

Abstract
The ability to target DNA specifically at any given position within the genome allows many intriguing possibilities and has inspired scientists for decades. Early gene-targeting efforts exploited chemicals or DNA oligonucleotides to interfere with the DNA at a given location in order to inactivate a gene or to correct mutations. We here describe an example towards correcting a genetic mutation underlying Pompe's disease using a nucleotide-fused nuclease (TFO-MunI). In addition to the promise of gene correction, scientists soon realized that genes could be inactivated or even re-activated without inducing potentially harmful DNA damage by targeting transcriptional modulators to a particular gene. However, it proved difficult to fuse protein effector domains to the first generation of programmable DNA-binding agents. The engineering of gene-targeting proteins (zinc finger proteins (ZFPs), transcription activator-like effectors (TALEs)) circumvented this problem. The disadvantage of protein-based gene targeting is that a fusion protein needs to be engineered for every locus. The recent introduction of CRISPR/Cas offers a flexible approach to target a (fusion) protein to the locus of interest using cheap designer RNA molecules. Many research groups now exploit this platform and the first human clinical trials have been initiated: CRISPR/Cas has kicked off a new era of gene targeting and is revolutionizing biomedical sciences.
This article is part of a discussion meeting issue ‘Frontiers in epigenetic chemical biology’.
Keywords: CRISPR/dCas, TALEs (transcription activator-like effectors), ZFPs (zinc finger proteins), polyamides, genome editing
1. Introduction
Most diseases are associated with genetic mutations or gene expression abnormalities. The ability to introduce or correct mutations and/or to restore gene expression of any given gene therefore would allow researchers to investigate the origins of diseases, and might even provide novel therapeutic options. The concept of introducing or correcting genetic mutations relies on the fact that double strand breaks in a living cell will induce mechanisms to repair the break. Depending on e.g. the cell cycle and chromatin context [1,2], the cell will choose to repair the damage by non homologous end joining, which can induce insertions/deletions often resulting in inactivation of the gene. The alternative repair pathway is via homologous recombination, in which the damaged allele is exchanged with a homologous template. By introducing DNA damage at a given location in the human genome, one can thus correct mutations or introduce sequences of interest.
Introduction of DNA damage specifically at one genomic locus requires programmable DNA-binding platforms, which can be used as gene-targeting devices to locate a nuclease to this genomic position. Next to fusing nucleases, several attempts have been made to modulate gene expression by fusing expression activators or repressors to these sites [3–6]. Both approaches generate research tools to address the functional effects of the many genetic and epigenetic differences that have been detected by numerous large scale -omics screening efforts for a broad spectrum of diseases. If specificity and effective delivery are established, these approaches open novel therapeutic possibilities. In this review, we provide a historic overview of the different gene-targeting platforms and describe an example of early day's gene correction for Pompe's disease. We will highlight in vivo successes of every platform and provide examples with respect to epigenetic editing. In epigenetic editing, DNA-binding platforms are engineered to target an epigenetic writer or erasers to a given location in the genome [4]. Rewriting the epigenetic landscape modulates gene expression [5,6] and some indications of sustained reprogramming has been achieved [7,8], although more research needs to be performed to understand the influence of the native chromatin context of the targeted gene [8,9]. Epigenetic reprogramming is less invasive compared to genome engineering, and it also allows re-expression of silenced genes. Altogether, epigenetic editing provides a promising exploitation of the gene targeting toolbox.
2. First generation: chemical (polyamides) and DNA (TFOs) gene-targeting platforms
Researchers have been engineering various platforms to target a given sequence within the human genome for decades. In the early days, prokaryotic DNA-binding domains were used in combination with their recognition sequences, artificially introduced into the genome (e.g. the LacO–LacR system) to obtain insights into e.g. gene expression regulation. As polyamides and triplex forming oligonucleotides (TFOs) allowed the binding of endogenous sequences [3], the engineering of these chemical agents provided new tools for biologists.
Polyamides (pAs) are small synthetic molecules that bind in the minor groove of DNA. The minor groove then widens and the major groove bends. This blocks transcription factor binding and thereby pAs can inhibit gene transcription by themselves. Stretches of polyamides (hydroxypyrrole (Hp), imidazole (Im) and pyrrole (Py)) can be engineered to target specific sequences by recognizing Watson–Crick base pairs: Py/Im to bind C–G; Py/Hp for A–T, Hp/Py for T–A and Im/Py pairs to bind G–C base pairs [10]. Hp degrades quickly in the presence of free radicals and acids, which limits the amount of Hp-containing pAs that are being investigated. Instead, a Py/Py pair can be used to bind both A–T and T–A pairs but it cannot distinguish the two base pairs, which further limits the applicability of pAs [11,12]. High molecular weight and a high concentration of Im decreased nuclear localization as shown by treating cells with fluorescently labelled pAs, which limits the targeting of C–G-rich promoter sequences in vivo [13,14]. Modifications, such as aryl-turns, can improve nuclear localization [13], and other improvements are being explored. If efficient delivery is assured, pAs can for example be fused to inhibitors of epigenetic enzymes, and as such successfully modulate gene expression [15]. Fusing protein effector domains to these agents, although technically challenging, is also possible [16], but to our knowledge no catalytic epigenetic modulators were fused to pAs.
The potency of pAs to modulate gene expression by themselves has been demonstrated in various animal studies. In one of such in vivo studies, a Py–Im polyamide targeting eight base pairs in the promoter of transforming growth factor (TGF)-β1 could reduce scar formation in a marmoset model [17]. Microarray analysis showed that treatment with a similar polyamide targeting seven base pairs in the promoter of TGF-β1 in rats changed the expression of only 3% of the 30 000 tested transcripts more than twofold, mostly associated with the injury model [18]. Other Py–Im polyamides tested in vivo were described to inhibit for example, ABCA1, TGF-β1, androgen response element driven and HIF-1 gene expression [19–22]. Despite such successes, engineering of polyamides requires specialized laboratories and difficulties of fusing effector domains limit their general applicability [23].
Triplex-forming oligonucleotides (TFOs) are relatively easy to design and effector domains can be fused, although this can also be challenging [24]. TFOs bind the oligopurine-rich strand in oligopurine–oligopyrimidine stretches of DNA via Hoogsteen hydrogen bonds in the major groove and as such can induce site-specific mutagenesis or influence gene expression [24]. The need to bind to oligopurine-rich sequences limits the choice of targets for TFOs [3,25]. However, there is a overrepresentation of TFO target sites in promoter regions of the human genome, which mitigates this limitation [26]. Rules to design oligonucleotides (TFO) forming triple helices have been well established. The corresponding nucleotide sequence is straightforward, while the choice of the chemical modifications of the nucleotide that best increase the stability of the triple helix needs to be further validated experimentally [25]. To exploit TFO for epigenetic editing, we fused a M.SssI variant to a TFO and demonstrated CpG-specific methylation [27]. Although fusion of peptide effectors to TFOs is technically challenging, few other examples have been reported, e.g. the transcriptional activator VP16 [28] and restriction enzymes, of which the activity was controlled by photocaging [29]. TFOs are thought to be very specific, as a single mismatch can abrogate the formation of a triple helix [30–32]. To our knowledge, large scale studies into the specificity of TFOs however have yet to be performed. Interestingly, naked TFOs designed to bind oestrogen receptor binding sites did not seem to be hampered by the chromatin status for triplex formation [33]. To overcome stability issues of TFOs, peptide nucleic acids (PNAs) have been developed as a variant to TFOs. PNAs have normal DNA bases, but these are coupled to an uncharged peptide backbone [34].
One of the first successful in vivo applications of TFOs for gene modification was shown by Vasquez et al. [35]. A TFO targeting the mouse supFG1 gene demonstrated a high specificity and affinity to the target and introduced site-specific mutations, while the control scrambled TFO did not. Although the efficiency of mutagenesis was quite low, these results do prove that TFOs can modify DNA in living organisms [35]. This TFO has also been proven to localize in the nucleus and induced mutations in supFG1 in multiple cell types upon intradermal administration [36]. In other studies, TFOs were successfully targeted to inhibit oncogene expression in mouse models of human cancer (targeting P53 to inhibit colon carcinoma [37], c-MYC in fusion with gemcitabine to inhibit colon cancer cells [38]). TFOs have also been used with success to inactivate gene expression by targeting DNA cleaving agents such as topoisomerase I and II inhibitors, to inactivate either oncogenes such as IGF1 and its receptor in prostate cancer cells models [39], or the MDR gene to impair drug resistance [40].
Apart from for gene inactivation, targeting DNA-damaging agents to DNA would allow the correction of genetic mutations, which for many genetic diseases would be a promising approach to a cure. A genetic disease gaining massive attention these days because of the costs of its treatment is Pompe disease. Pompe disease is a rare, though severe autosomal lysosomal storage disorder, caused by a mutation in the gene encoding the lysosomal enzyme acid-α-glucosidase (GAA). This autosomal recessive disorder shows a broad spectrum in clinical presentation as a result of glycogen storage in the lysosome, which can eventually cause early death of the patient. The only available therapy is enzyme replacement therapy (ERT), which is based on compensating the lack of GAA by administering recombinant produced enzyme intravenously to the patient leading to prolongation of life. This ERT costs between 400 000 and 700 000 Euros annually per patient. Although ERT ensures greater patient longevity, it does not cure the underlying genetic defect.
As GAA is a secreted enzyme, we reasoned that correction of the GAA mutation in secreting cells using the gene correction strategy (by introducing double strand breaks) would be a very promising strategy to actually cure the disease. For this purpose, we designed several experiments to explore the potential of TFOs coupled to DNA-damaging agents to induce homologous recombination (HR) for gene correction [41]. In short, TFOs were engineered to target the mouse GAA gene or the human GAA gene. The TFOs were coupled to a restriction endonuclease or to camptothecin. Co-transfection of a TFO-MunI fusion with a promoter-less luciferase reporter plasmid and a promoter-containing HR correction fragment revealed that we could introduce HR as luciferase activity was induced (figure 1). Binding affinity testing demonstrated that the TFO targeting the human GAA gene could bind its target with high affinity, but the TFO targeting the mouse GAA gene showed weak triplex binding (figure 2). Assessment of DNA damage (figures 3 and 4), indeed, could detect damage on nuclear DNA for the TFO coupled to camptothecin, which targeted the human GAA gene. Subsequent delivery of the human TFO-camptothecin construct in healthy human fibroblasts, however, failed to convincingly indicate successful targeting of the GAA gene since no lowering of GAA activity could be detected. Although GAA has a relatively long half-life, these observations prevented us from testing the constructs in patient material.
Figure 1.
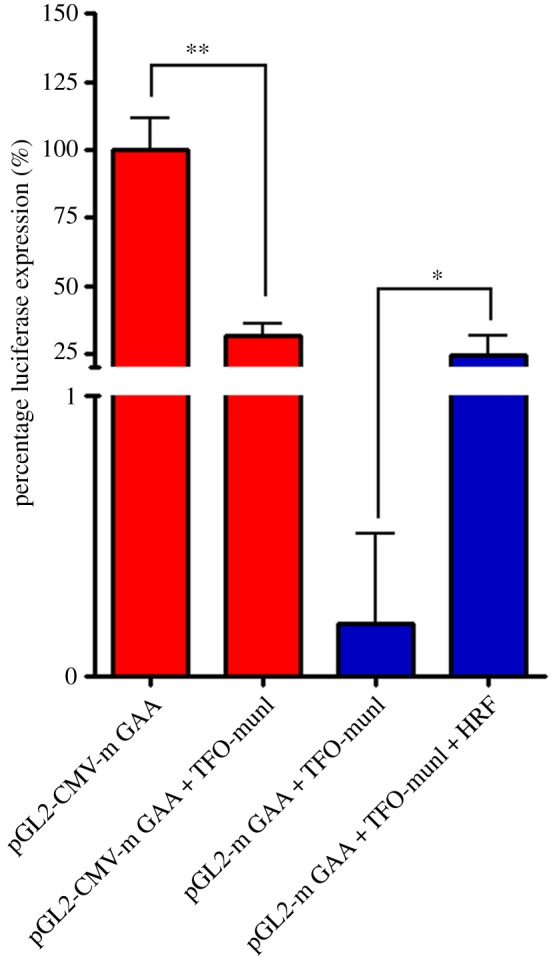
Co-transfection of reporter plasmids with mTFO-MunI allowed MunI digestion of the murine GAA (mGAA) gene and induced HR. A2780 cells were co-transfected with reporter plasmids and 0.25 µM TFO-conjugate in the absence or presence of a HR fragment. Total amount of DNA was 300 ng in each well. After 48 h, luciferase activity was determined. The luciferase expression of pGL2-CMV-mGAA was set at 100% for each independent experiment. Transfections were performed in triplicate for two independent experiments. One experiment is shown as representative (mean + s.d.). *, p < 0.05, **, p < 0.01. (Online version in colour.)
Figure 2.

Surface plasmon resonance (SPR analysis) of TFO binding to immobilized DNA targets at 37°C in MES buffer, pH 6.5. The sensorgrams show complex formation following injection of 35 ml 4 mM TFO (indicated by a black horizontal bar; (a) TFO-GU (targeting VEGF); (b) TFO-TM (targeting VEGF); (c) mTFO (targeting mGAA); (d) hTFO (targeting human GAA (hGAA)) over surfaces containing immobilized biotinylated hGAA (green line), mGAA (blue line) and VEGF (red line) target hairpin duplexes, after which dissociation of the complexes is monitored [41].
Figure 3.
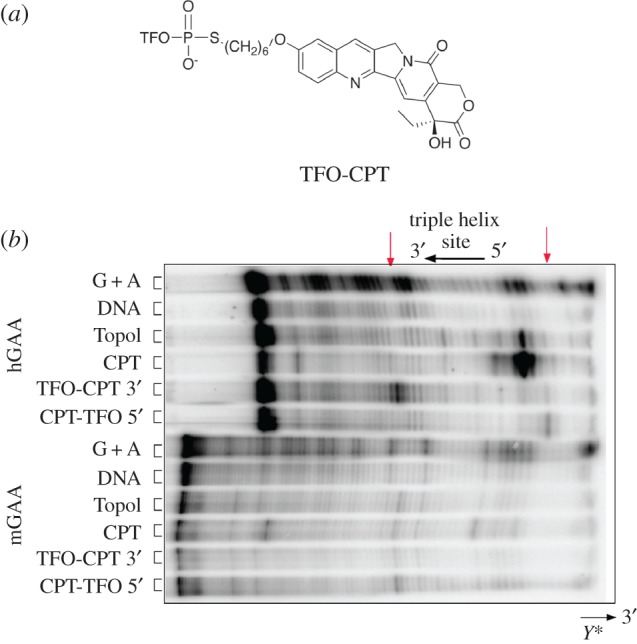
Topoisomerase I-mediated DNA cleavage to study the specificity of TFO-camptothecin (CPT) conjugates. (a) Structure of the TFO-CPT conjugate used in this study. (b) Cleavage products mediated by topoisomerase I (Topo I). Adenine/guanine-specific Maxam-Gilbert chemical cleavage reactions were used as markers (lane G+A). Target duplexes were incubated with 10 units of Topo I and in the presence of 50 µM CPT, or 5 µM TFO-CPT conjugates. The orientation of the triplex is indicated. Arrows indicate Topo I-mediated DNA cleavage at the target site (TFO-CPT 3′ cleaves at the 3′ end of the triple helix and TFO-CPT 5′ cleaves at the 5′ end of the triple helix). (Online version in colour.)
Figure 4.

Induction of DNA damage by TFO-CPT or scPvuII after transfection. A2780 cells were treated with 2 µM of the hTFO-CPT conjugate (and hTFO for control) or 1 µg of scPvuII. After 3 h, cells were collected and stained for phosphorylated H2AX (γH2AX) and analyzed by FACS. Transfections were performed in duplicate for three independent experiments. Data are represented as means of the means + s.e.m. Statistical comparison was performed between cells treated with Saint Mix only and cells treated with TFO only, hTFO-CPT or scPvuII. *, p < 0.05, **, p < 0.01. The chemical conjugation of the TFO to CPT has been described in detail elsewhere (see Vekhoff et al. [43]. In short, 3′ thiophosphorylated TFOs were conjugated to 10-(6-bromohexyloxy)-CPT and purified by reversed-phase high performance liquid chromatography (HPLC): hTFO-CPT: retention time 18.6 min; mTFO-CPT: retention time 18.4 min. The conjugates were characterized by UV spectroscopy, denaturing polyacrylamide gel electrophoresis and mass spectroscopy (ES-MS). ES-MS: hTFO-CPT: found [M-H] 4621.74; calculated: 4621.1. mTFO-CPT: found [M-H] 6988.24; calculated: 6988.8. SM, the delivery agent Saint Mix; scPvuII, single chain PvuII.
Correcting the genetic defect in Pompe disease at an early stage in development could actually provide a cure for Pompe disease, while correcting the gene in secreting cells at later stage would contribute to a decrease in the current ERT costs. Alternative to gene correction might be to upregulate the expression of GAA. Especially in patients with an adult onset phenotype who still have residual GAA enzyme activity this might be an alternative approach to explore. In this respect, Raben et al. [44] showed a dramatic increase in the levels of correctly spliced GAA mRNA after removal of approximately 90% of intron 1 (2.6 kb) sequence, indicating that the intron may contain a powerful transcriptional repressor. DNA targeting platforms, when optimized for delivery and specificity, thus provide two new avenues to be explored for Pompe's disease: correction of the genetic mutation or, for patients with residual activity, upregulation of GAA gene expression.
3. Second generation: protein-based gene-targeting platforms (ZFPs, TALEs)
As the chemical gene-targeting approaches also face difficulties, in fusing protein effector domains, these platforms are not developing rapidly and the majority of in vivo data have been obtained for the agents alone. However, to fully exploit the promise of gene targeting, fusion of either a nuclease or a transcriptional modulator would improve flexibility and effectivity. Protein-based gene-targeting approaches allow for straightforward fusion of any protein effector domain and have been explored both for introducing DNA damage as well as for gene expression modulation. The first platform in this respect is based on the most abundant family of mammalian transcription factors: zinc fingers proteins (ZFPs). ZFPs are relatively small proteins that facilitate effective delivery and heterochromatin access, and they can be based on human proteins, which further benefits in vivo applications as they would likely face low immunogenicity [45]. By the end of the past century, codes were developed to engineer ZFPs for almost every gene of interest [46]. As fusion of an effector is straightforward, this platform received attention from the biotechnological research community and several clinical trials have been initiated [47,48].
Although aspecific binding patterns were described for engineered ZFPs in ChIP-seq studies [49,50], some indications of single gene specificity have been reported for engineered zinc-finger proteins [51,52]. Interestingly, one of the first clinical ZFP trials explored the possibility of targeting a transcriptional activator to induce VEGF-A gene expression to promote revascularization [53]. This was tested in patients with peripheral diabetic neuropathy in two clinical trials. Phase II studies were halted as there was little difference between the treated and placebo group [53–55]. Although several animal models indicate effectivity of ZF-induced expression modulation [51,56,57], no other clinical ZF trial has been initiated to modulate gene expression. Many clinical trials, however, have been designed based on ZFP genome editing and involve for example HIV treatment. Here, a ZF-nuclease is designed to inactivate the human gene CCR5: HIV initially enters cells via this receptor, and infection is prevented if the gene is mutated [58]. Indeed, gene-modified cells were shown to have a survival advantage during treatment interruption [58]. As an effector, FokI is an often used nuclease that can induce double strand breaks upon dimerization: using FokI as a DNA damage inducer thus requires two DNA-binding domains to target neighbouring sites, which has the added advantage of increasing specificity of the approach [59].
Although the ZF community focused on targeting nucleases to specific genomic sites, the very first epigenetic editing study exploited this platform for targeting epigenetic enzymes (histone K9 methyltransferases Suvar and G9A) to a genomic locus (VEGF-A) [60]. It took ten years before this proof of concept study was followed up by us [50] and by others who even demonstrated efficacy in vivo [61]. In parallel, the first ZF-targeted DNA methylation studies also demonstrated feasibility and effective gene repression [62,63], although the sustainability of the effect is not clear [7,9].
Despite the genetic recognition code, ZFPs suffered from context dependency (the modular fingers do not bind independently of one another), which might explain gene aspecificity [49] and affects ease-of-design. The introduction of transcription activator-like effectors (TALEs) circumvented this complication as a TALE moiety is known for each of the four possible base pairs [64]. Indeed, TALEs seemed to surpass the use of ZFPs because of their straightforward one-to-one binding approach. However, TALEs are quite big and the high number of repeats can cause assembly problems and aggregation [65], although the latter can be prevented by the fusion of thioredoxin [66].
In vivo TALE studies are mainly based on viral delivery of the FokI-fusion constructs and have resulted in some therapeutic effects for animal models of e.g. cervical cancer, hepatitis and X-linked hyper-immunoglobulin M syndrome [67–69]. Interestingly, transcription activator-like effector nucleases (TALENs) were used to edit T cells from a HLA-mismatched healthy human donor ex vivo, to produce universal T cells. These cells were then used to successfully treat two infants with leukaemia until they could undergo allogeneic stem cell transplantation [70]. Also in vivo epigenetic editing using TALEs was recently shown in a mouse prostate cancer model. Here the methylation or demethylation of metastasis suppressor gene CRMP4 in the tumour cells respectively induced and repressed metastasis in mice [71]. Furthermore, in Drosophila, expression of the eve gene was repressed and activated by TALEs fused to different effector domains [72]. Interestingly, a clinical study is planned to start in 2018 on cervical cancer [73], and its outcome is of great interest to the field.
4. Third generation: RNA-based (CRISPR/Cas) gene-targeting platforms
In 2012, the CRISPR-Cas system was introduced as a highly efficient, easy, cheap and flexible method for genome editing, transcriptional perturbation, epigenetic modulation and genome imaging. CRISPR-Cas, first discovered in prokaryotes as an adaptive immunity system, is based on RNA-guides that target the CRISPR-associated nuclease (Cas9 protein) to specific DNA sequences. The Cas9 protein, serving as a cleavage mediator, forms a complex with the CRISPR RNA and a transactivating RNA, both fused to form a chimeric single guide RNA (sgRNA) that acts as the sequence recognizer [74,75]. Within the sgRNA, 20 nucleotides can be re-engineered in order to achieve efficient binding of any given stretch in eukaryotic genomes, allowing versatile targeted modifications [76]. CRISPR/Cas is often presented as a revolution in biomedical research and receives a lot of attention due to its promise regarding therapeutic potential [77–79].
Nowadays, many research groups have adapted CRISPR-Cas technology and demonstrated the potential of this RNA-guided platform in numerous in vivo studies: On one hand, CRISPR-induced gene disruptions allow relatively easy construction of (inducible) animal disease models [70]. On the other hand, successes in preclinical models, including metabolic disorders and muscular dystrophies, indicate therapeutic relevance [79]. Also for severe neurodegenerative conditions such as Huntington's disease, expression levels of the mutant allele could be reduced with therapeutically relevant efficiency [72]. The vast majority of the therapeutic attempts exploit adeno-associated viruses (AAV) for delivery. Nevertheless, to circumvent problems associated with this delivery technique (such as the packaging limit of 4.5 kb for transgenes), other delivery methods are exploited including direct delivery of CRISPR components to the liver via hydrodynamic transfection [80].
The CRISPR-Cas platform is the only DNA targeting platform that has intrinsic nuclease activity. For other applications such as epigenetic editing, this nuclease activity can be easily abolished while the resulting dead Cas (dCas) still functions as gene-targeting platform. Upon direct fusion of effector domains to dCas, imaging of genetic loci or gene expression modulation and reprogramming by epigenetic editing is feasible. The flexibility of the CRISPR-dCas systems also allows us to interrogate the effects of the microchromatin on sustained reprogramming and such insights are essential to obtain e.g. stable re-expression of epigenetically silenced genes [8]. Indirect recruiting of effector domains, through for example MS2 aptamers as extension to sgRNA molecules [69], is even further improving the flexibility of this platform. Within this third generation RNA-based platform, epigenetic editing has received widespread acceptance and many further improvements are to be expected, including (but not limited to) guide RNAs as scaffolds, Cas variants and delivery methods.
In this respect, strategies aiming at controlling gene expression in a spatial and temporal way by dCas9 constructs are of great interest. Some groups have recently exploited optogenetics by using light-induced peptide heterodimerization in order to create light-activated dCas9-effectors [81] that are able to recruit VP64 or p65AD to dCas9 by means of a cryptochrome-based blue light-sensing system. Other approaches exploit rapamycin-dependent dimerization of a split dCas9-VP64 as a chemically inducible system capable of activating endogenous loci [82]. These optogenetically and chemically inducible techniques expand dCas9-based regulation and can achieve accurate spatial and temporal gene expression programmes in in vivo models [74].
Although there is not yet extensive knowledge of manipulation of the epigenome in vivo [83,84], encouraging results using CRISPR-based systems have been obtained. Morita et al., for example, demonstrated targeted demethylation in in vivo mouse fetuses [85], while Liu et al. successfully induced DNA methylation in mice by zygote microinjection. Also upon lentiviral transduction of dCas9-TET tools in mouse brain, successful demethylation of an integrated reporter was obtained [86].
CRISPR-based systems still need to be improved in order to overcome some hurdles, e.g. off-target effects [87] and efficient and specific delivery. However, reported successes obtained for preclinical models induced by CRISPR-Cas and dCas [84] seem to outweigh these downsides that will hopefully be resolved soon. The ongoing progress in gene targeting allows the scientific community to look forward to a very bright future in respect of (epi)genome engineering.
5. Conclusion and perspectives
Gene targeting has been attempted for several decades using for example pAs or TFOs, but widespread application of these platforms was hampered by technical difficulties. Protein engineering facilitated the addition of effector domains, and zinc finger fusion proteins even made it into clinical trials. The TALE platform further facilitated DNA-binding codes, but seems outcompeted by the ease and low costs of sgRNA design, at least for research purposes. With the introduction of the CRISPR platform, the broad spectrum of possibilities of gene targeting can now be fully explored by any molecular biology laboratory. Although the translation to the clinic greatly depends on the outcome of clinical trials [73] and optimization of suitable delivery systems, gene-targeting efforts including epigenetic editing [88] are expected to open up avenues for various currently incurable diseases [79].
Acknowledgements
The authors wish to acknowledge P.M.J. McLaughlin, H.G. Kazemier, R.J. van der Lei of the University Medical Center Groningen, the Netherlands for their contributions. The COST action CM1406 (www.EpiChemBio.eu) is acknowledged for facilitating collaborations.
Data accessibility
This article has no additional data.
Competing interests
We declare we have no competing interests.
Funding
TFO data for Pompe disease were derived in the framework of an EU FP6 program, activity NEST (contract number 015509), in collaboration with A. Pingoud of Institute of Biochemistry, Justus-Liebig-University Giessen, Germany (who sadly passed away) and V. Siksnys, A. Šilanskas and M. Zaremba of the Institute of Biotechnology, Vilnius University, Lithuania.
References
- 1.Brandsma I, Gent DC. 2012. Pathway choice in DNA double strand break repair: observations of a balancing act. Genome Integr. 3, 9 ( 10.1186/2041-9414-3-9) [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Clouaire T, Legube G. 2015. DNA double strand break repair pathway choice: a chromatin based decision? Nucleus 6, 107–113. ( 10.1080/19491034.2015.1010946) [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Uil TG, Haisma HJ, Rots MG. 2003. Therapeutic modulation of endogenous gene function by agents with designed DNA-sequence specificities. Nucleic Acids Res. 31, 6064–6078. ( 10.1093/nar/gkg815) [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.de Groote ML, Verschure PJ, Rots MG. 2012. Epigenetic editing: targeted rewriting of epigenetic marks to modulate expression of selected target genes. Nucleic Acids Res. 40, 10 596–10 613. ( 10.1093/nar/gks863) [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Jurkowski TP, Ravichandran M, Stepper P. 2015. Synthetic epigenetics—towards intelligent control of epigenetic states and cell identity. Clin. Epigenetics 7, 18 ( 10.1186/s13148-015-0044-x) [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Park M, Keung AJ, Khalil AS. 2016. The epigenome: the next substrate for engineering. Genome Biol. 17, 183 ( 10.1186/s13059-016-1046-5) [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Stolzenburg S, Beltran AS, Swift-Scanlan T, Rivenbark AG, Rashwan R, Blancafort P. 2015. Stable oncogenic silencing in vivo by programmable and targeted de novo DNA methylation in breast cancer. Oncogene 34, 1–9. ( 10.1038/onc.2014.470) [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Cano-Rodriguez D, Gjaltema RAF, Jilderda LJ, Jellema P, Dokter-Fokkens J, Ruiters MHJ, Rots MG. 2016. Writing of H3K4Me3 overcomes epigenetic silencing in a sustained but context-dependent manner. Nat. Commun. 7, 12284 ( 10.1038/ncomms12284) [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Kungulovski G, Nunna S, Thomas M, Zanger UM, Reinhardt R, Jeltsch A. 2015. Targeted epigenome editing of an endogenous locus with chromatin modifiers is not stably maintained. Epigenetics Chromatin 8, 12 ( 10.1186/s13072-015-0002-z) [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.White S, Szewczyk JW, Turner JM, Baird EE, Dervan PB. 1998. Recognition of the four Watson–Crick base pairs in the DNA minor groove by synthetic ligands. Nature 391, 468–471. ( 10.1038/35106) [DOI] [PubMed] [Google Scholar]
- 11.Dervan PB, Edelson BS. 2003. Recognition of the DNA minor groove by pyrrole-imidazole polyamides. Curr. Opin. Struct. Biol. 13, 284–299. ( 10.1016/S0959-440X(03)00081-2) [DOI] [PubMed] [Google Scholar]
- 12.Blackledge MS, Melander C. 2013. Programmable DNA-binding small molecules. Bioorganic Med. Chem. 21, 6101–6114. ( 10.1016/j.bmc.2013.04.023) [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Meier JL, Montgomery DC, Dervan PB. 2012. Enhancing the cellular uptake of Py–Im polyamides through next-generation aryl turns. Nucleic Acids Res. 40, 2345–2356. ( 10.1093/nar/gkr970) [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Nishijima S, Shinohara K, Bando T, Minoshima M, Kashiwazaki G, Sugiyama H. 2010. Cell permeability of Py–Im-polyamide-fluorescein conjugates: influence of molecular size and Py/Im content. Bioorg. Med. Chem. 18, 978–983. ( 10.1016/j.bmc.2009.07.018) [DOI] [PubMed] [Google Scholar]
- 15.Pandian GN, et al. 2014. Distinct DNA-based epigenetic switches trigger transcriptional activation of silent genes in human dermal fibroblasts. Sci. Rep. 4, 3843 ( 10.1038/srep03843) [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Mapp AK, Ansari AZ, Ptashne M, Dervan PB. 2000. Activation of gene expression by small molecule transcription factors. Proc. Natl Acad. Sci. USA 97, 3930–3935. ( 10.1073/pnas.97.8.3930) [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Igarashi J, et al. 2015. Preclinical study of novel gene silencer pyrrole-imidazole polyamide targeting human TGF-β1 promoter for hypertrophic scars in a common marmoset primate model. PLoS ONE 10, 1–15. ( 10.1371/journal.pone.0125295) [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Matsuda H, et al. 2011. Transcriptional inhibition of progressive renal disease by gene silencing pyrrole–imidazole polyamide targeting of the transforming growth factor-β1 promoter. Kidney Int. 79, 46–56. ( 10.1038/ki.2010.330) [DOI] [PubMed] [Google Scholar]
- 19.Tsunemi A, et al. 2014. A novel gene regulator, pyrrole–imidazole polyamide targeting ABCA1 gene increases cholesterol efflux from macrophages and plasma HDL concentration. J. Mol. Med. 92, 509–521. ( 10.1007/s00109-013-1118-x) [DOI] [PubMed] [Google Scholar]
- 20.Serie K, Fukuda N, Nakai S, Matsuda H, Maruyama T, Murayama Y, Omata S. 2012. Pyrrole-imidazole polyamide targeting transforming growth factor 1 ameliorates encapsulating peritoneal sclerosis. Perit. Dial. Int. 32, 462–472. ( 10.3747/pdi.2011.00092) [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Hargrove AE, Martinez TF, Hare AA, Kurmis AA, Phillips JW, Sud S, Pienta KJ, Dervan PB. 2015. Tumor repression of VCaP xenografts by a pyrrole-imidazole polyamide. PLoS ONE 10, 9–13. ( 10.1371/journal.pone.0143161) [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Szablowski JO, Raskatov JA, Dervan PB. 2016. An HRE-binding Py-Im polyamide impairs hypoxic signaling in tumors. Mol. Cancer Ther. 15, 608–617. ( 10.1158/1535-7163.MCT-15-0719) [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Janssen B, van Ommeren S, Merkx M. 2015. Efficient synthesis of peptide and protein functionalized pyrrole-imidazole polyamides using native chemical ligation. Int. J. Mol. Sci. 16, 12 631–12 647. ( 10.3390/ijms160612631) [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Duca M, Vekhoff P, Oussedik K, Halby L, Arimondo PB. 2008. The triple helix: 50 years later, the outcome. Nucleic Acids Res. 36, 5123–5138. ( 10.1093/nar/gkn493) [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Vekhoff P, Ceccaldi A, Polverari D, Pylouster J, Pisano C, Arimondo PB. 2008. Triplex formation on DNA targets: how to choose the oligonucleotide. Biochemistry 47, 12 277–12 289. ( 10.1021/bi801087g) [DOI] [PubMed] [Google Scholar]
- 26.Wu Q, Gaddis SS, MacLeod MC, Walborg EF, Thames HD, DiGiovanni J, Vasquez KM. 2007. High-affinity triplex-forming oligonucleotide target sequences in mammalian genomes. Mol. Carcinog. 46, 15–23. ( 10.1002/mc.20261) [DOI] [PubMed] [Google Scholar]
- 27.van der Gun BTF, Maluszynska-Hoffman M, Kiss A, Arendzen AJ, Ruiters MHJ, McLaughlin PMJ, Weinhold E, Rots MG. 2010. Targeted DNA methylation by a DNA methyltransferase coupled to a triple helix forming oligonucleotide to down-regulate the epithelial cell adhesion molecule. Bioconjug. Chem. 21, 1239–1245. ( 10.1021/bc1000388) [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Kuznetsova S. 1999. Gene activation by triplex-forming oligonucleotide coupled to the activating domain of protein VP16. Nucleic Acids Res. 27, 3995–4000. ( 10.1093/nar/27.20.3995) [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Silanskas A, Foss M, Wende W, Urbanke C, Lagunavicius A, Pingoud A, Siksnys V. 2011. Photocaged variants of the MunI and PvuII restriction enzymes. Biochemistry 50, 2800–2807. ( 10.1021/bi2000609) [DOI] [PubMed] [Google Scholar]
- 30.Reither S, Jeltsch A. 2002. Specificity of DNA triple helix formation analyzed by a FRET assay. BMC Biochem. 3, 27 ( 10.1186/1471-2091-3-27) [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Eisenschmidt K, Lanio T, Simoncsits A, Jeltsch A, Pingoud V, Wende W, Pingoud A. 2005. Developing a programmed restriction endonuclease for highly specific DNA cleavage. Nucleic Acids Res. 33, 7039–7047. ( 10.1093/nar/gki1009) [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Arimondo PB, et al. 2006. Exploring the cellular activity of camptothecin-triple-helix-forming oligonucleotide conjugates. Mol. Cell. Biol. 26, 324–333. ( 10.1128/MCB.26.1.324-333.2006) [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Quintin J, et al. 2014. Dynamic estrogen receptor interactomes control estrogen-responsive trefoil factor (TFF) locus cell-specific activities. Mol. Cell. Biol. 34, 2418–2436. ( 10.1128/mcb.00918-13) [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Karkare S, Bhatnagar D. 2006. Promising nucleic acid analogs and mimics: characteristic features and applications of PNA, LNA, and morpholino. Appl. Microbiol. Biotechnol. 71, 575–586. ( 10.1007/s00253-006-0434-2) [DOI] [PubMed] [Google Scholar]
- 35.Vasquez KM, Narayanan L, Glazer PM. 2000. Specific mutations induced by triplex-forming oligonucleotides in mice. Science 290, 530–533. ( 10.1126/science.290.5491.530) [DOI] [PubMed] [Google Scholar]
- 36.Rogers FA, Hu R, Milstone LM. 2013. Local delivery of gene-modifying triplex-forming molecules to epidermis. J. Invest. Dermatol. 133, 685–691. ( 10.1038/jid.2012.351) [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Re RN, Cook JL, Giardina JF. 2004. The inhibition of tumor growth by triplex-forming oligonucleotides. Cancer Lett. 209, 51–53. ( 10.1016/j.canlet.2004.02.002) [DOI] [PubMed] [Google Scholar]
- 38.Boulware SB, Christensen LA, Thames H, Coghlan L, Vasquez KM, Finch RA. 2014. Triplex-forming oligonucleotides targeting c-MYC potentiate the anti-tumor activity of gemcitabine in a mouse model of human cancer. Mol. Carcinog. 53, 744–752. ( 10.1002/mc.22026) [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Oussedik K, Francois J-C, Halby L, Senamaud-Beaufort C, Toutirais G, Dallavalle S, Pommier Y, Pisano C, Arimondo PB. 2010. Sequence-specific targeting of IGF-I and IGF-IR genes by camptothecins. FASEB J. 24, 2235–2244. ( 10.1096/fj.09-132324) [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Stierle V, Duca M, Halby L, Senamaud-beaufort C, Capobianco ML, Laigle A, Jolles B, Arimondo PB. 2008. Targeting MDR1 gene: synthesis and cellular study of modified daunomycin-triplex-forming oligonucleotide conjugates able to inhibit gene expression in resistant cell lines. Mol. Pharmacol. 73, 1568–1577. ( 10.1124/mol.107.042010) [DOI] [PubMed] [Google Scholar]
- 41.Geel TM.2010. Chapter 7 Thesis University of Groningen. Pompe disease: Towards gene correction using targeted nucleases. See http://www.rug.nl/research/portal/files/2602788/14complete.pdf .
- 42.Vekhoff P, Ceccaldi A, Polverari D, Pylouster J, Pisano C, Arimondo PB. 2008. Triplex formation on DNA targets: how to choose the oligonucleotide. Biochemistry 47, 12 277–12 289. ( 10.1021/bi801087g) [DOI] [PubMed] [Google Scholar]
- 43.Vekhoff P, et al. 2009. Optimized synthesis and enhanced efficacy of novel triplex-forming camptothecin derivatives based on gimatecan. Bioconjug. Chem. 20, 666–672. ( 10.1021/bc800494y) [DOI] [PubMed] [Google Scholar]
- 44.Raben N, Nichols RC, Martiniuk F, Plotz PH. 1996. A model of mRNA splicing in adult lysosomal storage disease (glycogenosis type II). Hum. Mol. Genet. 5, 995–1000. ( 10.1093/hmg/5.7.995) [DOI] [PubMed] [Google Scholar]
- 45.Thakore PI, Black JB, Hilton IB, Gersbach CA. 2016. Editing the epigenome: technologies for programmable transcription and epigenetic modulation. Nat. Methods 13, 127–137. ( 10.1038/nmeth.3733) [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46.Klug A. 2010. The discovery of zinc fingers and their development for practical applications in gene regulation and genome manipulation. Q. Rev. Biophys. 43, 1–21. ( 10.1017/S0033583510000089) [DOI] [PubMed] [Google Scholar]
- 47.Jo Y-I, Kim H, Ramakrishna S. 2015. Recent developments and clinical studies utilizing engineered zinc finger nuclease technology. Cell. Mol. Life Sci. 72, 3819–3830. ( 10.1007/s00018-015-1956-5) [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48.Yin H, Kauffman KJ, Anderson DG. 2017. Delivery technologies for genome editing. Nat. Rev. Drug Discov. 16, 387–399. ( 10.1038/nrd.2016.280) [DOI] [PubMed] [Google Scholar]
- 49.Grimmer MR, Stolzenburg S, Ford E, Lister R, Blancafort P, Farnham PJ. 2014. Analysis of an artificial zinc finger epigenetic modulator: widespread binding but limited regulation. Nucleic Acids Res. 42, 10 856–10 868. ( 10.1093/nar/gku708) [DOI] [PMC free article] [PubMed] [Google Scholar]
- 50.Falahi F, Huisman C, Kazemier HG, van der Vlies P, Kok K, Hospers GAP, Rots MG. 2013. Towards sustained silencing of HER2/neu in cancer by epigenetic editing. Mol. Cancer Res. 11, 1029–1039. ( 10.1158/1541-7786.MCR-12-0567) [DOI] [PubMed] [Google Scholar]
- 51.Bustos F, et al. 2017. Epigenetic editing of the Dlg4/PSD95 gene improves cognition in aged and Alzheimer's disease mice. Brain 140, 3252–3268. ( 10.1093/brain/awx272) [DOI] [PMC free article] [PubMed] [Google Scholar]
- 52.Tan S, et al. 2003. Zinc-finger protein-targeted gene regulation: genomewide single-gene specificity. Proc. Natl Acad. Sci. USA 100, 11 997–12 002. ( 10.1073/pnas.2035056100) [DOI] [PMC free article] [PubMed] [Google Scholar]
- 53.Eisenstein M. 2012. Sangamo's lead zinc-finger therapy flops in diabetic neuropathy. Nat. Biotechnol. 30, 121–123. ( 10.1038/nbt0212-121a) [DOI] [PubMed] [Google Scholar]
- 54.US National Library of Medicine. 2016. Clinical trial of SB-509 in subjects with diabetic neuropathy. Clinicaltrials.gov. See https://www.clinicaltrials.gov/ct2/show/NCT01079325. [DOI] [PubMed]
- 55.US National Library of Medicine. 2012. Clinical trial of SB-509 in subjects with amyotrophic lateral sclerosis (ALS) (ALS). Clinicaltrials.gov. See https://www.clinicaltrials.gov/ct2/show/NCT00748501. [DOI] [PubMed]
- 56.Hamilton PJ, Burek DJ, Lombroso SI, Neve RL, Robison AJ, Nestler EJ, Heller EA. 2017. Cell-type-specific epigenetic editing at the Fosb gene controls susceptibility to social defeat stress. Neuropsychopharmacol 43, 272–284. ( 10.1038/npp.2017.88) [Epub ahead of print]. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 57.Bailus BJ, et al. 2016. Protein delivery of an artificial transcription factor restores widespread Ube3a expression in an Angelman syndrome mouse brain. Mol. Ther. 24, 548–555. ( 10.1038/mt.2015.236) [DOI] [PMC free article] [PubMed] [Google Scholar]
- 58.Tebas P, et al. 2014. Gene editing of CCR5 in autologous CD4 T cells of persons infected with HIV. N. Engl. J. Med. 370, 901–910. ( 10.1056/NEJMoa1300662) [DOI] [PMC free article] [PubMed] [Google Scholar]
- 59.Urnov FD, Rebar EJ, Holmes MC, Zhang HS, Gregory PD. 2010. Genome editing with engineered zinc finger nucleases. Nat. Rev. Genet. 11, 636–646. ( 10.1038/nrg2842) [DOI] [PubMed] [Google Scholar]
- 60.Snowden AW, Gregory PD, Case CC, Pabo CO. 2002. Gene-specific targeting of H3K9 methylation is sufficient for initiating repression in vivo. Curr. Biol. 12, 2159–2166. ( 10.1016/S0960-9822(02)01391-X) [DOI] [PubMed] [Google Scholar]
- 61.Heller EA, et al. 2014. Locus-specific epigenetic remodeling controls addiction- and depression-related behaviors. Nat. Neurosci. 17, 1720–1727. ( 10.1038/nn.3871) [DOI] [PMC free article] [PubMed] [Google Scholar]
- 62.Siddique AN, et al. 2013. Targeted methylation and gene silencing of VEGF-A in human cells by using a designed Dnmt3a–Dnmt3 L single-chain fusion protein with increased DNA methylation activity. J. Mol. Biol. 425, 479–491. ( 10.1016/j.jmb.2012.11.038) [DOI] [PubMed] [Google Scholar]
- 63.Rivenbark AG, Stolzenburg S, Beltran AS, Yuan X, Rots MG, Strahl BD, Blancafort P. 2012. Epigenetic reprogramming of cancer cells via targeted DNA methylation. Epigenetics 7, 350–360. ( 10.4161/epi.19507) [DOI] [PMC free article] [PubMed] [Google Scholar]
- 64.Mussolino C, Alzubi J, Fine EJ, Morbitzer R, Cradick TJ, Lahaye T, Bao G, Cathomen T. 2014. TALENs facilitate targeted genome editing in human cells with high specificity and low cytotoxicity. Nucleic Acids Res. 42, 6762–6773. ( 10.1093/nar/gku305) [DOI] [PMC free article] [PubMed] [Google Scholar]
- 65.Sun N, Abil Z, Zhao H. 2012. Recent advances in targeted genome engineering in mammalian systems. Biotechnol. J. 7, 1074–1087. ( 10.1002/biot.201200038) [DOI] [PubMed] [Google Scholar]
- 66.Ren R, et al. 2017. Visualization of aging-associated chromatin alterations with an engineered TALE system. Cell Res. 27, 483–504. ( 10.1038/cr.2017.18) [DOI] [PMC free article] [PubMed] [Google Scholar]
- 67.Bloom K, Ely A, Mussolino C, Cathomen T, Arbuthnot P. 2013. Inactivation of hepatitis B virus replication in cultured cells and in vivo with engineered transcription activator-like effector nucleases. Mol. Ther. 21, 1889–1897. ( 10.1038/mt.2013.170) [DOI] [PMC free article] [PubMed] [Google Scholar]
- 68.Hu Z, et al. 2015. TALEN-mediated targeting of HPV oncogenes ameliorates HPV-related cervical malignancy. J. Clin. Invest. 125, 425–436. ( 10.1172/JCI78206) [DOI] [PMC free article] [PubMed] [Google Scholar]
- 69.Hubbard N, et al. 2016. Targeted gene editing restores regulated CD40 L function in X-linked hyper-IgM syndrome. Blood 127, 2513–2522. ( 10.1182/blood-2015-11-683235) [DOI] [PubMed] [Google Scholar]
- 70.Qasim W, et al. 2017. Molecular remission of infant B-ALL after infusion of universal TALEN gene-edited CAR T cells. Sci. Transl. Med. 9, eaaj2013 ( 10.1126/scitranslmed.aaj2013) [DOI] [PubMed] [Google Scholar]
- 71.Li K, et al. 2015. Manipulation of prostate cancer metastasis by locus-specific modification of the CRMP4 promoter region using chimeric TALE DNA methyltransferase and demethylase. Oncotarget 6, 10 030–10 044. ( 10.18632/oncotarget.3192) [DOI] [PMC free article] [PubMed] [Google Scholar]
- 72.Crocker J, Stern DL. 2013. TALE-mediated modulation of transcriptional enhancers in vivo. Nat. Methods 10, 762–767. ( 10.1038/nmeth.2543) [DOI] [PMC free article] [PubMed] [Google Scholar]
- 73.US National Library of Medicine. 2017. A safety and efficacy study of TALEN and CRISPR/Cas9 in the treatment of HPV-related cervical intraepithelial neoplasia I. Clinicaltrials.gov. See https://clinicaltrials.gov/ct2/show/NCT03057912?term=Transcription+activatorlike+effector+nuclease&rank=1. [DOI] [PubMed]
- 74.Gasiunas G, Barrangou R, Horvath P, Siksnys V. 2012. Cas9-crRNA ribonucleoprotein complex mediates specific DNA cleavage for adaptive immunity in bacteria. Proc. Natl Acad. Sci. USA 109, E2579–E2586. ( 10.1073/pnas.1208507109) [DOI] [PMC free article] [PubMed] [Google Scholar]
- 75.Jinek M, Chylinski K, Fonfara I, Hauer M, Doudna JA, Charpentier E. 2012. A programmable Dual-RNA-guided DNA endonuclease in adaptive bacterial immunity. Science 337, 816–821. ( 10.1126/science.1225829) [DOI] [PMC free article] [PubMed] [Google Scholar]
- 76.Gasiunas G, Siksnys V. 2013. RNA-dependent DNA endonuclease Cas9 of the CRISPR system: holy grail of genome editing? Trends Microbiol. 21, 562–567. ( 10.1016/j.tim.2013.09.001) [DOI] [PubMed] [Google Scholar]
- 77.Hille F, Charpentier E. 2016. CRISPR-Cas: biology, mechanisms and relevance. Phil. Trans. R. Soc. B 371, 20150496 ( 10.1098/rstb.2015.0496) [DOI] [PMC free article] [PubMed] [Google Scholar]
- 78.Tschaharganeh DF, Lowe SW, Garippa RJ, Livshits G. 2016. Using CRISPR/Cas to study gene function and model disease in vivo. FEBS J. 283, 3194–3203. ( 10.1111/febs.13750) [DOI] [PMC free article] [PubMed] [Google Scholar]
- 79.Cornu TI, Mussolino C, Cathomen T. 2017. Refining strategies to translate genome editing to the clinic. Nat. Med. 23, 415–423. ( 10.1038/nm.4313) [DOI] [PubMed] [Google Scholar]
- 80.Weber J, et al. 2015. CRISPR/Cas9 somatic multiplex-mutagenesis for high-throughput functional cancer genomics in mice. Proc. Natl Acad. Sci. USA 112, 13 982–13 987. ( 10.1073/pnas.1512392112) [DOI] [PMC free article] [PubMed] [Google Scholar]
- 81.Polstein LR, Gersbach CA. 2015. A light-inducible CRISPR-Cas9 system for control of endogenous gene activation. Nat. Chem. Biol. 11, 198–200. ( 10.1038/nchembio.1753) [DOI] [PMC free article] [PubMed] [Google Scholar]
- 82.Zetsche B, Volz SE, Zhang F. 2015. A split-Cas9 architecture for inducible genome editing and transcription modulation. Nat. Biotechnol. 33, 139–142. ( 10.1038/nbt.3149) [DOI] [PMC free article] [PubMed] [Google Scholar]
- 83.Hamilton PJ, Lim CJ, Nestler EJ, Heller EA. 2018. Neuroepigenetic Editing. In Epigenome Editing: Methods and Protocols, Methods in Molecular Biology Series 1767 (eds Jeltsch A, Rots MG), pp. 113–136. ( 10.1007/978-1-4939-7774-1_5) [DOI] [PMC free article] [PubMed] [Google Scholar]
- 84.Waryah CB, Moses C, Arooj M, Blancafort P.. 2018. Zinc Fingers, TALEs and CRISPR systems: A comparison of tools for epigenome editing. In Epigenome editing: Methods and Protocols, Methods in Molecular Biology Series 1767 (eds Jeltsch A, Rots MG), pp. 19–63. ( 10.1007/978-1-4939-7774-1_5_2) [DOI] [PubMed] [Google Scholar]
- 85.Morita S, et al. 2016. Targeted DNA demethylation in vivo using dCas9–peptide repeat and scFv–TET1 catalytic domain fusions. Nat. Biotechnol. 34, 1060–1065. ( 10.1038/nbt.3658) [DOI] [PubMed] [Google Scholar]
- 86.Liu XS, et al. 2016. Editing DNA methylation in the mammalian genome. Cell 167, 233–247.e17. ( 10.1016/j.cell.2016.08.056) [DOI] [PMC free article] [PubMed] [Google Scholar]
- 87.Stella S, Montoya G. 2016. The genome editing revolution: a CRISPR-Cas TALE off-target story. Bioessays 38, S4–S13. ( 10.1002/bies.201670903) [DOI] [PubMed] [Google Scholar]
- 88.Wu D-D, Song J, Bartel S, Krauss-Etschmann S, Rots MG, Hylkema MN. 2017. The potential for targeted rewriting of epigenetic marks in COPD as a new therapeutic approach. Pharmacol. Ther. 182, 1–14. ( 10.1016/j.pharmthera.2017.08.007) [Epub ahead of print]. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Data Availability Statement
This article has no additional data.