

Abstract
Plateable human hepatocytes with human plasma were utilized to generate the uptake transporter kinetic data for pravastatin, an organic anion‐transporting polypeptide (OATP) transporter substrate. The active hepatic uptake of pravastatin was determined with a Jmax value of 134.4 pmol/min/million cells and Km of 76.77 µM in plateable human hepatocytes with human plasma. The physiologically‐based pharmacokinetic (PBPK) model with incorporation of these in vitro kinetic data successfully simulated the i.v. pharmacokinetic profile of pravastatin without applying scaling factor (the mean predicted area under the curve (AUC) is within 1.5‐fold of the observed). Furthermore, the PBPK model also adequately described the oral plasma concentration‐time profiles of pravastatin at different dose levels. The current investigation demonstrates an approach allowing us to build upon the translation of in vitro OATP uptake transporter data to in vivo, with a hope of utilizing the in vitro data for the prospective human pharmacokinetic (PK) prediction.
Study Highlights.
WHAT IS THE CURRENT KNOWLEDGE ON THE TOPIC?
☑ The PBPK models of pravastatin have been developed by various groups previously, and the OATP Clint,T input was either from the in vitro measurement with a requirement of a scaling factor or back calculated from in vivo data. It demonstrates a hurdle to the utilization of the in vitro data for prospective PK prediction of a compound that is an OATP substrate by PBPK.
WHAT QUESTION DID THIS STUDY ADDRESS?
☑ Is it possible to apply a PBPK model that incorporates the in vitro OATP hepatic uptake data generated in the developed system to directly predict the PK of pravastatin.
WHAT DOES THIS STUDY ADD TO OUR KNOWLEDGE?
☑ Incorporation of the OATP kinetic uptake data generated in plateable human hepatocytes with human plasma enabled the PBPK model to simulate the i.v. and oral PK profiles of pravastatin successfully without incorporating a scaling factor.
HOW MIGHT THIS CHANGE DRUG DISCOVERY, DEVELOPMENT, AND/OR THERAPEUTICS
☑ It demonstrates an approach allowing us to build upon the translation of in vitro OATP uptake transporter data to in vivo for pravastatin. It invites additional studies on more OATP substrates, with a hope of eventually utilizing the in vitro data for the prospective human PK prediction.
Active hepatic uptake is a critical step in drug disposition that may impact the pharmacokinetic (PK) properties of certain drugs. As illustrated extensively by examples of statin family, it is important to understand the role of the active hepatic uptake in drug clearance and its subsequent impact on drug disposition in order to understand the relationship between the intracellular concentration and observed efficacy.1 Organic anion‐transporting polypeptides (OATPs) are well known hepatic uptake transporters, and many statins are known to be substrates of these transporters.
Pravastatin is known to be a hepatic OATP uptake transporter substrate. Based on the human mass balance study after i.v. administration, 60% of the total dose of pravastatin was found in the urine and 34% was recovered in the feces (mainly through the biliary clearance).2 The fraction of oral dose of pravastatin absorbed is 34% and absolute bioavailability is 17%.2 There is a small amount of metabolism mediated by P450 enzymes, and their contribution to total clearance is minimal, such that cytochrome P450 inhibitory drug interactions have no real effect on the PK of pravastatin.3 Therefore, it is believed that the active hepatic uptake process plays an important role in pravastatin's clearance. The in vitro studies suggest that the hepatic uptake of pravastatin is mainly mediated by OATP1B1.4
The physiologically‐based pharmacokinetic (PBPK) approach has demonstrated its value in predicting the dynamic concentration‐time profile in the blood and tissues by combining the mechanistic understanding of the active hepatic uptake with the physiological, genetic, and demographic information. More importantly, PBPK provides a platform that allows testing of mechanistic hypotheses with respect to tissue concentrations in order to build pharmacokinetic/pharmacodynamic relationships for the active uptake substrates.5, 6 The Simcyp ® population‐based ADME simulator is one of the commercial PBPK software available to facilitate the PK profile prediction for compounds, such as pravastatin, that has PK disposition involving the active hepatic uptake transporters.7 The PBPK models of pravastatin have previously been developed by various groups5, 7, 8, 9 in order to predict the PK and transported mediated drug‐drug interactions. Inclusion of OATP transporter kinetic parameters is critical in predicting the PK profile of pravastatin. In the Simcyp default compound profile, in order to describe the clinical data, the OATP Clint,T input of hepatic uptake parameter for pravastatin was back calculated by fitting to the observed in vivo PK data. In the other studies,5, 7, 8, 9 the in vitro OATP Clint,T data generated in the suspended hepatocytes and the sandwich cultured hepatocytes were used. Nevertheless, when the in vitro data were incorporated into the PBPK model, a scaling factor was found to be necessary in order to predict the observed pravastatin PK profile accurately. It is evident that a different scaling factor is needed for different OATP substrates despite the in vitro data being generated in the same laboratory.8 This phenomenon demonstrates an important hurdle to the utilization of the in vitro data for prospective PK prediction of a new chemical entity (NCE) that is a substrate of OATP by PBPK modeling.
One of the approaches to overcome this challenge is to develop an in vitro model that can directly provide quantitative and translatable kinetic parameters for OATP substrates. Plateable human hepatocytes with human plasma are proposed as a promising in vitro tool for OATP transporter uptake assessment. Compared with the overexpressing cell lines, human hepatocytes provide an intact cell environment with relatively correct expression level of transporters, which mimics the physiological situation. In addition, compared to the medium without human plasma, the generated parameters in this system could inherently account for the in vivo impact of the binding activity on the active uptake process, including the plasma protein binding effect and potential interaction between plasma proteins and transporters. Ideally, if the in vitro generated active uptake parameters can successfully describe the PK of various OATP substrates when incorporated into the PBPK models, there will be higher confidence to utilize the in vitro data for prospective PK prediction for NCE as an OATP substrate.
In the present work, we applied the PBPK model that incorporates the hepatic uptake transporter data generated in the developed in vitro system (plateable human hepatocytes with human plasma) to directly predict the PK of pravastatin without applying a scaling factor. The performance of the PBPK model was assessed by simulating the i.v. PK of pravastatin and further verified by simulating the oral PK at different doses. Pravastatin was selected for this work because it has been studied by several laboratories using transporter data generated from different in vitro systems or by fitting of in vivo clinical data. The aim of this current work is to present an approach that could potentially improve our understanding of the translation of in vitro OATP transporter data to in vivo disposition. The limitations of the work and future studies will be discussed.
MATERIALS AND METHODS
Material
Pravastatin was purchased from Cayman Chemical Company (Ann Harbor, MI), and the rifampicin and tolbutamide were obtained from Sigma‐Aldrich (St. Louis, MO). Cryopreserved human hepatocytes and collagen coated 24‐well plates (CellAffix™) were obtained from A.P. Sciences (Columbia, MD). Universal cryopreserved cell recovery medium (UCRM™), hepatocyte induction medium (HIM™), modified human plasma medium (100% human plasma) (HPZ‐A™), hepatocyte rinse medium (HRM™), and hepatocyte incubation medium (HQM™) were obtained from In Vitro ADMET Laboratories (IVAL, Columbia, MD).
Active hepatic uptake transporter studies in plateable human hepatocytes with human plasma
Cryopreserved plateable human hepatocytes from one donor (lot #HH1057), a 33‐year‐old caucasian female, were thawed, and recovered using universal cryopreserved cell recovery medium. The cells were centrifuged at 100 × g for 10 minutes and, after viability determination by the trypan dye exclusion method, the cells were seeded in a collagen coated 24‐well plate at a density of 350,000 cells/well (500 µL of 0.7 × 106 cells/mL cell suspension) using hepatocyte induction medium. The cells were incubated in a humidified 95% balanced air/5% CO2 atmosphere in a 37°C incubator for 4 hours to facilitate the attachment. Following the attachment, the media was removed and the monolayer was washed to remove any unattached cells. Pravastatin at 10 concentrations (400, 300, 200, 100, 33.3, 11.1, 3.7, 1.2, 0.4, and 0.14 µM) was tested in modified human plasma medium, either alone or in the presence of uptake transporter inhibitor rifampicin at 125 µM. The uptake was initiated by the addition of pravastatin either alone or in combination with rifampicin. The reactions were terminated at 0 minutes and 3 minutes by washing the cells 4 times with ice‐cold rinse media‐HRM™ and 3 times with ice‐cold incubation media‐HQM. The incubation time of 3 minutes was selected based on the linear increase of the active uptake without the interference of passive uptake process. After rinse, the cells were lysed in HQM:organic mixture (acetonitrile and methanol in 3:1 ratio) containing internal standard (10 nM tolbutamide), centrifuged, and the supernatants were subjected to liquid chromatography tandem mass spectrometry analysis.
Bioanalysis of pravastatin in plated human hepatocytes
The liquid chromatography tandem mass spectrometry analysis was carried out with a Kinetex C18 column (Phenomenex, Torrance, CA) particle size 2.6 µM, 50 mm × 3 mm using the Acquity UPLC system coupled with an Applied Biosystems API5000 mass spectrometer. The separation was achieved by using a gradient of 0.1% formic acid in water (A) and 0.1% formic acid in acetonitrile (B) as mobile phases. The total mobile phase was delivered at 0.6 mL/min according to an elution program that started at 5% of B for 0.37 minutes and ramped to 95% of B over 1.13 minutes. The percentage of B was held at 95% for 0.2 minutes and returned to the initial 5% of B at 0.1 minutes. The column was allowed to re‐equilibrate for 1.3 minutes before the next injection.
The samples were analyzed in the negative ionization mode. The m/z for pravastatin and tolbutamide were 423.192 → 321.10 and 269.08 → 170.00, respectively. The lower and upper limits of quantification were 0.23 nM and 2 µM of pravastatin. Declustering potential was −160 V and collision energy was −22 V for pravastatin. Declustering potential was −115 V and collision energy was −26 V for the IS.
Modeling of the active hepatic uptake transporter kinetic parameters Jmax and Km of pravastatin
The following parameters were calculated using the measured pravastatin concentration.
Absolute intracellular pravastatin concentration
Mean of intracellular concentration at time 0 minutes for each tested concentration (A) was calculated, which was then subtracted from the corresponding measurement (B) at 3 minutes (X = B‐A). Mean of intracellular concentration in the presence of rifampicin at time 0 minutes for each tested concentration (C) was calculated, which was then subtracted from the corresponding measurement (D) at 3 minutes (Y = D‐C).
Net pravastatin uptake calculation: N = X‐Y
The calculated N (converted in the unit as pmol/min/million cells) and nominal concentrations were used for fitting by the Michaelis‐Menten kinetic model (GraphPad Prism 7), and the Jmax and the apparent Km were estimated.
Clinical pharmacokinetic data
A total of nine PK profiles of pravastatin from a literature search were identified and all utilized in the current PBPK model development and verification. Only one i.v. study was reported in the literature.2 Eight single oral dose PKs with different doses were reported in the literature: 0.0372 mg,10 19.2 mg,2 20 mg,11, 12 40 mg,13, 14, 15 and 60 mg.16 Clinical studies used for the PBPK model development: i.v. dose of 9.4 mg2 and oral dose of 40 mg13, 14, 15; clinical studies used for the PBPK model verification: oral dose of 0.0372 mg,10 19.2 mg,2 20 mg,11, 12 and 60 mg.16
Pravastatin PBPK model
Simcyp, version 15 (Certara, Princeton, NJ) PBPK model for pravastatin was used in this investigation. The input parameters for pravastatin model are summarized in Table 1. Information that has been previously disclosed in the Simcyp compound profile of pravastatin is indicated in Table 1. The basic model structure and details of model modification are described below.
Table 1.
Physiologically‐based pharmacokinetic model input parameters for pravastatin
| Input parameters | Value | Source | Input parameters | Value | Source |
|---|---|---|---|---|---|
| Physicochemical properties | Transporter | ||||
| MW | 424.5 | Default | Intestine | ||
| LogP | 2.2 | Default | CLint, MRP2 (µL/min/106 cells) | 1.2 | Default |
| Compound type | Monoprotic acid | Default | Scaling factor | 0.18 | Default |
| pKa | 4.55 | Default | Liver | ||
| Fraction unbound | 0.485 | Default | CLPD (mL/min/106 cells) | 0.0001 | Default |
| Blood/plasma ratio | 0.556 | Default |
Jmax, OATP
(pmol/min/106 cells) |
134.4 | Current investigation |
| Absorption | Km, OATP (uM) | 76.77 | Current investigation | ||
| Model | ADAM | fu,inc | 0.485 | Same as fu,p | |
| Intrinsic transcellular permeability Ptran,0 (10−6 cm/s) | 100 | Default model fitted as 800 | CLint, MRP2 (µL/min/106 cells) | 1.2 | Default |
| Input form | Solution | Scaling factor | 0.18 | Default | |
| Distribution | Kidney (OAT3/MATEs) | ||||
| Model | Full PBPK |
CLint,T (µL/min/106 cells) |
180/180 | Default | |
| Kp scalar | 1 | Scaling factor | 1/1 | Default | |
| Elimination | |||||
| Additional HLM CLint (µL/min/mg protein) | 4 | Default |
ADAM, advanced dissolution, absorption, and metabolism; CLint, intrinsic clearance; CLint,T, transporter intrinsic clearance; CLPD, passive diffusion clearance; fu,inc, unbound fraction in the incubation; fu,p, unbound fraction in human plasma; HLM, human liver microsome; Jmax, maximum rate of active uptake; Km, substrate concentration at 50% of maximum rate of active uptake; LogP, partition coefficient between aqueous and lipophilic phase; MATE, multidrug and toxin extrusion; MRP, multidrug resistance protein; MW, molecular weight; OATP, organic anion‐transporting polypeptide; OAT3, organic anion transporter‐3; PBPK, physiologically‐based pharmacokinetic.
Distribution
In the default compound profile, full PBPK with perfusion limited distribution model was used. The volume of distribution at steady state was calculated by the tissue partition coefficient using the equation described by the Rodgers et al.17, 18 and the Rodgers & Rowland19, 20 methods considering the rapid equilibrium between blood and tissues.
Clearance
In the default compound profile, the total clearance is assigned to the biliary clearance (40%), renal clearance (47%), and the metabolism clearance (13%) based on the clinical data and mass balance study data.2 The biliary clearance comprises (1) passive diffusion and OATP hepatic uptake transporter on the sinusoidal membrane and (2) multiple drug resistance‐associated protein 2 (MRP2) efflux transporter on the canalicular membrane.21, 22, 23 For the current investigation, the kinetic parameters of OATP uptake transporter generated in the plateable human hepatocytes with human plasma were used to replace the default model input in the model. Because the incubation medium is 100% human plasma, the fu,inc was assumed to be the same as the fu,p 0.485 in the model input. As the in vitro study is performed in human hepatocytes, it is challenging to distinguish the contribution between OATP1B1 and OATP1B3 on pravastatin. Therefore, the kinetic parameters are assigned to the OATP1B1 because this is the dominant transporter.4 No additional scaling factor was applied for the prediction of uptake by the OATP transporter. The input for passive diffusion is 0.0001 mL/min/106 cells determined in sandwich culture hepatocytes.8 The canalicular efflux by MRP2 was described using a measured Clint,T (1.2 µL/min/106 cells) in sandwich culture hepatocytes8 with a scaling factor of 0.18.9
The mechanistic kidney model was used to recover the observed renal clearance (26.6 L/h). The basal uptake was assigned to OAT324, 25 and MATE (used as surrogate for an unidentified renal apical efflux transporter) with Clint,T of 180 µL/min/106 cells for each transporter obtained from model fitting to the clinical PK data. An input of human liver microsome clearance (4 µL/min/mg protein) was used in the model as back calculation to account for the 13% metabolism clearance.
Absorption
In the default Simcyp compound profile, the absorption was predicted using the advanced dissolution, absorption, and metabolism (ADAM) model with pravastatin administered as a solution. A Clint,T (1.2 µL/min/cm2) was used for the intestinal MRP2 with the scaling factor of 0.18.8 The MechPeff model was used to predict the permeability.26 The value of intrinsic transcellular permeability is used for Peff,man prediction for the regions of the intestine, and it was updated in the current investigation as 100 × 10−6 cm/s in order to reflect the reported bioavailability (observed Fa and F as 0.34 and 0.17, and predicted Fa and F as 0.26 and 0.23, respectively).
PBPK model simulation
The prediction of distribution and clearance of pravastatin was evaluated by simulating the i.v. PK profile of pravastatin.2 Following the reported clinical study design, a single dose of 9.4 mg pravastatin was given as i.v. bolus over 2 minutes to healthy subjects with a body weight of 70 kg, age ranging from 21 to 39 years, and 50% as women. The simulation was conducted in a virtual population consisting of 10 trials of 8 individuals. The prediction of absorption of pravastatin was assessed by simulating the oral PK profile of pravastatin. The PK profiles of the 40 mg13, 14, 15 dose group were chosen because the reported concentration‐time profile covers a longer time (up to 12 hours) in comparison with other doses.
The model was further verified with additional oral PK data from doses of 0.0372 mg,10 19.2 mg,2 20 mg,11, 12 and 60 mg.16 The simulation of oral PK was conducted using virtual population of healthy subjects (10 trials of 23 individuals per trial) with body weight of 70 kg, age ranging from 18 to 25 years, with 53% women.
RESULTS
Active hepatic uptake of pravastatin by OATP transporter in plateable human hepatocytes with human plasma
Considering the plasma protein binding (fu,p 0.485) of pravastatin, to obtain the full saturation kinetic curve in plateable human hepatocytes with human plasma, a wide range of pravastatin concentration, ranging from 0.14–400 µM, were tested. In order to examine the active uptake process more closely, a high concentration of rifampicin (125 µM) was used in the current study with the intention to inhibit the OATP uptake process. The concentration of pravastatin associated with the cell was measured and the absolute intracellular pravastatin concentration was calculated (details described in the Methods section).The net active uptake of pravastatin is extracted from the incubation without and with rifampicin at 0 and 3 minutes across the tested concentrations of pravastatin. The Jmax and the apparent Km for pravastatin were determined as 134.4 ± 14.3 pmol/min/million cells and 76.77 ± 27.4 µM, respectively (Figure 1).
Figure 1.
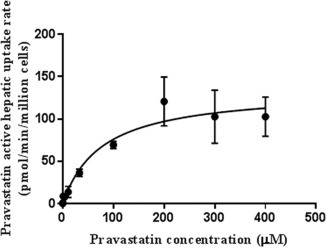
The rate of the hepatic active uptake against concentration of pravastatin in plateable human hepatocytes with human plasma (N = 3).
Prediction of pravastatin PK using the PBPK model
When the in vitro OATP transporter kinetic data were incorporated into the PBPK model, the model simulation captured the observed i.v. PK profile adequately (Figure 2 a), without applying any scaling factor on the measured kinetic data from the plateable human hepatocytes with human plasma. The predicted PK profile by the current model captured the shape of the observed PK profile well, particularly on the third distribution phase. The mean predicted pravastatin AUC (0.60 µM.hr) using the current model directly incorporating the in vitro kinetic data is within 1.5‐fold of that predicted (0.43 µM.hr) using the default input of Clint,T that was back calculated by fitting to the observed in vivo PK data (0.4 µM.hr; Figure 2 b).
Figure 2.
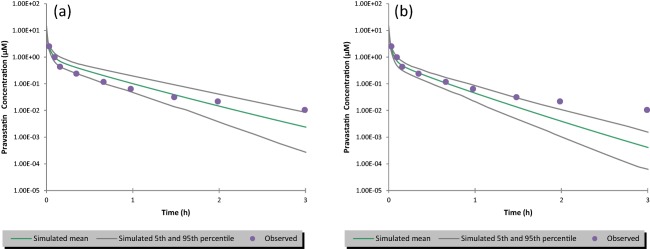
Simulated vs. observed plasma concentration‐time profiles of pravastatin after an i.v. bolus dose of 9.4 mg over 2 minutes. The dots are the observed clinical data. The simulated mean is the mean of 80 individuals (10 trials of 8 subjects per trial); the 5th and 95th percentile for 80 individuals simulated represents the 5th and 95th highest concentrations from the ranked concentrations. (a) The simulation using the parameters listed in Table 1; (b) the simulation using the default profile in Simcyp, the input is the same as listed in Table 1 except the Clint,T for organic anion‐transporting polypeptide (OATP)1B1 and OATP1B3 was set to be 14.057 and 1.343 uL/min/million cells.
When verifying the model's performance in simulating oral PK for pravastatin, the value of clearance and volume of distribution at steady state defined using the i.v. model was kept unchanged. The default model overpredicted Fa (0.76) and F (0.67). By modifying the intrinsic transcellular permeability, the model simulation provided values of Fa (0.26) and F (0.23) that closest represent the reported clinical data, Fa (34%) and F (18%).2 Thereafter, the PBPK model simulation captured the oral pravastatin PK profile from 0.0372 mg up to 60 mg doses of pravastatin (Figure 3 a‐e). The predicted and observed peak plasma concentration (Cmax) and AUC are shown in Supplementary Table S2. Although the model predicted mean Cmax and AUC are within twofold of the observed data, a trend of underprediction of Cmax was observed. Further refinement of the absorption model and/or investigation of variability between predicted and observed data are necessary when more clinical data become available.
Figure 3.
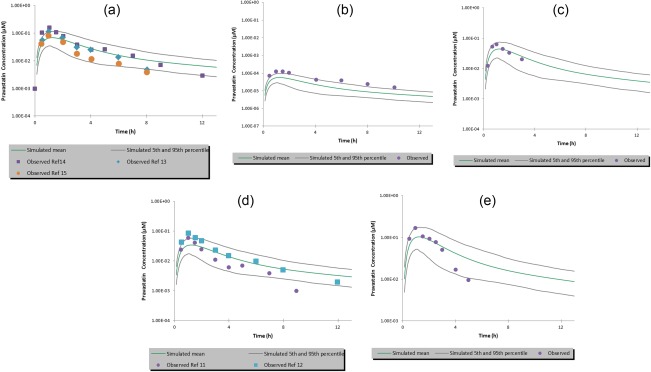
Simulated vs. observed plasma concentration‐time profiles of pravastatin after a single oral dose of (a) 40 mg; (b) 0.0372 mg; (c) 18.23 mg; (d) 20 mg, and (e) 60 mg. The dots are the observed clinical data. The simulated mean is the mean of 230 individuals (10 trials of 23 subjects per trial); the 5th and 95th percentiles for 230 individuals simulated represents the 5th and 95th highest concentrations from the ranked concentrations. The input parameters used in the simulation are listed in Table 1.
DISCUSSION
Pravastatin is well known as an in vivo OATP substrate, and there are several pioneer investigations on the PBPK work for pravastatin.5, 7, 8, 9 The platform provided by a software company, such as Simcyp, provided an opportunity for researchers to test and generate hypothesis via simulations as well as to further improve models as science evolves. In a recently published Simcyp compound profile7, the pravastatin PBPK model was built using a combination of the top‐down and bottom‐up approach. To capture the observed pravastatin PK profile, the Clint,T describing the OATP hepatic uptake were obtained through back calculation from in vivo data. For the other investigations, different in vitro assays have been explored to quantitatively assess the OATP uptake process for pravastatin. In the human suspended hepatocytes with an oil‐spin method, the Clint,T was determined as 4.5 and 0.7 µL/min/million cells at 1 µM and 100 µM of pravastatin.5 The scaling factor obtained in rats between in vitro hepatic uptake and in vivo observation was applied to the model in order to predict the observed human PK. In the sandwich culture hepatocytes, the Clint,T was determined to be 1.9 µL/min/million cell at 1 µM of pravastatin.8 When these in vitro data were directly used in the PBPK model, it failed to capture the curve of the time concentration profile of pravastatin administered intravenously.8, 9 Therefore, a scaling factor was applied in the model in order to fit the observed data. In addition, when multiple OATP substrates were tested in the same in vitro system, different scaling factors were reported for each substrate. It seems to be challenging to use such information for the prospective prediction of hepatic uptake by OATP for a NCE. Currently, a mean of the scaling factors were recommended to apply for an NCE, and the prediction outcome using a mean scaling factor is deemed to be further investigated.
Although overprediction of pravastatin concentration is the main observation reported previously when in vitro data were incorporated in a PBPK model without use of a scaling factor,8, 9 the current PBPK model with the hepatic uptake kinetic data generated from plateable human hepatocytes with human plasma is able to reasonably predict pravastatin PK without applying any scaling factor. In our approach, a systematic investigation of optimal experiment conditions for plateable human hepatocytes with human plasma assay was conducted before finalizing the study design. This optimization includes: the duration for the cell attachment, the uptake assay time point, the inclusion of the rifampicin, the final wash medium, and evaluation of the usefulness of monitoring medium concentration. Although there is no clear guidance on the uptake rate required, the initial screen and selection of the hepatocyte lot is critical for success. The expression level of OATP1B1 and OATP1B3 (18.3 and 1.7 fmol/million cells) in the hepatocyte lot used in the study is within the range of the literature reported level (3.4–23.2 for OATP1B1 and 1.5–6.5 for OATP1B3).27, 28, 29, 30 Because pravastatin undergoes only minimal metabolism and the incubation time was short, metabolism in hepatocyte is likely not an issue in this study. However, for drugs with considerable metabolism, a longer incubation time may be needed in order to monitor metabolite formation in addition to a short time point to characterize the active uptake process. Both parent and metabolite information needs to be integrated into the PBPK model. In the current investigation, instead of Clint,T, the hepatic uptake kinetic data, Jmax, and apparent Km (134.4 ± 14.34 pmol/min/million cells and 76.77 ± 27.43 µM, respectively) of pravastatin were characterized in plateable human hepatocytes with human plasma and used in the PBPK modeling. Sufficient concentrations of pravastatin were tested in vitro to allow generation of the reliable uptake kinetic parameters, and the inclusion of the OATP inhibitor rifampicin enables the in vitro data to reflect the active uptake process from the interference of the passive permeability. In general, the use of uptake transporter kinetic data, such as in vitro Km, enables the prediction of in vivo uptake clearance to be sensitive to the in vivo substrate concentration, which is more critical for substrates that have apparently nonlinear PK. This is important because the substrate concentration in hepatic inlet is the functional driver for the uptake transporters in the liver and this value usually changes dramatically with time. Moreover, when the in vitro transporter kinetic data were used for the PK prediction, the apparent Km was corrected with human plasma protein binding to combine with the free concentration as the functional systemic concentration utilized in the PBPK model. Having 100% human plasma as incubation medium may account for the in vivo impact of the human plasma protein on the active uptake process. This includes the impact of plasma protein binding on the active uptake parameter assessment, the potential interaction between plasma proteins and transporters at the functional level, and the interplay between the rate of the plasma protein binding and the rate of the active uptake process. The inclusion of human plasma in the incubation may benefit more the substrates with the high plasma protein binding by limiting the introduction of uncertainty associated with binding into the model prediction. Considering that pravastatin is not highly bound to the human plasma proteins (fu,p = 0.485), we also generated in vitro uptake kinetic data in plated human hepatocyte with the induction medium (not human plasma) and expected a similar prediction on pravastatin PK. Indeed, when the in vitro data (Jmax 28.49 ± 3.11 pmol/min/million cells and Km 11.72 ± 6.83 µM) corrected for hepatocyte binding of pravastatin (fu,inc 0.96)31 was incorporated into the same model, the simulations were able to capture the observed pravastatin PK without applying scaling factors (Supplementary Figure S1).
Although the current findings are encouraging, there are limitations and future studies are necessary in order to apply the approach broadly to all OATP substrates. First, it is known that the in vitro transporter data generated from different laboratories could be substantially different due to hepatocyte lots and experiment conditions used. Therefore, caution should be used when applying this in vitro approach across different laboratories. To increase the confidence in the prospective prediction, the in vitro‐in vivo translational relationship should be assessed and established under each laboratory's condition using a relevant known transporter substrate. It is anticipated that the relationship established could be different from one laboratory to another. Second, to further demonstrate the superiority of plasma medium over the traditional medium for the quantitative prediction, investigation using the same approach with more OATP substrates, which have different extent of plasma protein binding, is needed. Third, to broadly apply the current approach for prospective prediction, the investigation of the PBPK model's performance in predicting OATP transporter‐mediated drug‐drug interactions (Supplementary Table S3) using transporter inhibition data generated from plateable human hepatocytes with human plasma and the performance in simulating the pravastatin PK for clinical genetic polymorphism scenarios using the uptake data generated in overexpressed cell lines for OATP1B1 and OATP1B3 are necessary. All these could further improve our understanding of the in vitro to in vivo translation. Furthermore, a better understanding of other model parameters is also important for overall accurate prediction of PK for the OATP transporter substrates. The quantitative translation of in vitro transporter data variability to the predicted in vivo transporter clearance needs to be further studied once more clinical data become available.
It is fully recognized that even though the complete mechanism is yet to be understood, the present study result with pravastatin provides a first step toward bridging the gaps to the unknown. With more OATP substrates tested in this same way, the approach will allow us to build upon the translation of in vitro OATP uptake transporter data to in vivo disposition with a hope of utilizing the in vitro data for the prospective human PK prediction.
In conclusion, the OATP active hepatic uptake kinetic data generated in plateable human hepatocytes with human plasma enabled successful PBPK model simulation of pravastatin PK profile without applying a scaling factor.
Source of Funding
This study was supported by Genentech, A Member of the Roche Group.
Conflict of Interest
J.M., M.W., C.E.C.A.H., and Y.C. are employees of Genentech, a member of the Roche Group and also hold stock/shares in Genentech, a member of the Roche Group. U.D. was an employee of Vitro ADMET Laboratories Inc. A.P.L. is an employee of In Vitro ADMET Laboratories and holds shares in In Vitro ADMET Laboratories.
Author Contributions
J.M., U.D., M.W., C.E.C.A.H., A.P.L., and Y.C. wrote the manuscript. J.M., U.D., A.P.L., and Y.C. designed the research. J.M. and U.D. performed the research. J.M. and U.D. analyzed the data.
Supporting information
Supplementary Methods and Discussion
Supplementary Table S1 Key parameters in different PBPK models published in the literature
Supplementary Table S2 Observed and predicted AUC and Cmax of pravastatin
Supplementary Table S3 Observed and Predicted AUC ratio of pravastatin in the presence of OATP 1B1/3 inhibitor rifampicin and gemfibrozil
Supplementary Figure S1 The rate of the hepatic active uptake against concentration for pravastatin in plateable human hepatocytes with the induction medium (N = 3).
Supplementary Figure S2 Simulated vs. observed plasma concentration‐time profiles of pravastatin after an i.v. bolus dose of 9.4 mg over 2 minutes.
Supplementary Figure S3 Simulated vs. observed plasma concentration‐time profiles of pravastatin after a single oral dose of (a) 40 mg; (b) 0.0372 mg; (c) 18.23 mg; (d) 20 mg, and (e) 60 mg.
Acknowledgments
The authors thank Buyun Chen and Liling Liu for the OATP quantification, and Laurent Salphati, Eugene Chen, and Xingrong Liu for their review and helpful suggestions.
References
- 1. Shitara, Y. & Sugiyama, Y. Pharmacokinetic and pharmacodynamic alterations of 3‐hydroxy‐3‐methylglutaryl coenzyme A (HMG‐CoA) reductase inhibitors: drug‐drug interactions and interindividual differences in transporter and metabolic enzyme functions. Pharmacol. Ther. 112, 71–105 (2006). [DOI] [PubMed] [Google Scholar]
- 2. Singhvi, S.M. , Pan, H.Y. , Morrison, R.A. & Willard, D.A. Disposition of pravastatin sodium, a tissue‐selective HMG‐CoA reductase inhibitor, in healthy subjects. Br. J. Clin. Pharmacol. 29, 239–243 (1990). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3. Hatanaka, T. Clinical pharmacokinetics of pravastatin: mechanisms of pharmacokinetic events. Clin. Pharmacokinet. 39, 397–412 (2000). [DOI] [PubMed] [Google Scholar]
- 4. Nakai, D. et al Human liver‐specific organic anion transporter, LST‐1, mediates uptake of pravastatin by human hepatocytes. J. Pharmacol. Exp. Ther. 297, 861–867 (2001). [PubMed] [Google Scholar]
- 5. Watanabe, T. , Kusuhara, H. , Maeda, K. , Shitara, Y. & Sugiyama, Y. Physiologically based pharmacokinetic modeling to predict transporter‐mediated clearance and distribution of pravastatin in humans. J. Pharmacol. Exp. Ther. 328, 652–662 (2009). [DOI] [PubMed] [Google Scholar]
- 6. Watanabe, T. et al Investigation of the rate‐determining process in the hepatic elimination of HMG‐CoA reductase inhibitors in rats and humans. Drug Metab. Dispos. 38, 215–222 (2010). [DOI] [PubMed] [Google Scholar]
- 7.Simcyp. Summary of Simcyp compound profile for pravastatin (2016).
- 8. Jones, H.M. et al Mechanistic pharmacokinetic modeling for the prediction of transporter‐mediated disposition in humans from sandwich culture human hepatocyte data. Drug Metab. Dispos. 40, 1007–1017 (2012). [DOI] [PubMed] [Google Scholar]
- 9. Varma, M.V. et al Physiologically based modeling of pravastatin transporter‐mediated hepatobiliary disposition and drug‐drug interactions. Pharm. Res. 29, 2860–2873 (2012). [DOI] [PubMed] [Google Scholar]
- 10. Maeda, K. et al Identification of the rate‐determining process in the hepatic clearance of atorvastatin in a clinical cassette microdosing study. Clin . Pharmacol. Ther. 90, 575–581 (2011). [DOI] [PubMed] [Google Scholar]
- 11. Pan, H.Y. et al Pharmacokinetic interaction between propranolol and the HMG‐CoA reductase inhibitors pravastatin and lovastatin. Br. J. Clin. Pharmacol. 31, 665–670 (1991). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12. Keskitalo, J.E. et al No significant effect of ABCB1 haplotypes on the pharmacokinetics of fluvastatin, pravastatin, lovastatin, and rosuvastatin. Br. J. Clin. Pharmacol. 68, 207–213 (2009). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13. Neuvonen, P.J. , Kantola, T. & Kivisto, K.T. Simvastatin but not pravastatin is very susceptible to interaction with the CYP3A4 inhibitor itraconazole. Clin. Pharmacol. Ther. 63, 332–341 (1998). [DOI] [PubMed] [Google Scholar]
- 14. Kantola, T. , Backman, J.T. , Niemi, M. , Kivisto, K.T. & Neuvonen, P.J. Effect of fluconazole on plasma fluvastatin and pravastatin concentrations. Eur. J. Clin. Pharmacol. 56, 225–229 (2000). [DOI] [PubMed] [Google Scholar]
- 15. Aberg, J.A. et al Pharmacokinetic interaction between nelfinavir and pravastatin in HIV‐seronegative volunteers: ACTG Study A5108. AIDS 20, 725–729 (2006). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16. Igel, M. et al Impact of the SLCO1B1 polymorphism on the pharmacokinetics and lipid‐lowering efficacy of multiple‐dose pravastatin. Clin. Pharmacol. Ther. 79, 419–426 (2006). [DOI] [PubMed] [Google Scholar]
- 17. Rodgers, T. , Leahy, D. & Rowland, M. Physiologically based pharmacokinetic modeling 1: predicting the tissue distribution of moderate‐to‐strong bases. J. Pharm. Sci. 94, 1259–1276 (2005). [DOI] [PubMed] [Google Scholar]
- 18. Rodgers, T. , Leahy, D. & Rowland, M. Tissue distribution of basic drugs: accounting for enantiomeric, compound and regional differences amongst beta‐blocking drugs in rat. J. Pharm. Sci. 94, 1237–1248 (2005). [DOI] [PubMed] [Google Scholar]
- 19. Rodgers, T. & Rowland, M. Physiologically based pharmacokinetic modelling 2: predicting the tissue distribution of acids, very weak bases, neutrals and zwitterions. J. Pharm. Sci. 95, 1238–1257 (2006). [DOI] [PubMed] [Google Scholar]
- 20. Rodgers, T. & Rowland, M. Mechanistic approaches to volume of distribution predictions: understanding the processes. Pharm. Res. 24, 918–933 (2007). [DOI] [PubMed] [Google Scholar]
- 21. Yamazaki, M. , Kobayashi, K. & Sugiyama, Y. Primary active transport of pravastatin across the liver canalicular membrane in normal and mutant Eisai hyperbilirubinaemic rats. Biopharm. Drug Dispos. 17, 645–659 (1996). [DOI] [PubMed] [Google Scholar]
- 22. Yamazaki, M. , Akiyama, S. , Ni'inuma, K. , Nishigaki, R. & Sugiyama, Y. Biliary excretion of pravastatin in rats: contribution of the excretion pathway mediated by canalicular multispecific organic anion transporter. Drug Metab. Dispos. 25, 1123–1129 (1997). [PubMed] [Google Scholar]
- 23. Niinuma, K. et al Primary active transport of organic anions on bile canalicular membrane in humans. Am. J. Physiol. 276, G1153–G1164 (1999). [DOI] [PubMed] [Google Scholar]
- 24. Takeda, M. et al Evidence for a role of human organic anion transporters in the muscular side effects of HMG‐CoA reductase inhibitors. Eur. J. Pharmacol. 483, 133–138 (2004). [DOI] [PubMed] [Google Scholar]
- 25. Nakagomi‐Hagihara, R. , Nakai, D. & Tokui, T. Inhibition of human organic anion transporter 3 mediated pravastatin transport by gemfibrozil and the metabolites in humans. Xenobiotica 37, 416–426 (2007). [DOI] [PubMed] [Google Scholar]
- 26. Pade, D. , Jamei, M. , Rostami‐Hodjegan, A. & Turner, D.B. Application of the MechPeff model to predict passive effective intestinal permeability in the different regions of the rodent small intestine and colon. Biopharm. Drug Dispos. 38, 94–114 (2017). [DOI] [PubMed] [Google Scholar]
- 27. Bi, Y.A. et al In vitro evaluation of hepatic transporter‐mediated clinical drug‐drug interactions: hepatocyte model optimization and retrospective investigation. Drug Metab. Dispos. 40, 1085–1092 (2012). [DOI] [PubMed] [Google Scholar]
- 28. Badee, J. , Achour, B. , Rostami‐Hodjegan, A. & Galetin, A. Meta‐analysis of expression of hepatic organic anion‐transporting polypeptide (OATP) transporters in cellular systems relative to human liver tissue. Drug Metab. Dispos. 43, 424–432 (2015). [DOI] [PubMed] [Google Scholar]
- 29. Vildhede, A. et al Hepatic uptake of atorvastatin: influence of variability in transporter expression on uptake clearance and drug‐drug interactions. Drug Metab. Dispos. 42, 1210–1218 (2014). [DOI] [PubMed] [Google Scholar]
- 30. Kimoto, E. et al Characterization of organic anion transporting polypeptide (OATP) expression and its functional contribution to the uptake of substrates in human hepatocytes. Mol. Pharm. 9, 3535–3542 (2012). [DOI] [PubMed] [Google Scholar]
- 31. Menochet, K. , Kenworthy, K.E. , Houston, J.B. & Galetin, A. Use of mechanistic modeling to assess interindividual variability and interspecies differences in active uptake in human and rat hepatocytes. Drug Metab. Dispos. 40, 1744–1756 (2012). [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Supplementary Methods and Discussion
Supplementary Table S1 Key parameters in different PBPK models published in the literature
Supplementary Table S2 Observed and predicted AUC and Cmax of pravastatin
Supplementary Table S3 Observed and Predicted AUC ratio of pravastatin in the presence of OATP 1B1/3 inhibitor rifampicin and gemfibrozil
Supplementary Figure S1 The rate of the hepatic active uptake against concentration for pravastatin in plateable human hepatocytes with the induction medium (N = 3).
Supplementary Figure S2 Simulated vs. observed plasma concentration‐time profiles of pravastatin after an i.v. bolus dose of 9.4 mg over 2 minutes.
Supplementary Figure S3 Simulated vs. observed plasma concentration‐time profiles of pravastatin after a single oral dose of (a) 40 mg; (b) 0.0372 mg; (c) 18.23 mg; (d) 20 mg, and (e) 60 mg.