

Abstract
Cytosine (C) in DNA is often modified to 5-methylcytosine (m5C) to execute important cellular functions. Despite the significance of m5C for epigenetic regulation in mammals, damage to m5C has received little attention. For instance, almost no studies exist on erroneous methylation of m5C by alkylating agents to doubly or triply methylated bases. Owing to chemical evidence, and because many prokaryotes express methyltransferases able to convert m5C into N4,5-dimethylcytosine (mN4,5C) in DNA, mN4,5C is probably present in vivo. We screened a series of glycosylases from prokaryotic to human and found significant DNA incision activity of the Escherichia coli Nei and Fpg proteins at mN4,5C residues in vitro. The activity of Nei was highest opposite cognate guanine followed by adenine, thymine (T) and C. Fpg-complemented Nei by exhibiting the highest activity opposite C followed by lower activity opposite T. To our knowledge, this is the first description of a repair enzyme activity at a further methylated m5C in DNA, as well as the first alkylated base allocated as a Nei or Fpg substrate. Based on our observed high sensitivity to nuclease S1 digestion, we suggest that mN4,5C occurs as a disturbing lesion in DNA and that Nei may serve as a major DNA glycosylase in E. coli to initiate its repair.
This article is part of a discussion meeting issue ‘Frontiers in epigenetic chemical biology’.
Keywords: 5-methylcytosine methylation damage; DNA base excision repair; N4,5-dimethylcytosine; epigenetics
1. Introduction
The major epigenetic mark in mammalian DNA is 5-methylcytosine (m5C) [1]. The recent decade has provided substantial new knowledge on m5C function and turnover, especially the elucidation of its removal through oxidation of the 5-methyl group by the TET (ten–eleven translocation) family of dioxygenases followed by base excision repair (BER) initiated by the thymine-DNA glycosylase (TDG) [1–3]. In prokaryotic DNA, m5C shares importance with N4-methylcytosine (mN4C) and N6-methyladenine (mN6A) as canonical methyl-base modifications [4,5] which participate in biological functions such as protection against DNA cleavage in restriction–modification systems and DNA repair. Recently, mN6A was discovered as a widespread second epigenetic methylated base in eukaryotes [6,7]. All three base modifications are products of enzyme-catalysed transfer of a methyl group from S-adenosyl-l-methionine (SAM) to the cognate unmodified base by a DNA methyltransferase (MTase) [4,8].
Most human promoters contain CpG islands, which are genomic regions with significant overrepresentation of the CpG sequence. There is a strong correlation between promoter methylation and gene repression [1], and aberrant methylation is associated with many disease states including cancer [9,10]. Consequently, interference with m5C integrity may result in deregulation of genes in addition to mutagenicity and cytotoxicity.
It has been known for a long time that the CpG islands are mutational hotspots in eukaryotic cells [11]. The classical explanation is that 5-methylation of cytosine enhances the rate of spontaneous deamination under physiological conditions, and its deamination product, thymine, is repaired less efficiently than the deamination product of cytosine, uracil. However, most of the thymines generated from m5C deamination are replaced with cytosine prior to replication, where species-specific repair systems often are involved. In Escherichia coli, the very-short-patch repair system repairs the T · G mismatches, while in mammalian cells the BER pathway initiated by TDG is most important. These mechanisms are extensively investigated and are comprehensively described elsewhere [12–14].
In contrast to hydrolytic damage to m5C, few reports exist on damage inflicted by reactive oxygen species [15–19] and almost nothing is known about damage induced by methylation exposure or arising spontaneously in cells because of erroneous reaction with SAM or other methylating agents. Thus, the possible further methylation of m5C to di- or trimethylated bases either by methylating agents or by MTases has received little attention [20].
Many prokaryotes enzymatically modify cytosine at either the N4- or 5-position [5]. As m5C has been shown to be a substrate for enzymatic N4-methylation [20,21], we constructed model DNA substrates containing N4,5-dimethylcytosine (mN4,5C) and used them to screen a series of DNA glycosylases for their activity towards this dimethylated base in DNA. Here we report, for the first time to our knowledge, enzymatic excision of doubly methylated cytosine, by showing that the E. coli endonuclease VIII (Nei) [22] and formamidopyrimidine-DNA glycosylase (Fpg, also called MutM) [23] exhibit significant activity for removing mN4,5C from DNA in vitro.
2. Material and methods
(a). Preparation of mN4,5C-containing oligonucleotide substrate
Equimolar amounts of single-stranded forward (Fw) 5′-[Cy3]-C*G*G*TGAAGTAC[m5C]AGGAAGCGATTTCGA*C*C*C-3′ [synthetically incorporated Cy3 fluorophore] and reverse (Rev) 5′-G*G*G*TCGAAATCGCTTC[m5C]TGGTACTTC*A*C*CG-3′ oligodeoxyribonucleotides containing phosphorothioates (*) were annealed to form a 30 nucleotide (nt) DNA duplex. The duplex oligonucleotide (10 nmol) was incubated with 1.6 nmol of M.MvaI and 110 nmol of SAM in M.MvaI buffer (50 mM Tris-HCl (pH 9.0), 1 mM dithiothreitol (DTT)) at 37°C overnight. An aliquot of the reaction was analysed by digestion with MvaI restriction endonuclease to confirm complete methylation (electronic supplementary material, figure S1A). M.MvaI was removed by thermal denaturation (20 min at 65°C) followed by centrifugation. Denatured oligonucleotides (5 min at 95°C) were separated, analysed and purified (electronic supplementary material, figure S1B) on an integrated high-performance liquid chromatography/electrospray ionization/mass spectrometry system (Agilent 1290 Infinity) equipped with a Zorbax Eclipse Plus C18 column (15 cm × 2.1 mm) by elution with a linear gradient of solvents A (5 mM ammonium acetate, pH 7.0 in water) and B (5 mM ammonium acetate, pH 7.0 in methanol) at a flow of 0.17 ml min−1 at 55°C as follows: 0–13 min, 5–20% B; 13–20 min, 20–21% B; 20–25.5 min, 21–30% B; 25.5–27 min, 30–100% B. High-resolution mass spectra of samples were acquired on an Agilent Q-TOF 6520 mass analyser (100–3200 m/z range, negative ionization mode). The results were analysed with Agilent MassHunter Qualitative Analysis software (electronic supplementary material, figure S1C).
(b). Control oligonucleotide substrates
Synthetic 5′-labelled oligodeoxyribonucleotides 5′-[Cy3]-CCCTCGAGGTA[U]CATGGATCCGATCGATCCGATTTCGACCTCAAAACCTAGACGAATTCCG-3′ (Fw 60 nt; 11 nt incision product), 5′-[Cy3]-TAGACATTGCCCTCGAGGTA[U]CATGGATCCGATTTCGACCTCAAACCTAGACGAATTCCG-3′ (Fw 60 nt; 20 nt incision product) and 5′-[Cy3]-CCCTCGATGTA[U]CATGGATCCGATCGATCC-3′ (Fw 30 nt; 11 nt incision product), containing dU at a specific internal site (Sigma-Aldrich), were annealed to equimolar amounts of complementary (Rev) strands with G opposite U.
(c). Repair enzymes
Nei (2.1 µM), Fpg (13 µM), Nth (0.7 µM), Ung (1.95 µM), hSMUG1 (0.33 µM), Nfo (83 nM) and PseT (0.29 µM) were obtained from New England Biolabs. Mug (100 units ml−1) was obtained from Trevigen. hUNG (hUNGΔ84 with His-tag; 820 µM) was a gift from Bodil Kavli and Geir Slupphaug. hOGG1 (with His-tag; 5.4 µM) was produced as described [24]. hTDG (with His-tag; 15 µM) was a gift from David Schürmann and Primo Schär [25]. hNEIL1 (full length with His-tag; 4.5 µM), hNEIL2 (full length with His-tag; 5.3 µM) and hNEIL3 (residues 1–301 with His-tag; 8.6 µM) were purified as described [26,27]. More details are provided in ‘electronic supplementary material, Repair enzyme details'.
(d). Production and purification of M.MvaI
The mvaIM gene was cloned into the expression plasmid vector pET15b through the NdeI and BamHI sites, resulting in addition of an N-terminal His6 affinity tag. E. coli ER2566 cells carrying the pET15b-mvaIM plasmid and a T7 RNA polymerase gene on a separate plasmid, pACAR1 (G Vilkaitis 2004, unpublished data) were grown at 37°C in LB medium containing ampicillin (100 μg ml−1) and chloramphenicol (25 μg ml−1) to OD600 = 0.8. IPTG (isopropyl β-d-1-thiogalactopyranoside; 1 mM) was added and cells were grown at 16°C for 16 h. Cells were then sonicated and the supernatant was applied onto a HiTrap Chelating HP column (GE Healthcare). The protein was eluted with a 10–500 mM imidazole gradient, and pooled fractions were dialysed against a storage buffer (50 mM Tris-HCl (pH 8.0), 1 mM ethylenediaminetetraacetic acid (EDTA), 100 mM NaCl, 2 mM 2-mercaptoethanol, 50% glycerol). All purification steps were carried out at 4°C.
(e). Assays for excision of mN4,5C from DNA and incision of mN4,5C-containing DNA
Purified DNA glycosylases at varying concentrations were incubated with mN4,5C-single-stranded DNA (ssDNA; electronic supplementary material, figure S1D), or with mN4,5C-double-stranded DNA (dsDNA) where mN4,5C was placed opposite G, A, C or T (named mN4,5C · G-DNA, mN4,5C · A-DNA, mN4,5C · C-DNA and mN4,5C · T-DNA, respectively; figure 1a) at 37°C in a suitable reaction buffer (Nei buffer: 10 mM Tris-HCl (pH 8.0), 75 mM NaCl, 1 mM EDTA; Fpg buffer: 10 mM Bis-Tris-propane-HCl (pH 7.0), 10 mM MgCl2, 1 mM DTT, 0.1 mg ml−1 bovine serum albumin (BSA); for other glycosylases see figure legends of the respective electronic supplementary figures) in a final volume of 20 μl, if not otherwise stated. Reactions were terminated by the addition of 20 mM EDTA, 0.5% (w/v) sodium dodecyl sulfate and proteinase K (150 µg ml−1) followed by incubation at 37°C for 10 min, precipitation of DNA with ethanol and solubilization of the precipitate in water (if not otherwise stated, 10 μl) [28]. mN4,5C-DNA glycosylase activity was determined by NaOH-mediated (0.1 M final concentration) incision of the resulting abasic site (90°C for 10 min; electronic supplementary material, figure S2A), where incision of the DNA without alkaline treatment measured the mN4,5C-DNA incision activity (electronic supplementary material, figure S2B). Following addition of loading solution containing 80% (v/v) formamide, 1 mM EDTA and 1% (w/v) blue dextran (10 µl), and incubation at 95°C for 5 min to denature DNA, the vial was cooled on ice followed by quick centrifugation at 4°C. The samples (5 µl) were subjected to polyacrylamide gel electrophoresis (PAGE) using a gel (20% (w/v) polyacrylamide (acrylamide : bis-acrylamide 37.5 : 1)) containing 8 M urea. The PAGE was performed using a Tris–borate–EDTA buffer system (89 mM Tris base, 89 mM boric acid, 2 mM EDTA, pH 8.0). Visualization and quantification were performed by fluorescence imaging analysis using ImageQuant Software (Molecular Dynamics).
Figure 1.

E. coli Nei and Fpg proteins incise at mN4,5C in DNA. (a) Structure of 5′-labelled dsDNA substrates for the determination of DNA glycosylase activity at modified cytosines (X) placed against different opposing bases (Y). Stars (*) indicate nuclease-resistant phosphorothioate bonds; [Cy3], cyanine3 fluorophore. (b) Nei exhibits activity towards mN4,5C (but not C, mN4C or m5C) when placed opposite G in DNA. Nei (8.3 pmol) was incubated with the DNA substrate (1 pmol) at 37°C for 1 h (final volume, 20 µl). (c) Nei exhibits the highest activity towards mN4,5C when opposite G in DNA. For experimental details, see electronic supplementary material, figure S3. The average (±s.d.) of four independent experiments is presented. (d) Fpg exhibits the highest activity towards mN4,5C when placed opposite C, with lower activity opposite T, in DNA. DNA substrate (1 pmol) was incubated alone (lanes 1–5) or with Fpg (13 pmol; lanes 6–9) at 37°C 1 h. U · G-DNA (60 nt; 1 pmol) incubated without (lane 10) and with hUNG, the latter followed by NaOH/heat treatment (lane 11), was used as negative and positive control for active hUNG, respectively, which was used to convert U · G-DNA into AP-DNA to demonstrate active Fpg (i.e. lyase activity; lane 12). Reaction products were analysed by denaturing PAGE at 200 V for 2 h. The average (±s.d.) of three independent experiments is presented in (d)(ii). (Online version in colour.)
(f). Preparation of mN4,5C-containing plasmid DNA
The pBR322 plasmid DNA was isolated from E. coli strains ER2267 (dcm+, methylated at CC(T/A)G sites) or ER2566 (dcm−, unmodified DNA) using standard protocols. Plasmid DNA (15 µg) was incubated with 0.4 µM M.MvaI in 0.2 ml of reaction buffer containing 10 mM Tris-HCl (pH 8.5), 10 mM MgCl2, 100 mM KCl, 0.2 mg ml−1 BSA, 2 mM 2-mercaptoethanol and 0.5 mM SAM at 37°C for 4 h. DNA was then linearized with R.HindIII, extracted with phenol : chloroform (1 : 1) and chloroform, and precipitated with ethanol. The completeness of the M.MvaI-methylation in reaction aliquots was confirmed by R.MvaI restriction endonuclease digestion and agarose gel electrophoresis (see electronic supplementary material, figure S9).
(g). Nuclease S1 assay
Doubly methylated DNA (or mono-methylated/unmethylated controls) (0.5 µg) was incubated with 20 units S1 nuclease (Thermo Fisher Scientific) in supplier buffer for 1 h at 37°C. Reactions were stopped by proteinase K (Thermo Fisher Scientific) treatment and analysed by 1.5% agarose gel electrophoresis.
3. Results
To investigate possible enzymatic excision of mN4,5C from DNA, a 5′ fluorescently labelled oligodeoxyribonucleotide substrate with this di-methylated base inserted at a specific position was prepared (see ‘Material and methods’ for details). This ssDNA oligomer, mN4,5C-ssDNA, was alone, or annealed to a complementary strand with mN4,5C placed opposite either cognate G or non-cognate A, T or C (figure 1a), exposed to a series of available DNA glycosylases from E. coli or human (see below). Enzymatic base excision by so-called mono-functional DNA glycosylases results in an alkali-labile apurinic/apyrimidinic (AP) site, which can be monitored by the extent that NaOH cleaves this site by a β/δ-elimination reaction [29] (electronic supplementary material, figure S2A). Bi-functional DNA glycosylases like Nei and Fpg will in addition cleave the resulting AP-DNA, and these consecutive activities can be monitored by PAGE under denaturing conditions without NaOH/heat treatment (electronic supplementary material, figure S2B).
Interestingly, we observed that E. coli Nei exhibited significant activity for mN4,5C in dsDNA (figure 1b, lane 8), while no mN4,5C-DNA incision occurred without the enzyme (figure 1b, lane 4). Control experiments were carried out with identical DNA substrates where mN4,5C was replaced by m5C, mN4C or C, showing that Nei exhibited no activity towards the normal cytosines (figure 1b, lanes 5–7). The activity was highest when mN4,5C was placed opposite cognate G (figure 1c and and electronic supplementary material figure S3, lane 6), and substantially lower opposite non-cognate A and T (figure 1c and electronic supplementary material, figure S3, lanes 8 and 9, respectively), while close to zero opposite C (figure 1c and electronic supplementary material, figure S3, lane 7). Uracil-DNA glycosylase of E. coli (Ung) was used to form AP-DNA to confirm active Nei, due to the ability of the latter to recognize and incise AP sites (electronic supplementary material, figure S3, lane 12), while NaOH/heat treatment of AP-DNA was used to confirm active Ung (electronic supplementary material, figure S3, lanes 10 and 11). As the activity of Nei for mN4,5C · G was the highest, as well as this being the normal arrangement in vivo, further studies on Nei were only carried out using mN4,5C · G-DNA.
We also observed that E. coli Fpg exhibited significant activity for mN4,5C in DNA, although only when opposite pyrimidines on the complementary strand and with the highest activity opposite C (figure 1d, lanes 7 and 9). No mN4,5C-DNA incision occurred without enzyme (figure 1d, lanes 1–5) or when mN4,5C was placed opposite G or A (figure 1d, lanes 6 and 8, respectively). Surprisingly, Fpg also exhibited similar activity for m5C and mN4C, and even, although lower, for C itself when placed opposite C (AN Tesfahun, P Guragain, M Alexeeva, A Arshad, M Tomkuviené, JK Laerdahl, A Klungland, S Klimašauskas, S Bjelland 2017, unpublished results). This prompted us to conclude that the activity of Fpg on mN4,5C (figure 1d) is the manifestation of a hitherto unknown (methyl)C · C mismatch activity, rather than a specific mN4,5C repair function of Fpg. This activity is presently under investigation. All other enzymes tested (electronic supplementary material, figures S4–S6), that is, E. coli Ung, mismatch-specific uracil-DNA glycosylase (Mug) and endonuclease III (Nth) as well as human uracil-DNA glycosylase (hUNG), single-strand-selective mono-functional uracil-DNA glycosylase (hSMUG1), hTDG and 8-oxoguanine-DNA glycosylase (hOGG1) showed no detectable DNA glycosylase activity for mN4,5C at the concentrations and under the incubation conditions employed. Because of previously detected activity for base lesions placed opposite all normal bases and also in ssDNA for enzymes in the uracil-DNA glycosylase superfamily (UDG), Ung, hUNG, Mug and hSMUG1 were tested for mN4,5C removal in all these contexts without showing any activity (electronic supplementary material, figures S4A, S5A, S4B and S5B, respectively). Based on the known substrate preference, the family 2 UDG hTDG, and the bi-functional glycosylases Nth and hOGG1 [19], were only examined for DNA incision at the damage site in the dsDNA context, both showing no detectable activity (electronic supplementary material, figures S4C and S5C,D). As opposed to E. coli Nei, we also found no mN4,5C-specific activity by the human orthologues hNEIL1 (full length; electronic supplementary material, figure S6A), hNEIL2 (full length; electronic supplementary material, figure S6B) or hNEIL3 (truncated; amino acids 1–301; electronic supplementary material, figure S6C), neither opposite G, C, A or T nor placed in ssDNA, the latter only having been analysed for NEIL3. Active NEIL1 and NEIL2 were confirmed by their active AP lyase functions for dsDNA, while active NEIL3 was verified by active AP lyase function for ssDNA.
Determination of the mN4,5C·G-DNA incision activity as a function of protein concentration showed that the extent of incision follows the same equation that describes the dependence of V0 as a function of [S] in the Michaelis–Menten kinetic analysis (electronic supplementary material, figure S7A and B). However, it should be noted that the enzyme/substrate concentration ratio used was above 1:1, i.e. approaching single-turnover conditions. A similar profile was observed when the incision activity was determined as a function of time (electronic supplementary material, figure S7C and D), using an eightfold excess of enzyme over substrate. Kinetic analysis of the mN4,5C · G-DNA incision activity of Nei indicated Michaelis–Menten behaviour (electronic supplementary material, figure S8), resulting in an apparent Km of 311 ± 80 nM, a Vmax of 3.61 ± 0.46 nM min−1 and a kcat of 0.0087 ± 0.0011 min−1. These parameters were also determined with 1 mM of DTT in the incubation buffer, as often used in glycosylase assays, resulting in virtually identical values (data not shown). The obtained values are quite close to those determined for other Nei substrates [22,30,31].
To confirm the presence of a 3′-phosphorylated product following the incision of mN4,5C · G-DNA by Nei, we treated Nei-exposed DNA with T4 polynucleotide kinase (PseT), which specifically removes phosphate from the 3′-end [32,33], generating a slower-migrating product in PAGE (figure 2). As opposed to control incubation in the absence of enzyme or incubation with Nei alone which only formed 3′-phosphate, incubation with Nei followed by endonuclease IV (Nfo) [34] showed additional formation of a 3′-OH product (figure 2). A similarly efficient processing of the Nei product was achieved by addition of PseT, which converted the 3′-phosphate into the 3′-OH product. This confirms that the Nei-mediated incision of mN4,5C · G-DNA forms a 3′-phosphate, and accords with the presently accepted knowledge that Nfo may displace Nei from the resultant AP site as well as remove the 3′-phosphate [35], both resulting in the 3′-OH product necessary for repair synthesis.
Figure 2.
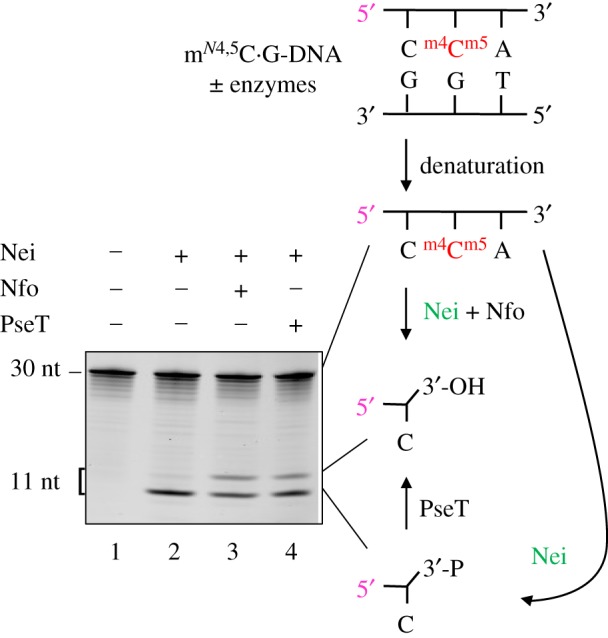
Definition and processing of the 3′-end following Nei-mediated incision of mN4,5 C · G-DNA. DNA substrate (figure 1a; 1 pmol) was incubated without (lane 1) or with Nei (2.1 pmol; lanes 2–4) at 37°C for 30 min, followed by no addition (lanes 1 and 2), addition of 0.083 pmol endonuclease IV (Nfo; lane 3) or addition of 0.29 pmol T4 polynucleotide kinase (PseT; lane 4) and incubation for an additional 30 min (final volume, 10 µl). Reaction products were analysed by denaturing PAGE at 500 V for 4 h. P, phosphate. (Online version in colour.)
To assess a possible impact of mN4,5C on the helical DNA structure (figure 3a), we also performed probing of modified plasmid DNA with nuclease S1, which is specific for ssDNA regions including DNA mismatches and lesions that disrupt dsDNA structure [38]. This DNA contained CmN4,5CWGG sites as a result of in vivo methylation by E. coli Dcm MTase followed by in vitro methylation with M.MvaI (electronic supplementary material, figure S9). The experiment showed extensive cleavage of the mN4,5C · G-DNA at CCWGG sites by S1, yielding a fragmentation pattern similar to that obtained by R.MvaI digestion of unmodified control DNA. These results clearly demonstrate that the presence of mN4,5C · G base pairs, as opposed to those involving mN4C or m5C, leads to local distortion of the helical DNA structure that is readily detectable by a structure-sensitive enzyme in vitro (figure 3b).
Figure 3.
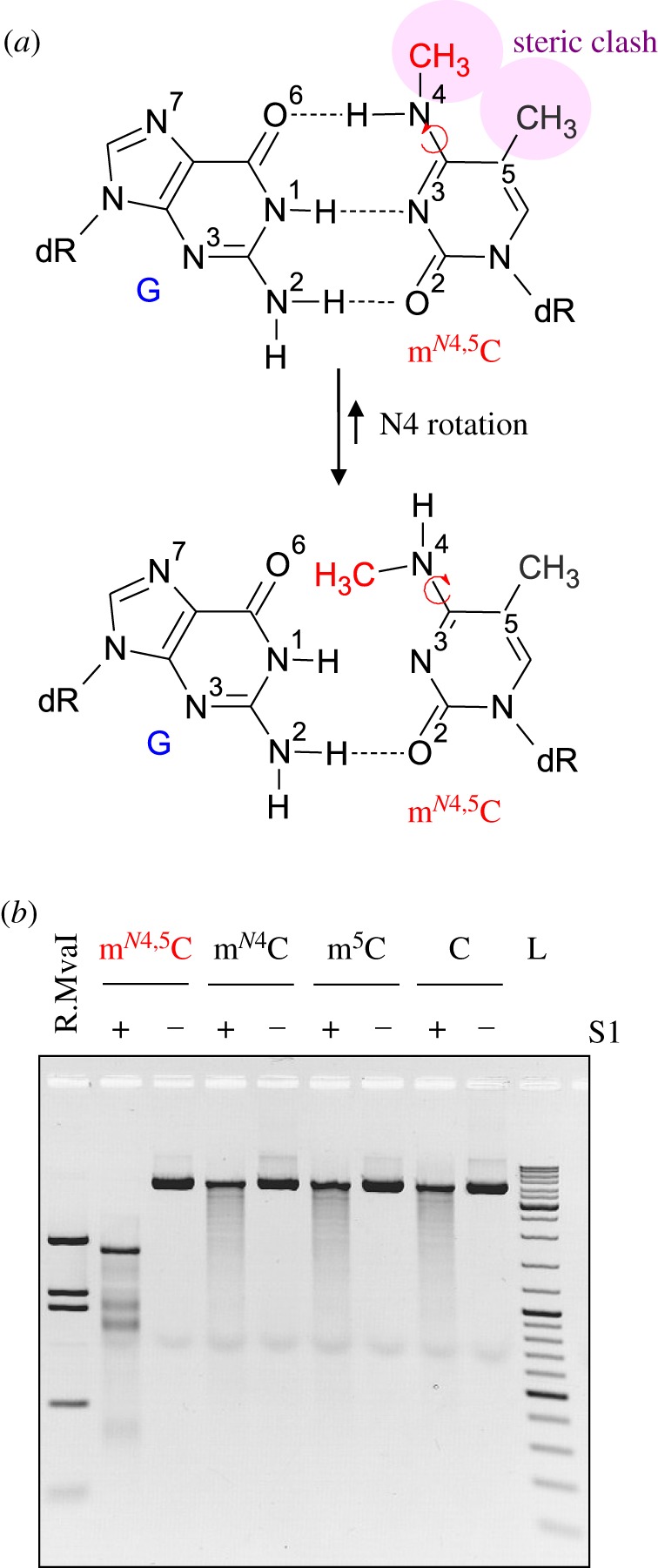
The mN4,5C residue disrupts normal DNA structure. (a) Structural considerations of the mN4,5C residue in dsDNA [20,36,37] show how rotation around the N4–C4 bond of mN4,5C relieves the steric clash between the methyl groups (upper panel) but at the same time disrupts the normal G · C hydrogen-bonding pattern (lower panel), precluding a normal Watson–Crick DNA structure. (b) Impact of mN4,5C on DNA structure probed by susceptibility to nuclease S1 digestion. pBR322 plasmid DNA containing mN4,5C, mono-methylated (either mN4C or m5C) or unmethylated cytosine (c), respectively, at CC(A/T)GG sites (electronic supplementary material, figure S9) was incubated with nuclease S1 and analysed by agarose gel electrophoresis. R.MvaI lane, commercial pBR322 digested with R.MvaI; L, GeneRuler DNA Ladder Mix.
4. Discussion
All three types of biological DNA methylation (i.e. mN4C, m5C and mN6A) occur in the major groove of the DNA helix where they can be readily sensed by cellular proteins and interpreted as biological signals. Importantly, these modifications do not alter or preclude the coding capacity of the target nucleobases, preserving the original content of the genome [8]. Here we present the first report to our knowledge describing a repair activity for a further methylated m5C residue in DNA, mN4,5C, in which two types of biological methylation occur in a single nucleobase. We show that the E. coli Nei DNA glycosylase/lyase initiates BER of mN4,5C in DNA by strand incision at the lesion site in vitro (figure 1b). The activity observed was highest when the lesion was placed opposite cognate G, and 3–4 times lower opposite A and T, while almost no activity was detected opposite C (figure 1c). We also show that E. coli Fpg exhibits a similar activity for mN4,5C in DNA, removing mN4,5C most efficiently opposite C followed by T (figure 1d). Although this appears to be the manifestation of a hitherto unknown activity acting on (methyl)C · C mismatches (AN Tesfahun, P Guragain, M Alexeeva, A Arshad, M Tomkuviené, JK Laerdahl, A Klungland, S Klimašauskas, S Bjelland 2017, unpublished observations) rather than a specific mN4,5C repair function of Fpg, it may nevertheless play a role in vivo. Altogether, our results suggest that Nei and Fpg may complement each other in the repair of mN4,5C, acting opposite different bases in DNA. In agreement with previous reports [12,35], we found that the DNA incised by Nei can be processed by downstream BER proteins (figure 2), reinforcing the role of Nei as a major DNA glycosylase in E. coli to initiate repair of mN4,5C in DNA (figure 4).
Figure 4.
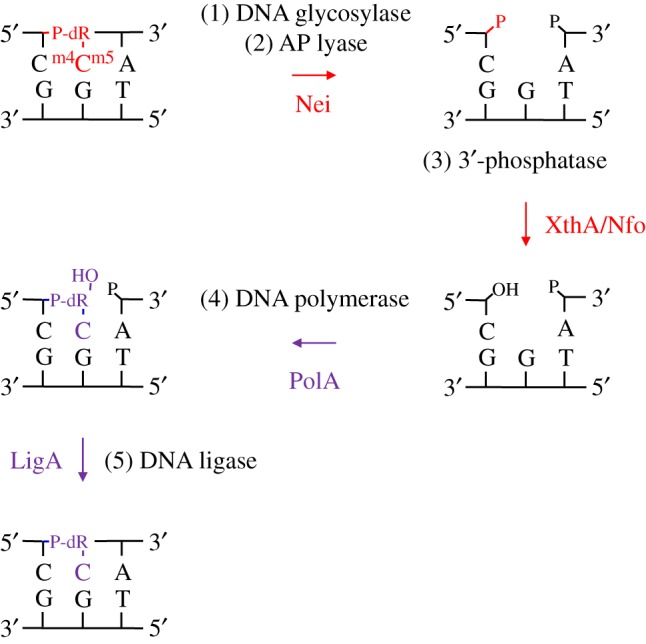
Proposed steps of the E. coli BER pathway for mN4,5C in DNA. Both the damaged base excision step 1 and the AP site incision step 2 are performed by the bi-functional DNA glycosylase Nei as reported here. The 3′-remnant must be removed (step 3) by a phosphatase, which in E. coli may be either a function of XthA or Nfo [35] (figure 2). The cleaned one-nucleotide gap in DNA is now ready for insertion of the correct dCMP (step 4) by the repair DNA polymerase I (PolA) [39] followed by nick-sealing (step 5) by DNA ligase (LigA) [40]. The excised target and newly inserted residues, along with their corresponding reaction arrows, are shown in red and violet, respectively; dR, deoxyribose.
The stability of the mN4,5C–deoxyribose bond was demonstrated 50 years ago by chemical synthesis of the N4,5-dimethylcytosine 2′-deoxynucleoside (mN4,5dC) and its 5′-monophosphate and 5′-triphosphate [41], indicating the chemical persistence of the mN4,5C residue in DNA. Molecular dynamics calculations and the crystal structure of mN4,5dC [42] show that, owing to a steric clash between the methyl groups in the cis-conformer, the N4-methyl group adopts a trans orientation with respect to the C5-methyl group. Although no structural study of the mN4,5C nucleotide in DNA has been reported yet, it is obvious that the trans N4-methyl group will preclude the formation of a proper base-pairing pattern with G on the opposite strand [36,37] (figure 3a). Indeed, our results presented here show that the mN4,5C residue specifically sensitizes DNA for S1 nuclease digestion (figure 3b), supporting a proposed irregular (open) structure of the mN4,5C · G base pair. In its cytidine form, mN4,5C is generally more stable to deamination and hydrolysis/depyrimidination than cytosine itself, m5C and mN4C [43], suggesting considerable in vivo stability and the need for active repair.
Our results indicate that the active site of at least two bacterial DNA repair enzymes is able to accommodate the mN4,5C residue during catalysis. This contrasts with several tested helix–hairpin–helix (HhH) or UDG DNA glycosylases of E. coli and human cells (electronic supplementary material, figures S4,S5) like Ung, Mug, Nth, hUNG, hSMUG1, hTDG and hOGG1, which showed no detectable activity under the incubation conditions employed. Curiously, this also applies to the human Nei orthologues hNEIL1, hNEIL2 and hNEIL3 (electronic supplementary material, figure S6). To our knowledge, mN4,5C is the first methylated base found to be a substrate for Nei. Nei, as well as Fpg, has a flexible and spacious active-site pocket that can bind a variety of nucleobase lesions, providing numerous direct or water-mediated atomic contacts [44]. Indeed, mN4,5C is quite similar to some other oxidized pyrimidine or purine Nei substrates [19]. Unlike Nei and Fpg, DNA glycosylases in the HhH superfamily, for example E. coli Nth and hOGG1, and the UDG superfamily, including E. coli Ung and Mug, as well as hUNG, hSMUG1 and hTDG, bind their target base lesions in deeply buried and relatively rigid recognition pockets [44], and these DNA glycosylases, therefore, appear to be mainly specialized for damaged bases of a single type and with little structural diversity.
Although relatively rare, the presence of cytosine-N4 MTases potentially acting at Dcm-methylated cytosines has been described in E. coli. For example, the M.EcoNI MTase produces mN4C at CCTNNNNNAGG sites (BP Anton, T Clark, M Boitano, J Korlach, RJ Roberts 2012, unpublished observations; http://rebase.neb.com/cgi-bin/erefget?5672), which may overlap with highly abundant Dcm Cm5C(T/A)GG sites in certain genomic contexts. Similar double methylation can also occur in other bacterial species or strains containing both types (m5C and mN4C) of MTases operating on overlapping targets in genomic DNA. Foreign MTase genes may be acquired by horizontal transfer of mobile elements of plasmid-borne restriction–modification systems or by viral infections. mN4,5C may also arise as a consequence of chemical insults to the cellular DNA.
Additional indirect evidence for its harmful effects and efficient removal of genomic mN4,5C in vivo comes from previous experiments that found negligible levels of mN4,5C in DNA of E. coli cells carrying active mvaIM and dcm genes, although persisting production of mN4,5C in DNA led to the induction of the SOS response [20]. Indeed, because of its helix-destabilizing features (figure 3), mN4,5C is probably also an efficient substrate for the nucleotide excision repair (NER) protein complex UvrABC in E. coli [12,45]. In particular, the SOS response induces several trans-lesion bypass polymerases probably able to insert e.g. C or T opposite mN4,5C in DNA [19]. We primarily expect Nei to initiate error-free BER of mN4,5C in DNA opposite cognate G (figure 4), which, as suggested above, may be backed up by NER. If mN4,5C escapes such repair, it may be bypassed by a replicative or trans-lesion polymerase to insert a non-cognate base opposite it. Then, Nei or Fpg-mediated error-prone repair can be a second line of defence if mN4,5C escapes the first Nei or maybe NER defence line and continues as a target for DNA synthesis. Because of high cellular toxicity, an ‘accepted’ price for mN4,5C removal is mutagenesis.
Supplementary Material
Acknowledgements
We thank G. Slupphaug, B. Kavli, D. Schürmann and P. Schär for gifts of enzymes, and S. Serva and G. Vilkaitis for valuable discussions and advice.
Data accessibility
The datasets supporting this article have been uploaded as part of the electronic supplementary material.
Authors' contributions
A.N.T. and M.A. designed and supervised the experiments, analysed the data and wrote parts of the manuscript. P.G. and M.T. designed and performed experiments, analysed the data and wrote parts of the manuscript. A.A. designed and performed experiments and analysed the data. R.G. designed and performed experiments. A.R. developed methods and designed and performed experiments. G.U. and M.B. produced experimental tools. J.K.L. wrote parts of the manuscript and contributed to important intellectual content. A.K. initiated the study and contributed to important intellectual content. S.K. initiated, supervised and managed the study, and wrote parts of the manuscript. S.B. initiated the study, designed the experiments, analysed the data, supervised and managed the study, and wrote the manuscript. All the authors read, commented on and revised the manuscript.
Competing interests
We declare we have no competing interests.
Funding
This research was supported by the University of Stavanger, Vilnius University and Oslo University Hospital/University of Oslo. The work was in part supported by NIH (grant HG007200 to S.K.) and COST action CM1406.
References
- 1.Klungland A, Robertson AB. 2017. Oxidized C5-methyl cytosine bases in DNA: 5-hydroxymethylcytosine; 5-formylcytosine; and 5-carboxycytosine. Free Radic. Biol. Med. 107, 62–68. ( 10.1016/j.freeradbiomed.2016.11.038) [DOI] [PubMed] [Google Scholar]
- 2.He YF, et al. 2011. Tet-mediated formation of 5-carboxylcytosine and its excision by TDG in mammalian DNA. Science 333, 1303–1307. ( 10.1126/science.1210944) [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Ito S, Shen L, Dai Q, Wu SC, Collins LB, Swenberg JA, He C, Zhang Y. 2011. Tet proteins can convert 5-methylcytosine to 5-formylcytosine and 5-carboxylcytosine. Science 333, 1300–1303. ( 10.1126/science.1210597) [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Jeltsch A, Jurkowska R. 2016. DNA methyltransferases–role and function. New York, NY: Springer. [Google Scholar]
- 5.Ehrlich M, Wilson GG, Kuo KC, Gehrke CW. 1987. N4-Methylcytosine as a minor base in bacterial DNA. J. Bacteriol. 169, 939–943. ( 10.1128/jb.169.3.939-943.1987) [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Luo GZ, Blanco MA, Greer EL, He C, Shi Y. 2015. DNA N6-methyladenine: a new epigenetic mark in eukaryotes? Nat. Rev. Mol. Cell Biol. 16, 705–710. ( 10.1038/nrm4076) [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Luo GZ, He C. 2017. DNA N6-methyladenine in metazoans: functional epigenetic mark or bystander? Nat. Struct. Mol. Biol. 24, 503–506. ( 10.1038/nsmb.3412) [DOI] [PubMed] [Google Scholar]
- 8.Kriukienė E, Liutkevičiūtė Z, Klimašauskas S. 2012. 5-Hydroxymethylcytosine—the elusive epigenetic mark in mammalian DNA. Chem. Soc. Rev. 41, 6916–6930. ( 10.1039/c2cs35104h) [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Dawson MA, Kouzarides T. 2012. Cancer epigenetics: from mechanism to therapy. Cell 150, 12–27. ( 10.1016/j.cell.2012.06.013) [DOI] [PubMed] [Google Scholar]
- 10.Ludwig AK, Zhang P, Cardoso MC. 2016. Modifiers and readers of DNA modifications and their impact on genome structure, expression, and stability in disease. Front. Genet. 7, 115 ( 10.3389/fgene.2016.00115) [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Pfeifer GP. 2006. Mutagenesis at methylated CpG sequences. Curr. Top. Microbiol. Immunol. 301, 259–281. ( 10.1007/3-540-31390-7_10) [DOI] [PubMed] [Google Scholar]
- 12.Friedberg EC, Walker GC, Siede W, Wood RD, Schultz RA, Ellenberger T. 2006. DNA repair and mutagenesis, 2nd edn Washington, DC: ASM Press. [Google Scholar]
- 13.Hashimoto H. 2014. Structural and mutation studies of two DNA demethylation related glycosylases: MBD4 and TDG. Biophysics (Nagoya-shi) 10, 63–68. ( 10.2142/biophysics.10.63) [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Iyer RR, Pluciennik A, Burdett V, Modrich PL. 2006. DNA mismatch repair: functions and mechanisms. Chem. Rev. 106, 302–323. ( 10.1021/cr0404794) [DOI] [PubMed] [Google Scholar]
- 15.Madugundu GS, Cadet J, Wagner JR. 2014. Hydroxyl-radical-induced oxidation of 5-methylcytosine in isolated and cellular DNA. Nucleic Acids Res. 42, 7450–7460. ( 10.1093/nar/gku334) [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Cao H, Jiang Y, Wang Y. 2009. Kinetics of deamination and Cu(II)/H2O2/ascorbate-induced formation of 5-methylcytosine glycol at CpG sites in duplex DNA. Nucleic Acids Res. 37, 6635–6643. ( 10.1093/nar/gkp615) [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Cao H, Wang Y. 2007. Quantification of oxidative single-base and intrastrand cr oss-link lesions in unmethylated and CpG-methylated DNA induced by Fenton-type reagents. Nucleic Acids Res. 35, 4833–4844. ( 10.1093/nar/gkm497) [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Gu C, Zhang Q, Yang Z, Wang Y, Zou Y, Wang Y. 2006. Recognition and incision of oxidative intrastrand cr oss-link lesions by UvrABC nuclease. Biochemistry 45, 10 739–10 746. ( 10.1021/bi060423z) [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Bjelland S, Seeberg E. 2003. Mutagenicity, toxicity and repair of DNA base damage induced by oxidation. Mutat. Res. 531, 37–80. ( 10.1016/j.mrfmmm.2003.07.002) [DOI] [PubMed] [Google Scholar]
- 20.Klimašauskas S, Gerasimaitė R, Vilkaitis G, Kulakauskas S. 2002. N4,5-Dimethylcytosine, a novel hypermodified base in DNA. Nucleic Acids Res. Suppl. 2, 73–74. ( 10.1093/nass/2.1.73) [DOI] [PubMed] [Google Scholar]
- 21.Butkus V, Klimašauskas S, Keršulytė D, Vaitkevičius D, Lebionka A, Janulaitis A. 1985. Investigation of restriction-modification enzymes from M. varians RFL19 with a new type of specificity toward modification of substrate. Nucleic Acids Res. 13, 5727–5746. ( 10.1093/nar/13.16.5727) [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Hazra TK, Izumi T, Venkataraman R, Kow YW, Dizdaroglu M, Mitra S. 2000. Characterization of a novel 8-oxoguanine-DNA glycosylase activity in Escherichia coli and identification of the enzyme as endonuclease VIII. J. Biol. Chem. 275, 27 762–27 767. ( 10.1074/jbc.M004052200) [DOI] [PubMed] [Google Scholar]
- 23.Boiteux S, Coste F, Castaing B. 2017. Repair of 8-oxo-7,8-dihydroguanine in prokaryotic and eukaryotic cells: properties and biological roles of the Fpg and OGG1 DNA N-glycosylases. Free Radic. Biol. Med. 107, 179–201. ( 10.1016/j.freeradbiomed.2016.11.042) [DOI] [PubMed] [Google Scholar]
- 24.Dalhus B, Forsbring M, Helle IH, Vik ES, Forstrøm RJ, Backe PH, Alseth I, Bjørås M. 2011. Separation-of-function mutants unravel the dual-reaction mode of human 8-oxoguanine DNA glycosylase. Structure 19, 117–127. ( 10.1016/j.str.2010.09.023) [DOI] [PubMed] [Google Scholar]
- 25.Hardeland U, Bentele M, Jiricny J, Schär P. 2000. Separating substrate recognition from base hydrolysis in human thymine DNA glycosylase by mutational analysis. J. Biol. Chem. 275, 33 449–33 456. ( 10.1074/jbc.M005095200) [DOI] [PubMed] [Google Scholar]
- 26.Vik ES, Alseth I, Forsbring M, Helle IH, Morland I, Luna L, Bjørås M, Dalhus B. 2012. Biochemical mapping of human NEIL1 DNA glycosylase and AP lyase activities. DNA Repair (Amst.) 11, 766–773. ( 10.1016/j.dnarep.2012.07.002) [DOI] [PubMed] [Google Scholar]
- 27.Krokeide SZ, Bolstad N, Laerdahl JK, Bjørås M, Luna L. 2009. Expression and purification of NEIL3, a human DNA glycosylase homolog. Protein Expr. Purif. 65, 160–164. ( 10.1016/j.pep.2008.11.014) [DOI] [PubMed] [Google Scholar]
- 28.Leiros I, et al. 2007. Structural basis for enzymatic excision of N1-methyladenine and N3-methylcytosine from DNA. EMBO J. 26, 2206–2217. ( 10.1038/sj.emboj.7601662) [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Bailly V, Verly WG. 1987. Escherichia coli endonuclease III is not an endonuclease but a β-elimination catalyst. Biochem. J. 242, 565–572. ( 10.1042/bj2420565) [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Dizdaroglu M, Burgess SM, Jaruga P, Hazra TK, Rodriguez H, Lloyd RS. 2001. Substrate specificity and excision kinetics of Escherichia coli endonuclease VIII (Nei) for modified bases in DNA damaged by free radicals. Biochemistry 40, 12 150–12 156. ( 10.1021/bi015552o) [DOI] [PubMed] [Google Scholar]
- 31.Kropachev KY, Zharkov DO, Grollman AP. 2006. Catalytic mechanism of Escherichia coli endonuclease VIII: roles of the intercalation loop and the zinc finger. Biochemistry 45, 12 039–12 049. ( 10.1021/bi060663e) [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Cameron V, Uhlenbeck OC. 1977. 3′-Phosphatase activity in T4 polynucleotide kinase. Biochemistry 16, 5120–5126. ( 10.1021/bi00642a027) [DOI] [PubMed] [Google Scholar]
- 33.Midgley CA, Murray NE. 1985. T4 polynucleotide kinase; cloning of the gene (pseT) and amplification of its product. EMBO J. 4, 2695–2703. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Warner HR, Demple BF, Deutsch WA, Kane CM, Linn S. 1980. Apurinic/apyrimidinic endonucleases in repair of pyrimidine dimers and other lesions in DNA. Proc. Natl Acad. Sci. USA 77, 4602–4606. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Doetsch PW, Cunningham RP. 1990. The enzymology of apurinic/apyrimidinic endonucleases. Mutat. Res. 236, 173–201. ( 10.1016/0921-8777(90)90004-O) [DOI] [PubMed] [Google Scholar]
- 36.Dattagupta JK, Saenger W, Bolewska K, Kulakowska I. 1977. X-ray study of 1,5,N(4),N(4)-tetramethylcytosine – an overcrowded molecule with planar structure. Acta Cryst. B 33, 85–89. ( 10.1107/S0567740877002635) [DOI] [Google Scholar]
- 37.Lesyng B, Saenger W. 1978. Theoretical investigations on the conformation of 1,5,N(4)-tetramethylcytosine. Biochim. Biophys. Acta 544, 215–224. ( 10.1016/0304-4165(78)90224-6) [DOI] [PubMed] [Google Scholar]
- 38.Viville S, Mantovani R. 1994. S1 mapping using single-stranded DNA probes. In Protocols for Gene Analysis 9 (ed. AJ Harwood) Methods Mol. Biol. 31, 299–305. [DOI] [PubMed] [Google Scholar]
- 39.Patel PH, Suzuki M, Adman E, Shinkai A, Loeb LA. 2001. Prokaryotic DNA polymerase I: evolution, structure, and ‘base flipping’ mechanism for nucleotide selection. J. Mol. Biol. 308, 823–837. ( 10.1006/jmbi.2001.4619) [DOI] [PubMed] [Google Scholar]
- 40.Chauleau M, Shuman S. 2016. Kinetic mechanism and fidelity of nick sealing by Escherichia coli NAD+-dependent DNA ligase (LigA). Nucleic Acids Res. 44, 2298–2309. ( 10.1093/nar/gkw049) [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.Kulikowski T, Żmudzka B, Shugar D. 1969. Preparation and properties of N4-alkyl analogues of 5-methyl-2′-deoxycytidine, their 5′-mono- and triphosphates, and N4-alkyl derivatives of 1,5-dimethylcytosine. Acta Biochim. Pol. 16, 201–217. [PubMed] [Google Scholar]
- 42.Audette GF, Kumar SVP, Gupta SV, Quail JW. 1998. N4,5-Dimethyl-2′-deoxycytidine. Acta Cryst. C 54, 1987–1990. ( 10.1107/S0108270198008026) [DOI] [Google Scholar]
- 43.Kusmierek J, Käppi R, Neuvonen K, Shugar D, Lönnberg H. 1989. Kinetics and mechanisms of hydrolytic reactions of methylated cytidines under acidic and neutral conditions. Acta Chem. Scand. 43, 196–202. ( 10.3891/acta.chem.scand.43-0196) [DOI] [Google Scholar]
- 44.Dalhus B, Laerdahl JK, Backe PH, Bjørås M. 2009. DNA base repair – recognition and initiation of catalysis. FEMS Microbiol. Rev. 33, 1044–1078. ( 10.1111/j.1574-6976.2009.00188.x) [DOI] [PubMed] [Google Scholar]
- 45.Kisker C, Kuper J, Van Houten B.. 2013. Prokaryotic nucleotide excision repair. Cold Spring Harb. Perspect. Biol. 5, a012591 ( 10.1101/cshperspect.a012591) [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Data Availability Statement
The datasets supporting this article have been uploaded as part of the electronic supplementary material.