

Abstract
The E-74 like factor 3 (ELF3) is a transcription factor induced by inflammatory factors in various cell types, including chondrocytes. ELF3 levels are elevated in human cartilage from patients with osteoarthritis (OA), and ELF3 contributes to the IL-1β-induced expression of genes encoding Mmp13, Nos2, and Ptgs2/Cox2 in chondrocytes in vitro. Here, we investigated the contribution of ELF3 to cartilage degradation in vivo, using a mouse model of OA. To this end, we generated mouse strains with cartilage-specific Elf3 knockout (Col2Cre:Elf3f/f) and Comp-driven Tet-off-inducible Elf3 overexpression (TRE-Elf3:Comp-tTA). To evaluate the contribution of ELF3 to OA, we induced OA in 12-week-old Col2Cre:Elf3f/f and 6-month-old TRE-Elf3:Comp-tTA male mice using the destabilization of the medial meniscus (DMM) model. The chondrocyte-specific deletion of Elf3 led to decreased levels of IL-1β- and DMM-induced Mmp13 and Nos2 mRNA in vitro and in vivo, respectively. Histological grading showed attenuation of cartilage loss in Elf3 knockout mice compared to wild type (WT) littermates at 8 and 12 weeks following DMM surgery that correlated with reduced collagenase activity. Accordingly, Elf3 overexpression led to increased cartilage degradation post-surgery compared to WT counterparts. Our results provide evidence that ELF3 is a central contributing factor for cartilage degradation in post-traumatic OA in vivo.
Introduction
Osteoarthritis (OA) is a whole joint disorder characterized by pathologic changes of multiple components of the joint1,2. Although OA has long been regarded as “wear and tear” arthritis, the involvement of inflammation in disease progression has been well recognized in recent years2,3. Inflammatory and mechanical stress in OA leads to abnormal activation of common signaling pathways in the cells resident in joint tissues4,5. Chondrocytes, constituting the unique cell type of articular cartilage, become activated in OA in response to the abnormal environment, leading to a plethora of transcriptional and non-transcriptional events that result in aberrant expression of genes contributing to the degradation of the extracellular matrix6.
The ETS transcription factors, defined by a highly conserved DNA binding domain7, are implicated in a diverse range of biological functions8,9. The E-74 like factor 3 (ELF3) is a member of the ELF subfamily with roles in epithelial cell differentiation and function10,11. ELF3 is abundantly expressed in epithelial tissues10. In non-epithelial cells, in which it is not expressed normally12,13, the induction of ELF3 by inflammatory stimuli drives the expression of genes such as cyclooxygenase 2 (COX2), nitric oxide synthase 2 (NOS2), angiopoietin-1, and matrix metalloproteinase 13 (MMP13)12,14–17.
ELF3 is expressed in the cartilage and synovium of patients with rheumatoid arthritis and OA12,17,18. We showed that ELF3 represses the expression of the type II collagen gene (COL2A1) by directly binding to the COL2A1 promoter18 and disrupting the activator functions of Sox9 and CBP/p30019. In addition, we showed that ELF3 binds to and transactivates the MMP13 promoter in chondrocytes, thereby mediating IL-1β- and TNFα-driven MMP13 gene expression17. Altogether, these results suggest that ELF3 plays a critical role in cartilage remodeling in OA disease.
In this study, we aimed to investigate the contribution of ELF3 to cartilage degradation in vivo in a model of post-traumatic OA. Using mice with Col2a1-driven Elf3 knockout (Elf3-cKO) or Comp-driven Tet-Off inducible overexpression of Elf3 (Elf3-Tg) subjected to destabilization of the medial meniscus (DMM), we show that Elf3 accounts, in large part, for the expression and activity of Mmp13 in articular chondrocytes in vivo.
Results
Generation and characterization of cartilage-specific Elf3 KO mice
To elucidate the role of Elf3 deficiency in articular cartilage remodeling in vivo, we first generated mice with cartilage-specific Elf3 KO. The Col2a1-Cre:Elf3f/f (Elf3-cKO) mice were born at the expected Mendelian ratio, with no gross abnormality, and with comparable size and weight to the Elf3f/f Cre-negative littermates (WT) and sex- and age-matched wild-type C57/BL6 mice (not shown). To assess knockout efficiency, total RNA was isolated from cartilage explants from femoral heads of 5- to 6-day-old cKO and WT littermates. Since Elf3 mRNA is barely detectable in articular chondrocytes and other non-epithelial cells12,15,17, we incubated the cartilage explants with 10 ng/ml IL-1β for 6 days. RTqPCR analyses confirmed increased Elf3 mRNA in IL-1β-treated WT cartilage explants, whereas IL-1β treatment did not induce Elf3 mRNA in cKO cartilage explants, verifying efficient Elf3 ablation in articular cartilage (Fig. 1A). Next, to verify the cartilage specificity of Elf3 deletion, we assessed Elf3 mRNA levels in tissues known to express Elf3 at baseline. As shown in Fig. 1B, Elf3 mRNA levels were similar in kidney, liver, and lung tissues obtained from cKO compared to WT littermates. Our preliminary in situ hybridization analyses showed the absence of Elf3 mRNA in the developing cartilage anlagen of C57/BL6 mice (not shown), suggesting that Elf3 does not contribute to growth plate development. To confirm this, we assessed whether the postnatal growth plate architecture was altered in the cKO animals. As shown in Fig. 1C, we observed no difference in the lengths of the post-natal proliferating and hypertrophic zones between WT and cKO mice at P7 or P21. Further, immunofluorescence analyses in P7 (Fig. 1D) and P21 (not shown) mice revealed comparable distribution of type X collagen protein in the hypertrophic zone of WT and cKO mice. Finally, Faxitron radiographic analyses revealed no difference in the lengths of long bones (tibiae and femora) of WT and cKO mice at P7 (not shown) and P21 (Fig. 1E). Together, these results indicate that Col2a1-driven Elf3 knockout does not lead to skeletal abnormalities, and that the adult Elf3 cKO mice are suitable for studying OA.
Figure 1.

Characterization of mice with cartilage-specific Elf3 deficiency. (A) IL-1β treatment significantly induced Elf3 mRNA in WT but not in cKO cartilage explants (n = 4/ea). Data are represented as the fold-change in mRNA levels relative to untreated controls (dotted line). (B) RTqPCR analyses of total RNA extracted from kidney, liver, and lung (n = 4/ea) showed no difference in Elf3 mRNA levels between cKO and WT samples. (C) Representative histological sections of growth plates from tibiae of cKO and WT mice at P7 and P21. Vertical lines indicate proliferative (PZ, white) and hypertrophic (HZ, black) zones. Quantification of the thickness of hypertrophic and proliferative zones is shown for P7 (n = 4/genotype) and P21 (n = 3/genotype). (D) Representative images of type X collagen (Col10) immunostaining in hypertrophic zones of cKO and WT mice at P7. The quantification of the Col10-positive area (n = 3/ea) is shown as mean signal intensity (red = Col10-positive, blue = DAPI). Scale bar = 100 μm. (E) Representative digital radiographs of tibiae and femora from WT and cKO mice at P21 (n = 3/ea). Quantification is shown on the right. Scale bar = 1.87 mm. Data are shown as mean ± S.D. *p < 0.05 by t-test.
Decreased expression of Elf3 target genes in primary chondrocytes from Elf3-deficient mice
We next assessed the functional impact of cartilage-specific Elf3 deletion by RTqPCR analyses of total RNA extracted from WT and cKO primary chondrocytes after treatment with 1 ng/ml of IL-1β for 6 hours in vitro. As shown in Fig. 2A, IL-1β-induced Elf3 expression was ablated in cKO cells without impacting the levels of Ehf and Elf5 mRNA, other members of the ELF subfamily (Fig. 2B,C). Importantly, while Elf3 KO did not impact the basal or IL-1β-driven induction of Mmp2, 3, 10, or 12, Ptgs2, or Adamts4 or 5 mRNA, we observed a significant reduction in the IL-1β-driven Nos2 and Mmp13 gene expression (Fig. 2D–L). Together, our results confirmed our previous findings in chondrocytes from global Elf3 KO mice17 and further suggested that ELF3 could contribute to cartilage remodeling in OA by driving collagenase gene expression.
Figure 2.

Decreased expression of IL-1β-induced Elf3 target genes in primary chondrocytes from Elf3-cKO mice. Primary chondrocytes isolated from 5- to 6-day-old WT and cKO littermates were left untreated (vehicle, PBS/BSA) or treated with IL-1β (1 ng/ml) for 6 h. The mRNA levels of Elf3 (A), Ehf (B), Elf5 (C), Mmp2 (D), Mmp3 (E), Mmp10 (F), Mmp12 (G), Mmp13 (H), Nos2 (I), Ptgs2 (J), Adamts4 (K), and Adamts5 (L) were analyzed by RTqPCR and are represented as fold-change vs. untreated controls (dotted line). Data are mean ± S.D. from at least 4 independent experiments. *p < 0.05, ** < 0.01 by t-test.
Cartilage-specific Elf3 deficiency attenuates the development of osteoarthritis in mice following destabilization of the medial meniscus
Next, to determine whether ELF3 contributes to OA progression in vivo, we compared cartilage degradation in WT and cKO mice after surgical induction of OA. Articular cartilage and joint morphology were not different in cKO mice compared to WT littermates at baseline, in unoperated knees (Supplementary Figure 2). At 4 weeks post-DMM, small fibrillations were observed in both WT and cKO articular cartilage, with no significant difference in the OA scores (Fig. 3A,B). However, at 8 and 12 weeks post-surgery, cartilage damage was significantly reduced in the cKO mice (Fig. 3C–F). We also analyzed the size and maturity of osteophytes, as described20. The unoperated knees did not form osteophytes, as expected, and we did not observe any difference in osteophyte formation, size, or maturity between genotypes at 8 weeks following surgery (Supplementary Figure 3). These results indicate that cartilage-specific Elf3 deletion impacts cartilage degradation without affecting osteophyte formation in a surgical model of post-traumatic OA.
Figure 3.

Cartilage-specific Elf3 deficiency attenuates the development of osteoarthritis in mice following destabilization of the medial meniscus (DMM). Representative Safranin O/Fast green-stained sections of the knee joints of DMM-operated 12-week-old male WT and cKO mice at (A) 4 weeks (WT = 7; cKO = 7), (C) 8 weeks (WT = 10; cKO = 11), and (E) 12 weeks (WT = 7; cKO = 8). Images show the medial femorotibial compartment of DMM-operated knees using 10X magnification. The arrows indicate sites of cartilage lesions. Quantification of cartilage damage in WT and cKO mice at 4 (B), 8 (D), and 12 (F) weeks following DMM is shown as OARSI SUM and MAX scores and represented as median and C.I. *p < 0.05 and **p < 0.01 by Mann-Whitney test.
Elf3 deficiency reduces collagenase activity in mice following destabilization of the medial meniscus
Elf3 mRNA levels were found to be increased in vivo in DMM-operated WT cartilage samples compared to unoperated WT controls, whereas Elf3 mRNA was virtually undetectable in both control and DMM-operated cKO cartilage samples (Fig. 4A), confirming that the protection against DMM-induced OA correlated with the loss of Elf3 gene expression. In addition, we observed significantly decreased Nos2 mRNA in cartilage samples obtained from DMM-operated cKO mice compared to WT counterparts (Fig. 4B). We did not detect significant changes in Mmp13 (Fig. 4B), Col2a1 (Supplementary Figure 4A), Acan (Supplementary Figure 4B) or Timp3 (Supplementary Figure 4C) mRNA levels between WT and cKO DMM-operated samples at 8 weeks post-surgery. We next assessed collagenase activity using the C1,2 C antibody, which detects collagenase-specific cleavage epitopes on type II collagen. Immunohistochemical analyses showed reduced C1,2 C immunostaining in cKO mice compared to WT counterparts post-DMM (Fig. 4C,D). Together, our results indicate that the Elf3 cKO mice are protected against DMM-induced cartilage loss and display reduced collagenase activity in vivo.
Figure 4.

Elf3 deficiency reduces collagenase activity in mice following destabilization of the medial meniscus (DMM). RT-qPCR analyses of total RNA isolated from articular cartilage from unoperated (Ctrl) and DMM-operated (DMM) joints of WT and cKO mice (n = 3/ea) at 8 weeks after surgery showing DMM-induced Elf3 mRNA in WT and cKO counterparts, represented as relative expression to Rpl13a (left) and fold-change vs. unoperated controls (right) (A). Nos2 and Mmp13 mRNA levels were assessed in cartilage isolated from DMM-operated limbs (n = 3/genotype) at 8 weeks after DMM. (B) Data is shown as fold change vs. WT levels (set at 1). (C) Representative images of unoperated (Control) or DMM-operated (8wk DMM) sections of WT and Elf3 cKO mice at 8 weeks after surgery stained with the C1, 2C antibody. (D) Quantification of the C1,2C-positive immunostaining at 8-weeks post-DMM revealed reduced collagenase activity in the cKO mice. Data are shown as fold-change over the immunopositive signal detected in the control knees, set at 1 (n = 3/ea). All data are shown as mean ± S.D. ***p < 0.001 by t-test (B), and *p < 0.05 and **p < 0.01 by ANOVA followed by Tukey’s post-hoc test (A and D). Comparisons with significant differences are indicated; all other comparisons were non-significant.
Generation and characterization of Comp-tTA;TRE-Elf3 mice
To determine the effects of enforced ELF3 overexpression in mouse knee joints on cartilage degradation, we generated a novel Tet-Off-inducible Elf3-overexpressing (Comp-tTA:TRE-Elf3, referred to as Elf3Tg) mouse strain. Pregnant females and litters were administered doxycycline until weaning, as described21, and post-natal Elf3 overexpression was induced by doxycycline withdrawal in Comp-expressing tissues. No doxycycline-induced defect or gross abnormality due to the transgene was observed in any of the lines (not shown). The ComptTA;TRE-Elf3 (Elf3Tg) and TRE-Elf3 (WT) control littermates displayed comparable size and weight to ComptTA and wild-type C57/B6 sex and age-matched mice at 1 and 3 months of age (not shown). We isolated total RNA from the articular cartilage of WT and Elf3Tg mice at 2, 5, and 8 months after doxycycline removal (3, 6, and 9 months of age, respectively). While we did not consistently detect changes in Elf3 mRNA in Elf3Tg mice at 2 months after doxycycline withdrawal (data not shown), increased Elf3 mRNA was observed in the articular cartilage of Elf3Tg mice compared to WT littermates at 6 and 9 months of age (Fig. 5A). Elf3 mRNA levels detected in kidney and liver were identical in WT and Elf3Tg mice (Fig. 5B), confirming that doxycycline withdrawal specifically upregulated Elf3 mRNA in Comp-expressing tissues. Finally, to assess whether increasing Elf3 expression postnatally in joint tissues would result spontaneously in early onset of OA-like pathology, we evaluated the knee joints of 6 and 9-month-old WT and Elf3Tg mice (5 and 8 months after doxycycline removal, respectively). Safranin O staining revealed comparable structural integrity in Elf3Tg and WT littermates at 6 (not shown) and 9 months of age (Fig. 5C), which was also confirmed by the lack of significant differences in OARSI cartilage degradation scores between genotypes (Fig. 5D).
Figure 5.

Characterization of the Elf3-Tg mice. (A) RT-qPCR analyses showed increased Elf3 mRNA in articular cartilage from Elf3Tg male mice after Dox withdrawal at 6 (WT = 4, Elf3Tg = 5) and 9 (n = 4/ea) months of age compared to WT littermates. Data are shown as mean ± S.D. (B) RTqPCR analysis of total RNA isolated from kidney and liver (n = 4/ea) showed no differences in Elf3 mRNA between WT and Elf3Tg mice at 9 months of age, after Dox withdrawal. Data are shown as mean ± S.D. (C) Representative images of histological sections from unoperated WT and Elf3Tg male mice at 9 months of age (8 months after Dox withdrawal) show no difference between genotypes. (D) OARSI cartilage degradation scoring, represented as median and C.I. (n = 5/ea), showing no significant difference between WT and Elf3Tg mice at 6 and 9 months of age (5 and 8 months after Dox withdrawal, respectively).
Forced expression of Elf3 increases cartilage degradation following destabilization of the medial meniscus
We next assessed whether increasing ELF3 postnatally in joint tissues would result in acceleration of post-traumatic OA by performing DMM surgeries in 6-month-old male WT and Elf3Tg mice (at 5 months after doxycycline withdrawal). Cartilage integrity was assessed in unoperated and DMM-operated WT and Elf3Tg mouse knee joints at 8 weeks post-DMM. Consistent with our previous findings (Fig. 5C,D), the Elf3Tg showed only a marginal increase in cartilage degradation scores in the unoperated limbs, further suggesting that Elf3 overexpression is not sufficient to drive cartilage degradation. However, the DMM-operated Elf3Tg mice exhibited significantly increased cartilage degradation at 8 weeks post-DMM compared to WT counterparts (Fig. 6A,B) without inducing significant changes in osteophyte size or maturity (Supplementary Figure 5). Taken together, our results demonstrate that ELF3 contributes to cartilage degradation in OA, in part by driving MMP13 gene expression and associated collagenase activity. Our results also show that enforced post-natal expression of Elf3 in cartilage is not sufficient to induce spontaneous OA, but increases the OA severity post-DMM, suggesting that additional biomechanical and/or inflammatory signals may be required to enable Elf3 to transactivate genes involved in cartilage degradation.
Figure 6.
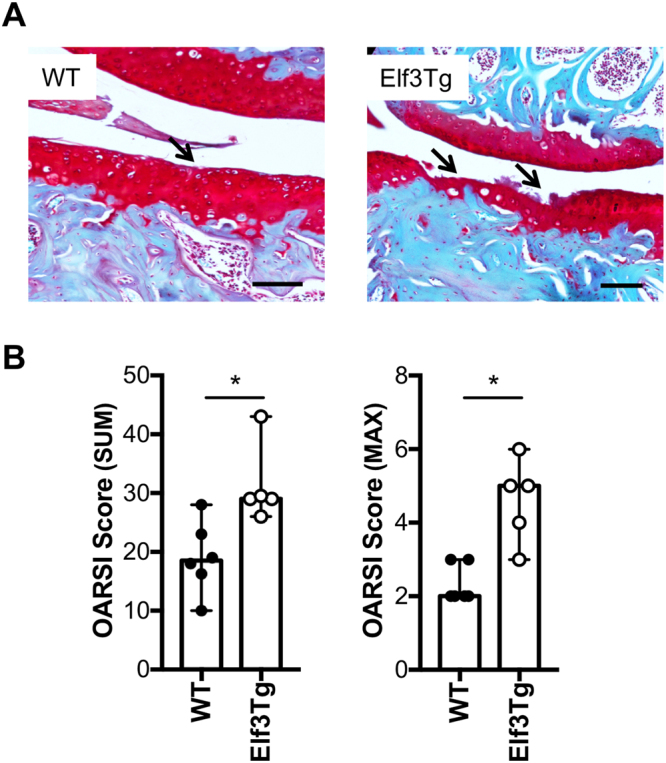
Forced expression of Elf3 increases cartilage degradation following destabilization of the medial meniscus (DMM). (A) Representative images of Safranin O-stained histological sections from DMM-operated 6-month-old WT and Elf3Tg male mice at 8 weeks post-DMM. The arrows indicate sites of cartilage lesions. (B) OARSI histological scores showing increased cartilage degradation in DMM-operated Elf3-overexpressing mice (WT = 6; Elf3Tg = 5). Data are shown as median and C.I. *p < 0.05 by Mann-Whitney test.
Discussion
OA disease involves the interplay of numerous regulators and signaling factors that in concert lead to deregulated remodeling of joint tissues. We showed previously that ELF3 mRNA and protein levels are increased in human OA cartilage, and our in vitro data in human and mouse primary chondrocytes provided evidence for the pivotal role of ELF3 in regulating MMP13 gene transcription17 and in disrupting chondrocyte anabolism18,19, suggesting its contribution to abnormal cartilage remodeling in OA disease. Here, we show that ELF3 contributes to increased collagenase activity and cartilage degradation in vivo, in a mouse model of post-traumatic OA, and that ELF3 overexpression accelerates cartilage degradation concomitant to post-traumatic OA disease.
To assess the effects of the Elf3 deletion in OA, WT and Elf3-cKO littermates were subjected to the DMM model, which mimics post-traumatic OA in humans22,23. The DMM-operated WT mice showed enhanced cartilage damage at 8 and 12 weeks compared to the cKO mice, which had significantly lower OA scores. The lack of difference observed between the genotypes at 4 weeks post-DMM may be due to the time it requires to upregulate sufficient Elf3 gene expression and/or activity of its target gene products, including matrix-degrading proteinases, which also require activation of pro-enzyme precursors24. In addition to progressive erosion of articular cartilage, OA is characterized by thickening of the subchondral bone and osteophyte formation25. Both Elf3-cKO mice and their WT littermates developed DMM-induced osteophytes, with no difference in size or maturity of osteophytes observed between genotypes, indicating that Elf3 does not have a significant role in osteophyte formation. This result is consistent with previous reports showing that Mmp13 knockout mice are protected from articular cartilage erosion, but not from osteophyte development26. The increased expression of Elf3 mRNA in the articular cartilage of the WT DMM-operated limbs suggests that Elf3 may be induced by mechanical stress in the absence of overt inflammation. DMM-induced expression of Elf3 and Mmp13 correlated with increased collagenase activity, which was reduced in cKO mice. The latter further suggests that ELF3 mediates cartilage degradation by specifically modulating MMP-13, in agreement with our previous in vitro results showing that other proteases are not affected by the absence of ELF3, and is consistent with the view that MMP-13 plays a central role in erosion of the collagen network, which marks OA progression27. However, while our observations in vitro support the notion that Elf3 drives cartilage degradation via MMP-13 induction and not through other collagenase or aggrecanase-mediated events, the contribution of Elf3 in driving the expression in vivo of other relevant inflammatory and catabolic genes that impact OA disease is yet to be elucidated. Similarly, with our experimental conditions we did not detect significant changes in selected anabolic genes. However, since changes in anabolism constitute a somewhat early event in OA disease and are sometimes difficult to detect reliably in cartilage with established pathology and evident structural damage, the impact of Elf3 in the anabolic responses of OA chondrocytes in vivo requires further investigation.
Our data showing increased ELF3 expression in human OA cartilage and in the DMM-operated mouse cartilage, and protection of Elf3-cKO mice from cartilage degradation post-DMM surgery, imply that increased Elf3 levels and/or activity may have a role in initiating or accelerating the progression of OA. To assess the effects of enforced expression of Elf3 in adult mouse articular cartilage, we generated the Comp-tTA:TRE-Elf3 (Elf3Tg) mouse strain, with inducible Elf3 expression. We chose the Tet-Off system because of the reported advantages over other regulated gene expression systems28,29. While removal of doxycycline induced Elf3 mRNA expression efficiently and specifically in the Elf3Tg cartilage, we did not observe significantly increased spontaneous or age-related cartilage degradation in the Elf3Tg mice at 6 and 9 months of age, indicating that increased Elf3 gene expression is not sufficient to drive OA disease. Interestingly, DMM-operated Elf3Tg mice showed increased cartilage degradation at 8 weeks after surgery.
Together, these observations suggest that biomechanical challenge may be required to enhance ELF3 activity via mechanotransduction events that induce activating kinases of ELF317, or to induce the expression or activity of other co-activators of ELF3, including AP-1 and NF-κB6,30–32. We showed previously that ELF3 synergizes with c-Fos/c-Jun to transactivate MMP13, and that the ELF3-induced transactivation of the MMP13 promoter in response to IL-1β in primary chondrocytes is partially mediated by activation of MEK/ERK17. In addition, ELF3 has been linked to NF-κB signaling in different gene promoters and cellular contexts, being a downstream target12,16,17,33, a co-factor14, and a modulator of canonical NF-κB signaling13. Thus, it is conceivable that mechanical stress activates upstream signaling pathways such as MAPKs to enhance ELF3 activity, but also upregulates other transcription factors, which could then directly or indirectly interact with ELF3 to drive gene transcription in chondrocytes. Future studies should aim to further elucidate the mechanisms of action by which ELF3 accelerates OA disease progression in vivo, by investigating whether stress-induced MAPK activation directly phosphorylates ELF3 and increases its stability, activity, subcellular location, and DNA binding capacity in vivo. Furthermore, mechanical stress could increase the expression and/or activity of ELF3’s interacting partners to drive transcription in a disease-, cell-, and/or promoter-specific manner. Our findings in vivo suggest that ELF3 is a critical transcriptional regulator of genes required for collagen erosion during the progression of OA disease. Thus, defining the downstream targets and interacting factors of ELF3 will provide valuable insight into disease mechanisms and potentially lead to the development of novel targeted therapies for OA.
Methods
See Supplementary Information for a detailed outline of the methods, procedures and specific materials used in this study.
Ethics statement
All experiments were performed according to the guidelines of the American Veterinary Association and were approved by the IACUC of the Hospital for Special Surgery, and all procedures are reported following the ARRIVE guidelines34.
Generation of genetically modified mice
For loss-of-function studies, we used a targeting vector obtained from the European Conditional Mouse Mutagenesis Program (EUCOMM, HTGRS6002_A_H11)35 to generate a novel Elf3f/f mouse strain (deposited in Jackson labs, Elf3tm1Mote Tyrc-2J/J; stock# 030056), in which exons 2–8 of the Elf3 gene are flanked by loxP sites. For deletion of the floxed alleles in chondrocytes, we crossed the Elf3f/f mice with Col2a1-Cre mice36,37. Additional details about the generation and genotyping of the Col2a1-Cre;Elf3f/f (Elf3-cKO) mice are provided in the Supplementary Methods. For gain-of-function studies, we generated the transgenic TRE-Elf3 mice (deposited in Jackson labs, C57BL/6-Tg(tetO-Elf3)1Mote/J; stock# 030058), that express Elf3 under the control of the tetracycline-responsive element (TRE). For this, a tet-responsive element (TRE; tetO) and a minimal CMV promoter were placed upstream of the full-length mouse Elf3 cDNA in a transgenic vector introduced in C57/BL6 embryos. To direct expression of Elf3 specifically in adult knee joints, we crossed the TRE-Elf3 strain with Comp-tTA mice21. Additional details about the generation and genotyping of ComptTA;TRE-Elf3 (Elf3Tg) mice are provided in the Supplementary Methods.
Destabilization of the Medial Meniscus (DMM) surgery and tissue processing post-DMM
DMM surgeries were performed, as described20,22, in 12-week-old male Elf3-cKO and WT littermates, or 6-month-old male Elf3Tg and WT littermates. The left knees were unoperated and served as contralateral controls. At 4, 8, and 12 weeks post-surgery, animals were euthanized and the knees collected and processed, as described20. Two blinded scorers assessed OA pathology38. The data are shown as sum and max scores. Sum scores were calculated by adding the scores of 10 sections per mouse. Osteophyte size and maturity were evaluated as described20,26.
Statistical analysis
Statistical analyses were performed using GraphPad Prism 7 Software (GraphPad Software, Sand Diego, CA). Data are reported as means ± S.D. or as median and 95% C.I. (non-normally distributed data) of at least three independent experiments. Data were assessed for approximation to the Gaussian distribution using the D’Agostino-Pearson omnibus test of normality. Distributions were considered to be Gaussian if the P-value for the null hypothesis was greater than 0.05. Unpaired Student t-test was used to establish statistical significance between two groups. Analysis of the histological scores was performed using Mann-Whitney test. For data involving multiple groups, one-way analysis of variance (ANOVA) was performed followed by Tukey’s post-hoc test. P < 0.05 was considered significant.
Data availability
The data that supports the findings of this study are available from the corresponding author on reasonable request.
Electronic supplementary material
Acknowledgements
We are grateful to Drs Carl P. Blobel and Gisela Weskamp for their help with the Southern blotting experiments, and to Dr. Yefu Li for providing the Comp-tTA mouse strain. We are also grateful to The Gene Targeting Resource Center at The Rockefeller University, and to The Sloan-Kettering Transgenic Mouse Facility for their help generating the Elf3f/f and TRE-Elf3 mouse strains, respectively. This work was supported by National Institutes of Health Grants R01-AG022021, R21-AR054887, and RC4 AR060546 (to M. B. G.) and R21-AG049980 (to M.O.).
Author Contributions
M.B.G. and M.O. contributed to the conception and design of the study. E.B.W., K.L.C., J.Q., J.C., C.L.D., D.A.P. and M.O. performed the collection of data. E.B.W., K.L.C., J.Q., J.C., C.L.D., D.A.P., M.B.G. and M.O. performed data analysis and interpretation. E.B.W., K.L.C., M.B.G. and M.O. drafted the article. E.B.W., K.L.C., J.Q., J.C., C.L.D., D.A.P., M.B.G. and M.O. reviewed and approved the manuscript.
Competing Interests
The authors declare no competing interests.
Footnotes
Electronic supplementary material
Supplementary information accompanies this paper at 10.1038/s41598-018-24695-3.
Publisher's note: Springer Nature remains neutral with regard to jurisdictional claims in published maps and institutional affiliations.
References
- 1.Man GS, Mologhianu G. Osteoarthritis pathogenesis - a complex process that involves the entire joint. J Med Life. 2014;7:37–41. [PMC free article] [PubMed] [Google Scholar]
- 2.Loeser RF, Goldring SR, Scanzello CR, Goldring MB. Osteoarthritis: a disease of the joint as an organ. Arthritis Rheum. 2012;64:1697–1707. doi: 10.1002/art.34453. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Goldring MB, Goldring SR. Osteoarthritis. J Cell Physiol. 2007;213:626–634. doi: 10.1002/jcp.21258. [DOI] [PubMed] [Google Scholar]
- 4.Goldring MB, Otero M. Inflammation in osteoarthritis. Curr Opin Rheumatol. 2011;23:471–478. doi: 10.1097/BOR.0b013e328349c2b1. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Goldring MB, et al. Roles of inflammatory and anabolic cytokines in cartilage metabolism: signals and multiple effectors converge upon MMP-13 regulation in osteoarthritis. Eur Cell Mater. 2011;21:202–220. doi: 10.22203/eCM.v021a16. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Marcu KB, Otero M, Olivotto E, Borzi RM, Goldring MB. NF-kappaB signaling: multiple angles to target OA. Curr Drug Targets. 2010;11:599–613. doi: 10.2174/138945010791011938. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Oikawa T, Yamada T. Molecular biology of the Ets family of transcription factors. Gene. 2003;303:11–34. doi: 10.1016/S0378-1119(02)01156-3. [DOI] [PubMed] [Google Scholar]
- 8.Sharrocks AD. The ETS-domain transcription factor family. Nat Rev Mol Cell Biol. 2001;2:827–837. doi: 10.1038/35099076. [DOI] [PubMed] [Google Scholar]
- 9.Sementchenko, V. I. & Watson, D. K. Ets target genes: past, present and future. Oncogene19, 6533-6548, 10.1038/sj.onc.1204034 (2000). [DOI] [PubMed]
- 10.Oliver JR, Kushwah R, Hu J. Multiple roles of the epithelium-specific ETS transcription factor, ESE-1, in development and disease. Lab Invest. 2012;92:320–330. doi: 10.1038/labinvest.2011.186. [DOI] [PubMed] [Google Scholar]
- 11.Oettgen P, et al. Genomic organization of the human ELF3 (ESE-1/ESX) gene, a member of the Ets transcription factor family, and identification of a functional promoter. Genomics. 1999;55:358–362. doi: 10.1006/geno.1998.5681. [DOI] [PubMed] [Google Scholar]
- 12.Grall F, et al. Responses to the proinflammatory cytokines interleukin-1 and tumor necrosis factor alpha in cells derived from rheumatoid synovium and other joint tissues involve nuclear factor kappaB-mediated induction of the Ets transcription factor ESE-1. Arthritis Rheum. 2003;48:1249–1260. doi: 10.1002/art.10942. [DOI] [PubMed] [Google Scholar]
- 13.Longoni N, et al. ETS transcription factor ESE1/ELF3 orchestrates a positive feedback loop that constitutively activates NF-kappaB and drives prostate cancer progression. Cancer Res. 2013;73:4533–4547. doi: 10.1158/0008-5472.CAN-12-4537. [DOI] [PubMed] [Google Scholar]
- 14.Rudders S, et al. ESE-1 is a novel transcriptional mediator of inflammation that interacts with NF-kappa B to regulate the inducible nitric-oxide synthase gene. J Biol Chem. 2001;276:3302–3309. doi: 10.1074/jbc.M006507200. [DOI] [PubMed] [Google Scholar]
- 15.Brown C, et al. ESE-1 is a novel transcriptional mediator of angiopoietin-1 expression in the setting of inflammation. J Biol Chem. 2004;279:12794–12803. doi: 10.1074/jbc.M308593200. [DOI] [PubMed] [Google Scholar]
- 16.Grall FT, et al. The Ets transcription factor ESE-1 mediates induction of the COX-2 gene by LPS in monocytes. FEBS J. 2005;272:1676–1687. doi: 10.1111/j.1742-4658.2005.04592.x. [DOI] [PubMed] [Google Scholar]
- 17.Otero M, et al. E74-like factor 3 (ELF3) impacts on matrix metalloproteinase 13 (MMP13) transcriptional control in articular chondrocytes under proinflammatory stress. J Biol Chem. 2012;287:3559–3572. doi: 10.1074/jbc.M111.265744. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Peng H, et al. ESE-1 is a potent repressor of type II collagen gene (COL2A1) transcription in human chondrocytes. J Cell Physiol. 2008;215:562–573. doi: 10.1002/jcp.21338. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Otero M, et al. ELF3 modulates type II collagen gene (COL2A1) transcription in chondrocytes by inhibiting SOX9-CBP/p300-driven histone acetyltransferase activity. Connect Tissue Res. 2017;58:15–26. doi: 10.1080/03008207.2016.1200566. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Culley KL, et al. Mouse models of osteoarthritis: surgical model of posttraumatic osteoarthritis induced by destabilization of the medial meniscus. Methods Mol Biol. 2015;1226:143–173. doi: 10.1007/978-1-4939-1619-1_12. [DOI] [PubMed] [Google Scholar]
- 21.Xu L, et al. Intact pericellular matrix of articular cartilage is required for unactivated discoidin domain receptor 2 in the mouse model. Am J Pathol. 2011;179:1338–1346. doi: 10.1016/j.ajpath.2011.05.023. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Glasson SS, Blanchet TJ, Morris EA. The surgical destabilization of the medial meniscus (DMM) model of osteoarthritis in the 129/SvEv mouse. Osteoarthritis Cartilage. 2007;15:1061–1069. doi: 10.1016/j.joca.2007.03.006. [DOI] [PubMed] [Google Scholar]
- 23.Little CB, Zaki S. What constitutes an “animal model of osteoarthritis”–the need for consensus? Osteoarthritis Cartilage. 2012;20:261–267. doi: 10.1016/j.joca.2012.01.017. [DOI] [PubMed] [Google Scholar]
- 24.Loeser RF, et al. Disease progression and phasic changes in gene expression in a mouse model of osteoarthritis. PLoS One. 2013;8:e54633. doi: 10.1371/journal.pone.0054633. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.van der Kraan PM, van den Berg WB. Osteophytes: relevance and biology. Osteoarthritis Cartilage. 2007;15:237–244. doi: 10.1016/j.joca.2006.11.006. [DOI] [PubMed] [Google Scholar]
- 26.Little CB, et al. Matrix metalloproteinase 13-deficient mice are resistant to osteoarthritic cartilage erosion but not chondrocyte hypertrophy or osteophyte development. Arthritis Rheum. 2009;60:3723–3733. doi: 10.1002/art.25002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Fosang AJ, Beier F. Emerging Frontiers in cartilage and chondrocyte biology. Best Pract Res Clin Rheumatol. 2011;25:751–766. doi: 10.1016/j.berh.2011.11.010. [DOI] [PubMed] [Google Scholar]
- 28.Furth PA, et al. Temporal control of gene expression in transgenic mice by a tetracycline-responsive promoter. Proc Natl Acad Sci USA. 1994;91:9302–9306. doi: 10.1073/pnas.91.20.9302. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Boger H, Gruss P. Functional determinants for the tetracycline-dependent transactivator tTA in transgenic mouse embryos. Mech Dev. 1999;83:141–153. doi: 10.1016/S0925-4773(99)00042-8. [DOI] [PubMed] [Google Scholar]
- 30.Chowdhury TT, Salter DM, Bader DL, Lee DA. Signal transduction pathways involving p38 MAPK, JNK, NFkappaB and AP-1 influences the response of chondrocytes cultured in agarose constructs to IL-1beta and dynamic compression. Inflamm Res. 2008;57:306–313. doi: 10.1007/s00011-007-7126-y. [DOI] [PubMed] [Google Scholar]
- 31.Nam J, Aguda BD, Rath B, Agarwal S. Biomechanical thresholds regulate inflammation through the NF-kappaB pathway: experiments and modeling. PLoS One. 2009;4:e5262. doi: 10.1371/journal.pone.0005262. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Sanz-Ramos P, Mora G, Ripalda P, Vicente-Pascual M, Izal-Azcarate I. Identification of signalling pathways triggered by changes in the mechanical environment in rat chondrocytes. Osteoarthritis Cartilage. 2012;20:931–939. doi: 10.1016/j.joca.2012.04.022. [DOI] [PubMed] [Google Scholar]
- 33.Wu J, et al. Regulation of epithelium-specific Ets-like factors ESE-1 and ESE-3 in airway epithelial cells: potential roles in airway inflammation. Cell Res. 2008;18:649–663. doi: 10.1038/cr.2008.57. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Kilkenny C, Browne WJ, Cuthill IC, Emerson M, Altman DG. Improving bioscience research reporting: the ARRIVE guidelines for reporting animal research. PLoS Biol. 2010;8:e1000412. doi: 10.1371/journal.pbio.1000412. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Friedel RH, Seisenberger C, Kaloff C, Wurst W. EUCOMM–the European conditional mouse mutagenesis program. Brief Funct Genomic Proteomic. 2007;6:180–185. doi: 10.1093/bfgp/elm022. [DOI] [PubMed] [Google Scholar]
- 36.Hall KC, et al. ADAM17 controls endochondral ossification by regulating terminal differentiation of chondrocytes. Mol Cell Biol. 2013;33:3077–3090. doi: 10.1128/MCB.00291-13. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Ovchinnikov DA, Deng JM, Ogunrinu G, Behringer RR. Col2a1-directed expression of Cre recombinase in differentiating chondrocytes in transgenic mice. Genesis. 2000;26:145–146. doi: 10.1002/(SICI)1526-968X(200002)26:2<145::AID-GENE14>3.0.CO;2-C. [DOI] [PubMed] [Google Scholar]
- 38.Glasson SS, Chambers MG, Van Den Berg WB, Little CB. The OARSI histopathology initiative - recommendations for histological assessments of osteoarthritis in the mouse. Osteoarthritis Cartilage. 2010;18(Suppl 3):S17–23. doi: 10.1016/j.joca.2010.05.025. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Data Availability Statement
The data that supports the findings of this study are available from the corresponding author on reasonable request.