

Abstract
The modification of synaptic strength produced by long-term potentiation (LTP) is widely thought to underlie memory storage. Indeed, given that hippocampal pyramidal neurons have > 10,000 independently modifiable synapses, the potential for information storage by synaptic modification is enormous. However, recent work suggests that CREB-mediated global changes in neuronal excitability also play a critical role in memory formation. Because these global changes have a modest capacity for information storage compared with that of synaptic plasticity, their importance for memory function has been unclear. Here we review the newly emerging evidence for CREB-dependent control of excitability and discuss two possible mechanisms. First, the CREB-dependent transient change in neuronal excitability performs a memory-allocation function ensuring that memory is stored in ways that facilitate effective linking of events with temporal proximity (hours). Second, these changes may promote cell-assembly formation during the memory-consolidation phase. It has been unclear whether such global excitability changes and local synaptic mechanisms are complementary. Here we argue that the two mechanisms can work together to promote useful memory function.
The elucidation of the molecular, cellular, and network mechanisms that underlie learning and memory has been a major goal of modern neuroscience. In an important early contribution, Donald Hebb proposed that the associations that constitute a memory are stored by means of activity-dependent changes in the strength of synapses1. Much subsequent work has shown that synapses in fact undergo activity-dependent strengthening as envisioned by Hebb, and do so via LTP (and the complementary long-term depression (LTD) process)2. In the canonical form of LTP found at CA1 hippocampal synapses, LTP induction depends on a particular type of glutamate receptor, NMDAR, and on a biochemical cascade initiated and sustained by the abundant synaptic protein calcium/calmodulin-dependent protein kinase II (CaMKII)3. Importantly, genetic modifications that interfere with NMDAR or CaMKII function not only block LTP, but also produce profound deficits in learning and memory storage4–6. Conversely, nearly all mutations that enhance memory also enhance LTP7. Other work has shown that LTP, once induced during learning7, can be bidi-rectionally modified by LTD/LTP-like stimulation, thereby leading to both reduction and re-emergence of memory-guided behavior8. Hippocampal pyramidal neurons have more than 10,000 synapses, and because each synapse can be independently modified by LTP9 (i.e., LTP is synapse specific), even a single neuron has an impressive information-storage capacity. Moreover, computational analysis shows that modification of synaptic strength by LTP is sufficient to produce distributed memory storage in neural networks10. Taken together, these findings have led to the widespread view that LTP mediates memory storage11.
There is, however, accumulating evidence that synapse-specific changes are not the only type of neuronal change necessary for memory functions. Notably, modification of global neuronal properties also has an important role in learning and memory. The evidence for such changes was initially obtained in invertebrate preparations used to study the presynaptic facilitation12 that underlies short-term behavioral sensitization. This facilitation involves an increase in presynaptic excitability caused by a reduction in K+ conductance13. Other work showed that conditioning of Hermissenda14 increased neuronal excitability by reducing K+ conductance. The investigation of learning-related changes in excitability was then extended to vertebrates15 and is now supported by multiple lines of evidence16–19. In this Perspective, we describe that evidence, as well as the critical role of the transcription factor CREB (cAMP-responsive element-binding protein) in this process. We then address the question of why vertebrate neurons that can store large amounts of information by modifying their numerous synapses also modify global cellular properties via transcriptional regulation. We describe two ideas about how synaptic and transcriptional modifications make different contributions necessary for the overall process of memory formation.
The role of the transcription factor CREB in memory
Early work in invertebrates pointed to the importance of transcriptional regulation in memory20. This led to interest in CREB because it undergoes phosphorylation-dependent activation that persists for hours in the vertebrate hippocampus after LTP induction21 and learning22. The importance of CREB for memory has now been demonstrated through bidirectional manipulation of CREB function23,24. Researchers have used a variety of methods to negatively modulate CREB, including the knockdown of CREB (specifically α/δ isoforms), antisense oligodeoxynucleotide-mediated CREB disruption, RNA interference, and targeted genetic mutation23,25–27. These manipulations invariably lead to memory deficits. Conversely, increases in levels of active CREB lead to memory enhancement28,29.
A second wave of progress in understanding CREB function arose from newly developed tools that allowed direct visualization and manipulation of the cells that mediate memory storage (‘memory trace’ cells). One of the resulting methods takes advantage of the fact that cells undergoing strong activity, as occurs during memory formation, synthesize elevated levels of a class of regulatory proteins referred to as immediate early genes (IEGs; for example, cFos and arc). These proteins have long been known to be expressed in cells activated during learning, and their expression can be used to identify memory trace cells30. Experiments showed that increasing the levels of CREB in a subset of cells increased the probability that those cells would be incorporated into the memory trace, whereas decreasing the levels of CREB had the opposite effect31,32. In trained animals, CREB-overexpressing cells have higher IEG expression than neighboring cells. Importantly, CREB-dependent increases in IEG expression do not occur in untrained mice31. These results demonstrate that relative CREB levels can affect which neurons are incorporated into a memory trace, a phenomenon referred to as memory allocation. Subsequent studies showed that inhibition of CREB-overexpressing cells negatively affects memory recall31,33–35, and thus demonstrated the necessity of these cells for memory retrieval.
Evidence that CREB modulates cell excitability
By what mechanism could CREB control memory allocation? Because LTP depends on the level of depolarization in the post-synaptic neurons, CREB might work by enhancing neuronal excitability and thereby increasing the incorporation of neurons into the memory trace. This possibility has now been tested in several ways. In one set of experiments, intracellular recordings were obtained from cells that overexpressed CREB. As shown in Fig. 1, the same magnitude of current pulse produced more action potentials in the CREB-overexpressing cells than in nearby neurons that did not overexpress CREB (also see refs.32,34,36,37). CREB overexpression also resulted in a smaller after-hyperpolarization (AHP) after a train of action potentials. Because such AHPs are generated by K+ channels38, it seems likely that the enhanced excitability of CREB-expressing cells is at least partly due to decreased K+ conductance. There may also be excitability changes that depend on changes in translation39, but these are outside the scope of this review because they do not involve CREB.
Fig. 1. CREB increases neuronal excitability.

a, Cultured hippocampal neurons were injected with a depolarizing current pulse. Cells transfected with CREB showed increased action potential firing compared with that in nontransfected cells. b–d, Acute lateral slices of rat amygdala were divided into three groups (HSV-CREB-transfected, HSV-LacZ-transfected, and nontransfected) and underwent whole-cell recordings 3 d after treatment. b, CREB-overexpressing neurons (transfected with HSV-CREB) fired more action potentials (right) than control neurons (CON; nontransfected and HSV-LacZ-transfected). c, Spike frequency adaptation was analyzed with a 400-pA, 600-ms current injection. Cells were classified as rapidly adapting (RA) if they fired between one and five spikes and then remained silent, or as slowly adapting (SA) if they fired six or more spikes. A greater fraction of HSV-CREB-transfected cells (compared with control cells) fired more than six times in response to current injection, which indicates that CREB reduces spike frequency adaptation and thus alters firing properties. d, Amplitude of post-burst AHP at the negative peak and 300 ms after current injection. There was no difference in amplitude at the negative peak, whereas at 300 ms, HSV-CREB cells showed a significant reduction in AHP amplitude (right; significant difference indicated by asterisk). All data are presented as mean ± s.e.m. *P < 0.05, unpaired t-test. Panel a reproduced with permission from ref. 32. Panels b and c reproduced with permission from ref. 34.
Another type of experiment was used to test directly whether manipulation of cell excitability is sufficient to affect a cell’s incorporation into the memory trace. In these studies, viral vectors were used to enhance excitability through reduction of K+ channel function (i.e., through expression of dominant-negative forms of two K+ channels involved in AHP: KCNQ2 and KCNQ332). Cells expressing mutant channels were indeed preferentially allocated to the memory trace, as indicated by increased levels of the IEG protein arc relative to those in neighboring uninfected neurons. In related experiments, cell excitability was reduced by the expression of Kir2.1, an inwardly rectifying K+ channel. Among Kir2.1 cells, the probability that cells were active was reduced approximately fivefold compared with cells that did not express the protein, and this led to decreased incorporation into the memory trace. Further experiments demonstrated the importance of excitability changes at the behavioral level: when a step function opsin was used to increase the excitability of a subset of amygdala neurons right before tone conditioning, subsequent behavioral experiments showed that these neurons were allocated to store the tone-shock association40.
Taken together, these results demonstrate that a major function of CREB is to enhance neuronal excitability41,42 and thereby modulate the allocation of neurons to the memory trace. This enhancement of excitability by strong neural activity stands in contrast to modifications of intrinsic and synaptic conductances that are homeostatic, that is, where strong neural activity leads to reduced excitability43. This raises the question of what function the enhancement of excitability by CREB might have. In neural network models, the enhancement of transmission by LTP is sufficient to produce memory function, so what does CREB-dependent enhancement of excitability add? One possibility is allocation, but what is the utility of allocation? These questions are addressed in the next section.
Functions of the cell-wide increase in excitability
Below, we first describe one hypothesis about the role of learning-dependent changes in global excitability that has substantial experimental support. We then put forward a second and more speculative possibility. These hypotheses are not mutually exclusive.
The allocate-to-link hypothesis
As described above, an increase in the amount of activated CREB enhances excitability and thereby biases neuron allocation into the memory trace. According to the ‘allocate-to-link’ hypothesis44, these changes form a linkage between memories of events that occur within hours of each other, and that linkage has an important function. As described above, an initial bout of learning leads to an increase in the amount of CREB in the memory-encoding neurons that lasts for hours. The resulting increase in excitability leads to the recruitment of many of these neurons to encode a new memory formed during the period of increased excitability. The net result is that two memories encoded close together in time are encoded by overlapping ensembles of neurons; thus, the two memories are linked, and that linkage may underlie the recall of separate events that occur during a several-hour period (Fig. 2a).
Fig. 2. Allocate-to-link hypothesis.

a, Memories that are encoded close in time are represented by overlapping neural populations as a result of learning-related increases in excitability45. This temporal window has been experimentally shown to last at least 5 h, but it is presumed that it can last as long as 1 d. This model provides a novel mechanism for temporal association and memory linking over time. b,c, Transfer of contextual fear provides support for temporal association via overlapping neural populations. b, Animals explored context A 7 d before context B, which was explored 5 h before context C. Calcium imaging data demonstrated greater overlap between neuronal ensembles activated during exploration of contexts B and C (5 h) than between those activated during exploration of contexts A and C (7 d). c, Transfer-of-fear experimental design. The context in which the mice were tested is outlined by a yellow rectangle and corresponds with the provided freezing assay data. There was little difference in freezing between contexts C and B, whereas there was significantly less freezing in D than in both C and B (data not shown). Imm, immediate; Cxt, context. Results are shown as mean ± s.e.m. **P < 0.01. Panel a reproduced with permission from ref. 33. Panels b and c reproduced with permission from ref. 45.
A recent study demonstrated that overlapping hippocampal neuronal ensembles do indeed capture memories of contexts explored close in time45. To directly determine whether overlapping cells encode the two contexts, the authors used a head-mounted miniature fluorescent microscope to monitor calcium transients within mouse hippocampal CA1 neurons as the mice explored different contexts. There was greater overlap between the neuronal ensembles activated by these contexts when the two contexts were explored within the same day (5 h apart) as opposed to on different days (7 d apart) (Fig. 2b). This provides direct support for the idea that overlapping neuronal ensembles encode memories formed close in time. A consequence of this neuronal overlap is that these memories become behaviorally linked; it was found that when one of the contexts induced a fear response, mice also became fearful of the linked context, even though they had never experienced anything aversive in that context (Fig. 2c).
Further support for the allocate-to-link hypothesis was obtained through manipulation of the specific fraction of shared neurons for two memories. These studies first demonstrated that a shared amygdala ensemble encodes two auditory fear memories that are acquired close in time (within 6 h) and that these memories are linked46. Researchers demonstrated the specific role of such shared neuronal ensembles by silencing them, which affected the behavioral interaction of two amygdala-dependent tasks but did not interfere with the retrieval of individual tasks47.
The allocate-to-link hypothesis assumes that the CREB-dependent increase in excitability increases the probability that a cell will become excited during temporally close encoding of other memories, thereby linking the memories by enhancing their synaptic connectivity. As noted, CREB-dependent increases in excitability are nonhomeostatic. Thus, there is the concern that this increase in excitability may enhance LTP and that the potentiated responses may make subsequent LTP more likely, potentially leading to runaway potentiation. However, synaptic strength is saturable48,49, and the resulting limit on LTP may obviate concerns of runaway excitation.
Assembly consolidation hypothesis
Many cells may represent similar information (for example, a place in the environment). During learning, these cells will fire together, and connections among them will be strengthened, thereby forming a stable memory ensemble. We now know that this strengthening will fade unless synapses undergo additional changes after learning, in a process termed consolidation. These consolidation processes, which include stabilization of synapses that were potentiated during learning (synaptic consolidation) and transfer of information from hippocampus to cortex (systems consolidation), occur during periods of rest and sleep that follow the learning events. During these periods, 100-ms-long events termed sharp-wave ripples (SWRs) take place in the hippocampus. Analysis of neural firing patterns during SWRs shows that they replay recent memory50–52. This replay is crucial for the formation of stable memory, as specific disruption of the SWR leads to strong memory deficits53–55. It would seem likely that a neuron’s involvement in SWRs would be enhanced by an increase in excitability (also see ref. 56). This leads us to suggest that another function of the CREB-dependent increase in excitability is to enhance the consolidation necessary for stable memory formation.
Mechanisms and selectivity of CREB activation
If CREB has an important role in memory allocation and consolidation, its activation should be largely restricted to cells that have been involved in learning and need to be incorporated into a memory ensemble. Action potentials are not a reliable indication of learning-related events because they can result from the activity of previously potentiated synapses. Similarly, LTP events at the synapse are not a reliable indicator that a cell should be part of a new ensemble because LTP can occur in a dendritic branch without somatic sodium spikes57,58. Making a cell fire, and thus able to be incorporated into an ensemble, may require that multiple branches undergo synaptic plasticity. Thus it may be desirable for CREB to be preferentially activated when there are both learning events in the dendrite and strong enough depolarization to cause firing. It is thus noteworthy that there is considerable complexity in the pathways that lead to CREB-dependent activation (Fig. 3): a calmodulin kinase cascade couples somatic action potentials to CREB activation59,60, whereas ERK diffusion from dendrite to soma couples synaptic plasticity to CREB activation61. One intriguing possibility is that these pathways perform the biochemical computation necessary to mark those cells that need to be incorporated into an ensemble.
Fig. 3. CREB-dependent enhancement of excitability is controlled both by dendritic LTP events and by somatic spiking, an enhancement that facilitates ensemble formation.
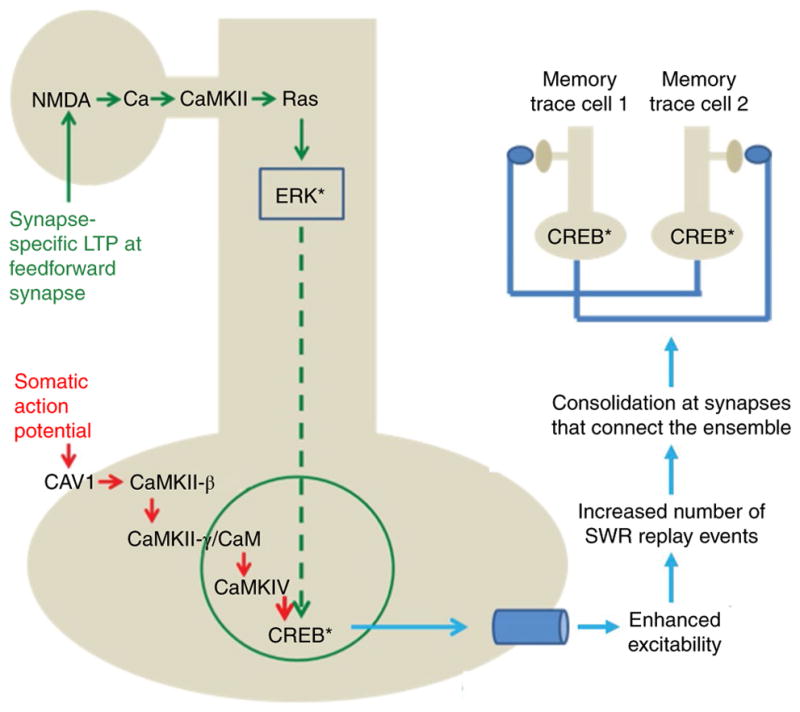
Induction of LTP at feedforward synapses results in CaMKII activation67, which then leads to extracellular signal-regulated protein kinase (ERK) activation at the synapse via synaptic Ras-GTPase-activating protein (synGAP) and Ras68 (see also refs. 69–71). Activated ERK (together with Jacob64) then moves to the soma61, leading to phosphorylation of CREB. CREB activation may occur by a second pathway: action potentials in the soma activate voltage-dependent Ca2+ channels. The resulting increase in Ca2+ levels initiates a complex cascade that leads to the entry of calmodulin (CaM) into the nucleus and the phosphorylation of CREB by calcium/calmodulin-dependent protein kinase IV (CaMKIV)59. Top right: two memory trace cells and their interconnections. The CREB-dependent increase in excitability in these cells enhances their participation in memory replay during SWR, leading to consolidation of the synaptic connections that link memory trace cells and thus the formation of a stable ensemble. Asterisks denote phosphorylation.
Discussion
The field of learning and memory has lacked a coherent view of why and how memory depends on both on synapse-specific changes in synaptic strength and global changes in neuronal function. Recent technological advances have allowed unprecedented visualization and control of circuit processes underlying memory, and the resulting findings support the view that global changes in excitability occur and make a critical contribution to the memory. These observations challenge standard models that attribute memory function solely to synaptic modification. We present two hypotheses of the specific role of the CREB-dependent changes in global excitability in memory that go beyond the traditional views; one (allocate-to-link) now has direct support, whereas the other (ensemble consolidation model) is built on experimental observations but has not yet been directly tested. Despite the conceptual differences between these models, they share a wide view of the overall process of memory—a view that includes events during encoding and consolidation, and thus goes beyond the processes that are directly responsible for ultimate memory storage. In the allocate-to-link model, CREB-dependent changes in excitability add an entirely new functionality to the memory system: the ability of one memory within a time frame to selectively associate with other memories within the same time frame. In the assembly consolidation model, the added functionality is the enhancement of consolidation—an enhancement that is specific to the memory trace cells and is ultimately necessary for the formation of a stable ensemble.
Neither of the proposed models posits that transcriptional changes actually underlie memory storage itself, and thus these models are consistent with the transient nature of CREB changes and learning and LTP. This is an important point because it is often suggested that transcriptional switching might allow for more stable long-term memory storage than synaptic switches that are dependent on only post-translational processes. We emphasize that the data on CREB do not support this suggestion; although CREB-dependent transcription appears to be necessary for the formation of stable memories (notably in the ensemble consolidation model), it is not itself a stable information-storage mechanism and thus cannot mediate long-term memory. That important function may rely on stable changes at the synapse (but see refs. 62,63) or on learning-related transcriptional changes other than those mediated by CREB64,65 (for the potential utility of hypothesized long-term changes in excitability, see ref. 66).
In summary, we argue that any overall model of the memory system must now include both persistent changes at synapses and transient changes in global excitability. Such dual mechanisms should not be viewed as contradictory. Rather, the CREB-dependent transcriptional changes function to promote stable synaptic modifications in a way that produces useful temporal linkages.
Acknowledgments
This work was supported by the NIH (grants U01NS090583, R56NS096710, R01DA043195, R01NS103168, U19NS104590, and NSF IOS-1526941 to J.L.; grants 2RF1AG013622-21 and R01MH113071 to A.J.S.) and the Dr. Miriam and Sheldon G. Adelson Medical Research Foundation (to A.J.S.).
Footnotes
Publisher’s note: Springer Nature remains neutral with regard to jurisdictional claims in published maps and institutional affiliations.
Competing interests
The authors declare no competing financial interests.
References
- 1.Hebb DO. The Organization of Behavior: A Neuropsychological Theory. Wiley; New York: 1949. [Google Scholar]
- 2.Bliss TV, Collingridge GL. A synaptic model of memory: long-term potentiation in the hippocampus. Nature. 1993;361:31–39. doi: 10.1038/361031a0. [DOI] [PubMed] [Google Scholar]
- 3.Bliss TVP, Collingridge GL. Expression of NMDA receptor-dependent LTP in the hippocampus: bridging the divide. Mol Brain. 2013;6:5. doi: 10.1186/1756-6606-6-5. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Nakazawa K, McHugh TJ, Wilson MA, Tonegawa S. NMDA receptors, place cells and hippocampal spatial memory. Nat Rev Neurosci. 2004;5:361–372. doi: 10.1038/nrn1385. [DOI] [PubMed] [Google Scholar]
- 5.Giese KP, Fedorov NB, Filipkowski RK, Silva AJ. Autophosphorylation at Thr286 of the alpha calcium-calmodulin kinase II in LTP and learning. Science. 1998;279:870–873. doi: 10.1126/science.279.5352.870. [DOI] [PubMed] [Google Scholar]
- 6.Rossetti T, et al. Memory erasure experiments indicate a critical role of CAMKII in memory storage. Neuron. 2017;96:207–216. doi: 10.1016/j.neuron.2017.09.010. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Lee YS, Silva AJ. The molecular and cellular biology of enhanced cognition. Nat Rev Neurosci. 2009;10:126–140. doi: 10.1038/nrn2572. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Nabavi S, et al. Engineering a memory with LTD and LTP. Nature. 2014;511:348–352. doi: 10.1038/nature13294. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Matsuzaki M, Honkura N, Ellis-Davies GCR, Kasai H. Structural basis of long-term potentiation in single dendritic spines. Nature. 2004;429:761–766. doi: 10.1038/nature02617. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Hopfield JJ. Neurons with graded response have collective computational properties like those of two-state neurons. Proc Natl Acad Sci USA. 1984;81:3088–3092. doi: 10.1073/pnas.81.10.3088. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Morris RGM, et al. Elements of a neurobiological theory of the hippocampus: the role of activity-dependent synaptic plasticity in memory. Philos Trans R Soc Lond B Biol Sci. 2003;358:773–786. doi: 10.1098/rstb.2002.1264. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Castellucci V, Kandel ER. Presynaptic facilitation as a mechanism for behavioral sensitization in Aplysia. Science. 1976;194:1176–1178. doi: 10.1126/science.11560. [DOI] [PubMed] [Google Scholar]
- 13.Siegelbaum SA, Camardo JS, Kandel ER. Serotonin and cyclic AMP close single K+ channels in Aplysia sensory neurones. Nature. 1982;299:413–417. doi: 10.1038/299413a0. [DOI] [PubMed] [Google Scholar]
- 14.Alkon DL. Changes of membrane currents during learning. J Exp Biol. 1984;112:95–112. doi: 10.1242/jeb.112.1.95. [DOI] [PubMed] [Google Scholar]
- 15.Disterhoft JF, Coulter DA, Alkon DL. Conditioning-specific membrane changes of rabbit hippocampal neurons measured in vitro. Proc Natl Acad Sci USA. 1986;83:2733–2737. doi: 10.1073/pnas.83.8.2733. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Josselyn SA, Köhler S, Frankland PW. Finding the engram. Nat Rev Neurosci. 2015;16:521–534. doi: 10.1038/nrn4000. [DOI] [PubMed] [Google Scholar]
- 17.Yu XW, Oh MM, Disterhoft JF. CREB, cellular excitability, and cognition: implications for aging. Behav Brain Res. 2017;322:206–211. doi: 10.1016/j.bbr.2016.07.042. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Kim JI, Cho HY, Han JH, Kaang BK. Which neurons will be the engram-activated neurons and/or more excitable neurons? Exp Neurobiol. 2016;25:55–63. doi: 10.5607/en.2016.25.2.55. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Oh MM, Disterhoft JF. Increased excitability of both principal neurons and interneurons during associative learning. Neuroscientist. 2015;21:372–384. doi: 10.1177/1073858414537382. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Kandel ER. Genes, nerve cells, and the remembrance of things past. J Neuropsychiatry Clin Neurosci. 1989;1:103–125. doi: 10.1176/jnp.1.2.103. [DOI] [PubMed] [Google Scholar]
- 21.Impey S, et al. Induction of CRE-mediated gene expression by stimuli that generate long-lasting LTP in area CA1 of the hippocampus. Neuron. 1996;16:973–982. doi: 10.1016/s0896-6273(00)80120-8. [DOI] [PubMed] [Google Scholar]
- 22.Bernabeu R, et al. Involvement of hippocampal cAMP/cAMP-dependent protein kinase signaling pathways in a late memory consolidation phase of aversively motivated learning in rats. Proc Natl Acad Sci USA. 1997;94:7041–7046. doi: 10.1073/pnas.94.13.7041. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Bourtchuladze R, et al. Deficient long-term memory in mice with a targeted mutation of the cAMP-responsive element-binding protein. Cell. 1994;79:59–68. doi: 10.1016/0092-8674(94)90400-6. [DOI] [PubMed] [Google Scholar]
- 24.Silva AJ, Kogan JH, Frankland PW, Kida S. CREB and memory. Annu Rev Neurosci. 1998;21:127–148. doi: 10.1146/annurev.neuro.21.1.127. [DOI] [PubMed] [Google Scholar]
- 25.Yin JC, Tully T. CREB and the formation of long-term memory. Curr Opin Neurobiol. 1996;6:264–268. doi: 10.1016/s0959-4388(96)80082-1. [DOI] [PubMed] [Google Scholar]
- 26.Guzowski JF, McGaugh JL. Antisense oligodeoxynucleotide-mediated disruption of hippocampal cAMP response element binding protein levels impairs consolidation of memory for water maze training. Proc Natl Acad Sci USA. 1997;94:2693–2698. doi: 10.1073/pnas.94.6.2693. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Peters M, et al. RNA interference in hippocampus demonstrates opposing roles for CREB and PP1α in contextual and temporal long-term memory. Genes Brain Behav. 2009;8:320–329. doi: 10.1111/j.1601-183X.2009.00474.x. [DOI] [PubMed] [Google Scholar]
- 28.Yin JC, Del Vecchio M, Zhou H, Tully T. CREB as a memory modulator: induced expression of a dCREB2 activator isoform enhances long-term memory in Drosophila. Cell. 1995;81:107–115. doi: 10.1016/0092-8674(95)90375-5. [DOI] [PubMed] [Google Scholar]
- 29.Josselyn SA, et al. Long-term memory is facilitated by cAMP response element-binding protein overexpression in the amygdala. J Neurosci. 2001;21:2404–2412. doi: 10.1523/JNEUROSCI.21-07-02404.2001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Guzowski JF, Worley PF. Cellular compartment analysis of temporal activity by fluorescence in situ hybridization (catFISH) Curr Protoc Neurosci. 2001;15:1.8.1–1.8.16. doi: 10.1002/0471142301.ns0108s15. [DOI] [PubMed] [Google Scholar]
- 31.Han JH, et al. Neuronal competition and selection during memory formation. Science. 2007;316:457–460. doi: 10.1126/science.1139438. [DOI] [PubMed] [Google Scholar]
- 32.Yiu AP, et al. Neurons are recruited to a memory trace based on relative neuronal excitability immediately before training. Neuron. 2014;83:722–735. doi: 10.1016/j.neuron.2014.07.017. [DOI] [PubMed] [Google Scholar]
- 33.Rogerson T, et al. Synaptic tagging during memory allocation. Nat Rev Neurosci. 2014;15:157–169. doi: 10.1038/nrn3667. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Zhou Y, et al. CREB regulates excitability and the allocation of memory to subsets of neurons in the amygdala. Nat Neurosci. 2009;12:1438–1443. doi: 10.1038/nn.2405. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Park S, et al. Neuronal allocation to a hippocampal engram. Neuropsychopharmacology. 2016;41:2987–2993. doi: 10.1038/npp.2016.73. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Lopez de Armentia M, et al. cAMP response element-binding protein-mediated gene expression increases the intrinsic excitability of CA1 pyramidal neurons. J Neurosci. 2007;27:13909–13918. doi: 10.1523/JNEUROSCI.3850-07.2007. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Dong Y, et al. CREB modulates excitability of nucleus accumbens neurons. Nat Neurosci. 2006;9:475–477. doi: 10.1038/nn1661. [DOI] [PubMed] [Google Scholar]
- 38.Storm JF. Potassium currents in hippocampal pyramidal cells. Prog Brain Res. 1990;83:161–187. doi: 10.1016/s0079-6123(08)61248-0. [DOI] [PubMed] [Google Scholar]
- 39.Gross C, Yao X, Pong DL, Jeromin A, Bassell GJ. Fragile X mental retardation protein regulates protein expression and mRNA translation of the potassium channel Kv4.2. J Neurosci. 2011;31:5693–5698. doi: 10.1523/JNEUROSCI.6661-10.2011. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Rogerson T, et al. Molecular and cellular mechanisms for trapping and activating emotional memories. PLoS One. 2016;11:e0161655. doi: 10.1371/journal.pone.0161655. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.Alberini CM. Transcription factors in long-term memory and synaptic plasticity. Physiol Rev. 2009;89:121–145. doi: 10.1152/physrev.00017.2008. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42.Kim J, Kwon JT, Kim HS, Han JH. CREB and neuronal selection for memory trace. Front Neural Circuits. 2013;7:44. doi: 10.3389/fncir.2013.00044. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43.Turrigiano G. Too many cooks? Intrinsic and synaptic homeostatic mechanisms in cortical circuit refinement. Annu Rev Neurosci. 2011;34:89–103. doi: 10.1146/annurev-neuro-060909-153238. [DOI] [PubMed] [Google Scholar]
- 44.Silva AJ, Zhou Y, Rogerson T, Shobe J, Balaji J. Molecular and cellular approaches to memory allocation in neural circuits. Science. 2009;326:391–395. doi: 10.1126/science.1174519. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45.Cai DJ, et al. A shared neural ensemble links distinct contextual memories encoded close in time. Nature. 2016;534:115–118. doi: 10.1038/nature17955. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46.Rashid AJ, et al. Competition between engrams influences fear memory formation and recall. Science. 2016;353:383–387. doi: 10.1126/science.aaf0594. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47.Yokose J, et al. Overlapping memory trace indispensable for linking, but not recalling, individual memories. Science. 2017;355:398–403. doi: 10.1126/science.aal2690. [DOI] [PubMed] [Google Scholar]
- 48.O’Connor DH, Wittenberg GM, Wang SSH. Graded bidirectional synaptic plasticity is composed of switch-like unitary events. Proc Natl Acad Sci USA. 2005;102:9679–9684. doi: 10.1073/pnas.0502332102. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49.Debanne D, Gähwiler BH, Thompson SM. Heterogeneity of synaptic plasticity at unitary CA3-CA1 and CA3-CA3 connections in rat hippocampal slice cultures. J Neurosci. 1999;19:10664–10671. doi: 10.1523/JNEUROSCI.19-24-10664.1999. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 50.Wilson MA, McNaughton BL. Reactivation of hippocampal ensemble memories during sleep. Science. 1994;265:676–679. doi: 10.1126/science.8036517. [DOI] [PubMed] [Google Scholar]
- 51.Nádasdy Z, Hirase H, Czurkó A, Csicsvari J, Buzsáki G. Replay and time compression of recurring spike sequences in the hippocampus. J Neurosci. 1999;19:9497–9507. doi: 10.1523/JNEUROSCI.19-21-09497.1999. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 52.Foster DJ, Wilson MA. Reverse replay of behavioural sequences in hippocampal place cells during the awake state. Nature. 2006;440:680–683. doi: 10.1038/nature04587. [DOI] [PubMed] [Google Scholar]
- 53.Girardeau G, Benchenane K, Wiener SI, Buzsáki G, Zugaro MB. Selective suppression of hippocampal ripples impairs spatial memory. Nat Neurosci. 2009;12:1222–1223. doi: 10.1038/nn.2384. [DOI] [PubMed] [Google Scholar]
- 54.Ego-Stengel V, Wilson MA. Disruption of ripple-associated hippocampal activity during rest impairs spatial learning in the rat. Hippocampus. 2010;20:1–10. doi: 10.1002/hipo.20707. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 55.Jadhav SP, Kemere C, German PW, Frank LM. Awake hippocampal sharp-wave ripples support spatial memory. Science. 2012;336:1454–1458. doi: 10.1126/science.1217230. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 56.Atherton LA, Dupret D, Mellor JR. Memory trace replay: the shaping of memory consolidation by neuromodulation. Trends Neurosci. 2015;38:560–570. doi: 10.1016/j.tins.2015.07.004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 57.Brandalise F, Carta S, Helmchen F, Lisman J, Gerber U. Dendritic NMDA spikes are necessary for timing-dependent associative LTP in CA3 pyramidal cells. Nat Commun. 2016;7:13480. doi: 10.1038/ncomms13480. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 58.Golding NL, Staff NP, Spruston N. Dendritic spikes as a mechanism for cooperative long-term potentiation. Nature. 2002;418:326–331. doi: 10.1038/nature00854. [DOI] [PubMed] [Google Scholar]
- 59.Ma H, et al. γCaMKII shuttles Ca2+/CaM to the nucleus to trigger CREB phosphorylation and gene expression. Cell. 2014;159:281–294. doi: 10.1016/j.cell.2014.09.019. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 60.Dudek SM, Fields RD. Somatic action potentials are sufficient for late-phase LTP-related cell signaling. Proc Natl Acad Sci USA. 2002;99:3962–3967. doi: 10.1073/pnas.062510599. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 61.Zhai S, Ark ED, Parra-Bueno P, Yasuda R. Long-distance integration of nuclear ERK signaling triggered by activation of a few dendritic spines. Science. 2013;342:1107–1111. doi: 10.1126/science.1245622. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 62.Ryan TJ, Roy DS, Pignatelli M, Arons A, Tonegawa S. Memory. Engram cells retain memory under retrograde amnesia. Science. 2015;348:1007–1013. doi: 10.1126/science.aaa5542. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 63.Chen S, Cai D, Pearce K, Sun PYW, Roberts AC, Glanzman DL. Reinstatement of long-term memory following erasure of its behavioral and synaptic expression in Aplysia. eLife. 2014;3:e03896. doi: 10.7554/eLife.03896. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 64.Zovkic IB, Guzman-Karlsson MC, Sweatt JD. Epigenetic regulation of memory formation and maintenance. Learn Mem. 2013;20:61–74. doi: 10.1101/lm.026575.112. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 65.Meadows JP, et al. Dynamic DNA methylation regulates neuronal intrinsic membrane excitability. Sci Signal. 2016;9:ra83. doi: 10.1126/scisignal.aaf5642. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 66.Titley HK, Brunel N, Hansel C. Toward a neurocentric view of learning. Neuron. 2017;95:19–32. doi: 10.1016/j.neuron.2017.05.021. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 67.Lisman J, Yasuda R, Raghavachari S. Mechanisms of CaMKII action in long-term potentiation. Nat Rev Neurosci. 2012;13:169–182. doi: 10.1038/nrn3192. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 68.Araki Y, Zeng M, Zhang M, Huganir RL. Rapid dispersion of SynGAP from synaptic spines triggers AMPA receptor insertion and spine enlargement during LTP. Neuron. 2015;85:173–189. doi: 10.1016/j.neuron.2014.12.023. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 69.Melgarejo da Rosa M, Yuanxiang P, Brambilla R, Kreutz MR, Karpova A. Synaptic GluN2B/CaMKII-α signaling induces synapto-nuclear transport of ERK and Jacob. Front Mol Neurosci. 2016;9:66. doi: 10.3389/fnmol.2016.00066. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 70.El Gaamouch F, et al. Interaction between αCaMKII and GluN2B controls ERK-dependent plasticity. J Neurosci. 2012;32:10767–10779. doi: 10.1523/JNEUROSCI.5622-11.2012. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 71.Tang S, Yasuda R. Imaging ERK and PKA activation in single dendritic spines during structural plasticity. Neuron. 2017;93:1315–1324. doi: 10.1016/j.neuron.2017.02.032. [DOI] [PMC free article] [PubMed] [Google Scholar]