

Summary
Metabolic reprogramming in cancer cells facilitates growth and proliferation. Increased activity of the serine biosynthetic pathway through the enzyme phosphoglycerate dehydrogenase (PHGDH) contributes to tumorigenesis. With a small substrate and a weak binding cofactor (NAD+), inhibitor development for PHGDH remains challenging. Instead of targeting the PHGDH active site, we computationally identified two potential allosteric sites and virtually screened compounds that can bind to these sites. With subsequent characterization, we successfully identified PHDGH non-NAD+ competing allosteric inhibitors that attenuate its enzyme activity, selectively inhibit de novo serine synthesis in cancer cells, and reduce tumor growth in vivo. Our study not only identifies novel allosteric inhibitors for PHGDH to probe its function and potential as a therapeutic target, but also provides a general strategy for the rational design of small molecule modulators of metabolic enzyme function.
In Brief
Wang et al. identify allosteric sites on PHGDH and discover inhibitors bound to these sites. These allosteric inhibitors are specific, bind to PHGDH in cells, reduce serine synthesis, and show activities in vivo.
Introduction
It has long been known that tumor cells exhibit altered glucose metabolism characterized by increased glucose uptake and incomplete oxidation to lactate in the presence of oxygen (Warburg, 1956; Zhao et al., 2016a). With the surge of interest in understanding cancer cell metabolism, it is now widely accepted that metabolic rearrangements accompanying malignant transformation also involve numerous other pathway alterations such as the increased flux of the pentose phosphate pathway (PPP), elevated rates of lipid biosynthesis, high glutamine consumption, maintenance of redox homeostasis, and alterations in autophagy (Pavlova and Thompson, 2016). Therefore, targeting the metabolic enzymes in these pathways provides a promising strategy for cancer therapy.
The gene encoding phosphoglycerate dehydrogenase (PHGDH), an enzyme that catalyzes the first committed step of serine biosynthesis, is also involved in metabolic reprogramming in cancer. PHGDH was identified as a focus of recurrent copy number gain across a large set of tumors (Beroukhim et al., 2010). The PHGDH gene that is located at chromosome 1p12 showed copy number gain in 16% of all cancers including 40% of melanoma and some triple negative breast cancers (Locasale et al., 2011; Possemato et al., 2011). Cancer cells with PHGDH amplifications are sensitive to PHGDH depletion, which indicates that the enzyme is required for the growth of certain tumor cells.
Recent studies have identified different regulatory mechanisms that can activate PHGDH through both transcriptional regulation and changes in its activity via posttranslational modifications (Ma et al., 2013; DeNicola et al., 2015; Ou et al., 2015; Ding et al., 2013). Additional studies have found several underappreciated functions for de novo synthesis of serine and the use of one-carbon metabolism including epigenetic maintenance and NADPH production that is important for biosynthesis and controlling the levels of reactive oxygen species (Fan et al., 2014; Mentch et al., 2015). Together these findings demonstrate that PHGDH is an attractive anti-cancer target, and that designing PHGDH inhibitors may be a fruitful enterprise.
Human PHGDH contains four domains: nucleotide-binding, substrate-binding, regulatory and intervening domains. Currently only the crystal structure containing the first two domains is available (PDB code: 2G76, Turnbull, 2006). The substrate-binding pocket of PHGDH is rather small, approximately 100-200 Å3, and the physiological concentration of its cofactor NAD+ is as high as 0.3 mM (Yamada et al., 2006). These properties likely increase the difficulties of the design of substrate-competitive inhibitors. Meanwhile, considering NAD+ or NADH is a widely used cofactor, which also easily causes the problem of specificity, we focused on designing allosteric inhibitors for PHGDH that do not compete with the native ligand. Allosteric regulation can be achieved by various effectors, ranging from small molecules to macromolecules (Merdanovic et al., 2013) and can have high specificity, as allosteric binding sites are usually not evolutionarily conserved. Computational methods for rational design of allosteric effectors were emerging (Wagner et al., 2016; Ma et al., 2016) and a number of successful application examples have been reported. For example, using the two-state Go model based allosteric site prediction method that we developed (Qi et al., 2012), we obtained novel allosteric inhibitors for Escherichia coli (E. coli) phosphoglycerate dehydrogenase (Wang et al., 2014). Novel enzymes activators were also found using combined computational and experimental approach (Meng et al., 2016), providing an alternative way to control disease-related molecular networks (Pei et al., 2014).
In the present study, we first computationally identified two potential allosteric sites in PHGDH and used them to virtually screen a compound library. Selected compounds were tested for their inhibition activities using recombinant enzyme, cancer cell-lines, and tumor xenograft models. Two distinct compounds with activity in cells were found. Their specificity was confirmed using CRISPR-Cas9 gene-targeting PHGDH, chemical compound pull-down in cancer cells, and metabolomics. Recently, three studies have reported compounds that have activity against PHGDH by using high-throughput experimental screening. One series of PHGDH inhibitors showed activities in enzymatic and cell-based assays, but the binding mechanism, selectivity towards PHGDH, and efficacy in vivo were unclear (Mullarky et al., 2016). Another series of inhibitors with bioactivities in enzymatic and cell-based assays, as well as a xenograft model, do not have clear binding sites (Pacold et al., 2016). The third series of inhibitors were found by fragment screen that bind to the adenine subsite with only millimolar protein binding affinities and no further biological activities were reported (Unterlass et al., 2016). To our knowledge, the present study is the first successful example of using a structure-based approach to discover allosteric inhibitors that directly and specifically target PHGDH.
Results
Allosteric Site Prediction and Identification of Novel Allosteric Inhibitors
Two potential allosteric sites, I and II, were identified computationally using a cavity detection algorithm based on defined geometric criteria (Yuan et al., 2013; Yuan et al., 2011) (Figure 1A). Site I is close to the active site and the NAD+/NADH-cofactor binding site, with a volume of 847 Å3 and a predicted maximal pKd of 8.71. It shares residues Gly 78, Val 79, Asp 80, Asn 81 and Val 82 with the active site. Site II is located in the substrate binding domain, with a volume of 463 Å3 and a predicted maximal pKd of 7.79. Molecular docking across a large virtual compound library was then conducted (Halgren et al., 2004; Friesner et al., 2004). Ninety-eight compounds were selected and then acquired to test their abilities to regulate PHGDH activity.
Figure 1. Identification of Novel Allosteric Inhibitors of PHGDH.
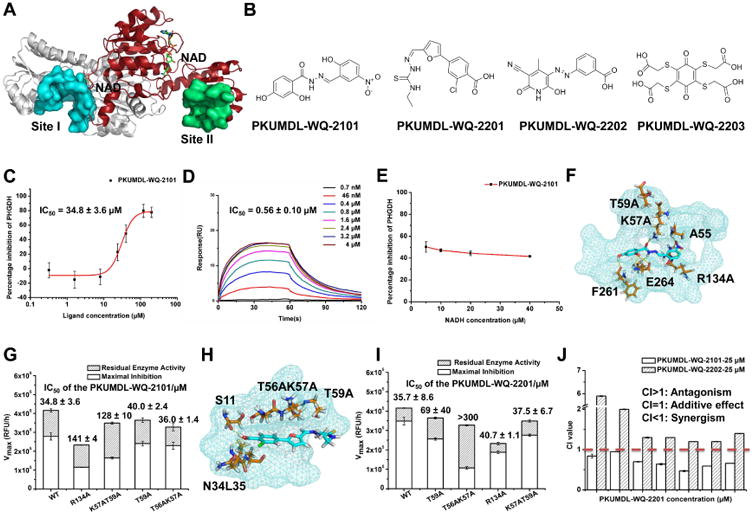
(A) Potential allosteric sites in PHGDH (PDB code: 2G76). The sites were predicted by the program of CAVITY and illustrated by the surface mode.The cofactor NAD+ was indicated in sticks. PHGDH forms a dimer in the crystal structure, site I and II exist in each monomer, and only one site I and one site II is shown in the figure for clarity. (B) Chemical structures of PHGDH inhibitors. (C) Enzyme inhibition dose-response curve of PKUMDL-WQ-2101. (D) SPR dose-response curve of PKUMDL-WQ-2101. (E) Cofactor competiton curve of PKUMDL-WQ-2101. The percentage inhibition did not obviously change along with the increase of NADH concentration, indicating that there are no significant interactions between PKUMDL-WQ-2101 and the cofactor binding site. (F-G) Predicted binding mode of PKUMDL-WQ-2101. The compound and key residues in sites I are shown in stick representation. Site I is shown in suface mode (F). Enzymatic activities and responses to PKUMDL-WQ-2101 of PHGDH mutants (G). (H-I) Predicted binding mode of PKUMDL-WQ-2201. The compound and key residues in sites II are shown in stick representation. Site II is shown in surface mode (H). Enzymatic activities and responses to PKUMDL-WQ-2201 of PHGDH mutants (I). (J) Inhibitors in different sites synergize to induce PHGDH inhibition. The concentration of PKUMDL-WQ-2101 and PKUMDL-WQ-2202 was kept at 25 μM for enzyme inhibition assay, while PKUMDL-WQ-2201 concentration varies. CI values <1 indicates synergistic interaction. Data shown represent the mean ± SD (n = 3). See Figure S1 for dose-response curves of PKUMDL-WQ-2201 to 2203 and Figure S2 for synergytic matrix.
PKUMDL-WQ-2101 in site I and PKUMDL-WQ-2201 to 2203 in site II were identified to significantly affect the PHGDH activity in a concentration dependent manner (Figures 1B, 1C, and S1A. SPECS IDs of these four compounds are shown in Table S1), and their KD values were determined by using Surface Plasmon Resonance (SPR) (Figures 1D, and S1B-S1G). SPR experiments also demonstrated these inhibitors did not aggregate under the experimental conditions.
To test whether the compounds indeed bind to site I and II, respectively, we selected PKUMDL-WQ-2101 in site I and PKUMDL-WQ-2201 in site II, and performed competition experiments and mutagenesis studies. The competition experiments between these compounds and cofactor NADH indicated that they did not bound in the cofactor site (Figures 1E and S1H). Inhibition ability of PKUMDL-WQ-2101 and PKUMDL-WQ-2201 for the C-terminal truncated PHGDH containing only the substrate binding domain and the nucleotide binding domain demonstrated that the C-terminal regulatory and intervening domains did not contribute to binding and the compounds bound to the N-terminal domains used for virtual screening (Figure S1I and S1J). Based on the docking structures, mutants R134A and K57AT59A for PKUMDL-WQ-2101, and mutants T59A and T56AK57A for PKUMDL-WQ-2201 were made and tested (Figures 1F-1I). All these mutants retained their secondary structures (Figure S1K) and exhibited reduced responses to the corresponding inhibitors. For PKUMDL-WQ-2101, the PHGDH-inhibiting activities were dramatically reduced for mutants R134A (IC50 = 141 ± 4 μM, max inhibition = 49%) and K57AT59A (IC50 = 128 ± 10 μM, max inhibition = 47%) compared to that for WT PHGDH (IC50 = 34.8 ± 3.6 μM, max inhibition = 67%) (Figure 1G). For PKUMDL-WQ-2201, its inhibition abilities for PHGDH mutants were also significantly decreased. The IC50 values for T59A and T56AK57A were 69 ± 40 and > 300 μM, respectively, while the IC50 value for WT PHGDH was 35.7 ± 8.6 μM (Figure 1I). In addition, the inhibition ability of PKUMDL-WQ-2101 to T59A and T56AK57A (key residues in site II) or the inhibition ability of PKUMDL-WQ-2201 to R134A and K57AT59A (key residues in site I) were also measured to verify the specificity of inhibitor binding (Figures 1G and 1I). These results support that PKUMDL-WQ-2101 and PKUMDL-WQ-2201 bound in site I and site II, respectively.
Combination therapy is emerging as a promising strategy to generate synergistic therapeutic effects, reduce side effects of monotherapy, overcome multidrug resistance (MDR), and reduce dose of each drug require (Yang et al., 2015; Botham et al., 2014), which can also be used to demonstate whether two compounds bind to the same site or to different sites. Synergism can be quantified through the calculation of Combination Indices (CI) (Chou, 2006). When the concentration of PKUMDL-WQ-2101 was kept at 25 μM (about IC50 value), synergistic interactions were observed with PKUMDL-WQ-2201 concentration ranging from 1 to 200 μM (Figures 1J and S2A). In contrast, when the concentration of PKUMDL-WQ-2202 was kept at 25 μM, PKUMDL-WQ-2201 and PKUMDL-WQ-2202 showed antagonism rather than synergy, indicating that they compete for a single site (Figures 1J and S2B). Together, these synergistic results further confirm our docking results about the binding sites of the compounds.
Cellular Effects of PKUMDL-WQ-2101 and PKUMDL-WQ-2201
The effects of the compounds against a panel of cancer cell lines along with one immortalized human breast epithelial cell line were evaluated. PKUMDL-WQ-2101 and PKUMDL-WQ-2201 showed dose-dependent suppression effects on the cell viability at micromolar concentrations, with good selectivity for PHGDH amplified breast cancer cell lines (Figures 2A and 2B), while PKUMDL-WQ-2202 and PKUMDL-WQ-2203 showed weak bioactivity in cell based assays with EC50 values more than 200 μM (Figure S3). The antitumor activities of PKUMDL-WQ-2101 in the two PHGDH amplified breast cancer cell lines (MDA-MB-468 and HCC70) were 7.70 and 10.8 μM, which were 3- to 4-, 8- to 12-, and 14- to 20-fold more active than its antitumor activities in PHGDH non-dependent cell lines, MDA-MB-231, ZR-75-1 and MCF-7 cell lines, respectively. For PKUMDL-WQ-2201, the EC50 values were 6.90 μM in MDA-MB-468 and 10.0 μM in HCC70 cell lines, which were 13- to 18-fold more active than that of ZR-75-1. No bioactivities in the other three PHGDH non-amplified breast cancer cell lines tested were measurable. Meanwhile, PKUMDL-WQ-2101 and PKUMDL-WQ-2201 exerted weak cytotoxic effects on the MCF-10A cell line, which was consistent with previous observations of PHGDH requirements using genetic approaches (Locasale et al., 2011). The antitumor activities of PKUMDL-WQ-2101 and PKUMDL-WQ-2201 to MDA-MB-468 cells may be caused by their influence on cell cycle (Figures 2C and 2D).
Figure 2. Bioactivities of PKUMDL-WQ-2101 and PKUMDL-WQ-2201 in cell based assays.
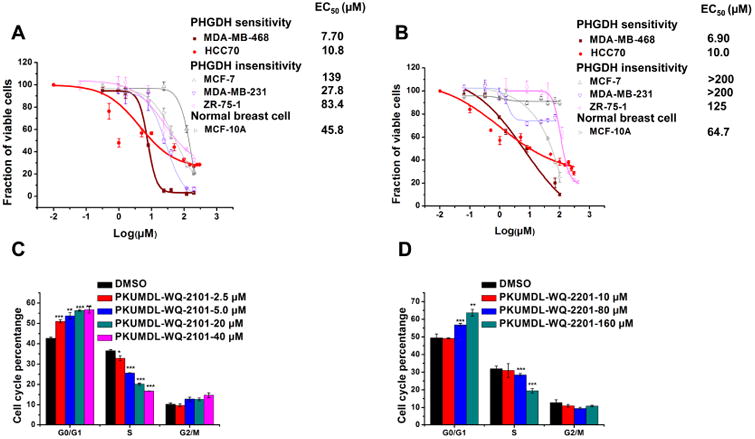
(A-B) Growth inhibition activity of PKUMDL-WQ-2101 (A) and PKUMDL-WQ-2201 (B) in MDA-MB-468, HCC70, MCF-7, MDA-MB-231, ZR-75-1 and MCF-10A cells, respectively. Cells were exposed to vehcile or various concentrations of PKUMDL-WQ-2101 for 72h followed by MTT assay. The EC50 value of PKUMDL-WQ-2201 for MCF-7 and MDA-MB-231 was larger than 200 μM, so the corresponding dose-response curve was not presented here. (C-D) Percentage of MDA-MB-468 cells in different phases of the cell cycle after respectively treatment with 2.5, 5.0, 20 and 40 μM PKUMDL-WQ-2101 (C), and 10, 80 and 160 μM PKUMDL-WQ-2201 (D) for 24 hours. DMSO was used as vehcile. Data represent the mean ± SD independent experiments. Difference is significant by two-tailed multiple t-test, *p < 0.05, **p < 0.01, ***p < 0.001. See Figure S3 for cell bioactivities of PKUMDL-WQ-2202 and 2203.
Compound Activity is Selective for PHGDH
To further evaluate the activity and selectivity of the compounds, we developed a clustered regularly interspaced short palindromic repeats (CRISPR)-CRISPR-associated protein 9 (Cas9) mediated PHGDH gene knockout (KO). We designed a single-guide RNA (sgRNA) with a protospacer adjacent motif (PAM) sequence specifically targeting a coding region in exon 8 of the PHGDH gene, predicted to result in a frame shift mutation and loss-of-function (Shalem et al., 2014; Mali et al., 2013) (Figure 3A). A clonal population of SKOV3 ovarian cancer cells was obtained and able to grow in the absence of PHGDH. Complete knockout was confirmed with immunoblotting in reference to a cell line created by targeting a sgRNA against GFP (Figure 3B). A 6-day growth curve revealed the ability of PHGDH KO cells to grow, albeit more slowly than the GFP KO control cells (p<0.01, two-tailed multiple t-test) (Figure 3C). We then evaluated the compounds PKUMDL-WQ-2101 and PKUMDL-WQ-2201 on these cell lines. The GFP KO cells exhibited sensitivity to PKUMDL-WQ-2101 (IC50= 37.3 μM) (Figure S4A) and, albeit to a lesser extent to PKUMDL-WQ-2201 (IC50 = 291.5.3 μM) (Figure S4B). To further understand the specificity of these compounds, 6-day proliferation assays were carried out in SKOV3 control and PHGDH KO cells. GFP KO cell growth was significantly suppressed after treatment with PKUMDL-WQ-2101 (p<10-3, two-tailed student's t-test) (Figure 3D), whereas PHGDH KO cells were able to proliferate in the presence of the compound (Figure 3E). Similarly, albeit to a lesser extent, proliferation in SKOV3 control cells was suppressed after 6 days in the presence of PKUMDL-WQ-2201 (p<0.05, two-tailed student's t-test) (Figure 3F), whereas PHGDH KO cell growth remained unaffected (p>0.99, two-tailed student's t-test) (Figure 3G). Chemical compound pull down assays were also carried out to verify PKUMDL-WQ-2101 with the best binding affinity was specifically bind to PHGDH in MDA-MB-468 cells (Figures S4C-4E). These results indicated that the cytotoxicity to these compounds appears to a large extent specific to PHGDH and serine synthesis.
Figure 3. CRISPR-Cas9 mediated PHGDH KO and PHGDH inhibition by PKUMDL-WQ-2101 and PKUMDL-WQ-2201.
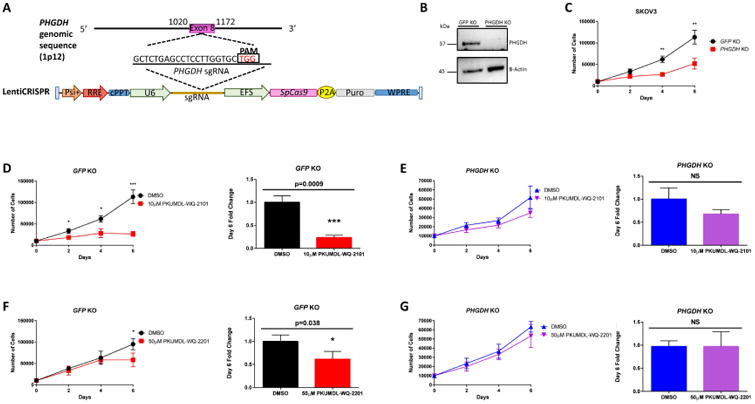
(A)Overview of LentiCRISPR system and sgRNA design generated for targeted PHGDH deletion in SKOV3 ovarian cancer cells. (B) Western blot analysis for SKOV3 GFP KO control and SKOV3 PHGDH KO cells with actin as a loading control. (C) Growth curve comparing SKOV3 GFP KO control and PHGDH KO over 6 days. (D) Growth curves of SKOV3 GFP KO control and (E) PHGDH KO cells after 6 days of treatment with vehicle or 10μM PKUMDL-WQ-2101 followed by cell counting. (F) Growth curves of SKOV3 GFP KO control or (G) PHGDH KO cells after 6 days of treatment with vehicle or 50μM PKUMDL-WQ-2201 followed by cell counting. All values represent the mean ± SEM from n=3 biological replicates. P values were obtained from a two-tailed student's t-test, *P< 0.05, **p<0.01, ***P<0.001. See Figure S4 for PKUMDL-WQ-2101 and 2201 bioactivies on SKOV3 GFP KO cells and results of PKUMDL-WQ-2101 pull down assays.
We then investigated the effects of PHGDH KO on serine metabolism. Liquid chromatography coupled to high resolution mass spectrometry (LC-HRMS) and stable isotope labeling were used to monitor the conversion of uniformly labeled U-13C-glucose to metabolites in the serine metabolic network in both SKOV3 GFP KO control and SKOV3 PHGDH KO cells (Figure 4A). We detected 13C-glucose incorporation in both serine and glycine in SKOV3 GFP KO cells, but not in SKOV3 PHGDH KO cells, confirming that the knockout fully abrogated de novo serine synthesis (Figure 4B). Both compounds (about IC50) also produced comparable metabolic effects on the serine metabolic network and reduced glucose incorporation into serine and glycine metabolites by more than 50% (Figures 4C and 4D). We further investigated pathways downstream of serine upon inhibition of PHGDH with PKUMDL-WQ-2101 and PKUMDL-WQ-2201. Given that serine is essential for nucleotide synthesis (Locasale, 2013), we investigated whether 13C-glucose consumption into nucleotides was altered after treatment. We analyzed the mass isotopomer distribution (MID) of one pyrimidine and purine synthesis by measuring uridine triphosphate (UTP) and adenosine triphosphate (ATP), and determined whether a difference in the mass shift of 1 or 2 (m+1 or m+2), known to result from incorporation of serine or glycine, was observed. We also monitored any changes in m+6 or m+7, which correspond to labeling from both the pentose phosphate and serine biosynthesis pathways. Upon treatment with both PKUMDL-WQ-2101 and PKUMDL-WQ-2201, decreases in m+2, m+6, and m+7 glucose labeling were observed in UTP and ATP, indicating a direct effect of PHGDH inhibition on nucleotide synthesis (Figure 4E and 4F). A sharp decrease in the m+5 peak was also observed due to a decrease in ribose labeling from the pentose phosphate pathway, suggesting that PHGDH ablation likely exerts effects on nucleotide synthesis through affecting glycolysis or occurs indirectly as a product of on-target cytotoxicity of the compound. We excluded interpretation of the m+1 peak due to the confounding influence of natural abundance isotopes. We also analyzed glucose incorporation into glutathione, another metabolite belonging to a pathway downstream of serine and glycine synthesis, and decreases in m+2 were found in cells treated with both compounds (Figure 4G). All together, these data suggest that PHGDH inhibition by PKUMDL-WQ-2101 and PKUMDL-WQ-2201 decreases de novo serine synthesis and metabolism downstream of the serine synthesis pathway, with effects comparable to PHGDH genetic deletion.
Figure 4. PKUMDL-WQ-2101 and PKUMDL-WQ-2201 inhibit the serine biosynthesis pathway in cells.
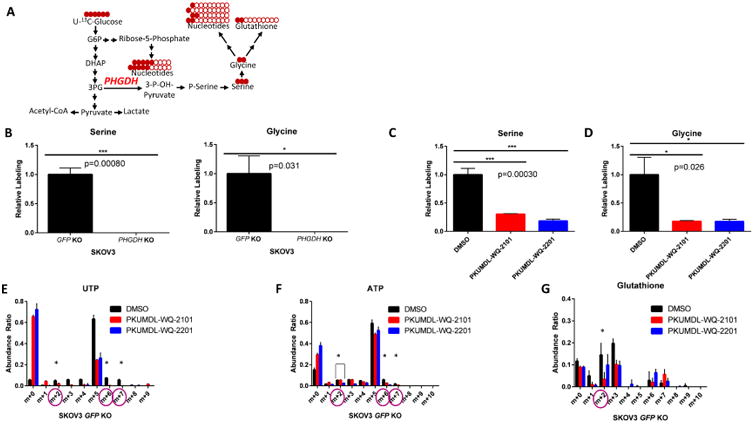
(A) Schematic of U-13C-glucose stable isotope labeling used to detect carbon labeling from glucose (red) in metabolites part of the serine metabolic network. (B) 13C-serine and 13C-glycine labeling from glucose in SKOV3 GFP KO control cells compared to SKOV3 PHGDH KO cells after 24 hours. (C)13C-serine and (D) 13C-glycine labeling from glucose in SKOV3 GFP KO cells after 24 hour treatment with 37 μM PKUMDL-WQ-2101 and 291 μM PKUMDL-WQ-2201, followed by subsequent U-13C-glucose labeling. (E) Mass isotopomer distribution (MID) of UTP and (F) ATP after 24 hour treatment with 37 μM PKUMDL-WQ-2101 and 291 μM PKUMDL-WQ-2201, followed by subsequent U-13C-glucose labeling. (G) Glutathione after 24 hour treatment with 37 μM PKUMDL-WQ-2101 and 291 μM PKUMDL-WQ-2201, followed by subsequent U-13C-glucose labeling. All values represent the mean ± SEM from n=3 biological replicates. Difference is significant by One-Way ANOVA, *P< 0.05, **p<0.01, ***P<0.001.
PKUMDL-WQ-2101 and PKUMDL-WQ-2201 Inhibits Tumor Growth of Amplified Cell Lines in vivo
Previous studies have questioned whether PHGDH inhibition is required for longer term tumor maintenance (Chen et al., 2013). To further understand the role of PHGDH in tumor growth and maintenance in vivo, MDA-MB-468 and MDA-MB-231 cells were injected into the fourth mammary pad of NOD.CB17 Scid/J mice. Tumor volumes were monitored every 2 days. We found that both PKUMDL-WQ-2101 and PKUMDL-WQ-2201 exhibited substantial inhibitory effects on MDA-MB-468 xenografts compared with vehicle-treated mice after 30 days of drug delivery (Figure 5). For MDA-MB-231 xenografts, neither PKUMDL-WQ-2101 nor PKUMDL-WQ-2201 affected tumor growth (Figure S5A and S5B) further confirming the specificity of the compounds. The compounds appeared to also be tolerated as all mice were able to maintain normal body weight over the course of the experiments (Figure S5C-S5E). Though the two compounds synergized each other to potently induce enzyme activity inhibition and the death of cell line in culture, the combination strategy was not applied to the mice model, due to complicated pharmacokinetic issues of in vivo compound concentration and clearance time, which may not exactly match. Nevertheless, these findings confirm the bioactivity, tolerability, and selectivity for PHGDH in vivo.
Figure 5. Bioactivities of PKUMDL-WQ-2101 and PKUMDL-WQ-2201 in vivo.
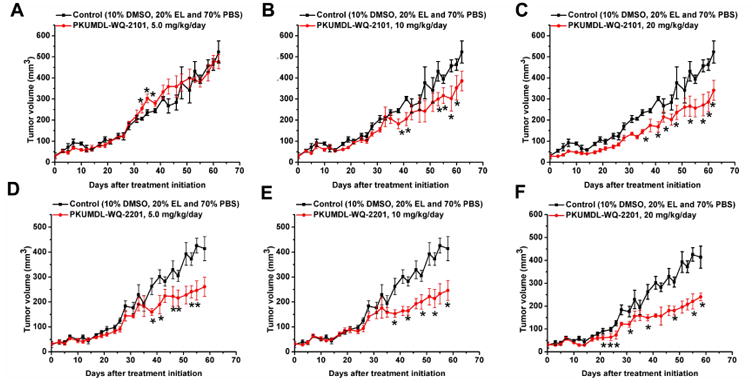
(A-F) After 30 days of drug delivery, treatment with PKUMDL-WQ-2101 (A-C) or PKUMDL-WQ-2201 (D-F) significantly suppressed the growth of tumors compared with control-treated group. Data represent the mean ± SEM independent experiments. Difference is significant by two-tailed multiple t-test, *p < 0.05. See Figure S5 for PKUMDL-WQ-2101 and 2201 biactivities on MDA-MB-231 xenografts and mice growth curves.
Discussion
Using a structure-based drug design approach, we successfully identified compounds that bound to the predicted allosteric sites and effectively inhibited the enzyme activity of PHGDH. These compounds exhibited sub-micromolar to micromolar binding affinities and inhibited cancer cell growth in the micromolar range. PKUMDL-WQ-2101 and PKUMDL-WQ-2201 showed good activity and selectivity to PHGDH over-expression breast cancer cells. The use of CRISPR-Cas9 mediated PHGDH KO in SKOV3 cells provided a genetic evaluation of the relative on-and off- target effects of each compound, whereby PKUMDL-WQ-2101 had high selectivity for PHGDH control but not KO cells. PKUMDL-WQ-2101 and PKUMDL-WQ-2201 were proven to suppress PHGDH amplified breast cancer cell growth in mice. Our study provides the first successful example of PHGDH allosteric inhibitor discovery using a structure-based approach.
The identified PKUMDL-WQ-2101 and PKUMDL-WQ-2201 compounds are novel allosteric inhibitors for PHGDH with unique structures. No biological activities for these compounds have been reported before. Though PKUMDL-WQ-2101 was predicted as pan-assay interference compounds (PAINS) (Baell and Holloway, 2010), due to the hydroxyl-phenyl-hydrazone group in its structure, the promiscuity has been eliminated by changing ionic strength or adding DTT in the enzymatic assay, SPR, and mutagenesis experiments, for the hydroxyl-phenyl-hydrazone group was generally believed to have the tendency for aggregation (McGovern et al., 2002), spectroscopic absorption (Auld et al., 2008), chelation (Ainscough et al., 1999) and reactivity, thus inactive proteins. In addition, the catalytic process of PHGDH or PSAT1 does not need the participation of metal ions, and PKUMDL-WQ-2101 showed good selectivity to PHGDH amplified breast cancer cells. Furthermore, PKUMDL-WQ-2101 showed bioactivity in vivo, confirming that PHGDH is required for tumor maintenance. These experimental results confirmed that PKUMDL-WQ-2101 specifically bound to PHGDH and inhibited its enzymatic activity.
Allosteric regulation needs communication between the allosteric site and the distant functional site. Therefore, high binding affinity of allosteric ligands may not necessarily cause strong influence on protein function (Nussinov and Tsai, 2014). In the case of PKUMDL-WQ-2101, while its Kd value is 0.56 ± 0.10 μM, its PHGDH inhibition activity IC50 value is 34.8 ± 3.6 μM. Previous studies showed that a large number of protein conformations in solution pre-exist and can be characterized by the energy landscape (Kar et al., 2010). Allosteric effectors may change the distribution of these conformations. PKUMDL-WQ-2101 and PKUMDL-WQ-2201 inhibited PHGDH activity mainly by forming hydrogen-bond networks with site I and II, respectively, limiting the movement of the rigid domains, preventing the active sites from closing, thus stabilizing PHGDH in the inactive conformation. Nevertheless, further experimental or computational studies are needed to better understand this inhibitory mechanism.
Three recent studies have reported compounds with activities against PHGDH. One study reported an example of a PHGDH inhibitor by screening a library of 800, 000 drug-like compounds (Mullarky et al., 2016). The best compound, CBR-5884 inhibited PHGDH enzymatic activity with an IC50 of 33 ± 12 μM in a time-dependent manner. CBR-5884 was speculated as a covalent inhibitor binding to a Cys in the non-active site and disrupting the enzyme oligomerization state. At 30 μM, CBR-5884 inhibited the growth of MDA-MB-468 cells by 35% to 60% in serine-replete media, and by 80% to 90% in serine-deplete media. Neither a direct binding test nor postulated binding site was reported. CBR-5884 was unstable in mouse plasma and could not be used for in vivo testing. Another study reported three PHGDH inhibitors by first screening a 400,000-compound NIH Molecular Libraries Small Molecule Repository (MLSMR) library and then optimizing the lead compounds (Pacold et al., 2016). The best compound, NCT-503, exhibited an IC50 value of 2.5 ± 0.6 μM and showed some selectivity in PHGDH amplified breast cancer cell lines and had bioactivities in a xenograft model. Although NCT-503 was found not substrate competitive, its specific binding site remains unknown. The third study reported 15 fragments with PHGDH inhibition activities by first screening a library of 600 fragments, then validating the fragments by using the thermal shift assay, isothermal titration calorimetry (ITC) competition experiments and X-ray crystallography (Unterlass et al., 2016). All the 15 fragments bound in the adenine subsite with millimolar binding affinities. However, fragment activities in cells and tumors were not reported. In the present study, we successfully discovered novel allosteric inhibitors for PHGDH using structure-based design approach with the best IC50 of 28.1 ± 1.3 μM for enzyme inhibition. PKUMDL-WQ-2101 and PKUMDL-WQ-2201 were confirmed to specifically bind to PHGDH in PHGDH amplified breast cancer cells with EC50 values less than 10 μM in serine-replete media, which was better than that of CBR-5884 and similar to that of NCT-503. Furthermore, PKUMDL-WQ-2101 and PKUMDL-WQ-2201 also suppressed tumor growth in mice. We started from purposely designing allosteric inhibitors for the predicted allosteric sites, while CBR-5884 and NCT-503 were found from high-throughput screening. Nevertheless, all the compounds inhibit PHGDH by an allosteric effect, demonstrating that allosteric inhibition is a promising strategy to suppress its activity. More allosteric inhibitors for PHGDH can be expected in the future.
In the past decade, considerable efforts have been devoted to identify agents to suppress oncogenesis and tumor progression (Hanahan and Weinberg, 2011), and then develop drugs to selectively kill cancer cells based on their metabolic alterations. Several drug candidates were successfully discovered and entered into clinic trials, such as AZD3965 (Birsoy et al., 2013; Sonveaux et al., 2008) and TCD-717 (Clem et al., 2011). Some anti-metabolite agents even have been used in clinic for a long time, such as 5-fluorouracil, methotrexate, and gemcitabine (Galluzzi et al., 2013). We are hopeful that PKUMDL-WQ-2101 and PKUMDL-WQ-2201 may be an additional starting point for further targeting cancer metabolism. In conclusion, we have successfully discovered PHGDH allosteric inhibitors targeting the predicted allosteric sites by using virtual screening and experimental validation. The compounds reported can be further optimized and developed for next-generation anti-cancer therapies.
Significance
Cancer cells reprogram metabolism to support their growth and proliferation. During the past decades, targeting cancer metabolism has emerged as a promising strategy for the development of selective anti-cancer agents. The gene encoding phosphoglycerate dehydrogenase (PHGDH), an enzyme that catalyzes the first critical step of serine biosynthesis is involved in metabolic reprogramming in cancer. The PHGDH gene that is located at chromosome 1p12 showed copy number gain in 16% of all cancers including 40% of melanoma and some triple negative breast cancers. Cancer cells with PHGDH amplifications are sensitive to PHGDH depletion, which indicates that the enzyme is required for the growth of certain tumor cells. Although the importance of PHGDH as a cancer target has been proposed, the lack of small molecules inhibitors hinders further exploration. The high cellular concentration and widely used of its cofactor (NAD+) and small size of the active site make inhibitor discovery targeting the active site difficult. We used a computational approach to scan for possible allosteric sites and used them to virtually screen for allosteric inhibitors. Two novel allosteric sites on PHGDH were identified. Compounds that directly bind to these sites, inhibit PHGDH enzyme activity, suppress cancer cell proliferation and in vivo tumor growth were found. The best compound binds to PHGDH with a dissociation constant of 0.56 μM, which selectively inhibits PHGDH amplified breast cancer cell line with EC50s less than 7.7 μM. The inhibitors were also characterized using metabolomics on PHGDH-amplified, CRISPR-Cas9-generated PHGDH knockout cell lines, and mice to demonstrate the specificity and activity in vivo. Our study not only identifies novel allosteric inhibitors for PHGDH with in vivo activity to probe its function and potential as a therapeutic target, but also provides a general strategy for the rational design of small molecule modulators of metabolic enzyme function.
Experimental Procedures
Allosteric site prediction and virtual screening
Potential allosteric sites in PHGDH N-terminal fragment structure containing the substrate binding and the nucleotide binding domains (PDB code: 2G76) were identified using the CAVITY program (Yuan et al., 2013; Yuan et al., 2011) and then applied to screen for potential allosteric inhibitors. The program Glide Standard Precise (SP) mode and Extra Precise (XP) mode were used to do the molecular docking studies and screen the SPECS library (Friesner et al., 2004; Halgren et al., 2004). The top 5% compounds from the XP mode were chosen for manual selection and purchased from SPECS for experimental testing.
Molecular cloning, protein expression and purification
The full-length PHGDH or PSAT1 open reading frame (Seajet Scientific, Beijing, China) was amplified by polymerase chain reaction (PCR), ligated into the pET21a(+) vector, transformed to the BL21 (DE3) strain of Escherichia coli (E. coli), and purified using a nickel-nitrilotriacetic column (HisTrap HP; GE Healthcare) and then a gel-filtration column (Sephacryl S-200 HR, GE Healthcare). For details, see the Supplemental Experimental Procedures.
Enzyme assay
Due to the unavailability of PHGDH direct-substrate phosphohydroxypyruvate (PHP), the enzyme activity of PHGDH was measured accompanied with the upstream of PSAT1 catalytic reaction (Hart et al., 2007). For details, see the Supplemental Experimental Procedures.
Surface Plasmon Resonance (SPR) experiments
The binding affinities of compounds towards PHGDH were assayed using the SPR-based Biacore T200 instrument (GE Healthcare). PHGDH was immobilized on a CM5 sensor chip by using standard amine-coupling at 25°C with 1× running buffer PBS-P (GE Healthcare), as described previously (Wang et al., 2014). For details, see the Supplemental Experimental Procedures.
Competition experiments
To investigate competition effects between the compounds and the cofactor NADH, we performed compound-cofactor competition experiments as follows:
Before Pser was added to start the reaction, the enzyme sample was pre-incubated with cofactor and the compound for 10 min at 25°C. The compound was kept at a constant inhibitory concentration (50 μM), while NADH concentration was gradually increased from 5 to 40 μM. At these concentrations, the compounds inhibited PHGDH activity by ∼50% when the NADH concentration was 150 μM.
Mutagenesis experiments
All mutagenesis experiments were carried out according to the instructions of the QuikChangeSite-Directed Mutagenesis (SBS Genetech Co., Beijing, China). The plasmid pET-21a(+)-containing wild-type (WT) PHGDH was mutated to obtain the mutants. The DNA sequences of all mutants were verified by DNA sequencing. The protein expression and activity assays of the mutants were performed as described for the WT.
Cell culture
MDA-MB-468, MDA-MB-231, and ZR-75-1 from China Infrastructure Cell Line Resources, and SKOV3 and HCC70 from ATCC were maintained in RPML-1640 culture medium (Gibco) supplemented with 10% fetal bovine serum and 1% penicillin/ml/streptomycin. MCF-7 from China Infrastructure Cell Line Resources and HEK293T from ATCC were maintained in Dulbecco's modified Eagle's medium (DMEM, Gibco) supplemented with 10% fetal bovine serum and 1% penicillin/ml/streptomycin. MCF-10A from China Infrastructure Cell Line Resources was maintained in DMEM/F12 (1:1) medium (Gibco) and supplemented with 5% horse serum, 10 μg/ml insulin, 0.1 μg/ml cholera toxin, 0.5 μg/ml hydrocortisone, and 0.02 μg/ml epidermal growth factor (EGF).
Proliferation Assays
SKOV3 GFP KO and PHGDH KO cells (10,000 cells/well) were plated into 24-well culture plates in triplicate. After 24 hours, cells were treated with DMSO or compound. Each day, cells were counted by trypan-blue exclusion test for cell viability at a 1:1 ratio using a hemocytometer.
MTT assays
MDA-MB-468 (5000 cells/well), HCC70 (5000 cells/well), MCF-7 (3000 cells/well), MDA-MB-231 (2000 cells/well), ZR-75-1 (4000 cells/well), and MCF-10A (3000 cells/well) in exponential growth were plated into 96-well culture plates and allowed to adhere overnight. The number of viable cells was assessed by spectrophotometry at 490 nm using a BioTek Synergy4 microplate reader after 3-days treatment, and calculated as the percentage of absorbance of treated cells relative to that of solvent controls.
For SKOV3 WT/KO cells (30,000 cells/well) were plated in a 96-well plate. The following day, media was aspirated and replaced with 100 μl phenol-red free RPMI-1640 (Gibco) and 12mM Methyl thiazolyldiphenyl-tetrazoliumbromide (MTT, Thermo Fisher Scientific) was added to the cells. After 4 hours, the media containing MTT was aspirated and 50 μl DMSO was added to dissolve the formazan and read at 540nm.
Synergistic experiments between PKUMDL-WQ-2101 and PKUMDL-WQ-2201 in enzymatic assays and cell-based assays
For enzymatic assay, one concentration among 0, 1, 5, 12.5, 25, 50, 100, 200 μM of PKUMDL-WQ-2101 was successively mixed with different concentrations of PKUMDL-WQ-2201 (0, 1, 5, 12.5, 25, 50, 100, 200 μM), and the mixture was then pre-incubated with enzyme samples to test their effects on PHGDH activity.
For cell based assay, MDA-MB-468 cells (5000 cells/well) were plated in 96-well plates, allowed to adhere overnight and incubated with the different combinations of PKUMDL-WQ-2101 (0, 0.1, 0.5, 1, 2.5, 5, 7.5, 10 μM) and PKUMDL-WQ-2201 (0, 0.1, 0.5, 1, 2.5, 5, 7.5, 10 μM) for three days. The EC50 values of the combinations were measured by MTT methods.
Flow cytometric analysis of cell cycle
MDA-MB-468 Cells (300, 000 cells/well) in exponential growth were plated into 6-well culture plates and then treated in triplicate with or without various concentrations of PKUMDL-WQ-2101 and PKUMDL-WQ-2201. After 24 h, cells were harvested by trypsinization and centrifugation, and then washed twice with 1× PBS, fixed in 70% ice-cold ethanol, and kept at 4°C overnight. The fixed cells were afterwards washed in 1× PBS and resuspended in 1× PBS containing 0.5% triton-x-100, 50 μg/ml Prodiumiodide (PI) and 50 μg/ml DNase-free RNase A. The cell suspension was incubated in the dark for 30 min at 37°C and analyzed using a BD FACSCanto™ cytometer.
Generation of CRISPR-Cas9 PHGDH Knockout Cells
LentiCRISPR transfer plasmid (Addgene Plasmid 49535), LentiCRISPR- EGFP sgRNA 1 (Addgene Plasmid 51760), PMD2.G VSV-G envelope expressing plasmid (Addgene Plasmid 12259), and PsPAX.2 lentiviral packaging plasmid (Addgene Plasmid 12260) were purchased. The target sequence of the sgRNA is GCTCTGAGCCTCCTTGGTGC (exon 8 of PHGDH). The plasmids were virally transfected into HEK293T cells using polyethylemine (PEI) (Polysciences, Inc) and transduced into SKOV3 cells as previously described (Shalem et al. 2014). Single-cell colonies of puromycin-resistant cells were selected and validated by western blotting.
Immunoblotting
Protein was extracted from cells using 1X RIPA buffer (Rockland Immunochemicals, Inc.) and centrifuged at 2000 rpm for 30 minutes at 4°C. Protein concentrations were measured using Bradford Protein Assay (Bio-Rad) and loaded onto 7.5% SDS-PAGE gels transferred to PVDF membranes. Membranes were blocked in 5% dry milk in TBST and incubated with anti-β-actin (Cell Signaling 8H10D10) 1:2000 or anti-PHGDH (Sigma-Aldrich WH0026227M1) 1:1000. Horseradish peroxidase conjugated anti-mouse (Rockland 611G4302), 1:2000 was used as secondary antibody. Chemiluminescent signals were detected with Clarity Western ECL Detection Kit (Bio-Rad) and imaged using a ChemiDoc MP System (Bio-Rad).
U-13C-glucose stable isotope labeling
SKOV3 cells (300,000 cells/well) were plated in a 6-well plate and allowed to adhere to the plate. Cells were then replaced with RPMI-1640 media containing 11mM U13C-glucose (Cambridge Isotope Laboratories, Inc.) and incubated for 24 hours. For U13C-glucose tracing with drug treatments, cells were first treated with their corresponding compounds for 24 hours, followed by media replacement with 11mM U 13C-glucose and corresponding drug treatment. Metabolites were then extracted.
Metabolite extraction
Metabolite extraction and subsequent Liquid-Chromatography coupled to High-Resolution Mass Spectrometry (LC-HRMS) for polar metabolites of HCT116 cells were carried out using a Q-ExactiveOrbitrap Plus as previously described(Liu et al., 2014). For details, see the Supplemental Experimental Procedures.
Peak extraction and data analysis
Raw data collected from LC-Q Exactive Plus MS is processed on Sieve 2.0 (Thermo Scientific). Peak alignment and detection are performed according to the protocol described by Thermo Scientific. For a targeted metabolite analysis, the method “peak alignment and frame extraction” is applied. An input file of theoretical m/z and detected retention time of 197 known metabolites is used for targeted metabolite analysis with data collected in positive mode, while a separate input file of 262 metabolites is used for negative mode. m/z width is set to 10 ppm. The output file including detected m/z and relative intensity in different samples is obtained after data processing. If the lowest integrated mass spectrometer signal (MS intensity) is less than 1000 and the highest signal is less than 10,000, then this metabolite is considered below the detection limit and excluded for further data analysis. If the lowest signal is less than 1000, but the highest signal is more than 10,000, then a value of 1000 is imputed for the lowest signals. Serine and glycine samples were normalized by comparing relative labeling of glucose-derived labeled metabolites from treated with vehicle samples. For all other samples, mass isotopomer distributions (MID) were calculated and samples were normalized by comparing the ratio of glucose-derived labeled metabolites to unlabeled metabolites within each sample. Quantitation and statistics were calculated using Microsoft Excel and GraphPad Prism 6.
MDA-MB-468 and MDA-MB-231 xenograft mouse models
PKUMDL-WQ-2101 and PKUMDL-WQ-2201 bioactivity assay in vivo - All animal experiments were performed in compliance with guidelines of the Animal Welfare Act and the guide for the care and use of laboratory animals following protocols approved by the Institutional Animals Care and Use Committee (IACUC). MDA-MB-468 or MDA-MB-231 cells were injected into the fourth mammary fat pad of NOD.CB17 Scid/J mice at 2×105 or 5×105 cells per injection site, respectively (Vital River Laboratory Animal Technology Co., Ltd., Beijing, China). For MDA-MB-468, when the average tumor volume reached 30 mm3, the mice were randomized into 7 groups (n=5): vehicle control (10%DMSO, 20% EL and 70% PBS, IP); 20, 10, and 5 mg/kg/day PKUMDL-WQ-2101 or PKUMDL-WQ-2201 (IP), respectively. For MDA-MB-231, after the tumor was palpable, the mice were randomized into 3 groups (n=5): vehicle control (10%DMSO, 20% EL and 70% PBS, IP); 20 mg/kg/day PKUMDL-WQ-2101 (IP); 20 mg/kg/day PKUMDL-WQ-2201 (IP). The tumor volume was calculated using the formula width (mm)2× length (mm) × 0.5.
Supplementary Material
Highlights.
Two previously unknown allosteric sites on PHGDH were identified.
Inhibitors that bind to these sites were discovered.
Inhibitors bound to PHGDH in cells and reduced the synthesis of serine and glycine.
Inhibitors suppressed tumor growth in mice.
Acknowledgments
This project was supported in part by the Ministry of Science and Technology and the National Natural Science Foundation of China (Grants 2015CB910300, 2016YFA0502303 and 21633001 to L.L.); by the National Institutes of Health (Grants R00CA168997 and R01CA193256 to J.W.L.), and a predoctoral fellowship award from the National Science Foundation (to M.V.L.) in the United States of America. The authors thank Dr. Min Fang in the College of Life Sciences, Peking University for his support and discussions on the cell based studies.
Footnotes
Author Contributions: L.L. and Q.W. conceived the project. Q.W., M.V.L., J.W.L. and L.L. designed the study and analyzed data. Q.W. performed allosteric site prediction and virtual screening studies, molecular cloning, mutagenesis, protein expression and purification experiments, enzymatic assays, SPR assays, cell based assays and in vivo studies. M.V.L. conducted CRISPR-Cas9 experiments, MTT assays, and breast cancer cell metabolomics. P.L., X.D., Y.L. contributed to the chemical synthesis and purity analysis of the compounds. Q.W., M.V.L., J.W.L. and L.L. wrote the manuscript. All the authors discussed the results and contributed to the writing and editing of the manuscript.
Publisher's Disclaimer: This is a PDF file of an unedited manuscript that has been accepted for publication. As a service to our customers we are providing this early version of the manuscript. The manuscript will undergo copyediting, typesetting, and review of the resulting proof before it is published in its final citable form. Please note that during the production process errors may be discovered which could affect the content, and all legal disclaimers that apply to the journal pertain.
References
- Ainscough EW, Brodie AM, Denny WA, Finlay GJ, Gothe SA, Ranford JD. Cytotoxicity of salicylaldehyde benzoylhydrazone analogs and their transition metal complexes: quantitative structure-activity relationships. J Inorg Biochem. 1999;77:125–133. doi: 10.1016/s0162-0134(99)00131-2. [DOI] [PubMed] [Google Scholar]
- Auld DS, Southall NT, Jadhav A, Johnson RL, Diller DJ, Simeonov A, Austin CP, Inglese J. Characterization of chemical libraries for luciferase inhibitory activity. J Med Chem. 2008;51:2372–2386. doi: 10.1021/jm701302v. [DOI] [PubMed] [Google Scholar]
- Baell JB, Holloway GA. New substructure filters for removal of pan assay interference compounds (PAINS) from screening libraries and for their exclusion in bioassays. J Med Chem. 2010;53:2719–2740. doi: 10.1021/jm901137j. [DOI] [PubMed] [Google Scholar]
- Beroukhim R, Mermel CH, Porter D, Wei G, Raychaudhuri S, Donovan J, Barretina J, Boehm JS, Dobson J, Urashima M, et al. The landscape of somatic copy-number alteration across human cancers. Nature. 2010;463:899–905. doi: 10.1038/nature08822. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Birsoy K, Wang T, Possemato R, Yilmaz OH, Koch CE, Chen WW, Hutchins AW, Gultekin Y, Peterson TR, Carette JE, et al. MCT1-mediated transport of a toxic molecule is an effective strategy for targeting glycolytic tumors. Nat Genet. 2013;45:104–108. doi: 10.1038/ng.2471. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Botham RC, Fan TM, Im I, Borst LB, Dirikolu L, Hergenrother PJ. Dual small-molecule targeting of procaspase-3 dramatically enhances zymogen activation and anticancer activity. J Am Chem Soc. 2014;136:1312–1319. doi: 10.1021/ja4124303. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Chen J, Chung F, Yang G, Pu M, Gao H, Jiang W, Yin H, Capka V, Kasibhatla S, Laffitte B, et al. Phosphoglycerate dehydrogenase is dispensable for breast tumor maintenance and growth. Oncotarget. 2013;4:2502–2511. doi: 10.18632/oncotarget.1540. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Chou TC. Theoretical basis, experimental design, and computerized simulation of synergism and antagonism in drug combination studies. Pharmacol Rev. 2006;58:621–681. doi: 10.1124/pr.58.3.10. [DOI] [PubMed] [Google Scholar]
- Clem BF, Clem AL, Yalcin A, Goswami U, Arumugam S, Telang S, Trent JO, Chesney J. A novel small molecule antagonist of choline kinase-alpha that simultaneously suppresses MAPK and PI3K/AKT signaling. Oncogene. 2011;30:3370–3380. doi: 10.1038/onc.2011.51. [DOI] [PMC free article] [PubMed] [Google Scholar]
- DeNicola GM, Chen PH, Mullarky E, Sudderth JA, Hu Z, Wu D, Tang H, Xie Y, Asara JM, Huffman KE, et al. NRF2 regulates serine biosynthesis in non-small cell lung cancer. Nat Genet. 2015;47:1475–1481. doi: 10.1038/ng.3421. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ding J, Li T, Wang X, Zhao E, Choi JH, Yang L, Zha Y, Dong Z, Huang S, Asara JM, Cui H, et al. The histone H3 methyltransferase G9A epigenetically activates the serine-glycine synthesis pathway to sustain cancer cell survival and proliferation. Cell Metab. 2013;18:896–907. doi: 10.1016/j.cmet.2013.11.004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Fan J, Ye J, Kamphorst JJ, Shlomi T, Thompson CB, Rabinowitz JD. Quantitative flux analysis reveals folate-dependent NADPH production. Nature. 2014;510:298–302. doi: 10.1038/nature13236. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Friesner RA, Banks JL, Murphy RB, Halgren TA, Klicic JJ, Mainz DT, Repasky MP, Knoll EH, Shelley M, Perry JK, et al. Glide: a new approach for rapid, accurate docking and scoring. 1 Method and assessment of docking accuracy. J Med Chem. 2004;47:1739–1749. doi: 10.1021/jm0306430. [DOI] [PubMed] [Google Scholar]
- Galluzzi L, Kepp O, Vander Heiden MG, Kroemer G. Metabolic targets for cancer therapy. Nat Rev Drug Discov. 2013;12:829–846. doi: 10.1038/nrd4145. [DOI] [PubMed] [Google Scholar]
- Halgren TA, Murphy RB, Friesner RA, Beard HS, Frye LL, Pollard WT, Banks JL. Glide: a new approach for rapid, accurate docking and scoring. 2 Enrichment factors in database screening. J Med Chem. 2004;47:1750–1759. doi: 10.1021/jm030644s. [DOI] [PubMed] [Google Scholar]
- Hanahan D, Weinberg RA. Hallmarks of cancer: the next generation. Cell. 2011;144:646–674. doi: 10.1016/j.cell.2011.02.013. [DOI] [PubMed] [Google Scholar]
- Hart CE, Race V, Achouri Y, Wiame E, Sharrard M, Olpin SE, Watkinson J, Bonham JR, Jaeken J, Matthijs G, et al. Phosphoserine aminotransferase deficiency: a novel disorder of the serine biosynthesis pathway. Am J Hum Genet. 2007;80:931–937. doi: 10.1086/517888. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Heigwer F, Kerr G, Boutros M. E-CRISP: fast CRISPR target site identification. Nat Met. 2014;11:122–123. doi: 10.1038/nmeth.2812. [DOI] [PubMed] [Google Scholar]
- Kar G, Keskin O, Gursoy A, Nussinov R. Allostery and population shift in drug discovery. Curr Opin Pharmacol. 2010;10:715–722. doi: 10.1016/j.coph.2010.09.002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Liu X, Ser Z, Locasale JW. Development and quantitative evaluation of a high-resolution metabolomics technology. Anal Chem. 2014;86:2175–2184. doi: 10.1021/ac403845u. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Locasale JW. Serine, glycine and one-carbon units: cancer metabolism in full circle. Nat Rev Cancer. 2013;13:572–583. doi: 10.1038/nrc3557. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Locasale JW, Grassian AR, Melman T, Lyssiotis CA, Mattaini KR, Bass AJ, Heffron G, Metallo CM, Muranen T, Sharfi H, et al. Phosphoglycerate dehydrogenase diverts glycolytic flux and contributes to oncogenesis. Nat Genet. 2011;43:869–874. doi: 10.1038/ng.890. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ma L, Tao Y, Duran A, Llado V, Galvez A, Barger JF, Castilla EA, Chen J, Yajima T, Porollo A, Medvedovic M, et al. Control of nutrient stress-induced metabolic reprogramming by PKCzeta in tumorigenesis. Cell. 2013;152:599–611. doi: 10.1016/j.cell.2012.12.028. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ma X, Meng H, Lai L. Motions of Allosteric and Orthosteric Ligand-Binding Sites in Proteins are Highly Correlated. J Chem Inf Model. 2016;56:1725–1733. doi: 10.1021/acs.jcim.6b00039. [DOI] [PubMed] [Google Scholar]
- Mali P, Yang L, Esvelt KM, Aach J, Guell M, DiCarlo JE, Norville JE, Church GM. RNA-guided human genome engineering via Cas9. Science. 2013;339:823–826. doi: 10.1126/science.1232033. [DOI] [PMC free article] [PubMed] [Google Scholar]
- McGovern SL, Caselli E, Grigorieff N, Shoichet BK. A common mechanism underlying promiscuous inhibitors from virtual and high-throughput screening. J Med Chem. 2002;45:1712–1722. doi: 10.1021/jm010533y. [DOI] [PubMed] [Google Scholar]
- Meng H, McClendon CL, Dai Z, Li K, Zhang X, He S, Shang E, Liu Y, Lai L. Discovery of Novel 15-Lipoxygenase Activators To Shift the Human Arachidonic Acid Metabolic Network toward Inflammation Resolution. J Med Chem. 2016;59:4202–4209. doi: 10.1021/acs.jmedchem.5b01011. [DOI] [PubMed] [Google Scholar]
- Mentch SJ, Mehrmohamadi M, Huang L, Liu X, Gupta D, Mattocks D, Gomez Padilla P, Ables G, Bamman MM, Thalacker-Mercer AE, et al. Histone Methylation Dynamics and Gene Regulation Occur through the Sensing of One-Carbon Metabolism. Cell Metab. 2015;22:861–873. doi: 10.1016/j.cmet.2015.08.024. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Merdanovic M, Monig T, Ehrmann M, Kaiser M. Diversity of allosteric regulation in proteases. ACS Chem Biol. 2013;8:19–26. doi: 10.1021/cb3005935. [DOI] [PubMed] [Google Scholar]
- Mullarky E, Lucki NC, Beheshti Zavareh R, Anglin JL, Gomes AP, Nicolay BN, Wong JC, Christen S, Takahashi H, Singh PK, et al. Identification of a small molecule inhibitor of 3-phosphoglycerate dehydrogenase to target serine biosynthesis in cancers. Proc Natl Acad Sci USA. 2016;113:1778–1783. doi: 10.1073/pnas.1521548113. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Nussinov R, Tsai CJ. Unraveling structural mechanisms of allosteric drug action. Trends Pharmacol Sci. 2014;35:256–264. doi: 10.1016/j.tips.2014.03.006. [DOI] [PubMed] [Google Scholar]
- Ou Y, Wang SJ, Jiang L, Zheng B, Gu W. p53 Protein-mediated regulation of phosphoglycerate dehydrogenase (PHGDH) is crucial for the apoptotic response upon serine starvation. J Biol Chem. 2015;290:457–466. doi: 10.1074/jbc.M114.616359. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Pacold ME, Brimacombe KR, Chan SH, Rohde JM, Lewis CA, Swier LJ, Possemato R, Chen WW, Sullivan LB, Fiske BP, et al. A PHGDH inhibitor reveals coordination of serine synthesis and one-carbon unit fate. Nat Chem Biol. 2016;12:452–458. doi: 10.1038/nchembio.2070. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Pavlova NN, Thompson CB. The Emerging Hallmarks of Cancer Metabolism. Cell Metab. 2016;23:27–47. doi: 10.1016/j.cmet.2015.12.006. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Pei J, Yin N, Ma X, Lai L. Systems biology brings new dimensions for structure-based drug design. J Am Chem Soc. 2014;136:11556–11565. doi: 10.1021/ja504810z. [DOI] [PubMed] [Google Scholar]
- Possemato R, Marks KM, Shaul YD, Pacold ME, Kim D, Birsoy K, Sethumadhavan S, Woo HK, Jang HG, Jha AK, et al. Functional genomics reveal that the serine synthesis pathway is essential in breast cancer. Nature. 2011;476:346–350. doi: 10.1038/nature10350. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Qi Y, Wang Q, Tang B, Lai L. Identifying Allosteric Binding Sites in Proteins with a Two-State Go Model for Novel Allosteric Effector Discovery. J Chem Theory Comput. 2012;8:2962–2971. doi: 10.1021/ct300395h. [DOI] [PubMed] [Google Scholar]
- Shalem O, Sanjana NE, Hartenian E, Shi X, Scott DA, Mikkelsen TS, Heckl D, Ebert BL, Root DE, Doench JG. Genome-scale CRISPR-Cas9 knockout screening in human cells. Science. 2014;343:84–87. doi: 10.1126/science.1247005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sonveaux P, Vegran F, Schroeder T, Wergin MC, Verrax J, Rabbani ZN, De Saedeleer CJ, Kennedy KM, Diepart C, Jordan BF, et al. Targeting lactate-fueled respiration selectively kills hypoxic tumor cells in mice. J Clin Invest. 2008;118:3930–3942. doi: 10.1172/JCI36843. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Turnbull AP, Salah E, Savitsky P, Gileadi O, von Delft F, Edwards A, Arrowsmith C, Weigelt J, Sundstrom M, Oppermann U. Crystal structure of human 3-phosphoglycerate dehydrogenase. 2006 [Online]. Available: http://www.rcsb.org/pdb/explore/explore.do?structureId=2g76.
- Unterlass JE, Basle A, Blackburn TJ, Tucker J, Cano C, Noble ME, Curtin NJ. Validating and enabling phosphoglycerate dehydrogenase (PHGDH) as a target for fragment-based drug discovery in PHGDH-amplified breast cancer. Oncotarget. 2016 doi: 10.18632/oncotarget.11487. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wagner JR, Lee CT, Durrant JD, Malmstrom RD, Feher VA, Amaro RE. Emerging Computational Methods for the Rational Discovery of Allosteric Drugs. Chem Rev. 2016;116:6370–6390. doi: 10.1021/acs.chemrev.5b00631. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wang Q, Qi Y, Yin N, Lai L. Discovery of novel allosteric effectors based on the predicted allosteric sites for Escherichia coli D-3-phosphoglycerate dehydrogenase. PLoS One. 2014;9:e94829. doi: 10.1371/journal.pone.0094829. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Warburg O. On the origin of cancer cells. Science. 1956;123:309–314. doi: 10.1126/science.123.3191.309. [DOI] [PubMed] [Google Scholar]
- Yamada K, Hara N, Shibata T, Osago H, Tsuchiya M. The simultaneous measurement of nicotinamide adenine dinucleotide and related compounds by liquid chromatography/electrospray ionization tandem mass spectrometry. Anal Biochem. 2006;352:282–285. doi: 10.1016/j.ab.2006.02.017. [DOI] [PubMed] [Google Scholar]
- Yang Q, Yang Y, Li L, Sun W, Zhu X, Huang Y. Polymeric nanomedicine for tumor-targeted combination therapy to elicit synergistic genotoxicity against prostate cancer. ACS Appl Mater Interfaces. 2015;7:6661–6673. doi: 10.1021/am509204u. [DOI] [PubMed] [Google Scholar]
- Yuan Y, Pei J, Lai L. LigBuilder 2: a practical de novo drug design approach. J Chem Inf Model. 2011;51:1083–1091. doi: 10.1021/ci100350u. [DOI] [PubMed] [Google Scholar]
- Yuan Y, Pei J, Lai L. Binding site detection and druggability prediction of protein targets for structure-based drug design. Curr Pharm Des. 2013;19:2326–2333. doi: 10.2174/1381612811319120019. [DOI] [PubMed] [Google Scholar]
- Zhao E, Ding J, Xia Y, Liu M, Ye B, Choi JH, Yan C, Dong Z, Huang S, Zha Y, et al. KDM4C and ATF4 Cooperate in Transcriptional Control of Amino Acid Metabolism. Cell Rep. 2016a;14:506–519. doi: 10.1016/j.celrep.2015.12.053. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.