488 nm and 25 mW 561 nm laser lines
- Exposure: 100 ms (488nm, LAM-1∷GFP), 200 ms (561 nm, mCh∷PLCδPH)
- Gain: 0
- Binning: 2 × 2
- Z-step size: 1 μm
- Total # of steps: 7 (covering 6 μm at 1 μm z-steps)
- Time intervals: 60 sec
- Time-lapse duration: 90 min
▲CRITICAL STEP Use the same settings for quantitative measurements. Set exposure time and laser intensity to ensure proper image quality, but without photobleaching. Minimize EM gain for an EM-CCD camera to achieve high signal-noise ratios (SNR). Settings need to be optimized according to different experimental contexts.
-
12
Place the slide with the immobilized worms on the stage of a spinning disk confocal microscope.
-
13
Identify healthy L3 larvae by their sizes under a 10× objective lens.
▲CRITICAL STEP It is important to select healthy worms for imaging. We avoid worms that have lysed cells (apparent by clear regions) in the head or body for imaging. We also do not image worms that are not at the standard lateral or ventral view position as non-standard orientations can cause difficulty for data analysis after 3D reconstruction.
-
14
Determine the developmental stage with respect to the number of primary vulval cells (P6.p one-, two-, or four-cell stage; Supplementary Fig. 1, Supplementary Table 1) using a 100× oil objective lens. Choose P6.p two-cell stage worms for time-lapse imaging as wild-type ACs initiate BM breaching at this stage (approximately 30 hours after hatching when grown at 20°C).
-
15
Start time-lapse imaging and re-adjust the focus plane during the imaging process if using a microscope with manual focus adjustment function.
▲CRITICAL STEP Correction for drift in the Z-axis (axial movements, which may result from the agar pad drying slightly or the worms settling into the agar) will need to be performed routinely every 5 minutes for the first 20 minutes of your time-lapse. After 20 minutes this Z-drift usually stabilizes. Correcting for this drift can be done between time points by manually adjusting the Z-position of the microscope stage to keep the AC within the limits of your set Z-stack, without needing to pause the acquisition or adjust the stage piezo. Minor movements of the worm in the X- and Y-axis (planar movements) can be corrected for later during image analysis (see below, IMAGE ANALYSIS).
? TROUBLESHOOTING
-
16
End image acquisition and save files. We typically save acquired images as .TIFF or .IPM (iVision format) files, which are accessible by a wide variety of software. PAUSEPOINT You can proceed to analysis of data at the time of your choosing.
Importing and visualizing 4D data in Imaris ● TIMING 20 min
-
17|. Open the original time-series dataset in Imaris. Ensure that the specifications for the X, Y, and Z dimensions (at this step X, Y, and Z should each have a value of one; note these are not the voxel dimensions, see below), as well as T (number of time points) and C (number of channels) of the dataset are correct [File > Open… > Settings… > File Names without Delimiter] (see Supplementary Video 5 for a detailed video of steps 13-18).
? TROUBLESHOOTING
-
18
Adjust the voxel dimensions. Ideally, this information will be contained within the metadata of your files and will not need to be manually entered.
▲CRITICAL STEP The geometry size of a pixel and the image binning should match image acquisition settings. The pixel size of our imaging system is 0.062 μm × 0.062 μm or 0.15 μm × 0.15 μm when binning is set to 1×1 on the left and right imaging systems shown in Figure 4, respectively. If z-stack distance interval sets to 0.5 μm, the voxel size is then 0.062 × 0.062 × 0.5 μm3. A stage micrometer should be used to independently verify calculated voxel dimensions. You should be suspicious if the X, Y, and Z voxel dimensions of the imported data are 1 μm × 1 μm × 1 μm or proportionately 1:1:1.
? TROUBLESHOOTING
-
19
Adjust intensity levels and pseudocolor channels as appropriate [Edit > Show Display Adjustments].
-
20
Use the spot-tracking tool in Imaris to correct natural drift that occurs over the course of the time lapse (See Supplementary Video 5). To do this click on the “spots” icon in Imaris. Select Segment only a Region of Interest and de-select Process Entire Image Finally and click the next icon.
-
21
Set the ROI to include all of the X- and Y-axes, but only a single plane in the Z-axis and select next.
-
22
Select the channel that includes the AC marker fluorescence and set the Estimated Diameter to 10 μm, then click next twice. Choose the algorithm Connected Components; click next, and then the finish icon. Select the entire track over time, choose Correct Drift > Include Entire Result > OK.
-
23
Crop the volume in 3D to include only the information needed for analysis [Edit > Crop 3D…]. This will decrease the file size.
▲CRITICAL STEP Depending on the object(s) you are trying to measure, you may need to realign the dataset such that the X-, Y- and Z-axes of the camera square up with the anterior-posterior (A-P), dorsal-ventral (D-V) and left-right (L-R) axes of the worm (Fig. 4b). This will facilitate partitioning of cellular domains for analysis (e.g., the AC’s invasive cell membrane). This can be done using the free-rotation function in Imaris [Image Processing > Free Rotate…] (Fig. 5a).
-
24
Save the dataset as a new file (as an .IMS file in Imaris) to serve as the working dataset for subsequent analyses [File > Save as… > .IMS]. For presentation or publication purposes videos can be generated to display particular aspects of your dataset [View > Animation]. Within Imaris you may also capture snapshots of a 3D volume from multiple perspectives [View > Snapshot] (Fig. 5b).
Figure 5.
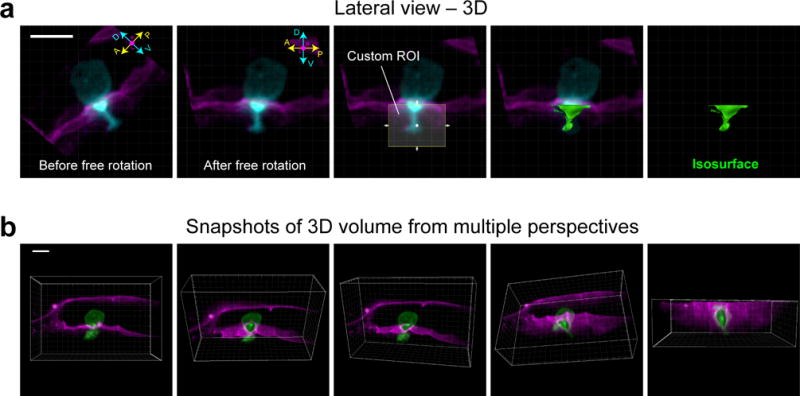
Realignment by free rotation and the snapshot tool in Imaris. (a) Images show lateral view of a 3D reconstruction of the AC (cyan) as it invades through the BM (magenta). Prior to free rotation (left-most panel), the anterior-posterior (A-P) and dorsal-ventral (D-V) axes of the AC do not line up with the X- and Y-axis of the camera (white-lined grid). After free rotation, however, the AC and the camera area align. After realignment, a custom region of interest (ROI, which can only be drawn within the axes of the camera) can be used to generate an isosurface (see below, IMAGE ANALYSIS, Step 2.2) in place of the portion of the AC that has extended through BM. (b) Images of a 3D reconstruction were captured from multiple perspectives using the snapshot tool in Imaris. Scale bars represent 5 μm.
Quantifying the spatio-temporal dynamics of AC invasion ● TIMING variable
-
25Use option A to open a ventral view dataset containing an invadopodia marker and use the spot function in Imaris to quantify AC-invadopodia dynamics (see Fig 6., Supplementary Video 6). Use option B to measure invasive protrusion volume with isosurfaces (Fig. 7, Supplementary Video 7). Use option C to quantify the rate of BM hole expansion (Fig. 8, Supplementary Video 8).
-
(A)Using the spot function in Imaris to quantify AC-invadopodia dynamics
- Click on the Surfaces icon. Deselect Smooth and click on Next. Set a threshold that accurately reflects the invadopodia you want to measure and click Next. Take note of the threshold value for future reference and click Next three times and then Finish.
-
Choose the add new spots icon. Make sure to have Segment only a Region of Interest deselected before hitting next. Deselect Background Subtraction and enter “0.5” in the box labeled Estimated Diameter before selecting next. Set the filter type to Intensity Mean Ch-1 and enter the intensity threshold noted in (A) above. Select Next twice. Choose Autoregressive Motion in the Algorithm options. Under Parameters enter ‘0.5’ in Max Distance and ‘0’ in Max Gap Score. Click Next. Select and delete the default track. Click Finish.▲CRITICAL STEP At 15-sec intervals, we allow a drift distance of ‘0.7 μm’ and use a frame gap of ‘0’. By our criterion, an object must be present in at least two consecutive frames to be identified as an AC-invadopodia (if a spot disappears and then reappears, it will be counted as a new AC-invadopodia, see Fig. 6C). Although the software will do a reasonable job generating tracks by connecting spots in consecutive frames, you will need to verify that tracks are properly assigned. This will require you to manually edit individual tracks, which will be time-consuming and limit the number of animals that can be examined.
- Click on the statistics tool icon to see data values resulting from the analysis. Click on the save icon to export the statistical data to an Excel file.
- Determine descriptive data for the following AC-invadopodia dynamics using the exported numbers (see Fig. 6d):
-
●Invadopodia Size. Individual invadopodia sizes (in μm, see Fig. 6A) are listed in the Diameter tab in the Excel spreadsheet.
-
●Invadopodia Lifetimes. Refer to Track Duration values, which represent the number of time points an invadopodia persisted. This number must be multiplied by the duration of each time point in order to get an actual value in minutes. In wild-type animals the number of AC-invadopodia during the hour leading up to invasion does not appear to significantly change over time.
-
●Average rate of invadopodia formation. Divide the Number of Tracks by the duration of the time-lapse. For example, if 100 AC-invadopodia (tracks) are detected in a 20-minute time-lapse then the average rate of AC-invadopodia formation for that animal is 100/20 = 5 AC-invadopodia per minute.
-
●Invadopodia numbers over time. Use values listed as Number of Spots per Time Point.
-
●
-
BMeasure invasive protrusion volume with isosurfaces
- Open a lateral view Imaris file containing a fluorescent marker that labels the cell membrane protrusion (see Table 1 and Supplementary Table 2).
-
Click on the New Isosurface icon. Deselect the box Process Entire Image Finally and click Next. Adjust the region of interest box so that it only includes the area beneath the AC directly adjacent the uterine-vulval BM (see Fig. 5a for an example). Click Next twice. Select a Threshold value that closely fits the volume of the protrusion over the entire time course. Document the Threshold value, click Next twice and select Finish. Click on the statistics tab to export data for the volume of the protrusion at each time point to an Excel file.▲CRITICAL STEP Apply the same threshold to generate isosurfaces for comparison. Use the function of ‘cut surface’ for spatial segmentation of an isosurface. There may be instances (e.g., BM is curved or fluorescence intensity is too variable for proper thresholding) where you need to manually outline the protrusion in each frame instead of using the ROI tool. After clicking the New Isosurface icon, click on Skip automatic creation, edit manually. Then, use the paintbrush tool to outline the surfaces you would like to include in the analysis for each frame and click Finish.
-
B)Quantify the rate of BM hole expansion (Fig. 8, Supplementary Video 8).
- Import an Imaris file containing a worm expressing a BM marker imaged from the ventral perspective that has been drift corrected and realigned by free rotation to display the BM from a straight-on perspective (See steps 20-23, Fig. 5b right-most panel).
-
Export a movie (.MOV) or .TIFF series (as maximum image projections) in Imaris and then open the file in ImageJ.▲CRITICAL STEP Include a scale bar in the movie exported from Imaris, which will allow you to convert the data from an area of pixels to an area of μm2. Use the Straight (line) tool to draw a line along the scale bar and note the number of pixels that correspond to the μm in the error bar. This information will be necessary in (G) below.
- Use the Rectangular selection in ImageJ to select a ROI that excludes the black margins outlining the video data and select Image > Crop.
- Adjust the threshold [Image > Adjust > Threshold] until the BM gap is precisely fitted during the duration of the movie. Select Apply and OK to create a binary image.
- Choose Analyze > Analyze Particles. Input ‘3000-Infinity’ in the Size and choose Outlines in the Show option and click OK. Select Yes when asked, “Process all… images.”
- Copy the results and paste these data into an Excel file. Exclude any outlines that do not correspond to the area of hole opening).
- Calculate the area of BM removal at each time point with the following formula:
-
(A)
Figure 6.
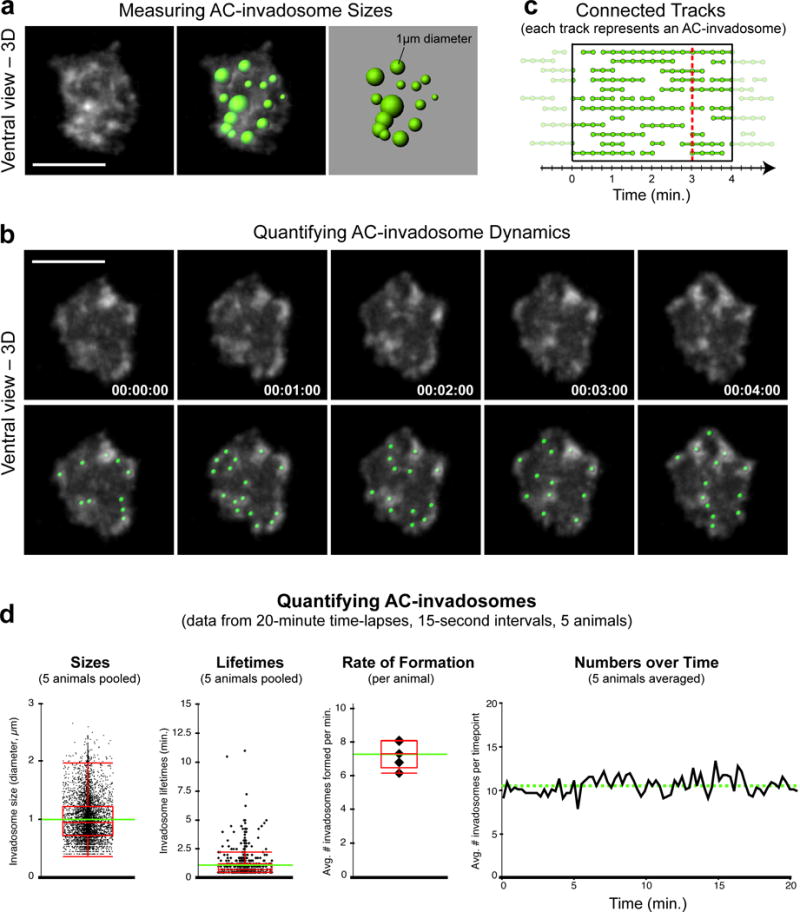
Quantifying AC-invadopodia dynamics. (a) Images show a single time point, ventral view perspective of AC-invadopodia, enriched in the F-actin probe mCh∷moeABD (left). Variably-sized spots (right) representing AC-invadopodia sizes that were generated in Imaris based on the mCh∷moeABD signal (right) are overlaid onto the fluorescent image (middle).(b) Time series montage shows five frames of the F-actin-rich AC-invadopodia at 1-minute intervals (top). Green spots were used to track AC-invadopodia dynamics (bottom). Scale bars represent 5 μm. (c) Diagram shows several tracks (connected spots) within the highlighted 4-minute window. Each track represents the lifetime of a single AC-invadopodia. (d) Graphs display quantitative measurements of wild-type AC-invadopodia that were acquired using the spot-tracking function in Imaris. Intra-animal variability of pooled data was negligible. Images in (b) and graphs in (d) were adapted from Hagedorn et al., 2013.
Figure 7.
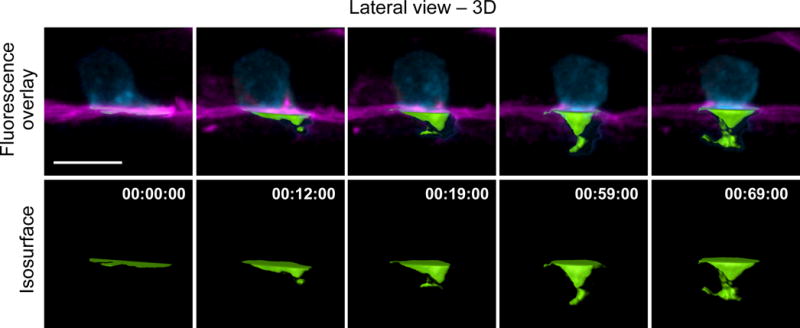
Using isosurfaces to make spatial measurements. Time-series montage shows 3D reconstructions at several time points over the course of AC invasion. The AC is expressing the PI(4,5)P2 sensor mCherry∷PLCδPH (cyan); the BM is labeled with LAM-1∷GFP (magenta). Isosurface renderings (green) were used to measure the volume of the AC’s invasive protrusion that had extended through the BM. Scale bar represents 5 μm.
Figure 8.
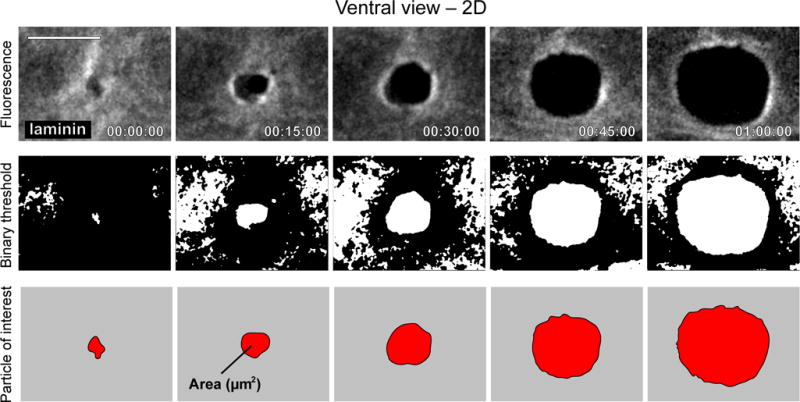
Quantification of BM hole expansion over time. Time series montage shows ventral view of the expanding hole in the BM that results from AC invasion. Top row shows grayscale fluorescence of laminin (LAM-1∷GFP) in the BM. Second row shows the same images after a binary threshold was applied in ImageJ. Bottom row shows the hole in the BM as a particle in ImageJ, after smaller objects and those touching the edges of the image were filtered away. Images in top row were adapted from Hagedorn et al., 2012. Scale bar represents 5 μm.
● TIMING
Steps 1-4, Synchronize worm cultures for imaging: 15 min
Steps 5-10, Mounting worms on agarose pad: 20 min
Steps 11-16, Image acquisition: 20-180 min
Step 17-24, Image processing: 20 min
Step 25-27, Quantifying invasion dynamic and data analysis: variable
ANTICIPATED RESULTS
Our time-lapse protocol allows for imaging the cell invasion process through BM at single-cell resolution in living animals. By analyzing the data resulting from the images captured, quantitative in vivo traits can be obtained of invadopodia dynamics, plasma membrane protrusion volume, and BM removal. 4D data analysis on marker proteins for subcellular structures of interest, such as F-actin-based invadopodia, can reveal spatial and temporal features of these structures during AC invasion, including turnover rate, lifetime, and dynamic localization. We can consistently measure AC-invadopodia dynamics in wild-type worms and have been successful in characterizing the role of proteins that regulate invadopodia dynamics through genetic mutant and RNAi knockdown studies. These include several cytoskeletal proteins including the F-actin severing protein cofilin that promotes invadopodial turnover, the Rho GTPase Cdc42 that initiates the seeding of new invadopodia, and the Rab dissociation inhibitor GDI-1 that targets the endolysosome-derived invadopodial membrane to the invasive cell membrane to form invadopodia15,18 (Fig. 6, Table 1). In addition, this protocol has the sensitivity to identify changes in the volume of the invasive protrusion that follows invadopodia formation (Fig. 7). We have identified the netrin receptor UNC-40 (vertebrate DCC) as a key receptor that traffics to the BM breach site and directs the creation of the invasive protrusion through its F-actin regulatory effectors UNC-34 (Ena/VASP) and MIG-2 (RhoG)43,44. Finally, the range/shape/size, and dynamic patterns of BM removal can be precisely measured by following the steps outlined above16,42,45,53 (Fig 8). Using these methods we identified several proteins that regulate BM clearing under the AC including hemicentin, a conserved extracellular member of the immunoglobulin superfamily, which physically links the two juxtaposed BM under the AC to optimize BM breaching and hole opening45. In addition, we discovered that overexpression of SPARC, a matricellular protein that is upregulated in many if not most metastatic cancers, promotes AC invasion by perturbing deposition of type IV collagen, thus thinning the BM16. In summary, this protocol can be coupled with a multiplicity of combinations of fluorescently tagged proteins, varying genetic backgrounds, and conditions (pharmaceutical treatment, starvation, etc) to uncover the molecular and cellular mechanisms that drive the invasion process in vivo in both development and disease states such as cancer.
Supplementary Material
Supplementary Video 1 Invasive protrusion growth by the AC (green) during basement membrane (magenta) transmigration (62 min; Scale bar, 5 μm.)
Supplementary Video 2 Dynamic expansion of a BM opening generated by the AC (70 min; Scale bar, 5 μm).
Supplementary Video 3 Anesthetizing worms, preparing an agar pad, and mounting anesthetized worms.
Supplementary Video 4 Time-lapse image acquisition.
Supplementary Video 5 Importing and visualizing 4D data in Imaris.
Supplementary Video 6 Using spots to quantify AC-invadopodia dynamics.
Supplementary Video 7 Measuring invasive protrusion volumes with isosurfaces.
Supplementary Video 8 Quantifying the rate of BM hole expansion.
Supplementary Figure 1| (a) The schematic depicts the developmental time points during anchor cell (AC) invasion in C. elegans. The AC invades in tight synchrony with the 10 fated vulval precursor cell P6.p cell divisions during the L3 larval stage. During the P6.p one-cell stage, F-actin/endolysosomal membrane based invadopodia form along the invasive cell membrane (orange) within the AC (green) at the AC-basement membrane (BM) interface (magenta). AC invasion initiates during the late P6.p two-cell stage, when one or two invadopodium breaches the BM. A single breaching invadopodium transitions to an invasive protrusion that shuts down further invadopodia formation. As the invasive protrusion enlarges, it clears a large gap in the basement membrane and moves between the central P6.p vulval cells by the P6.p four-cell stage. (b) AC invasion through BM. During invasion (0-53 minutes), the AC (F-actin; green) generates a protrusion that breaches the BM (laminin; magenta). While the BM opening expands, the protrusion grows, and reaches a mature size. Scale bar, 5 μm.
{kind=link}
TABLE 2.
Troubleshooting table
| Step | Problem | Possible reason | Solution |
|---|---|---|---|
| 9 | Too many bubbles around worms | Too little anesthetic liquid left on slide | Stop removing anesthetic with the mouth pipette once the liquid no longer easily fills the capillary glass without applying suction |
| Worms too widely distributed on the agarose pad | Too much anesthetic liquid is left on the pad | Try to minimize the amount of liquid transferred with the worms by gently tapping the glass spot plate (the worms will coalesce at the bottom of the well (see Fig. 3c), allowing you to draw them up in a minimal amount of liquid) | |
| 10 | Worms become sick or die during imaging | Too much OP50 is transferred with worms Worms are injured while they are anesthetized, transferred, and covered by a coverslip The interface between the coverslip and the agarose pad is not properly sealed |
Use a minimal amount of bacteria to transfer worms Use care when handling worms at all points Use a small paintbrush to apply VALAP to four sides of the coverslip and leave a small gap for gas exchange |
| 14 | Worms are not at the correct stage in development to image | Wrong sized animals are picked Worms cultured for too short or long after syncing Worms not kept at an optimal temperature |
Sync animals by bleaching instead of trying to pick from mixed aged plates Optimize the correct incubation time and temperature for each strain |
| 15 | Specimens with low fluorescence signal will result in grainy images | EM Gain might be too high while exposure time is likely too short | Adjust the settings until high-quality images are acquired |
| Worms start moving during time-lapse | Worms are too crowded or the incubation in anesthetic was too short | Lengthen incubation time in anesthetic, avoid mounted worms that are touching each other, as they tend to move more during time-lapses | |
| Photobleaching occurs during imaging | Worms are exposed to too much cumulative laser power | Reduce laser power, exposure time | |
| 17 | Imaging data cannot be loaded into Imaris | The format of data files is not properly selected in Imaris | Try to correctly match file format selected in Imaris with that of raw data files |
| 18 | Disproportionate 3D rendering | Geometry set in Imaris does not match acquired raw images | Verify correct voxel dimensions. If voxel size was determined by calculation, confirm with a stage micrometer |
EDITORIAL SUMMARY.
This protocol describes how to use multi-channel time-lapse confocal imaging of anchor cell invasion in live C. elegans to monitor cell invasion through basement membranes.
Acknowledgments
We thank Paul Maddox for advice and reagents during the initiation of this project Nicolas Devos for help with video editing, Meghan Morrissey, Dan Keeley, and Kaleb Naegeli for comments on the manuscript and all former and current members of the Sherwood laboratory for their support. This work was supported by The Pew Scholars Program in the Biomedical Sciences, NIGMS R01 GM079320 and NIGMS R35 MIRA GM118049 to D.R.S.
Footnotes
TWEET @dukeU and #celegans and @SherwoodElegans
AUTHOR CONTRIBUTIONS
E.H. preformed the experiments and designed the protocol. L.K., E.H., Z.W. and D.S. contributed to writing the manuscript. L.K., E.H., and Z.W. prepared the figures and tables. S.J. advised on imaging. L.W., W.S., and S.L. provided supplementary materials.
COMPETING FINANCIAL INTERESTS The authors declare no competing financial interests.
References
- 1.Yurchenco PD. Basement membranes: cell scaffoldings and signaling platforms. Cold Spring Harb Perspect Biol. 2011;3 doi: 10.1101/cshperspect.a004911. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Pozzi A, Yurchenco PD, Iozzo RV. The nature and biology of basement membranes. Matrix Biol. 2017;57–58:1–11. doi: 10.1016/j.matbio.2016.12.009. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Madsen CD, Sahai E. Cancer dissemination–lessons from leukocytes. Dev Cell. 2010;19:13–26. doi: 10.1016/j.devcel.2010.06.013. [DOI] [PubMed] [Google Scholar]
- 4.Kelley LC, Lohmer LL, Hagedorn EJ, Sherwood DR. Traversing the basement membrane in vivo: a diversity of strategies. J Cell Biol. 2014;204:291–302. doi: 10.1083/jcb.201311112. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Rowe RG, Weiss SJ. Breaching the basement membrane: who, when and how? Trends Cell Biol. 2008;18:560–574. doi: 10.1016/j.tcb.2008.08.007. [DOI] [PubMed] [Google Scholar]
- 6.Hagedorn EJ, Sherwood DR. Cell invasion through basement membrane: the anchor cell breaches the barrier. Curr Opin Cell Biol. 2011;23:589–596. doi: 10.1016/j.ceb.2011.05.002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Christofori G. New signals from the invasive front. Nature. 2006;441:444–450. doi: 10.1038/nature04872. [DOI] [PubMed] [Google Scholar]
- 8.Paul CD, Mistriotis P, Konstantopoulos K. Cancer cell motility: lessons from migration in confined spaces. Nat Rev Cancer. 2017;17:131–140. doi: 10.1038/nrc.2016.123. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Schoumacher M, Louvard D, Vignjevic D. Cytoskeleton networks in basement membrane transmigration. Eur J Cell Biol. 2011;90:93–99. doi: 10.1016/j.ejcb.2010.05.010. [DOI] [PubMed] [Google Scholar]
- 10.Albini A, Noonan DM. The “chemoinvasion” assay, 25 years and still going strong: the use of reconstituted basement membranes to study cell invasion and angiogenesis. Curr Opin Cell Biol. 2010;22:677–689. doi: 10.1016/j.ceb.2010.08.017. [DOI] [PubMed] [Google Scholar]
- 11.Wang Z, Sherwood DR. Dissection of genetic pathways in C. elegans. Methods Cell Biol. 2011;106:113–157. doi: 10.1016/B978-0-12-544172-8.00005-0. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Beerling E, Ritsma L, Vrisekoop N, Derksen PWB, van Rheenen J. Intravital microscopy: new insights into metastasis of tumors. J Cell Sci. 2011;124:299–310. doi: 10.1242/jcs.072728. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Schindler AJ, Sherwood DR. Morphogenesis of the caenorhabditis elegans vulva. Wiley Interdiscip Rev Dev Biol. 2013;2:75–95. doi: 10.1002/wdev.87. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Sherwood DR, Sternberg PW. Anchor cell invasion into the vulval epithelium in C. elegans. Dev Cell. 2003;5:21–31. doi: 10.1016/s1534-5807(03)00168-0. [DOI] [PubMed] [Google Scholar]
- 15.Lohmer LL, et al. A Sensitized Screen for Genes Promoting Invadopodia Function In Vivo: CDC-42 and Rab GDI-1 Direct Distinct Aspects of Invadopodia Formation. PLoS Genet. 2016;12:e1005786. doi: 10.1371/journal.pgen.1005786. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Morrissey MA, et al. SPARC Promotes Cell Invasion In Vivo by Decreasing Type IV Collagen Levels in the Basement Membrane. PLoS Genet. 2016;12:e1005905. doi: 10.1371/journal.pgen.1005905. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Matus DQ, et al. In vivo identification of regulators of cell invasion across basement membranes. Sci Signal. 2010;3:ra35. doi: 10.1126/scisignal.2000654. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Hagedorn EJ, et al. ADF/cofilin promotes invadopodial membrane recycling during cell invasion in vivo. J Cell Biol. 2014;204:1209–1218. doi: 10.1083/jcb.201312098. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Hagedorn EJ, et al. Integrin acts upstream of netrin signaling to regulate formation of the anchor cell’s invasive membrane in C. elegans. Dev Cell. 2009;17:187–198. doi: 10.1016/j.devcel.2009.06.006. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Sherwood DR, Butler JA, Kramer JM, Sternberg PW. FOS-1 promotes basement-membrane removal during anchor-cell invasion in C. elegans. Cell. 2005;121:951–962. doi: 10.1016/j.cell.2005.03.031. [DOI] [PubMed] [Google Scholar]
- 21.Hohenester E, Yurchenco PD. Laminins in basement membrane assembly. Cell Adh Migr. 2013;7:56–63. doi: 10.4161/cam.21831. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Randles MJ, Humphries MJ, Lennon R. Proteomic definitions of basement membrane composition in health and disease. Matrix Biol. 2016 doi: 10.1016/j.matbio.2016.08.006. [DOI] [PubMed] [Google Scholar]
- 23.Carreon TA, Edwards G, Wang H, Bhattacharya SK. Segmental outflow of aqueous humor in mouse and human. Exp Eye Res. 2016 doi: 10.1016/j.exer.2016.08.001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Benton G, Arnaoutova I, George J, Kleinman HK, Koblinski J. Matrigel: from discovery and ECM mimicry to assays and models for cancer research. Adv Drug Deliv Rev. 2014;79–80:3–18. doi: 10.1016/j.addr.2014.06.005. [DOI] [PubMed] [Google Scholar]
- 25.Schoumacher M, Glentis A, Gurchenkov VV, Vignjevic DM. Basement membrane invasion assays: native basement membrane and chemoinvasion assay. Methods Mol Biol. 2013;1046:133–144. doi: 10.1007/978-1-62703-538-5_8. [DOI] [PubMed] [Google Scholar]
- 26.Deryugina EI, Quigley JP. Chick embryo chorioallantoic membrane model systems to study and visualize human tumor cell metastasis. Histochem Cell Biol. 2008;130:1119–1130. doi: 10.1007/s00418-008-0536-2. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Chen MB, et al. On-chip human microvasculature assay for visualization and quantification of tumor cell extravasation dynamics. Nat Protoc. 2017;12:865–880. doi: 10.1038/nprot.2017.018. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Hiramatsu R, et al. External mechanical cues trigger the establishment of the anterior-posterior axis in early mouse embryos. Dev Cell. 2013;27:131–144. doi: 10.1016/j.devcel.2013.09.026. [DOI] [PubMed] [Google Scholar]
- 29.Nakaya Y, Sukowati EW, Sheng G. Epiblast integrity requires CLASP and Dystroglycan-mediated microtubule anchoring to the basal cortex. J Cell Biol. 2013;202:637–651. doi: 10.1083/jcb.201302075. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Nakaya Y, Sukowati EW, Alev C, Nakazawa F, Sheng G. Involvement of dystroglycan in epithelial-mesenchymal transition during chick gastrulation. Cells Tissues Organs (Print) 2011;193:64–73. doi: 10.1159/000320165. [DOI] [PubMed] [Google Scholar]
- 31.Srivastava A, Pastor-Pareja JC, Igaki T, Pagliarini R, Xu T. Basement membrane remodeling is essential for Drosophila disc eversion and tumor invasion. Proc Natl Acad Sci U S A. 2007;104:2721–2726. doi: 10.1073/pnas.0611666104. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Pastor-Pareja JC, Grawe F, Martín-Blanco E, García-Bellido A. Invasive cell behavior during Drosophila imaginal disc eversion is mediated by the JNK signaling cascade. Dev Cell. 2004;7:387–399. doi: 10.1016/j.devcel.2004.07.022. [DOI] [PubMed] [Google Scholar]
- 33.Morin X, Daneman R, Zavortink M, Chia W. A protein trap strategy to detect GFP-tagged proteins expressed from their endogenous loci in Drosophila. Proc Natl Acad Sci U S A. 2001;98:15050–15055. doi: 10.1073/pnas.261408198. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Guha A, Lin L, Kornberg TB. Regulation of Drosophila matrix metalloprotease Mmp2 is essential for wing imaginal disc:trachea association and air sac tubulogenesis. Dev Biol. 2009;335:317–326. doi: 10.1016/j.ydbio.2009.09.005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Parton RM, Vallés AM, Dobbie IM, Davis I. Live cell imaging in Drosophila melanogaster. Cold Spring Harb Protoc. 2010 doi: 10.1101/pdb.top75. pdb.top75.2010. [DOI] [PubMed] [Google Scholar]
- 36.Stoletov K, et al. Visualizing extravasation dynamics of metastatic tumor cells. J Cell Sci. 2010;123:2332–2341. doi: 10.1242/jcs.069443. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Stoletov K, Montel V, Lester RD, Gonias SL, Klemke R. High-resolution imaging of the dynamic tumor cell vascular interface in transparent zebrafish. Proc Natl Acad Sci U S A. 2007;104:17406–17411. doi: 10.1073/pnas.0703446104. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Kedrin D, et al. Intravital imaging of metastatic behavior through a mammary imaging window. Nat Methods. 2008;5:1019–1021. doi: 10.1038/nmeth.1269. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Pittet MJ, Weissleder R. Intravital imaging. Cell. 2011;147:983–991. doi: 10.1016/j.cell.2011.11.004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Antinucci P, Hindges R. A crystal-clear zebrafish for in vivo imaging. Sci Rep. 2016;6:29490. doi: 10.1038/srep29490. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.Hastie EL, Sherwood DR. A new front in cell invasion: The invadopodial membrane. Eur J Cell Biol. 2016;95:441–448. doi: 10.1016/j.ejcb.2016.06.006. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42.Ihara S, et al. Basement membrane sliding and targeted adhesion remodels tissue boundaries during uterine-vulval attachment in Caenorhabditis elegans. Nat Cell Biol. 2011;13:641–651. doi: 10.1038/ncb2233. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43.Wang Z, et al. UNC-6 (netrin) stabilizes oscillatory clustering of the UNC-40 (DCC) receptor to orient polarity. J Cell Biol. 2014;206:619–633. doi: 10.1083/jcb.201405026. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44.Hagedorn EJ, et al. The netrin receptor DCC focuses invadopodia-driven basement membrane transmigration in vivo. J Cell Biol. 2013;201:903–913. doi: 10.1083/jcb.201301091. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45.Morrissey MA, et al. B-LINK: a hemicentin, plakin, and integrin-dependent adhesion system that links tissues by connecting adjacent basement membranes. Dev Cell. 2014;31:319–331. doi: 10.1016/j.devcel.2014.08.024. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46.Chen X, Feng X, Guang S. Targeted genome engineering in Caenorhabditis elegans. Cell Biosci. 2016;6:60. doi: 10.1186/s13578-016-0125-3. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47.Hochbaum D, Ferguson AA, Fisher AL. Generation of transgenic C. elegans by biolistic transformation. J Vis Exp. 2010 doi: 10.3791/2090. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48.Berkowitz LA, Knight AL, Caldwell GA, Caldwell KA. Generation of stable transgenic C. elegans using microinjection. J Vis Exp. 2008 doi: 10.3791/833. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49.Pettitt J, Wood WB, Plasterk RH. cdh-3, a gene encoding a member of the cadherin superfamily, functions in epithelial cell morphogenesis in Caenorhabditis elegans. Development. 1996;122:4149–4157. doi: 10.1242/dev.122.12.4149. [DOI] [PubMed] [Google Scholar]
- 50.Inoue T, et al. Gene expression markers for Caenorhabditis elegans vulval cells. Mech Dev. 2002;119(Suppl 1):S203–9. doi: 10.1016/s0925-4773(03)00117-5. [DOI] [PubMed] [Google Scholar]
- 51.Matus DQ, et al. Invasive Cell Fate Requires G1 Cell-Cycle Arrest and Histone Deacetylase-Mediated Changes in Gene Expression. Dev Cell. 2015;35:162–174. doi: 10.1016/j.devcel.2015.10.002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 52.McKay SJ, et al. Gene expression profiling of cells, tissues, and developmental stages of the nematode C. elegans. Cold Spring Harb Symp Quant Biol. 2003;68:159–169. doi: 10.1101/sqb.2003.68.159. [DOI] [PubMed] [Google Scholar]
- 53.McClatchey ST, et al. Boundary cells restrict dystroglycan trafficking to control basement membrane sliding during tissue remodeling. elife. 2016;5 doi: 10.7554/eLife.17218. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 54.Bettinger JC, Euling S, Rougvie AE. The terminal differentiation factor LIN-29 is required for proper vulval morphogenesis and egg laying in Caenorhabditis elegans. Development. 1997;124:4333–4342. doi: 10.1242/dev.124.21.4333. [DOI] [PubMed] [Google Scholar]
- 55.Carisey A, Stroud M, Tsang R, Ballestrem C. Fluorescence recovery after photobleaching. Methods Mol Biol. 2011;769:387–402. doi: 10.1007/978-1-61779-207-6_26. [DOI] [PubMed] [Google Scholar]
- 56.Chang CW, Sud D, Mycek MA. Fluorescence lifetime imaging microscopy. Methods Cell Biol. 2007;81:495–524. doi: 10.1016/S0091-679X(06)81024-1. [DOI] [PubMed] [Google Scholar]
- 57.Gligorijevic B, Kedrin D, Segall JE, Condeelis J, van Rheenen J. Dendra2 photoswitching through the Mammary Imaging Window. J Vis Exp. 2009 doi: 10.3791/1278. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 58.Zou W, Yadav S, DeVault L, Nung Jan Y, Sherwood DR. RAB-10-Dependent Membrane Transport Is Required for Dendrite Arborization. PLoS Genet. 2015;11:e1005484. doi: 10.1371/journal.pgen.1005484. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 59.Schindler AJ, Baugh LR, Sherwood DR. Identification of late larval stage developmental checkpoints in Caenorhabditis elegans regulated by insulin/IGF and steroid hormone signaling pathways. PLoS Genet. 2014;10:e1004426. doi: 10.1371/journal.pgen.1004426. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 60.Clay MR, Sherwood DR. Basement Membranes in the Worm: A Dynamic Scaffolding that Instructs Cellular Behaviors and Shapes Tissues. Curr Top Membr. 2015;76:337–371. doi: 10.1016/bs.ctm.2015.08.001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 61.Ben-Yakar A, Chronis N, Lu H. Microfluidics for the analysis of behavior, nerve regeneration, and neural cell biology in C. elegans. Curr Opin Neurobiol. 2009;19:561–567. doi: 10.1016/j.conb.2009.10.010. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 62.Chronis N, Zimmer M, Bargmann CI. Microfluidics for in vivo imaging of neuronal and behavioral activity in Caenorhabditis elegans. Nat Methods. 2007;4:727–731. doi: 10.1038/nmeth1075. [DOI] [PubMed] [Google Scholar]
- 63.Keil W, Kutscher LM, Shaham S, Siggia ED. Long-Term High-Resolution Imaging of Developing C. elegans Larvae with Microfluidics. Dev Cell. 2017;40:202–214. doi: 10.1016/j.devcel.2016.11.022. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 64.Chai Y, et al. Live imaging of cellular dynamics during Caenorhabditis elegans postembryonic development. Nat Protoc. 2012;7:2090–2102. doi: 10.1038/nprot.2012.128. [DOI] [PubMed] [Google Scholar]
- 65.Knobel KM, Jorgensen EM, Bastiani MJ. Growth cones stall and collapse during axon outgrowth in Caenorhabditis elegans. Development. 1999;126:4489–4498. doi: 10.1242/dev.126.20.4489. [DOI] [PubMed] [Google Scholar]
- 66.McGee-Russell SM, Allen RD. Reversible stabilization oflabile microtubules in the reticulopodial network of Allogromia. Adv Cell Mol Biol. 1971;1:153–184. [Google Scholar]
- 67.Wang L, Audhya A. In vivo imaging of C. elegans endocytosis. Methods. 2014;68:518–528. doi: 10.1016/j.ymeth.2014.03.028. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 68.Kim E, Sun L, Gabel CV, Fang-Yen C. Long-term imaging of Caenorhabditis elegans using nanoparticle-mediated immobilization. PLoS ONE. 2013;8:e53419. doi: 10.1371/journal.pone.0053419. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 69.Sulston J, Hodgkin J. In: The NematodeCaenorhabditis elegans. Wood WB, editor. Cold Spring Harbor Laboratory; 1988. pp. 587–606. [Google Scholar]
- 70.Baugh LR. To grow or not to grow: nutritional control of development during Caenorhabditis elegans L1 arrest. Genetics. 2013;194:539–555. doi: 10.1534/genetics.113.150847. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 71.Maxwell CS, Antoshechkin I, Kurhanewicz N, Belsky JA, Baugh LR. Nutritional control of mRNA isoform expression during developmental arrest and recovery in C. elegans. Genome Res. 2012;22:1920–1929. doi: 10.1101/gr.133587.111. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 72.Park EC, Horvitz HR. Mutations with dominant effects on the behavior and morphology of the nematode Caenorhabditis elegans. Genetics. 1986;113:821–852. doi: 10.1093/genetics/113.4.821. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 73.Stiernagle T. Maintenance of C. elegans. WormBook. 2006:1–11. doi: 10.1895/wormbook.1.101.1. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Supplementary Video 1 Invasive protrusion growth by the AC (green) during basement membrane (magenta) transmigration (62 min; Scale bar, 5 μm.)
Supplementary Video 2 Dynamic expansion of a BM opening generated by the AC (70 min; Scale bar, 5 μm).
Supplementary Video 3 Anesthetizing worms, preparing an agar pad, and mounting anesthetized worms.
Supplementary Video 4 Time-lapse image acquisition.
Supplementary Video 5 Importing and visualizing 4D data in Imaris.
Supplementary Video 6 Using spots to quantify AC-invadopodia dynamics.
Supplementary Video 7 Measuring invasive protrusion volumes with isosurfaces.
Supplementary Video 8 Quantifying the rate of BM hole expansion.
Supplementary Figure 1| (a) The schematic depicts the developmental time points during anchor cell (AC) invasion in C. elegans. The AC invades in tight synchrony with the 10 fated vulval precursor cell P6.p cell divisions during the L3 larval stage. During the P6.p one-cell stage, F-actin/endolysosomal membrane based invadopodia form along the invasive cell membrane (orange) within the AC (green) at the AC-basement membrane (BM) interface (magenta). AC invasion initiates during the late P6.p two-cell stage, when one or two invadopodium breaches the BM. A single breaching invadopodium transitions to an invasive protrusion that shuts down further invadopodia formation. As the invasive protrusion enlarges, it clears a large gap in the basement membrane and moves between the central P6.p vulval cells by the P6.p four-cell stage. (b) AC invasion through BM. During invasion (0-53 minutes), the AC (F-actin; green) generates a protrusion that breaches the BM (laminin; magenta). While the BM opening expands, the protrusion grows, and reaches a mature size. Scale bar, 5 μm.