

ABSTRACT
Most organisms, from Bacteria to Eukarya, synthesize UDP–N-acetylglucosamine (UDP-GlcNAc) from fructose-6-phosphate via a four-step reaction, and UDP–N-acetylgalactosamine (UDP-GalNAc) can only be synthesized from UDP-GlcNAc by UDP-GlcNAc 4-epimerase. In Archaea, the bacterial-type UDP-GlcNAc biosynthetic pathway was reported for Methanococcales. However, the complete biosynthetic pathways for UDP-GlcNAc and UDP-GalNAc present in one archaeal species are unidentified. Previous experimental analyses on enzymatic activities of the ST0452 protein, identified from the thermophilic crenarchaeon Sulfolobus tokodaii, predicted the presence of both a bacterial-type UDP-GlcNAc and an independent UDP-GalNAc biosynthetic pathway in this archaeon. In the present work, functional analyses revealed that the recombinant ST2186 protein possessed an glutamine:fructose-6-phosphate amidotransferase activity and that the recombinant ST0242 protein possessed a phosphoglucosamine-mutase activity. Along with the acetyltransferase and uridyltransferase activities of the ST0452 protein, the activities of the ST2186 and ST0242 proteins confirmed the presence of a bacterial-type UDP-GlcNAc biosynthetic pathway in S. tokodaii. In contrast, the UDP-GlcNAc 4-epimerase homologue gene was not detected within the genomic data. Thus, it was expected that galactosamine-1-phosphate or galactosamine-6-phosphate (GalN-6-P) was provided by conversion of glucosamine-1-phosphate or glucosamine-6-phosphate (GlcN-6-P). A novel epimerase converting GlcN-6-P to GalN-6-P was detected in a cell extract of S. tokodaii, and the N-terminal sequence of the purified protein indicated that the novel epimerase was encoded by the ST2245 gene. Along with the ST0242 phosphogalactosamine-mutase activity, this observation confirmed the presence of a novel UDP-GalNAc biosynthetic pathway from GlcN-6-P in S. tokodaii. Discovery of the novel pathway provides a new insight into the evolution of nucleotide sugar metabolic pathways.
IMPORTANCE In this work, a novel protein capable of directly converting glucosamine-6-phosphate to galactosamine-6-phosphate was successfully purified from a cell extract of the thermophilic crenarchaeon Sulfolobus tokodaii. Confirmation of this novel activity using the recombinant protein indicates that S. tokodaii possesses a novel UDP-GalNAc biosynthetic pathway derived from glucosamine-6-phosphate. The distributions of this and related genes indicate the presence of three different types of UDP-GalNAc biosynthetic pathways: a direct pathway using a novel enzyme and two conversion pathways from UDP-GlcNAc using known enzymes. Additionally, Crenarchaeota species lacking all three pathways were found, predicting the presence of one more unknown pathway. Identification of these novel proteins and pathways provides important insights into the evolution of nucleotide sugar biosynthesis, as well as being potentially important industrially.
KEYWORDS: thermostable enzymes, UDP-GalNAc, UDP-GlcNAc, Sulfolobus tokodaii, GlcN-6-phosphate/GalN-6-phosphate epimerase, phosphoglucosamine-mutase, phosphogalactosamine-mutase, glutamine:fructose-6-phosphate amidotransferase, UDP-GalNAc biosynthetic pathway, evolution, pathway-bridging enzyme
INTRODUCTION
Carbohydrate molecules play many crucial roles in most organisms as sources of energy, as part of heredity molecules, in immunological recognition, and in the separation of internal and external cellular environments. Among carbohydrate molecules, N-acetylglucosamine (GlcNAc) and N-acetylgalactosamine (GalNAc) are found conjugated to a variety of molecules, including peptidoglycans (1), lipopolysaccharides (2), chitin (3), and glycans that are N and O linked to proteins (4, 5) in microorganisms and also participate in N- and O-linked glycosylation (6) in eukaryotic cells. In order to incorporate these molecules into polymer structures, they need to be converted to their activated forms, namely, UDP–N-acetylglucosamine (UDP-GlcNAc) and UDP–N-acetylgalactosamine (UDP-GalNAc). In bacteria, UDP-GlcNAc is synthesized from fructose-6-phosphate (Frc-6-P) through a four-step reaction as shown in Fig. 1: reaction I, conversion of Frc-6-P to glucosamine-6-phosphate (GlcN-6-P); reaction II, isomerization of GlcN-6-P to GlcN-1-P; reaction III, acetylation of GlcN-1-P to GlcNAc-1-P; and reaction IV, combination of GlcNAc-1-P with UTP to form UDP-GlcNAc (7, 8). In eukaryotes, the acetylation of GlcN-6-P occurs prior to the conversion of the phosphate position (9). Conversely, in most organisms, UDP-GalNAc is known to be synthesized solely from UDP-GlcNAc by the action of UDP-GlcNAc 4-epimerase (reaction c in Fig. 1) (10–12). In Archaea, the third domain of life, which was established by a comparison of 16S rRNA gene sequences around 40 years ago (13), it was reported for UDP-GlcNAc biosynthetic pathway only that the MMP1680 protein and the MMP1077 protein, identified from Methanococcus maripaludis, exhibited catalyzing activity for reactions I and II, respectively, and that the MJ1101 protein, identified from Methanococcus jannaschii, exhibited activity catalyzing both reactions III and IV (Fig. 1) (14). However, the complete biosynthetic pathway for UDP-GlcNAc present in one archaeal species has not been characterized, especially for Crenarchaeota, and the UDP-GalNAc biosynthetic pathway has not been characterized in any archaeon.
FIG 1.

Outline of the predicted and confirmed UDP-GlcNAc and UDP-GalNAc biosynthetic pathways. The chemical names within boxes are sugar compounds in these metabolic pathways. The enzymatic activity required for each step is shown within parentheses as a roman numeral. Arrows labeled a and b are the predicted reactions required for the construction of the distinct UDP-GalNAc biosynthetic pathway (shown on the left) and analyzed in this work. The dashed arrow labeled c indicates the known UDP-GalNAc biosynthetic pathway. The stippled background indicates the UDP-GalNAc biosynthetic pathway present in S. tokodaii identified in this study. Each cofactor used for each reaction is shown alongside the reaction. The ST designations indicate the protein catalyzing the corresponding reaction identified from S. tokodaii, and the underlined designations indicate the proteins characterized in this study.
Sulfolobus tokodaii, an acido- and thermophilic crenarchaeon isolated from a Beppu hot spring in Japan in the early 1980s (15), possesses unusual properties, including utilization of the nonphosphorylated Entner-Doudoroff (ED) pathway for energy production, the presence of introns in its tRNA genes, and a sulfur utilization pathway. Although its entire genomic sequence was determined 15 years ago (16), the UDP-GlcNAc biosynthetic pathway could not be predicted from its genomic data. The ST0452 protein, encoded by the gene that was originally predicted to encode glucose-1-phosphate thymidylyltransferase by similarity analysis, is a bifunctional protein with multiple sugar-1-phosphate nucleotidylyltransferase activities, including N-acetylglucosamine-1-phosphate uridyltransferase (GlcNAc-1-P UTase) (reaction IV in Fig. 1) (17) and N-acetylgalactosamine-1-phosphate uridyltransferase (GalNAc-1-P UTase) (reaction VII in Fig. 1) activities, and multiple amino-sugar-1-phosphate acetyltransferase activities, including glucosamine-1-phosphate acetyltransferase (GlcN-1-P AcTase) (reaction III in Fig. 1) and galactosamine-1-phosphate acetyltransferase (GalN-1-P AcTase) (reaction VI in Fig. 1) activities (18). Its confirmed activities (reactions III and IV in Fig. 1) resulted in the proposal of the existence of a bacterial-type UDP-GlcNAc biosynthetic pathway (18).
In this study, we attempted to characterize the biosynthetic pathways for both the UDP-GlcNAc and UDP-GalNAc molecules in S. tokodaii. We experimentally confirmed the UDP-GlcNAc biosynthetic pathway predicted from experimental data for the ST0452 protein using the genomic data and in addition identified a novel and direct biosynthetic pathway for UDP-GalNAc from GlcN-6-P through the isolation of novel isomerase activity from cell extracts of S. tokodaii. These observations provide important information concerning the evolution and development of nucleotide sugar molecule biosynthetic pathways, suggest future studies focused on understanding the reaction mechanism and three-dimensional structure of the novel enzyme, and in addition provide a tool for the industrial production of sugar molecules that are currently not commercially available.
RESULTS AND DISCUSSION
Confirmation of bacterial-type UDP-GlcNAc biosynthetic pathway.
The previously confirmed GlcN-1-P AcTase and GlcNAc-1-P UTase activities (reactions III and IV in Fig. 1) of the ST0452 protein suggest that the bacterial-type UDP-GlcNAc biosynthetic pathway is present in S. tokodaii (18). The ST2186 gene was detected in the S. tokodaii genomic data as being homologous to a gene encoding a glutamine:fructose-6-phosphate amidotransferase (GF6PAT), an enzyme catalyzing the first reaction of the UDP-GlcNAc biosynthetic pathway converting Frc-6-P to GlcN-6-P (reaction I in Fig. 1). To confirm the expected enzymatic activity, the ST2186 gene was expressed in Escherichia coli. The recombinant ST2186 protein produced from the first ATG codon (underlined codon in Fig. 2) (ST2186L) was successfully expressed in E. coli as a thermostable and soluble protein, as shown in Fig. 3. Its GF6PAT activity was then analyzed. During the GF6PAT reaction, the amino group of glutamine is transferred to Frc-6-P to produce GlcN-6-P (Fig. 1), and glutamine is therefore converted to glutamate. A linear increase in glutamate concentration with time means that the GF6PAT reaction is proceeding. When the ST2186L protein was added to the reaction mixture and incubated at 80°C, glutamate production was not observed, revealing that the GF6PAT activity was not present in the ST2186L protein (Fig. 4A).
FIG 2.

Sequence alignment of the N-terminal regions of the ST2186 protein and homologous bacterial proteins. Alignment of amino acid sequences was performed using the ClustalW program provided from DDBJ. ST2186n and Ec_n indicate the nucleotide sequences corresponding to the N-terminal regions of the ST2186 protein and the GF6PAT protein from Escherichia coli (locus, AE005174), respectively. ST2186p, Hi, Bs, and Ec_p indicate the amino acid sequences of the N-terminal regions of the ST2186 protein and the GF6PAT proteins from Haemophilus influenzae (locus, AAC22088), Bacillus subtilis (locus, GLMS_BACPZ), and E. coli (locus, AE005604_9), respectively. The underlined codon is the start codon (ATG) used for construction of the expression vector for the ST2186L protein. The double-underlined minor start codon (GTG) was used for construction of the expression vector for the ST2186S protein after conversion to the ATG codon. Gaps are shown by dashes.
FIG 3.
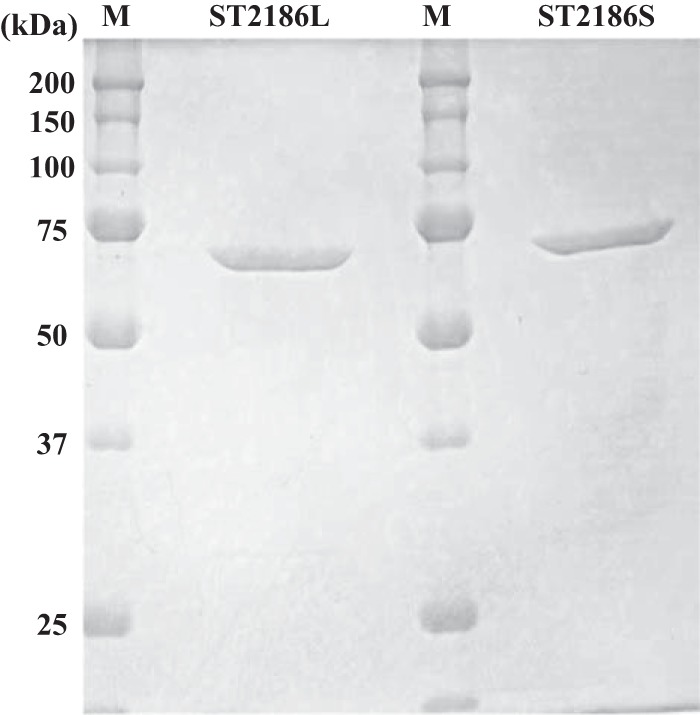
SDS-PAGE separation patterns of the purified recombinant ST2186 proteins. SDS-PAGE analyses of the ST2186 protein synthesized from the first ATG codon (ST2186L) and from the third TGT codon (ST2186S) are shown. The separated proteins were stained with Coomassie brilliant blue R-250. Lanes M, standard molecular mass marker proteins. The molecular mass of the ST2186 protein is estimated to be 65.5 kDa.
FIG 4.

UPLC elution profile of Gln and the products produced by the action of the two ST2186 proteins. The top panels show the elution profiles of the Gln and Glu standard molecules, and the other panels show the elution profiles of products formed by incubation with the ST2186L (A) and ST2186S (B) proteins for the indicated periods at 80°C.
As GF6PAT activity was not detected in the ST2186L protein, the N-terminal amino acid sequence of the ST2186 protein was carefully compared with those of bacterial GF6PAT enzymes. As shown in Fig. 2, the ST2186L protein was found to contain an additional three amino acid residues (Met-Ser-Leu) upstream of the usual translation initiation position of the bacterial GF6PAT enzymes. Thus, the fourth codon, GTG, in the ST2186 gene, an alternative start codon but at a position usually used as the translation initiation codon in bacterial enzymes, was used for the translation start position for expression in E. coli by conversion to ATG. The three-residue-shorter form of the protein (ST2186S) was also successfully expressed as a soluble and thermostable protein in E. coli, as shown in Fig. 3, after which its GF6PAT activity was analyzed. When the recombinant ST2186S protein was added to the reaction mixture and incubated at 80°C, a clear, time-dependent production of glutamate was observed (Fig. 4B). This observation revealed that the ST2186S protein can catalyze the GF6PAT reaction. This is the experimental evidence that the ST2186S protein can catalyze the first step of the UDP-GlcNAc biosynthetic pathway from Frc-6-P in S. tokodaii.
From the experimental data on these two forms of the ST2186 protein, it can be suggested that the N-terminal three residues completely inhibit the ST2186 GF6PAT activity. It was previously shown that the N-terminal Cys residue has a crucial role in GF6PAT glutaminase catalysis (19–21), because the GF6PAT activity was completely lacking on the mutant protein after replacement of the first Cys residue by Ala. The analyses of the crystal structure of the bacterial wild-type GF6PAT protein indicate that interaction of the first Cys residue with the C terminus of another subunit is essential for the catalytic activity (22, 23). The crystal structure of the C1A mutant protein indicated that the normal dimeric form was changed to an inactive hexamer form by this substitution (24). Also, with addition of a His6 tag at the N-terminal end of the Candida albicans GF6PAT enzyme, GF6PAT activity was completely lost (25). This observation supports our data that when a 200-fold excess of the synthesized peptide (Met-Ser-Leu) was added to the reaction mixture containing the ST2186S protein, no inhibition of the GF6PAT activity was observed (data not shown). Although prediction of the three-residue structure was not successful, our observations accompanied by the previous data suggest that the three residues covalently associated with the N terminus of the GF6PAT enzyme may inhibit construction of the proper interaction of the first Cys residue with the C-terminal domain of another subunit. To analyze whether the longer form of the ST2186 protein is synthesized in living S. tokodaii cells, purification of the native form of the protein and determination of its N-terminal amino acid sequence will be necessary.
In contrast, a gene homologous to the phosphoglucosamine-mutase (PGlcNM) gene, encoding the enzyme capable of catalyzing the second step (reaction II in Fig. 1) in the bacterial-type UDP-GlcNAc biosynthetic pathway from Frc-6-P, was not found in the S. tokodaii genomic data. Only the ST0242 gene, which was originally predicted to encode the phosphomannomutase/phosphoglucomutase (PMM/PGM) by similarity searching, was detected within the S. tokodaii genomic data as the sole gene homologous to a phospho-sugar mutase (PSugM) gene. Although PGlcNM and PMM/PGM are known to be independent proteins in most bacteria (26), because no other homologous gene encoding a protein with the PSugM feature was detected within the S. tokodaii genomic data, the ST0242 gene product was expected to possess PGlcNM activity.
To test this expectation, the recombinant ST0242 protein was expressed in E. coli, and it was found in the aqueous fraction as soluble protein after incubation for 20 min at 80°C (Fig. 5). The PGlcNM activity was then analyzed for the purified recombinant ST0242 protein. Direct detection of the conversion between GlcN-1-P and GlcN-6-P was not analyzed, because the equipment detecting these two amino sugar molecules was not available in our laboratory at the time of this analysis. Therefore, a coupling reaction system for detecting PGlcNM activity was constructed using the ST0452 protein as the coupling enzyme. The ST0452 protein can catalyze an acetyl-transfer reaction only for GlcN-1-P to produce GlcNAc-1-P and not for GlcN-6-P (reaction III in Fig. 1) (18). The same ST0452 protein then can combine the produced GlcNAc-1-P with UTP to produce UDP-GlcNAc (17). When GlcN-6-P is used as the sole substrate for mutase activity, if the protein in the reaction mixture can convert GlcN-6-P to GlcN-1-P, it will be catalyzed by the ST0452 protein, a coupling enzyme in this system, to UDP-GlcNAc in the presence of UTP and acetyl coenzyme A (acetyl-CoA) (reaction IV in Fig. 1). In this coupling system, production of the UDP-GlcNAc is evidence for the presence of the GlcNM activity. To evaluate the PGlcNM activity of the candidate ST0242 protein, the final product was analyzed by high-pressure liquid chromatography (HPLC). Before incubation, no UDP-GlcNAc was detected in the reaction mixture (Fig. 6A). In contrast, after 5 min of incubation, UDP-GlcNAc was clearly detected (Fig. 6B), revealing that the ST0242 protein possessed PGlcNM activity.
FIG 5.
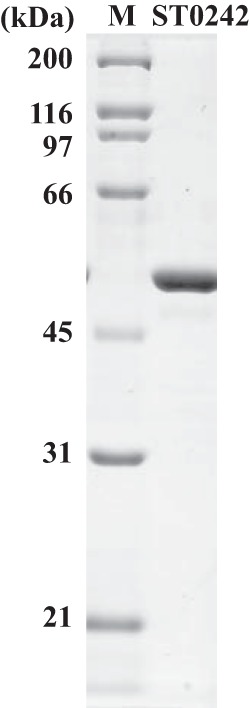
SDS-PAGE separation patterns of the purified recombinant ST0242 protein. SDS-PAGE analysis of the purified ST0242 protein after treatment at 80°C for 20 min and staining with Coomassie brilliant blue R-250 is shown. Lane M, standard molecular mass marker proteins. The molecular mass of the ST0242 protein is estimated to be 50.1 kDa.
FIG 6.
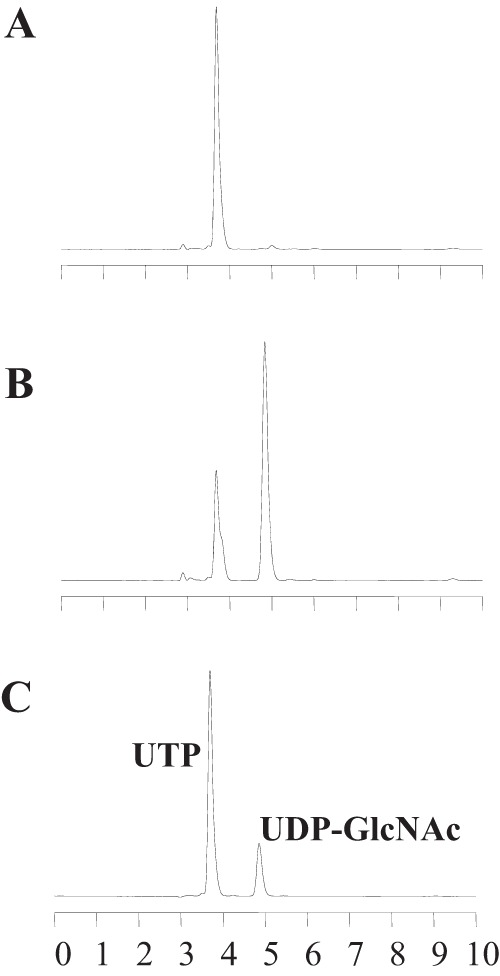
HPLC elution profile of the product of the ST0242 protein. HPLC elution profiles of UTP and UDP-GlcNAc molecules, products of the coupling reaction with ST0242 and ST0452 activity, are shown. (A and B) Elution profiles of the products formed before (A) and after (B) 5 min of incubation with the ST0242 and ST0452 proteins at 80°C. (C) Elution profiles of the UTP and UDP-GlcNAc standard molecules.
In addition to the PGlcNM activity, PMM/PGM activity was detected in the ST0242 protein (data not shown). To understand the multiple substrate recognition mechanism of the ST0242 protein, a more detailed analysis of its activity, including substitution of the amino acid residues located within its reaction center and determination of the three-dimensional structure of a complex form of the ST0242 protein with a substrate(s), will be required.
The identification of GF6PAT activity in the recombinant ST2186 protein and PGlcNM activity in the ST0242 protein confirmed the presence of the bacterial-type UDP-GlcNAc biosynthetic pathway from Frc-6-P in S. tokodaii.
Identification of a novel UDP-GalNAc biosynthetic pathway.
From the previous observations that the GalN-1-P AcTase and GalNAc-1-P UTase activities (reactions VI and VII in Fig. 1) could be identified in the ST0452 protein (18) and that the UDP-GlcNAc 4-epimerase gene could not be found within the S. tokodaii genomic data, we predicted that the common UDP-GalNAc biosynthetic pathway leading from UDP-GlcNAc, catalyzed by UDP-GlcNAc 4-epimerase, was not functional in S. tokodaii (see reaction c in Fig. 1). Therefore, an unknown and distinct UDP-GalNAc biosynthetic pathway was expected to be present in S. tokodaii. If the UDP-GalNAc biosynthetic pathway utilizes steps similar to those in the parallel UDP-GlcNAc biosynthetic pathway, in the step upstream from galactosamine-6-phosphate (GalN-6-P), i.e., the step corresponding to reaction I in the UDP-GlcNAc biosynthetic pathway, tagatose-6-phosphate (Tag-6-P) should be utilized as starting material. However, it is known that the reverse direction of this reaction is used to supply Tag-6-P in bacteria (27). Therefore, it was expected that an alternative pathway capable of converting GlcN-1-P to GalN-1-P or GlcN-6-P to GalN-6-P (reaction a or b in Fig. 1) might be present in S. tokodaii.
To explore this possibility, a cell extract of S. tokodaii was prepared, concentrated 20-fold, and incubated separately with GlcN-1-P or GlcN-6-P. The absence of endogenous sugar molecules, which influenced the following analyses, was confirmed by chromatography analysis of concentrated cell extract alone. Although no product was detected when GlcN-1-P was used as the substrate, an unidentified peak was detected after incubation of the S. tokodaii cell extract with GlcN-6-P at 80°C (Fig. 7, line d). This observation indicated the presence of a novel phosphorylated amino sugar isomerase activity capable of converting GlcN-6-P to GalN-6-P in S. tokodaii. However, since GalN-6-P is not commercially available, it was not possible to compare the elution position of standard GalN-6-P with that of the product produced from GlcN-6-P by the S. tokodaii cell extract. The elution position of the unidentified peak was compared with those of other available singly phosphorylated sugar molecules; however, the same elution position was not detected, revealing that the unidentified peak contained an unavailable compound including GalN-6-P.
FIG 7.

Elution profiles of standards and the phosphorylated amino sugar molecules. Lines a and b, ion chromatography elution patterns of the two standard molecules, GalN-1-P (a) and GlcN-6-P (b). Lines c and d, elution profiles of the products obtained from GalN-1-P following incubation with the ST0242 protein at 80°C (c) and from GlcN-6-P following incubation with the S. tokodaii cell extract (d). Line e, elution profile of the mixture of the two samples used for lines c and d. The arrows indicate the peaks for GalN-1-P, GlcN-6-P, and the product from GlcN-6-P with the S. tokodaii cell extract or from GalN-1-P with the ST0242 protein.
Therefore, a different approach was required to obtain evidence that the product in the unidentified peak was actually GalN-6-P. As shown in Fig. 1, when GalN-6-P is produced from GlcN-6-P, the mutase enzyme must convert GalN-6-P to GalN-1-P (reaction V in Fig. 1) for this distinct UDP-GalNAc biosynthetic pathway. GalN-1-P was available, and the previously described ST0242 protein was the sole PSugM candidate in this archaeon. Therefore, if the ST0242 protein could catalyze the conversion of GalN-1-P to the unidentified product (reaction V in Fig. 1) and the product peak could migrate to the same position as that of the unidentified product formed from GlcN-6-P by the S. tokodaii cell extract, it should be evident that the unidentified peak is GalN-6-P. To explore this notion, GalN-1-P was incubated with the purified recombinant ST0242 protein. In the analysis of this reaction, an unidentified peak was detected at the same position as that of the product formed from GlcN-6-P by the cell extract of S. tokodaii, as shown by line c in Fig. 7. To evaluate whether these two unidentified products were exactly same, these two reaction solutions were mixed and analyzed. As shown in line e in Fig. 7, the two unidentified products migrated at the exactly the same position, revealing that the same product was produced by two independent reactions using GlcN-6-P and GalN-1-P as substrates. Thus, we concluded that the unidentified product produced by these two independent reactions was GalN-6-P. Confirmation of this GalN-6-P product by different approaches, such as nuclear magnetic resonance (NMR), should be done. We are attempting this analysis; however, it has not yet succeeded.
The observations above suggest that an uncharacterized novel phosphorylated amino sugar isomerase enzymatic activity converting GlcN-6-P to GalN-6-P is present in the S. tokodaii cell extract and that the ST0242 protein possesses phosphogalactosamine-mutase activity.
We therefore attempted to purify this uncharacterized protein with the novel phosphorylated amino sugar isomerase activity converting GlcN-6-P to GalN-6-P from the S. tokodaii cell extract. After a five-step purification described in Materials and Methods, a fraction containing two main protein bands was recovered as the fraction indicating the expected novel phosphorylated amino sugar isomerase activity from the S. tokodaii cell extract (Fig. 8A, lane F). The N-terminal amino acid sequences of these proteins were then determined. The sequence determined for the stronger band, XNNPXENXXN, was compared with the amino acid sequences predicted from all the open reading frames (ORFs) present in the S. tokodaii genomic data. The N-terminal sequence of the ST2245 protein, MNNPYENWIN, matched the obtained sequence. The nucleotide sequence of the ST2245 gene was used to design PCR primers to amplify the gene, and the amplified fragment was cloned into an expression vector. As a result, the recombinant ST2245 protein (Fig. 8B) was successfully produced in E. coli as a soluble and thermostable protein, after which its enzymatic activity was analyzed. As shown in Fig. 9, the recombinant ST2245 protein had a higher enzymatic activity than the purified fraction, revealing that the recombinant ST2245 protein possessed the ability to convert GlcN-6-P to GalN-6-P. This is therefore the first report of the identification of a GlcN-6-P/GalN-6-P epimerase activity.
FIG 8.
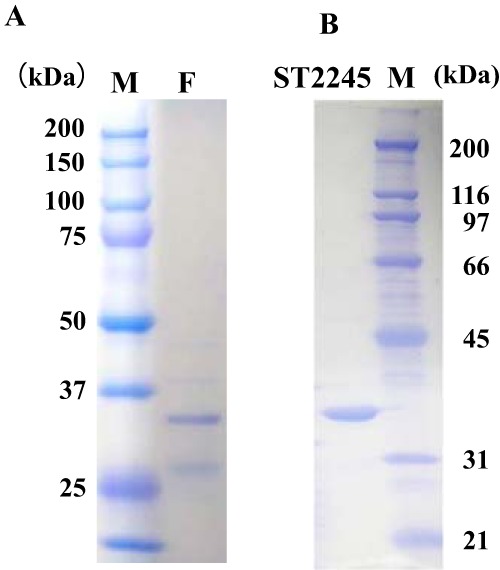
SDS-PAGE separation patterns of the purified fraction and the recombinant ST2245 protein. SDS-PAGE patterns of the final fraction (F) of purified S. tokodaii cell extract and the recombinant ST2245 protein (ST2245) stained with Coomassie brilliant blue R-250 are shown. Lane M, standard molecular mass marker proteins. The ST2245 protein showed a molecular mass of ca. 35.1 kDa.
FIG 9.
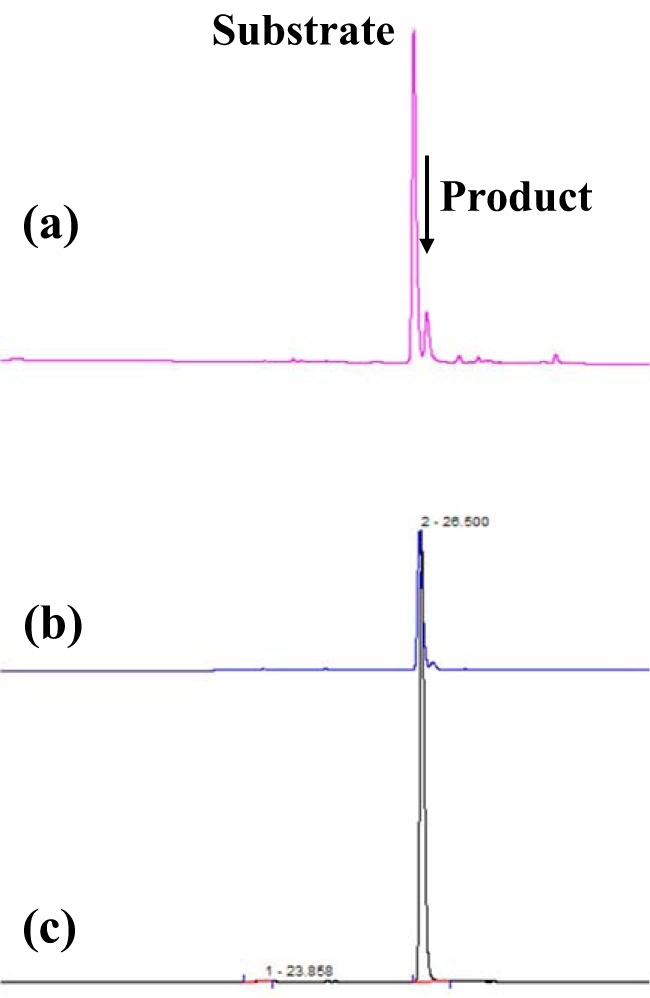
Elution profiles of the substrate and the phosphorylated amino sugar molecules produced by the purified fraction and the recombinant ST2245 protein. Ion chromatography elution profiles of the product produced from GlcN-6-P by the recombinant ST2245 protein (line a), the product produced from GlcN-6-P by the final fraction of the native purified enzyme from the S. tokodaii cell extract (line b), and the standard GlcN-6-P substrate molecule (line c) are shown. The arrow indicates the product (GalN-6-P) peak produced from GlcN-6-P.
This novel enzyme should provide important information for understanding the catalytic mechanism capable of interconversion of GlcN-6-P and GalN-6-P, because previously characterized enzymes capable of interconverting of Glc and Gal are able to catalyze only the conversion of nucleotide sugar substrates, such as UDP-Glc or UDP-GlcNAc. In order to fully understand the nature of the ST2245 protein, its three-dimensional structure should also be determined.
From the observations described above, we can conclude that S. tokodaii possesses a bacterial-type UDP-GlcNAc biosynthetic pathway comprised of the ST2186, ST0242, and ST0452 proteins (I to IV in Fig. 1). It also is shown that S. tokodaii does not use the common pathway for UDP-GalNAc formation from UDP-GlcNAc by the action of UDP-GlcNAc 4-epimerase but instead forms UDP-GalNAc via a distinct biosynthetic pathway comprised of the ST2245, ST0242, and ST0452 proteins (a, V, VI, and VII in Fig. 1). To the best of our knowledge, this is the first report of a distinct UDP-GalNAc biosynthetic pathway in any organism.
Evolutionary analysis of UDP-GalNAc biosynthetic pathways.
Because the novel enzyme capable of converting GlcN-6-P to GalN-6-P and the distinct UDP-GalNAc biosynthetic pathway were identified only in the thermophilic archaeon S. tokodaii, it was of interest to analyze the evolution and development of this pathway in archaea and other microorganisms. To determine how many microorganisms may utilize this distinct UDP-GalNAc biosynthetic pathway, the distribution of genes homologous to the ST2245 gene was examined.
For UDP-hexose 4-epimerases, classification into three groups was proposed based on their substrate specificity (28). Among these three groups, enzymes in group 2, which can epimerize UDP-Glc/Gal and UDP-GlcNAc/GalNAc, and in group 3, which preferably epimerize between UDP-GlcNAc and UDP-GalNAc, can catalyze the reaction of UDP-GalNAc biosynthesis from UDP-GlcNAc. Thus, typical enzymes in these two groups were also chosen as queries for analysis of the gene distribution. The ST2245 protein, the bacterial UDP-GlcNAc 4-epimerase (accession no. Q8GGB8) (29), and bacterial UDP-GlcNAc/Glc 4-epimerase (accession no. ABA29504) (30) were used as queries to search for the distribution of their homologous genes in microorganisms.
As shown in Table 1, genes homologous to ST2245 were detected only within the Crenarchaeota species; Euryarchaeota and bacteria did not possess such genes. All Euryarchaeota species analyzed and one bacterial species were found to possess genes homologous the to bacterial UDP-GlcNAc 4-epimerase gene, and all the bacterial species analyzed possessed genes homologous to the UDP-GlcNAc/Glc 4-epimerase gene.
TABLE 1.
Distribution of three genes in different microorganisms
| Organism | Presence of gene fora: |
||
|---|---|---|---|
| ST2245 | UDP-GlcNAc 4-Ep | UDP-Glc 4-Ep | |
| Crenarchaeota | |||
| Sulfolobus acidocaldarius | + | ND | ND |
| Sulfolobus islandicus | + | ND | ND |
| Sulfolobus solfataricus | + | ND | ND |
| Metallosphaera sedula | + | ND | ND |
| Desulfurococcus kamchatkensis | ND | ND | ND |
| Caldivirga maquilingensis | ND | ND | ND |
| Aeropyrum pernix | ND | ND | ND |
| Thermofilum pendens | ND | ND | ND |
| Euryarchaeota | |||
| Pyrococcus horikoshii | ND | + | ND |
| Pyrococcus furiosus | ND | + | ND |
| Pyrococcus abyssi | ND | + | ND |
| Methanococcus jannaschii | ND | + | ND |
| Bacteria | |||
| Rhodothermus obamensis | ND | + | ND |
| Hydrogenobaculum sp. strain Y04AAS1 | ND | ND | + |
| Desulforudis audaxviator | ND | ND | + |
| Chloroherpeton thalassium | ND | ND | + |
| Anaerocellum thermophilum | ND | ND | + |
| Caldicellulosiruptor saccharolyticus | ND | ND | + |
| Hydrogenivirga sp. strain 128-5-R1-1 | ND | ND | + |
| Dictyoglomus turgidum | ND | ND | + |
+, present; ND, not detected. UDP-GlcNAc 4-Ep, UDP-GlcNAc 4-epimerase; UDP-Glc 4-Ep, UDP-GlcNAc/Glc 4-epimerase.
These data indicate that each species utilizes one of these three systems, except for four Crenarchaeota species, i.e., Aeropyrum pernix, Caldivirga maquilingensis, Desulfurococcus kamchatkensis, and Thermofilum pendens, that were found not to possess any of the three homologous genes. The observation that species both with and without the ST2245-homologous genes are present in Crenarchaeota indicates that the distinct UDP-GalNAc biosynthetic pathway might have been established after the independence of Crenarchaeota.
As listed in Table 1, four Crenarchaeota species were found not to possess any of the three genes, indicating that these species could not form UDP-GalNAc via the common or distinct UDP-GalNAc biosynthetic pathway. This observation indicates the possible presence of another distinct UDP-GalNAc biosynthetic pathway in these four Crenarchaeota species.
The ST2245 protein might also provide important information on the evolution of the UDP-GalNAc biosynthetic pathway. To obtain more knowledge, a more detailed analysis, including solving the actual activities of genes detected in the other species, is required. Finally, the ST2245 and ST0242 proteins might have potential for the industrial production of GalN-6-P, which is not commercially available at present.
MATERIALS AND METHODS
General materials and microorganisms.
UTP, phosphorylated sugars, and all chemical compounds used in this work were purchased from Sigma-Aldrich Co. (St. Louis, MO). The restriction enzymes and a DNA ligation kit (Mighty Mix) were obtained from TaKaRa Bio Inc. (Shiga, Japan). KOD-plus-Neo DNA polymerase was purchased from Toyobo Co., Ltd. (Osaka, Japan). The thermoacidophilic crenarchaeon S. tokodaii (JCM 10545) was obtained from the Japan Collection of Microorganisms. Growth of S. tokodaii and extraction of its genomic DNA were carried out according to a previously described protocol (17). Escherichia coli strains JM109 for DNA manipulation and BL21-CodonPlus(DE3)-RIPL and RIL for protein expression were purchased from TaKaRa Bio Inc. and Stratagene (La Jolla, CA), respectively. The expression plasmid vectors pET21a, pET23b, and pET28a were purchased from Novagen (Madison, WI). The His SpinTrap Talon column and SpinHis Trap column were obtained from GE Healthcare Life Sciences Corp. (Piscataway, NJ), and the Amicon Ultra 0.5-ml Ultracel-30K filter and Amicon Ultra-0.5 centrifugal filter devices were purchased from Merck Millipore, Ltd. (Darmstadt, Germany).
Construction of expression vectors.
To construct the expression vector for the S. tokodaii glutamine:fructose-6-phosphate amidotransferase candidate (ST2186), translation of which was started from the first Met codon, the primers pST2186LF (GGCCATATGTCTCTTGTGTGCGG) and pST2186R (CCCCCCCGAATTCTTATTATTCTACAGTAACTGTTTTAG) were designed from the 5′- and 3′-terminal regions of the ST2186 ORF, respectively. To construct the expression vector which expressed the ST2186 protein from the third codon, the primer pST2186SF (CATATGTGCGGAATAATTGGTATAGTATCACTAAG) was designed from the third-codon region with conversion of the TGT codon to ATG. PCR products were digested with NdeI and EcoRI and cloned into the pET21a vector digested with the same restriction enzymes. After confirmation by sequencing, the constructed plasmids were designated pETST2186L and pETST2186S.
In order to construct the expression vector for the phospho-sugar mutase candidate (ST0242), primers pST0242F (TTAATTCCATATGGGTAAGCTTTTTGGTACTGAC) and pST0242R (ATATACTCGAGTCATTTACCCTCTACAATC) were designed from the 5′ and 3′ regions of the ST0242 ORF, respectively, and used for PCR amplification of the ST0242 ORF region. The amplified PCR product was digested with NdeI and XhoI and ligated with the pET28a DNA digested with the same restriction enzymes. The nucleotide sequence of the expression vector was confirmed and shown to encode a histidine tag at the N-terminal end of the encoded protein. The plasmid was designated pETST0242NHis.
For construction of the expression vector encoding the protein with novel phosphorylated-amino-sugar epimerase activity, the determined N-terminal sequence was used to search for ORFs in S. tokodaii. The ST2245 ORF was detected as a candidate novel epimerase, and the nucleotide sequences of its 5′ and 3′ regions were used to design primers for PCR amplification of this ORF. The primers pST2245F (CATGCCATGGATAATCCATATGAGAATTGG) and pST2245RH (CCGCTCGAGTAAATTCTGATCTGCATTAAT) were used for PCR amplification of the ORF region. The amplified PCR fragment digested with NcoI and XhoI was inserted into the pET23b vector digested with the same restriction enzymes. After confirmation by sequencing, the expression vector possessing the ST2245 ORF with a histidine tag at the N-terminal end of its product was designated pETST2245His.
Expression and purification of recombinant proteins.
BL21-CodonPlus(DE3)-RIPL cells harboring one of the constructed expression vectors were incubated at 37°C until the absorbance at 600 nm reached 0.7. After addition of IPTG (isopropyl-β-d-thiogalactopyranoside) to a final concentration of 50 μg/ml, incubation at 25°C was continued for a further 3 h. The harvested E. coli cells were resuspended into 1/20 volume of 50 mM MOPS (morpholinepropanesulfonic acid) buffer (pH 7.5) and then ruptured by sonication with a Bioruptor (CosmoBio, Tokyo, Japan). After centrifugation at 4,000 × g for 20 min, the collected supernatant was treated at 80°C for 20 min to remove E. coli proteins. The recombinant proteins with a 6×His tag, the ST2245 and ST0242 proteins, were purified using a SpinHis Trap column, and the proteins without a 6×His tag, the ST2186L and ST2186S proteins, were purified by affinity chromatography and gel filtration. The concentrations of the purified proteins were determined using a Pierce bicinchoninic acid (BCA) protein assay kit (Thermo Scientific, Waltham, MA).
Purification of the protein with novel GlcN-6-P epimerase activity. (i) Preparation of crude extract.
For preparation of the cell extract, S. tokodaii was cultured in 5 liters of Sulfolobus culture medium (31) supplemented with 1 g/liter glucose and 1 g/liter Casamino Acids at 80°C for 3 days at 150 rpm, and the grown cells were collected by centrifugation. After washing with 500 ml of 20 mM Tris-HCl (pH 7.5), the cells were suspended in 250 ml of the same solution. The suspended cells were ruptured by sonication with a Vibra-Cell (Sonics, Newton, CT), using 60 cycles of 20-s pulses followed by a 10-s rest on ice; the resulting lysate was centrifuged at 10,000 × g for 15 min at 4°C. The supernatant was collected and used as the crude extract.
(ii) Ammonium sulfate precipitation.
Solid ammonium sulfate was gradually added to the crude extract to a final concentration of 30%. After incubating the extract for 30 min with stirring at room temperature, the precipitate was separated by centrifugation. Solid ammonium sulfate was added to the supernatant fraction to a concentration of 60%. After incubation of this mixture for 30 min with stirring at room temperature, the precipitate was removed by centrifugation. The expected activity was detected in the supernatant fraction. This fraction was then subjected to the further purification with a chromatography column, using AKTAprime (GE Healthcare, Piscataway, NJ).
(iii) Weak anion-exchange chromatography on Toyopearl DEAE-650.
After dialysis of the fraction against 20 mM Tris-HCl (pH 8.0) buffer solution, the fraction was applied to a Toyopearl DEAE-650 ion-exchange chromatography column (Tosoh, Tokyo, Japan) equilibrated with the same buffer solution. After washing with the same solution, proteins were eluted with a solution containing 20 mM Tris-HCl (pH 8.0) and 500 mM NaCl. The fractions with the expected activity were collected and used for the next purification step.
(iv) Anion-exchange chromatography on HiTrap Q.
After dialysis against 20 mM Tris-HCl (pH 8.0) buffer solution, the fraction was loaded onto a HiTrap Q anion-exchange chromatography column (GE Healthcare, Piscataway, NJ) equilibrated with the same solution. After washing with the same buffer solution, bound proteins were eluted with a linear gradient of 0 to 0.5 M NaCl in 20 mM Tris-HCl (pH 8.0) buffer solution. The active fractions with the expected activity were pooled and used for the next step of purification.
(v) Hydrophobic interaction chromatography on HiTrap Phenyl.
The fractions demonstrating the expected activity were collected and dialyzed against a solution containing 20 mM Tris-HCl (pH 8.0) and 30% ammonium sulfate to adjust the condition of the sample for hydrophobic interaction purification using a HiTrap phenyl column (GE Healthcare, Piscataway, NJ). After a washing with the same buffer solution, bound proteins were eluted with a linear gradient of 30 to 0% ammonium sulfate in 20 mM Tris-HCl (pH 8.0) solution. The proteins in the fraction with the expected activity were used for N-terminal amino acid sequencing.
Determination of the N-terminal sequences of proteins.
For determining the N-terminal amino acid sequences of the proteins included in the final purified fraction, the proteins were separated by SDS-polyacrylamide gel electrophoresis and transferred onto a polyvinylidene difluoride (PVDF) membrane after staining with Ponceau S solution (0.1% [wt/vol] Ponceau S in 5% [vol/vol] acetic acid). The PVDF membrane including each protein was separated and applied to a Shimadzu Biotech PPSQ-31A protein sequencer (Kyoto, Japan). The phenylthiohydantoin (PTH) mixture of amino acid standards (Wako Pure Chemicals, Osaka, Japan) was used for setting up the standard condition for amino acid determination. The operation was performed according to the manufacturer's manual. The determined amino acid sequences were used for similarity searching against the all of the ORFs in the genomic data for S. tokodaii.
Analyses of enzymatic activities. (i) GF6PAT.
Based on the catalytic mechanism of glutamine:fructose-6-phosphate amidotransferase (GF6PAT) activity, an amino group is transferred from glutamine to Frc-6-P, and as a result, glutamine is converted to glutamate. Thus, in order to detect the enzymatic activity, glutamate production was monitored by ultraperformance liquid chromatography (UPLC).
Assay of the GF6PAT activity was performed in a 100-μl reaction mixture containing 50 mM HEPES buffer (pH 7.5), 200 mM KCl, 2 mM dithiothreitol (DTT), 0.5 mM Frc-6-P, 0.5 mM glutamine, and 6.55 ng of the purified recombinant ST2186L or ST2186S protein. After 5 min of preincubation at 80°C, the reaction was initiated by the addition of the recombinant protein and allowed to progress at 80°C for 20 min. Io order to terminate the reaction, 100 μl of 10% trichloroacetic acid (TCA) was added. For quantitative analysis of glutamate, amino acid labeling was used. A 25-μl aliquot of the final reaction solution was mixed with 50 μl of 2% Boc-Cys-OH, 2% o-phthalaldehyde methanol solution, and 175 μl of borate buffer (pH 10.4) and incubated for 2 min at 25°C in the dark. Immediately after completion of labeling, the sample was analyzed on an Acquity UPLC TUV system (Waters, Milford, MA) with an AccQ-Tag Ultra RP column (2.1 by 100 mm). The flow rate and column temperature were maintained at 0.25 ml/min and 30°C, respectively. The UPLC gradient profile used was 15 to 21% B for 7 min, 21 to 40% B for 1 min, 40% B for 1 min, and 40 to 15% B for 0.01 min (A, 50 mM sodium acetate buffer [pH 5.9]; B, methanol). The excitation and emission wavelengths for the detection of fluorescent amino acids were 350 nm and 450 nm, respectively.
(ii) PGlcNM.
For detection of phosphoglucosamine-mutase (PGlcNM) activity, a coupling reaction with the ST0452 protein was used. The ST0452 protein can catalyze the acetylation reaction only for GlcN-1-P to form GlcNAc-1-P, which is then combined with UTP to form UDP-GlcNAc by the function of the same ST0452 protein. If GlcN-6-P is provided as a substrate and acetyl-CoA, UTP, and the ST0452 protein are added, and if ST0242 could catalyze mutase activity on GlcN-6-P, then the product GlcN-1-P could be used in the following reaction with the coupling enzyme, the ST0452 protein, to yield UDP-GlcNAc. Thus, PGlcNM activity can be monitored by the production of UDP-GlcNAc.
The coupling reaction was carried out in a 20-μl reaction solution containing 50 mM MOPS (pH 7.6), 2 mM MgCl2, 2 mM GlcN-6-P, 20 μM glucosec-1,6-bis-phosphate (GlcBP), 2 mM acetyl-CoA, 0.5 mM UTP, 10 ng of the purified ST0452 protein, and 2 ng of the recombinant ST0242 protein. After pretreatment at 80°C for 1 min, the reaction was initiated by the addition of GlcN-6-P. After the reaction was allowed to progress for 5 min at 80°C, the reaction solution was immediately cooled on ice to terminate the reaction, and then 180 μl of 500 mM KH2PO4 was added to adjust the solution content to that used in HPLC separation. A 50-μl aliquot of the solution was analyzed on a Hitachi HPLC LaChrom Elite (Hitachi High-Technologies, Tokyo, Japan) with a 0.46- by 25-cm Wakosil 5C18-200 column (Wako, Tokyo, Japan). The flow rate of the 500 mM KH2PO4 solution was maintained at 1 ml/min. The product, UDP-GlcNAc, was monitored by absorbance at 257 nm.
(iii) Glucosamine-6-phosphate epimerase.
The glucosamine-6-phosphate epimerase activity assay was carried out in a 500-μl reaction solution containing 50 mM Tris-HCl (pH 8.0), 1 mM GlcN-6-P, 1 mM MgCl2, 0.5 mM EDTA, and 50 μl of the crude extract or fraction solution. After 2 min of pretreatment at 80°C, the reaction was initiated by the addition of the protein in the fraction and allowed to progress for the appropriate period at 80°C. After completion of the reaction, it was terminated by immediately placing the mixture on ice. The reaction solution was passed through Amicon Ultra-0.5 centrifugal filter devices to remove the remnant proteins and particles in the reaction solution. Phosphorylated sugar molecules contained in a 5-μl aliquot of the reaction solution were then analyzed using a Thermo Scientific Dionex ICS-5000+ ion chromatography system equipped with an amperometry detector and a CarboPac PA10 column (Thermo Scientific, Waltham, MA). The flow rate and column temperature were maintained at 1 ml/min and 30°C, respectively. The ion chromatography gradient profile used was 10 to 20% B for 20 min and 20 to 50% B for 10 min (A, 100 mM NaOH; B, 1 M sodium acetate in 100 mM NaOH).
(iv) Phosphogalactosamine-mutase.
The phosphogalactosamine-mutase reaction was carried out in a 500-μl reaction mixture containing 50 mM Tris-HCl (pH 7.6), 5 mM MgCl2, 1 mM GalN-1-P, 100 μM GlcBP, 2.5 mM DTT, and 0.6 μg of the recombinant ST0242 protein. After pretreatment at 80°C for 2 min, the reaction was initiated by addition of the ST0242 protein and allowed to progress at 80°C. After incubation for the appropriate period, the reaction was terminated by chilling on ice. To remove the proteins from the solution, the reaction mixture was passed through Amicon Ultra-0.5 centrifugal filter devices. A 5-μl aliquot of the treated solution was then analyzed using a Thermo Scientific Dionex ICS-5000+ ion chromatography system equipped with an amperometry detector and a CarboPac PA10 column under the same conditions referred to above.
ACKNOWLEDGMENTS
This work was partly supported by the Institute for Fermentation, Osaka (IFO).
We thank Katsumi Doi for help with the analysis of the N-terminal amino acid sequences of the proteins and Toshihisa Ohshima for stimulating discussions.
REFERENCES
- 1.Barbosa MD, Young G, Fang J, Kurilla MG, Pompliano DL. 2002. Development of a whole-cell assay for peptidoglycan biosynthesis inhibitors. Antimicrob Agents Chemother 46:943–946. doi: 10.1128/AAC.46.4.943-946.2002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Whitfield C, Trent MS. 2014. Biosynthesis and export of bacterial lipopolysaccharides. Annu Rev Biochem 83:99–128. doi: 10.1146/annurev-biochem-060713-035600. [DOI] [PubMed] [Google Scholar]
- 3.Zhang D, Wang PG, Qi Q. 2007. A two-step fermentation process for efficient production of penta-N-acetyl-chitopentaose in recombinant Escherichia coli. Biotechnol Lett 29:1729–1733. doi: 10.1007/s10529-007-9462-y. [DOI] [PubMed] [Google Scholar]
- 4.Niemann MC, Bartrina I, Ashikov A, Weber H, Novak O, Spichal L. 2015. Arabidopsis ROCK1 transports UDP-GlcNAc/UDP-GalNAc and regulates ER protein quality control and cytokinin activity. Proc Natl Acad Sci U S A 112:291–296. doi: 10.1073/pnas.1419050112. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Mohorko E, Glockshuber R, Aebi M. 2011. Oligosaccharyltransferase: the central enzyme of N-linked protein glycosylation. J Inherit Metab Dis 34:869–878. doi: 10.1007/s10545-011-9337-1. [DOI] [PubMed] [Google Scholar]
- 6.Hanisch F. 2001. O-glycosylation of the mucin type. J Biol Chem 382:143–149. [DOI] [PubMed] [Google Scholar]
- 7.Mengin-Lecreulx D, Heijeenoort J. 1993. Identification of the glmU gene encoding N-acetylglucosamine-1-phophate uridyltransferase in Escherichia coli. J Bacteriol 175:6150–6157. doi: 10.1128/jb.175.19.6150-6157.1993. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Mengin-Lecreulx D, Heijeenoort J. 1994. Copurification of glucosamine-1-phosphate acetyltransferase and N-acetylglucosamine-1-phosphate uridyltransferase activities of Escherichia coli: characterization of the glmU gene product as a bifunctional enzyme catalyzing two subsequent steps in the pathway for UDP-N-acetylglucosamine synthesis. J Bacteriol 176:5788–5795. doi: 10.1128/jb.176.18.5788-5795.1994. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Gooday GW. 1977. Biosynthesis of the fungal wall—mechanisms and implications. The first Fleming Lecture. J Gen Microbiol 99:1–11. doi: 10.1099/00221287-99-1-1. [DOI] [PubMed] [Google Scholar]
- 10.Stingele F, Vincent SJF, Faber EJ, Newell JW, Kamerling JP, Neeser JR. 1999. Introduction of the exopolysaccharide gene cluster from Streptococcus thermophiles Sfi6 into Lactococcus lactis MG1363: production and characterization of an altered polysaccharide. Mol Microbiol 32:1287–1295. doi: 10.1046/j.1365-2958.1999.01441.x. [DOI] [PubMed] [Google Scholar]
- 11.Guo H, Li L, Wang PG. 2006. Biochemical characterization of UDP-GlcNAc/Glc 4-epimerase from Escherichia coli O86:B7. Biochemistry 45:13760–13768. doi: 10.1021/bi0612770. [DOI] [PubMed] [Google Scholar]
- 12.Schulz JM, Ross KL, Malmstrom K, Krieger M, Fridovich-Keil JL. 2005. Mediators of galactose sensitivity in UDP-galactose 4′-epimerase-impaired mammalian cells. J Biol Chem 280:13493–13502. doi: 10.1074/jbc.M414045200. [DOI] [PubMed] [Google Scholar]
- 13.Woese CR, Mangrum LJ, Fox GE. 1978. Archaebacteria. J Mol Evol 11:245–252. doi: 10.1007/BF01734485. [DOI] [PubMed] [Google Scholar]
- 14.Namboori SC, Graham DE. 2008. Acetamido sugar biosynthesis in the Euryarchaea. J Bacteriol 190:2987–2996. doi: 10.1128/JB.01970-07. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Suzuki T, Iwasaki T, Hara K, Kon T, Ueki T, Yamagishi A, Oshima T. 2002. Sulfolobus tokodaii sp. nov. (f. Sulfolobus sp. strain 7), a new member of the genus Sulfolobus isolated from Beppu Hot Springs, Japan. Extremophiles 6:39–44. [DOI] [PubMed] [Google Scholar]
- 16.Kawarabayasi Y, Hino Y, Horikawa H, Jin-no K, Takahashi M, Sekine M, Baba S, Ankai A, Kosugi H, Hosoyama A, Fukui S, Nagai Y, Nishijima K, Otsuka R, Nakazawa H, Takamiya M, Kato Y, Yoshizawa T, Tanaka T, Kudoh Y, Yamazaki J, Kushida N, Oguchi A, Aoki K, Masuda S, Yanagii M, Nishimura M, Yamagishi A, Oshima T, Kikuchi H. 2001. Complete genome sequence of an aerobic thermoacidophilic crenarchaeon, Sulfolobus tokodaii strain 7. DNA Res 8:123–140. doi: 10.1093/dnares/8.4.123. [DOI] [PubMed] [Google Scholar]
- 17.Zhang Z, Tsujimura M, Akutsu J, Sasaki M, Tajima H, Kawarabayasi Y. 2005. Identification of an extremely thermostable enzyme with dual sugar-1-phosphate nucleotidylyltransferase activities from an acidothermophilic archaeon, Sulfolobus tokodaii strain 7. J Biol Chem 280:9698–9705. doi: 10.1074/jbc.M411211200. [DOI] [PubMed] [Google Scholar]
- 18.Zhang Z, Akutsu J, Kawarabayasi Y. 2010. Identification of novel acetyltransferase activity on the thermostable protein ST0452 from Sulfolobus tokodaii strain 7. J Bacteriol 192:3287–3293. doi: 10.1128/JB.01683-09. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Badet B, Vermoote P, Haumont PY, Lederer F, Le Goffic F. 1987. Glucosamine synthetase from Escherichia coli: purification, properties, and glutamine-utilizing site location. Biochemistry 26:1940–1948. doi: 10.1021/bi00381a023. [DOI] [PubMed] [Google Scholar]
- 20.Badet B, Vermoote P, Le Goffic F. 1988. Glucosamine synthetase from Escherichia coli: kinetic mechanism and inhibition by N3-fumaroyl-L-2,3-diaminopropionic derivatives. Biochemistry 27:2282–2287. doi: 10.1021/bi00407a006. [DOI] [PubMed] [Google Scholar]
- 21.Valerio-Lepiniec M, Aumont-Nicaise M, Roux C, Raynal B, England P, Badet B, Badet-Denisot MA, Desmadril M. 2010. Analysis of the Escherichia coli glucosamine-6-phosphate synthase activity by isothermal titration calorimetry and differential scanning calorimetry. Arch Biochem Biophys 498:95–104. doi: 10.1016/j.abb.2010.04.010. [DOI] [PubMed] [Google Scholar]
- 22.Isupov MN, Obmolova G, Butterworth S, Badet-Denisot MA, Badet B, Polikarpov I, Littlechild JA, Teplyakov A. 1996. Substrate binding is required for assembly of the active conformation of the catalytic site in Ntn amidotransferases: evidence from the 1.8 Å crystal structure of the glutaminase domain of glucosamine 6-phosphate synthase. Structure 4:801–810. doi: 10.1016/S0969-2126(96)00087-1. [DOI] [PubMed] [Google Scholar]
- 23.Mouilleron S, Badet-Denisot MA, Badet B, Golinelli-Pimpaneau B. 2011. Dynamics of glucosamine-6-phosphate synthase catalysis. Arch Biochem Biophys 505:1–12. doi: 10.1016/j.abb.2010.08.008. [DOI] [PubMed] [Google Scholar]
- 24.Mouilleron S, Badet-Denisot MA, Pecqueur L, Madiona K, Assrir N, Badet B, Golinelli-Pimpaneau B. 2012. Structural basis for morpheein-type allosteric regulation of Escherichia coli glucosamine-6-phosphate synthase: equilibrium between inactive hexamer and active dimer. J Biol Chem 287:34533–34546. doi: 10.1074/jbc.M112.380378. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Olchowy J, Kur K, Sachadyn P, Milewski S. 2006. Construction, purification, and functional characterization of His-tagged Candida albicans glucosamine-6-phosphate synthase expressed in Escherichia coli. Protein Expr Purif 46:309–315. doi: 10.1016/j.pep.2005.07.030. [DOI] [PubMed] [Google Scholar]
- 26.Mengin-Lecreulx D, van Heijenoort J. 1996. Characterization of the essential gene glmM encoding phosphoglucosamine mutase in Escherichia coli. J Biol Chem 271:32–39. doi: 10.1074/jbc.271.1.32. [DOI] [PubMed] [Google Scholar]
- 27.Shiota T, Bluementhal H, Disraely MN, McCann MP. 1962. Tagatose 6-phosphate formation from galactosamine 6-phosphate by cell-free extracts of Lactobacillus casei. Arch Biochem Biophys 96:143–146. doi: 10.1016/0003-9861(62)90462-9. [DOI] [PubMed] [Google Scholar]
- 28.Ishiyama N, Creuzenet C, Lam JS, Berghuis AM. 2004. Crystal structure of WbpP, a genuine UDP-N-acetylglucosamine 4-epimerase from Pseudomonas aeruginosa: substrate specificity in UDP-hexose 4-epimerases. J Biol Chem 279:22635–22642. doi: 10.1074/jbc.M401642200. [DOI] [PubMed] [Google Scholar]
- 29.Creuzenet C, Belanger M, Wakarchuk WW, Lam JS. 2000. Expression, purification, and biochemical characterization of WbpP, a new UDP-GlcNAc C4 epimerase from Pseudomonas aeruginosa serotype O6. J Biol Chem 275:19060–19067. doi: 10.1074/jbc.M001171200. [DOI] [PubMed] [Google Scholar]
- 30.Bernatchez S, Szymanski CM, Ishiyama N, Li J, Jarrell HC, Lau PC, Berghuis AM, Young NM, Wakarchuk WW. 2005. A single bifunctional UDP-GlcNAc/Glc 4-epimerase supports the synthesis of three cell surface glycoconjugates in Campylobacter jejuni. J Biol Chem 280:4792–4802. doi: 10.1074/jbc.M407767200. [DOI] [PubMed] [Google Scholar]
- 31.Brock TD, Brock KM, Belly RT, Weiss RL. 1972. Sulfolobus: a new genus of sulfur oxidizing bacteria living at low pH and high temperature. Arch Mikrobiol 84:54–68. doi: 10.1007/BF00408082. [DOI] [PubMed] [Google Scholar]