

Abstract
Sepsis commonly results in both acute and chronic brain dysfunction, which dramatically increase the morbidity associated with this common disease. Chronic brain dysfunction in animal models of sepsis survival is linked to persistent neuroinflammation and expression of multiple cytokines. We have previously found, however, that microglia predominantly upregulate the damage associated molecule S100A8/A9 after sepsis. Here, we show that S100A8/A9 is increased in the brains of patients who died of sepsis, and that S100A8 is expressed in astrocytes and myeloid cells. Using a mouse model of sepsis survival, we show that S100A8/A9 is persistently expressed in the brain after sepsis. S100A9 expression is necessary for recruitment of neutrophils to the brain, and priming production of reactive oxygen species and TNFα secretion in microglia and macrophages. Despite improving these indices of chronic inflammation, however, S100A9 deficiency also results in worsened anxiety-like behavior 2 weeks after sepsis. Taken together, these results indicate that S100A8/A9 contributes to several facets of neuroinflammation in sepsis survivor mice, including granulocyte recruitment and priming of microglial reactive oxygen species and cytokine production, and that these processes may be protective against anxiety behavior in sepsis survivors.
Keywords: microglia, reactive oxygen species, priming, TNFα
Introduction
Sepsis, a severe inflammatory response to infection, is a leading cause of death in the United States and results in both acute and chronic brain dysfunction (1, 2). As treatment of sepsis has improved, over 1.3 million patients survive sepsis in the United States alone each year (3–6). Sepsis survivors frequently suffer long-term functional sequelae of their critical illness, and have markedly increased risk of developing chronic brain dysfunction, including cognitive and affective disorders (7–12). Both acute and chronic brain injury may be due to insults occurring during severe sepsis and critical illness, including hypoxia, hypotension and oxidative stress (12, 13). Patients who die of sepsis exhibit diverse forms of neuropathology, including hemorrhage, ischemia, elevated brain expression of multiple cytokines, and microglial activation (14–16). The link between the severe forms of neuropathology observed in patients who are dying of sepsis and the pathophysiology of long-term brain injury are unclear. However, in animal models, chronic brain dysfunction after sepsis is associated with ongoing neuroinflammation during the period of recovery from critical illness (17–19).
While pathogen associated molecules serve as profound stimuli of the innate immune system and neuroinflammation, prolonged expression of endogenous damage associated molecular pattern (DAMP) molecules perpetuates systemic and neuroinflammation (20, 21). For example, systemic neutralization of the DAMP HMGB1 in sepsis survivor mice reduces neuroinflammation and improves sepsis-associated behavioral deficits (19). In addition, neutralization of the receptor for advanced glycated endproducts (RAGE) within the hippocampus ameliorates memory impairment and amyloidogenesis in sepsis survivor mice (22). We previously found that the genes S100A8 and S100A9, components of the heterodimeric DAMP S100A8/A9, are highly upregulated in microglia of sepsis survivor mice two weeks after undergoing cecal ligation and puncture (CLP) (17, 23).
In the central nervous system (CNS), S100A8/A9 has been shown to contribute to deleterious inflammation. In a model of antibiotic-treated pneumococcal meningitis, deficiency of S100A9 reduced cerebrospinal fluid pleocytosis, cerebral edema, and chemokine expression without worsening bacterial clearance or mortality (24). S100A8 and S100A9 have been associated with non-infectious CNS pathology, as well. Neutralization of S100A8 decreased acute postoperative neuroinflammation and sickness behavior in a tibial fracture model (25). S100A8 and S100A9 expression are also increased in aging and in Alzheimer’s disease, and are intrinsically amyloidogenic (26–30).
In this study, we demonstrate that the DAMP S100A8/A9 is elevated in the brains of patients who die of sepsis and is produced by multiple cell types within the human brain. Using a murine model of survival after sepsis, we find that S100A8/A9 is persistently expressed in the mouse brain for at least two weeks following sepsis. S100A8/A9 expression contributes both to infiltration of peripheral immune cells into the CNS after sepsis, and to priming of microglial activation in sepsis survivor mice. Deficiency of S100A8/A9, however, resulted in worsened behavioral outcomes in sepsis survivor mice. These findings suggest that S100A8/A9 is highly expressed in the acute systemic inflammatory response and is an important driver of long-term neuroinflammation after sepsis, but may be functionally protective in the setting of infection.
Materials and Methods
Post-mortem human brain tissue
Patient samples were obtained from the Michigan Alzheimer’s Disease Center brain bank. Chart review was performed to determine if brain bank donors had died while hospitalized at the University of Michigan, and if so, whether they had died of sepsis or a clear noninfectious cause (Table S1). Brain tissue associated with the Michigan Alzheimer’s Disease Center brain bank was either formalin fixed followed by paraffin embedding at the time of autopsy, or frozen at the time of autopsy and stored at −20C. For this study, frozen brain tissue was obtained from the parietal cortex. Fixed tissue sections were obtained from parietal or temporal cortex and sectioned at 5 μm. This type of study involving only deceased subjects is not considered human subjects research, and formal consent is not required.
Mice
Both male and female mice were used in experimental and control groups at 8–12 weeks of age. In cases where both male and female mice were used in a single experiment, sex was balanced among all treatment and control groups. C57BL/6 mice were obtained from Jackson Laboratory. S100A9−/− mice were a gift of Maria Castro (University of Michigan) with permission of Thomas Vogl (University of Munster) (31). TNFα−/− mice were of the strain B6.129S-Tnftm1Gkl/J, originally obtained from Jackson labs (32). CCR2RFP/+∷CX3CR1GFP/+ mice were based on individual knock-in mice and bred in cis by Richard Ransohoff, and were a gift of Richard Ransohoff and Dolores Hambardzumyan (33–35). S100A9−/− and TNFα−/− were bred as homozygotes, and were compared to non-littermate control C57BL/6 mice. CCR2RFP/+∷CX3CR1GFP/+ mice were compared to littermate controls. All applicable international, national, and/or institutional guidelines for the care and use of animals were followed. All procedures performed in studies involving animals were in accordance with the ethical standards of the institution or practice at which the studies were conducted. The study protocol was approved by the University Committee on the Use and Care of Animals of the University of Michigan (protocol #PRO00007115).
Quantification of S100A8/A9 in brain tissue
Human brain tissue (200–400 mg) was homogenized in modified RIPA buffer (140 mM NaCl, 10 mM Tris-HCl, 1% Triton X-100, 0.1% SDS, 0.1% Na deoxycholate) supplemented with protease inhibitor (Roche) using a rotor-stator homogenizer (Tissue-Tearor, Biospec Products). Lysates were sonicated briefly (20 seconds at 62 W) and then agitated intermittently for 1 hour. Particulates were cleared by centrifugation. S100A8/A9 was measured by ELISA according to the manufacturer’s instructions (Biolegend). All samples were diluted 10-fold for ELISA and normalized to total protein as measured by bicinchoninic acid assay (Thermo Scientific). Mouse brain protein extracts were prepared from mouse brain hemispheres in the same manner, and S100A8/A9 was measured by a heterodimer specific ELISA (R&D Systems).
Immunofluorescent staining
Sections of human brain tissue (5 μm) were mounted on Superfrost Plus slides and dried overnight at 37°C. Slides were deparaffinized and washed in Tris-buffered saline (TBS). Endogenous peroxidases were quenched in 1% hydrogen peroxide. Heat mediated epitope retrieval was performed in alkaline buffer (10 mM Tris, 1 mM EDTA, 0.05% Tween, pH 9) in a pressure cooker at 15 psi of pressure for 10 minutes. Slides were cooled, washed with TBS and blocked in blocking buffer (10% normal horse serum, 3% BSA, 1% glycine, 0.4% Triton-X in TBS). For single stained sections, sections were incubated overnight at 4°C with rabbit anti-S100A8 antibody (Abcam, clone EPR3554, 2.5 μg/mL). Sections were then washed and incubated in biotinylated anti-rabbit secondary antibody (Vector Labs) for 30 minutes at room temperature, followed by amplification with avidin-biotin-horseradish peroxidase (HRP) complex reagent (Vector Labs). Staining was then visualized with fluorescein-tyramide (Perkin-Elmer). For double staining with S100A8 and cell type markers, sections were deparaffinized, treated with hydrogen peroxide, and underwent epitope retrieval. Sections were then incubated overnight at at 4°C with either goat anti-Iba1 (Abcam, polyclonal), goat anti-GFAP (Abcam, polyclonal), mouse anti-ZO1 (Thermo Fisher, clone ZO1-1A12), goat polyclonal IgG, or rabbit polyclonal IgG. They were then incubated with the appropriate secondary antibody, avidin-biotin amplification, and visualization with fluorescein-tyramide. The antibody complex was then eluted by microwave heating in citrate solution (10 mM citrate, pH 6.0) until boiling, and residual HRP activity was quenched by treatment in 1% peroxide. Sections were then re-blocked in blocking buffer and incubated overnight at 4°C with rabbit anti-S100A8 antibody. Sections were then incubated with secondary antibody, avidin-biotin-HRP, and visualized using Alexa 350 tyramide (Life Technologies) as above. Alexa 350 was utilized due to intense tissue autofluorescence at wavelengths above 600 nM. Sections were washed extensively with TBS between all steps. The specificity of multiplex staining was confirmed by replacing primary antibodies with the appropriate species of IgG and verifying that there was no cross-reaction labeling with fluorescein or Alexa-350.
Confocal Imaging
Sections were examined under a confocal laser scanning microscope where indicated (Leica SP5 Inverted 2-Photon, Manheim, DE). Images were acquired using a white light laser exciting at 495 nm (Alexa Fluor 488) or 594 nm (Alexa 594) and multiphoton excitation at 720nm (Alexa Fluor 350). Alexa 488 emission was detected at 500-580 nm, Alexa 594 emission was detected at 604-750 nm and Alexa 350 emission was detected at 360-470 nm. Images were obtained on six sequential scans. Images acquired as z-stacks and presented using orthogonal projections or maximum intensity projections. Merging of images on co-stained slides was performed using FIJI/ImageJ (36). Red pseudocolor was applied to Alexa 350 images to improve visualization of co-staining.
Cecal ligation and puncture
Cecal ligation and puncture was performed as previously described (17, 37). Animal suffering and distress were minimized with local lidocaine infiltration as well as anesthesia with ketamine and xylazine. Briefly, under aseptic conditions, a 1–2 cm laparotomy was performed. The cecum was ligated with a silk suture and punctured through-and-through with a 19-gauge needle. The incision was closed with surgical clips. Sham operation consisted of laparotomy with mobilization of the cecum, but without ligation-puncture procedure. Imipenem/cilastatin (Merck, 0.5 mg/mouse in 200 μl of normal saline) and normal saline (0.5 mL) were administered subcutaneously to all mice at the time of surgery. All mice also received buprenorphine (0.05 mg/kg) for analgesia two times a day for 48 hours post-operatively. The University of Michigan policy for humane endpoints was followed.. Deaths occurred within 5 days of CLP, were attributed to the procedure, and were considered expected.
Isolation of whole-brain RNA
Mice were euthanized by CO2 inhalation and rapidly trancardially perfused with ice cold Hank’s Balanced Salt Solution (HBSS). Brains were grossly free of blood. Brain hemispheres were then homogenized in 3 mL of Trizol reagent and RNA was isolated according to the manufacturer’s instructions (Qiagen).
Flow cytometry and cell sorting
Blood was prepared by ACK lysis. Single cell suspensions of brain were prepared as previously described (17, 38). Mice were euthanized with CO2 and transcardially perfused with ice-cold HBSS. Brains were grossly free of blood. Whole brain was minced, and either mechanically triturated (in experiments for determination of relative proportions of infiltrating cell types) or enzymatically homogenized (in cell sorting experiments) with Neural Tissue Dissociation Kit with Papain, Miltenyi) and passed through a 70 μm cell strainer to form a single cell suspension. Cells were enriched by centrifugation over a discontinuous gradient of HBSS, 37% and 70% Percoll and collecting the fraction at the 37%/70% boundary. Cells were then washed and stained with fluorophore conjugated antibodies prior to analysis on a FACSAria II flow cytometer and cell sorter (BD). Antibodies include anti-CD11b (clone M1/70, BD), anti-CD45 (clone 30-F11, BD), anti-CD64 (clone X54-5/7.1, BD), anti-Ly6G (clone 1A8, Biolegend), anti-Ly6C (clone HK1.4, Biolegend). Propidium iodide was used as a live/dead marker. Gating was performed as described in Figure S1.
Isolation of RNA from cell populations
Cells were sorted into PBS supplemented with 10% FBS and 25 mM HEPES, and were centrifuged at 500g to remove FACS fluid prior to lysis in RLT buffer. Between 100,000 and 200,000 cells were sorted per sample. RNA was extracted using RNAeasy mini columns (Qiagen) according to the manufacturer instructions, followed by re-precipitation in sodium acetate and ethanol to remove residual buffer salts.
Gene expression analysis
RT-PCR was performed on a StepOnePlus thermocycler (Applied Biosystems) as previously described (39). All primers were purchased from Integrated DNA Technologies. Fold changes were calculated using the ΔΔCt method relative to the geometric mean of unoperated wild-type controls.
In situ hybridization
Mice were euthanized by CO2 inhalation and rapidly transcardially perfused with cold 1× DEPC-PBS and fixed with 4% PFA in DEPC-PBS. Brains were sectioned at 2mm thick in a brain matrix, post-fixed and cryoprotected in 20% sucrose-DEPC-PBS. Tissue sections were embedded and sectioned at 15μm on a cryostat, and stored at −20°C until use.
Oligonucleotide probe sequences were determined using Unique Probe Selector 2.0 software (http://array.iis.sinica.edu.tw/ups/, (40), Table S2) and synthesized with amide modifications (Integrated DNA Technologies). Probes were labeled with digoxigenin (DIG) NHS-esters (Roche) using the manufacturer’s protocol (Sigma). Briefly, sequential probes for each gene were combined equally into a single cocktail and diluted to 8μg/mL in 100mM sodium borate. Probes were combined with 14mM DIG-esters in anhydrous dimethylformamide and mixed rotating overnight at room temperature. Labeled probes were precipitated using ethanol (EtOH) and 3M sodium acetate. DIG-ester labeling was confirmed via dot blot using the DIG Nucleic Acid Detection Kit (Roche) per manufacturer’s instructions.
Purified probe mixtures (3μl per slide) were combined with carrier RNA (1:1 salmon sperm SS DNA 10μg/μl, Escherchia coli tRNA 100μg/μl), speed vacuum concentrated for 5 minutes at 30°C and added to 80% deionized formamide . Probes were denatured at 85°C for 5 minutes and added to the hybridization mixture (19.5% dextran sulfate, 4mg/mL BSA, 20mM ribonucleoside vanadyl complex, 2× SSC, 0.1× PBS) with a final concentration of 40% deionized formamide. Prepared slides were equilibrated for 20 minutes at room temperature and rinsed with PBS, DEPC water, and 0.1M triethanolamine (TEA) pH 8.0, followed by a 10-minute incubation in 0.1M TEA with 0.25% acetic anhydride. Samples were dehydrated and delipidated. The probe mixture was placed directly onto each tissue section and covered in parafilm. Slides were incubated overnight at 40°C in a humidified RNAse free chamber. Following hybridization, slides were brought slowly to room temperature and rinsed with 1× SSC and then washed three times with 1× SSC at 50°C. Permeabilization was performed using 0.3% Triton-X-100 in PBS. Slides were incubated overnight with sheep anti-DIG Fab fragments (Roche, polyclonal) and rabbit anti-GFAP antibody (Dako, Z0334) in blocking buffer. Endogenous peroxidases were quenched with 1% hydrogen peroxide. Sections were incubated with biotinylated donkey anti-sheep (Invitrogen, polyclonal) secondary antibody in blocking buffer for 30 minutes, followed by amplification with avidin-biotin-horseradish peroxidase (HRP) complex reagent (Vector Labs). Staining was then visualized with fluorescein-tyramide (Perkin-Elmer) and anti-rabbit Alexa 594 (Molecular Probes, polyclonal) secondary antibody.
Sections were examined under a confocal laser scanning microscope (Leica SP5 Inverted 2-Photon, Manheim, DE). Non-specific background staining was identified using scramble probe and no probe hybridization controls. Background fluorescence was minimized using black level balancing (Smart Offset) with lowest background set using scramble probe control slides in post-CLP mice. This black level balance was maintained for all subsequent slides.
Colony forming unit assays
Peritoneal lavage was performed by closed injection and withdrawal of 3 mL sterile PBS with EDTA into the abdominal cavity. Brain hemispheres or whole spleens were homogenized in sterile PBS. Aerobic culture of serial lysate dilutions was performed at 37°C on tryptic soy agar plates.
Respiratory burst assay
Single cell suspensions were isolated from whole brain as described above, using enzymatic digestion and myelin removal over a Percoll gradient. Cells were stained with CD11b-BV650 and CD45-BV421 on ice for 30 minutes and then washed and resuspended in media (DMEM, 2% FBS, 25 mM HEPES) and warmed at 37°C in 5% CO2 for 15 minutes. Concentrated phorbol 12-myristate 13-acetate (PMA) (Sigma) was added to a final concentration of 5 μM and incubated for 15 minutes. For unstimulated cells, an equal volume of media was added. Dihydrorhodamine-1,2,3 (DHR) was added to a final concentration of 5 μg/mL and incubated for a further 10 minutes (41). The DHR reaction was then quenched on ice. Cells were analyzed immediately on an Attune cytometer (Applied Biosystems). Microglia/macrophages were identified as CD45mid/CD11b+ cells. DHR fluorescence was measured with excitation at 488 nm and emission at 530/30 nm. The difference between the distribution of DHR fluorescence in stimulated and unstimulated control cells from the same animal was quantified using the chi-squared goodness of fit T(x) statistic as implemented in FlowJo software (Treestar):
where O is the frequency of events within a given bin of the fluorescence intensity histogram. T(x) was calculated on 300 consecutive events in each sample to minimize variability due to event number.
Ex vivo microglial secretion assays
Single cell suspensions were isolated from whole brains of age and sex matched wild-type and S100A9−/− mice using enzymatic digestion, and myelin was removed using a Percoll gradient. Cells were stained for CD45, CD11b, and CD64 and live microglia/macrophages were identified as CD45mid/CD11b+/CD64+ cells negative for propidium iodide staining (Fig. S1). 30,000 cells per well were sorted using a 100 μm nozzle directly into 96 well plates containing either basal media (DMEM with 10% FBS and 25 mM HEPES) or media with 100-500 ng/ml LPS (E. coli O111:B4, Sigma), though 100 ng/ml appeared to elicit a maximal response. Cells were then incubated for 18 hours in 5% CO2 at 37°C. Supernatants were centrifuged at 1000g for 10 minutes to remove any residual cells. TNFα was measured in the supernatants by AlphaLISA (Perkin Elmer) according to the manufacturer’s instructions.
Behavior
Young adult wild-type and S100A9−/− male and female mice were used for these studies. Unoperated controls were used to control for any effect of repeated testing. Mice underwent one, five-minute exploration of an open field 72 × 72 cm in dimension. Mice then underwent CLP and recovery as previously described. 14 days after CLP, mice underwent one additional 5-minute exploration of the open field. Behavior was analyzed to quantify time in center of the open field and total distance moved using Ethovision 11.5 software (Noldus Information Technology, Inc., Leesburg, VA). Analysis was performed by 2-way ANOVA of baseline and post-CLP locomotion. Post CLP changes in each measure were calculated as a fractional change from baseline: (M14 days – Mbaseline) / Mbaseline.
Experimental design
The study of human brain expression of S100A8/A9 was a retrospective study based on a convenience sample determined by reviewing all cases in the University of Michigan Alzheimer’s Disease Center brain bank. The individual performing ELISA analysis of human brain tissue was blinded to subjects’ cause of death. Analysis of immunostaining was done without blinding, as quantification of relative expression of S100A8/A9 in patients with and without sepsis was accomplished via the ELISA. In murine studies, tissues and cells were isolated without blinding, as the experimental treatment of the animals was obvious from physical inspection. Quantification of gene or protein expression, however, was performed by an investigator blinded to treatment group. Based on our prior work examining neuroinflammation in CLP, we anticipated that while measures of inflammation in unoperated or sham operated animals would be relatively homogeneous, the response to CLP would be heterogeneous and lead to non-normal distributions of responses. We therefore planned a sample size of at least 5 animals per group, and all data here are presented with individual values in addition to aggregate descriptive statistics.
Data analysis
Analyses included ANOVA as indicated in the text, followed by post-hoc testing. Post hoc tests were adjusted for multiple comparisons using Tukey’s (1-way ANOVA) or Sidak’s (2-way ANOVA) tests. In order to minimize spurious comparisons, we prespecified post-hoc comparisons only among CLP/sham and CLP/unoperated at each time point, and among CLP operated animals of different genotypes. All figures show mean and standard error unless otherwise specified. Statistical analyses were carried out in Prism (Graphpad).
Results
S100A8/A9 is expressed by multiple cell types in the brains of patients who died of sepsis
Given that elevated levels of S100A8/A9 heterodimer have been demonstrated in the serum of patients with sepsis, we sought to determine whether this DAMP is expressed within the CNS of patients with sepsis (42). Donors to the University of Michigan brain bank who had clearly died of sepsis or non-infectious causes were identified by chart review (Table S1). S100A8/A9 protein is significantly increased in parietal cortex lysates of patients who died of sepsis compared to patients who died of non-infectious causes (Fig. 1A, t-test, p=0.0054). Given that the measurement of S100A8/A9 in brain tissue also includes residual endovascular protein, we then examined S100A8 expression by immunohistochemistry.
Fig. 1.
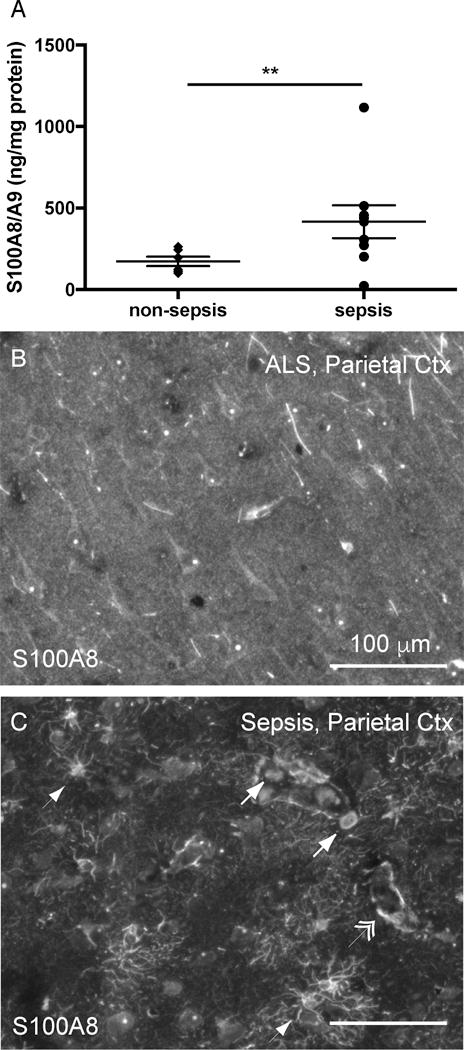
S100A8 is increased in the brains of patients with sepsis. S100A8/A9 protein is increased in the parietal cortex of patients who die of sepsis compared to patients who die of non-infectious causes (t-test, p=0.005) (A). S100A8 immunoreactivity is markedly increased in a representative patient who died of sepsis (C) compared to a patient who died of respiratory failure consequent to amyotrophic lateral sclerosis (B). S100A8 immunoreactivity appears in cells of multiple morphologies, including perivascular mononuclear cells (arrow), cells with stellate morphology (arrowhead) and along the vasculature (double arrowhead). ** p = 0.005
S100A8 immunoreactivity was present in cellular patterns with multiple morphologies in the brain, and notably increased in patients who died of sepsis compared to controls (Fig. 1B and 1C). Immunoreactivity was observed in cells with stellate morphology (arrowhead), endovascular and perivascular mononuclear cells (arrows), as well as surrounding the vasculature (double arrowheads) (Fig. 1C). We carried out multiplex immunohistochemistry to characterize these cells further. S100A8 did not co-localize with the myeloid cell marker Iba1 in cells with microglial morphology (Fig. 2A), but did co-localize with Iba1 expressing intravascular, perivascular and infiltrating cells (Fig. 2B, E). S100A8 staining was most consistently found in stellate cells co-expressing the astrocyte marker GFAP (Fig. 2C, F). Perivascular S100A8 immunoreactivity did not co-localize with the tight junction protein ZO-1, suggesting that perivascular S100A8 is not expressed in endothelial cells, but rather in astrocyte foot processes (Fig 2D, G).
Fig. 2.
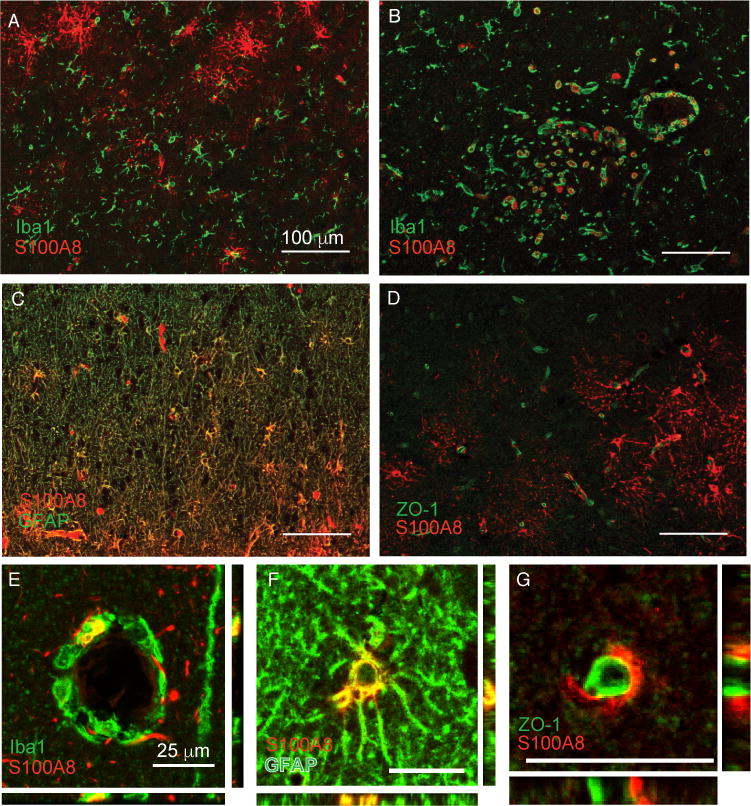
S100A8 is expressed in multiple cell types in the parietal cortex of patients with sepsis. S100A8 immunoreactivity does not colocalize with Iba1+ microglia (A) but does strongly colocalize with perivascular Iba1+ cells, likely infiltrating myeloid cells (B, E). S100A8 is also expressed in astrocytes (C, F). Apparent perivascular S100A8 immunoreactivity is adjacent to, but does not colocalize with the endothelial marker ZO-1 (D, G).
Long term expression of S100A8/A9 in microglia and infiltrating myeloid cells in murine sepsis survivors
Characterizing the long-term expression of S100A8/A9 in patients surviving sepsis would require a cohort of brain donors who survived a recent episode of sepsis and then went on to die of noninfectious causes. Given that no such cohort exists, we employed CLP as a murine model of treated sepsis to study S100A8/A9 expression in sepsis survivors (17). We found that S100A8/A9 protein expression was significantly elevated in whole brain lysates at 5 days and 14 days after CLP compared to unoperated controls (Fig. 3A, 2-way ANOVA, p=0.017 for effect of intervention). S100A8/A9 was specifically elevated in CLP survivor mice compared to sham controls 14 days after sepsis (p=0.039), but not 5 days following CLP compared to sham controls (Fig. 3A). In contrast, S100A8 gene expression was significantly increased both 5 days and 14 days after CLP as compared to sham controls (Fig. 3B; 2-way ANOVA, p=0.018 for effect of intervention, post-hoc p=0.008 at 5 days and p=0.001 at 14 days for sham vs. CLP).
Fig. 3.
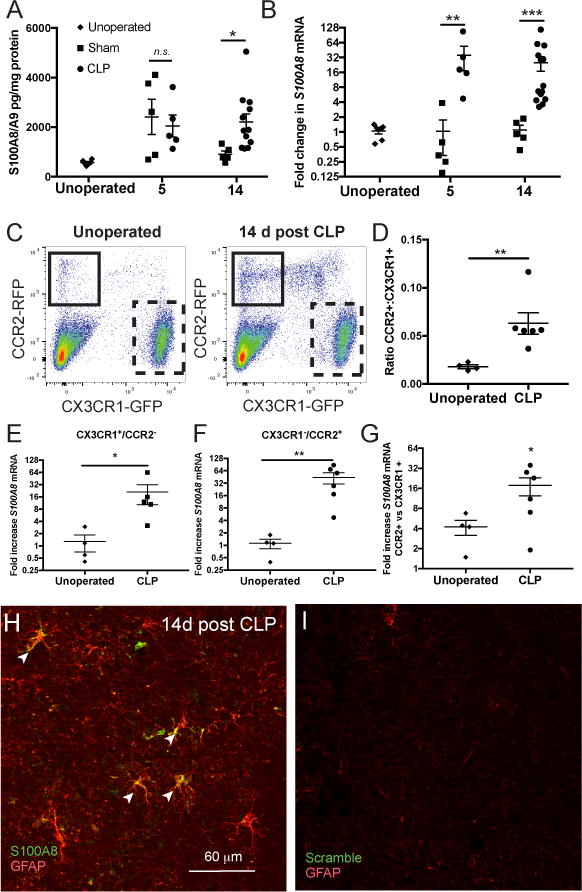
In whole brain homogenates, S100A8/A9 expression is nonspecifically elevated 5 days after CLP, and remains elevated 14 days after CLP in sepsis survivor mice but not in sham controls (A, 2-way ANOVA, p=0.017 for effect of intervention and Sidak’s test p= 0.039 at for CLP vs sham at 14 days). S100A8 gene expression, however, is increased exclusively in sepsis survivor mice both 5 and 14 days after CLP (B, 2-way ANOVA, p=0.018 for effect of intervention, Sidak’s test p<0.01 for CLP vs sham at 5 days and 14 days). S100A8/A9 is expressed persistently in microglia and monocytes in murine sepsis survivors. CX3CR1+/CCR2- microglia/macrophages and CX3CR1-/CCR2+ monocytes are differentiated by GFP and RFP expression in reporter mice (C). Relative abundance of monocytes in whole-brain single cell suspension is increased 14 days after CLP compared to naïve controls (C, D, Mann-Whitney U test, p<0.01). S100A8 gene expression is increased 14 days after CLP in both microglia (E, Mann-Whitney U test, p=0.015) and monocytes (F, Mann-Whitney U test, p<0.01), though monocytes express higher levels of S100A8 mRNA than microglia (G, Wilcoxon test, p=0.03 for ratio >1). S100A8 is expressed in murine astrocytes after sepsis. S100A8 mRNA colocalizes with GFAP+ astrocytes by in situ hybridization 14 days after CLP (H). In situ hybridization of the same tissue with scramble probes were used as a threshold control (I). *p < 0.05, ** p < 0.01, *** p < 0.001
Given that we had originally identified S100A8 and S100A9 gene expression after sepsis in microglia identified by surface marker expression, but had not found substantial microglial S100A8 immunoreactivity in the human brain, we confirmed microglial expression of S100A8 using CCR2RFP/+∷CX3CR1GFP/+ reporter mice (17, 34). Microglia/macrophages were identified by flow cytometry as a clear GFP+RFP- population, while monocytes were identified as a small GFP-RFP+ population (Fig. 3C). While the CX3CR1GFP/+ reporter does not establish that identified cells are microglia of yolk-sac origin and does not exclude monocyte-derived CNS macrophages, it does identify CNS-resident cells. We found that GFP expressing cells in sepsis survivor mice were within the brain parenchyma, while RFP expressing cells were present in the meninges, perivascular locations, and choroid plexus, as expected (Fig. S2A–C). While we did observe a GFPlowRFP+ population by flow cytometry, these cells were excluded from analysis and were not readily observed histologically. Furthermore, GFPlowRFP+ cells in the circulation were readily distinguished from GFP+RFP- cells derived from the brain (Fig. S2D–E), and it is thus unlikely that cells identified as microglia/macrophages are simply infiltrating or intravascular monocytes.
The proportion of monocytes increased dramatically 14 days after CLP in sepsis survivor mice (Fig. 3C, 3D, Mann-Whitney, p=0.009). Cells from 2 animals were pooled to ensure that at least 200,000 cells could be sorted for each point presented. S100A8 gene expression was significantly increased in microglia/macrophages (Fig. 3E, Mann-Whitney p=0.015) and monocytes (Fig. 3F, Mann-Whitney p=0.009) 14 days after CLP compared to unoperated controls. Of note, given that we did not observe S100A8 immunoreactive microglia but did find abundant S100A8 immunoreactive infiltrating myeloid cells in the brains of patients who died of sepsis, we compared monocyte and microglial S100A8 expression. Monocytes expressed 16-fold greater S100A8 relative to microglia/macrophages following CLP (Fig. 2G, Wilcoxon signed-rank test, p=0.03 for ratio >1), suggesting that microglial expression of S100A8 may fall below the threshold of detection by immunohistochemistry.
In addition to expression by microglia and infiltrating myeloid cells, we found by in situ hybridization that murine astrocytes are capable of expressing S100A8 mRNA in sepsis survivor mice (Fig. 3H).
S100A9 deficiency does not change mortality, but does impair bacterial clearance after CLP
The effect of S100A8/A9 deficiency on mortality and severity of illness in murine models of infectious illness varies widely among reported models (43–46). S100A8/A9 deficiency is modeled by S100A9 deletion due to embryonic lethality of S100A8 knockouts and lack of S100A8 secretion in S100A9−/− mice (47). In our implementation of CLP, we observe long term survival of approximately 80% in both wild-type and S100A9−/− mice (Fig. 4A, n = 28 wild-type mice and n = 40 S100A9−/− mice in multiple cohorts over more than 6 months, Gehan-Breslow-Wilcoxon test, p = 0.5). There is no significant effect of genotype on weight loss or weight regain over 1 week following CLP (Fig. 4B, 2-way ANOVA with p=0.0005 for effect of time after CLP, p=0.28 for effect of genotype, and p=0.96 for genotype/time interaction). Early burden of intraperitoneal infection with aerobic bacteria was not different among wild-type and S100A9−/− sepsis survivors 3 days after CLP. However, by 1 week after CLP, wild-type mice cleared intraperitoneal infection, while S100A9−/− mice demonstrated persistent bacterial burden (Fig. 4C, peritoneal lavage, 2-way ANOVA with p=0.0026 for interaction of time and genotype, with post-hoc p=0.41 at 3 days and p=0.003 at 7 days among genotypes). Dissemination of culturable bacteria to the spleen and brain was low and did not differ among genotypes at either time point (Fig 4C, 2-way ANOVA with 0.35 for effect of genotype in spleen and 0.08 for effect of genotype in brain).
Fig. 4.
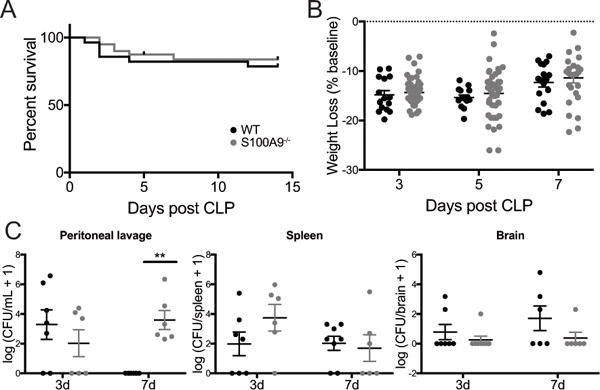
S100A9 deficiency does not change survival or weight change after CLP, but does impair local bacterial clearance. There was no significant difference in survival after CLP in wild-type and S100A9−/− mice (A, Gehan-Breslow-Wilcoxon test, p=0.5). S100A9−/− mice lost and regained a similar percentage of baseline body weight after CLP as wild-type controls (B, 2-way ANOVA, p=0.0005 for effect of time, p=0.28 for effect of genotype). While there was no significant difference in intraperitoneal bacterial burden 3 days after CLP, S100A9−/− mice failed to clear intraperitoneal infection 7 days after CLP (C, 2-way ANOVA p<0.01 for interaction of time and genotype, Sidak’s p=0.003 at 7 days for differences among genotypes). Low levels of disseminated bacteria were detected in the spleen (D, 2-way ANOVA p=0.35) and brain (E, 2-way ANOVA p=0.08) but did not differ by genotype. ** p <0.01
S100A9 deficiency decreases PMN recruitment to the brain
While S100A8/A9 is produced by multiple cell types within the CNS, we also expect it to have pleiotropic effects on neuroinflammation after CLP. We have previously found that the CNS is infiltrated by myeloid cells for several weeks following CLP (17). S100A9 deficient mice are also known to have less neutrophil recruitment to the CNS after direct intracisternal injection of bacteria (24). We therefore compared the relative abundance of neutrophils in whole brain derived cell suspensions in wild-type and S100A9−/− mice after CLP or sham operation. Infiltrating cells were quantified 19 days post CLP, as 3 weeks post CLP has been previously reported as the time of maximal cellular infiltration in the spleen (48). Neutrophils were identified as CD45+/CD11b+/Ly6Cmid/Ly6G+ cells (Fig. 5A). Both wild-type and S100A9−/− male mice had an increased proportion of brain neutrophils 19 days after CLP compared to sham-operated controls. The proportion of neutrophils in S100A9−/− mice was significantly reduced compared to wild-type mice, however (Fig. 5B, t-test, p 0.023). Of note, the 30-40% proportion of Ly6G+ myeloid cells in this preparation is far above what is observed in situ. Cell suspensions were prepared after transcardiac perfusion by mechanical tissue dissociation, which biases the resulting suspension toward infiltrating, perivascular, or adherent endovascular cells and is less efficient at isolating microglia and other parenchymal cells.
Fig. 5.
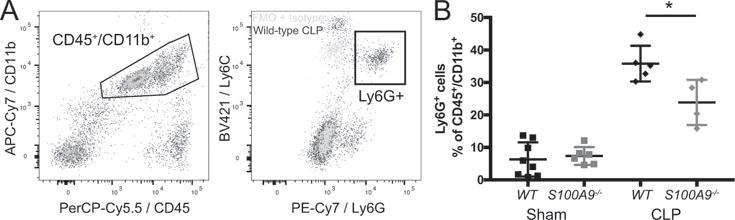
S100A9 contributes to myeloid cell infiltration in the brain. Neutrophils are identified as CD45+/CD11b+ cells expressing Ly6G above the level of an isotype stained control (A). S100A9−/− mice have a decreased proportion of neutrophils in whole brain lysates 19 days after CLP (B, t test). * p < 0.025
Microglial priming after sepsis depends on S100A9
In addition to its action on neutrophil recruitment, we asked whether S100A8/A9 influenced microglial function after sepsis. Unlike neutrophils and other myeloid cells, quiescent microglia are not known to produce significant reactive oxygen species (ROS) (49). Nevertheless, brain dysfunction after endotoxemia is known to depend on nitric oxide synthase expression, suggesting that ROS are important mediators of brain injury after sepsis (18). We therefore examined microglial ROS production evoked by PMA stimulation ex vivo using dihydrorhodamine 123 (DHR), a fluorescent indicator of peroxynitrite and peroxide formation (50, 51). As expected, stimulation with PMA did not result in significant changes in DHR fluorescence compared to treatment with basal media in microglia isolated from naïve mice (Fig. 6A). Microglia isolated from sepsis survivor mice 5 days after CLP, however, showed a significant shift in DHR fluorescence upon stimulation with PMA (Fig. 6A). These changes were abolished in S100A9−/− mice (Fig. 6A). The difference in DHR fluorescence in basal conditions and with PMA stimulation was quantified using the χ2 statistic for distributions (Fig. 6B). There was a significant difference among groups (2-way ANOVA, p = 0.0045 for effect of genotype), with microglia derived from wild-type sepsis survivors having significantly greater ROS response to PMA stimulation than both naïve wild-type (p = 0.0015) and S100A9−/− sepsis survivor mice (p = 0.0008). This impairment in primed ROS production is not due to baseline defects in ROS production. Splenic CD11b+/Ly6Cmid/Ly6G+ neutrophils isolated from unoperated S100A9−/− mice had preserved ROS production (Fig 6C,D). Unoperated CD11b+/Ly6Chi/Ly6G- monocytes did not demonstrate a significant ROS response to PMA stimulation using this technique (Fig. 6E,F).
Fig. 6.
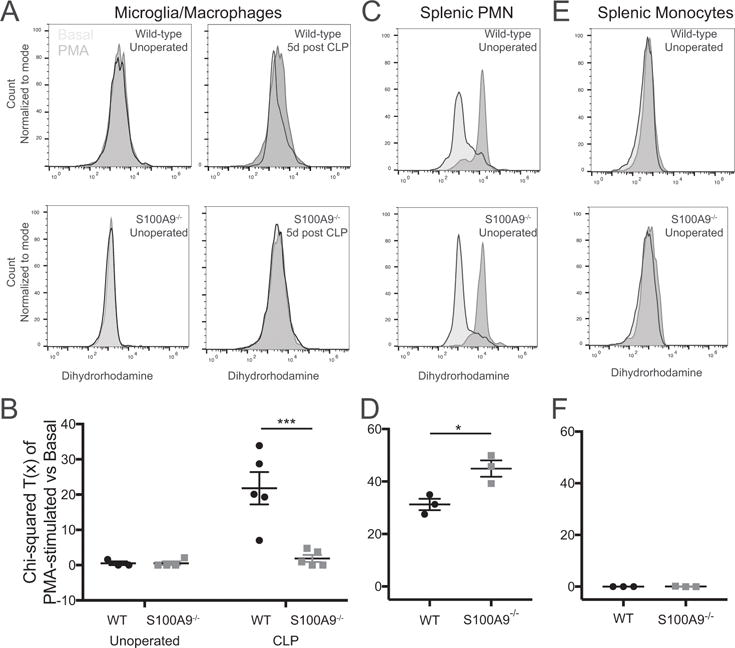
S100A9 deficiency abolishes priming of microglial ROS production after CLP. Microglia isolated from sepsis survivor mice 5 days after CLP produce increased ROS, as measured by dihydrorhodamine fluorescence, when stimulated ex vivo with PMA (A,). Microglia isolated from either naïve mice or S100A9−/− sepsis survivor mice show no response to PMA stimulation (A). The PMA response, quantified by the chi-squared difference in fluorescence between basal and PMA stimulated microglia, is significantly greater in wild-type sepsis survivor mice than in S100A9−/− sepsis survivor mice or naïve controls (B, 2-way ANOVA, p=0.045 for effect of genotype). ROS response of splenic neutrophils is preserved at baseline in S100A9−/− mice (C,D). By this method, naïve splenic monocytes did not produce a measurable ROS response (E,F). * p < 0.05, *** p < 0.001
We have previously found that CLP results in long-term expression of multiple cytokines and chemokines in the brains of sepsis survivor mice, including TNFα (17). Given that S100A8/A9 is known to facilitate TNFα release from bone marrow derived cells in response to endotoxemia, we sought to determine whether S100A8/A9 and TNFα might form a feed-forward circuit that sustains neuroinflammation (47). Both 5 days and 14 days after CLP, TNFα−/− mice exhibit decreased whole-brain expression of S100A8 (Fig. 7A, 5 day p = 0.033, 14 day p=0.027). We then examined the specific effect of S100A9 deficiency on microglial expression of TNFα. Microglia/macrophages were isolated from wild-type or S100A9−/− mice by fluorescence activated cell sorting, identified at CD11b+/CD45mid/CD64+ cells. Fourteen days after CLP, basal microglia/macrophage expression of TNFα mRNA is modestly increased compared to naïve controls (Fig. 7B, p=0.009). When cultured ex vivo in basal media, microglia/macrophages isolated from sepsis survivor mice do not secrete more TNFα than microglia/macrophages from controls (Fig. 7C). However, ex vivo LPS stimulation revealed that microglia/macrophages from sepsis survivor mice are primed to produce TNFα. Increased TNFα secretion was observed in microglia/macrophages derived from wild-type sepsis survivor mice 5 days after CLP with LPS stimulation compared to those derived from naïve mice (Fig 7C, 2-way ANOVA with p<0.0001 for effect of genotype and p=0.003 for effect of CLP, post-hoc p=0.012 for effect of CLP in wild-type cells), but no priming effect was seen in microglia/macrophages derived from S100A9−/− sepsis survivor mice (Fig. 7C, post-hoc p=0.22 for effect of CLP in S100A9−/− cells). In a separate experiment, we observed that microglial/macrophage priming persists to at least 2 weeks post-CLP in wild-type mice (Fig. 7C, right, p=0.0026).
Fig. 7.
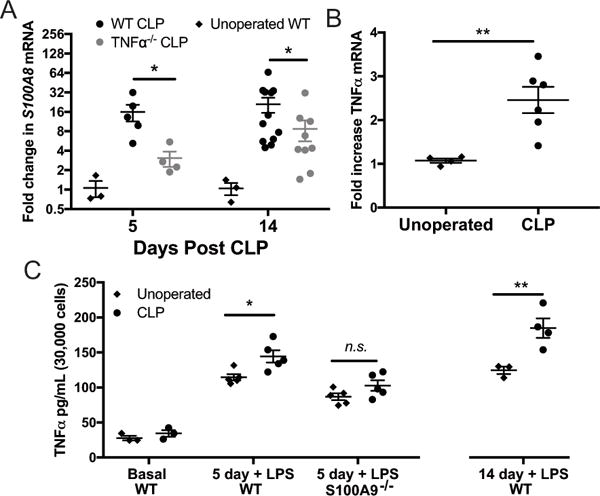
Whole-brain S100A8 expression after sepsis is reduced by TNFα deficiency (A). S100A9 deficiency abolishes priming of microglial TNFα production after CLP. Fourteen days after CLP, microglia express modestly elevated levels of TNFα mRNA (B, t test p=0.009). When cultured ex vivo, microglia isolated from sepsis survivor mice do not secrete increased amounts of TNFα (C). After stimulation with LPS, however, microglia isolated from sepsis survivor mice 14 days after CLP exhibit marked potentiation of TNFα secretion (C). Microglia isolated from S100A9−/− mice 5 days after CLP do not exhibit potentiation of LPS-stimulated TNFα secretion, compared to wild-type sepsis survivors (C, 2-way ANOVA, p<0.0001 for effect of genotype). * p < 0.05, ** p < 0.01
Deficiency of S1009 worsens behavior outcomes in sepsis survivor mice
We sought to determine whether S100A9 might play a role in brain function after recovery from sepsis. We used a five-minute open field test to assay locomotion and anxiety-like behavior before and after CLP in male and female wild-type and S100A9 knockout mice. As the results were similar for both sexes, they were grouped together for the analysis. Analysis of total distance traveled revealed an effect of genotype on baseline locomotion, with decreased total distance traveled by S100A9−/− mice compared to wild-type (Fig. 8A, ANOVA p = 0.004, post-hoc p = 0.003 for difference among S100A9−/− and wild-type groups). In 2-way repeated measures ANOVA, there was an overall effect of time (either pre/post CLP or repeated testing), genotype, and time/genotype interaction (2-way ANOVA, effect of group p < 0.001, effect of time p < 0.001, interaction p = 0.0013). All groups including the wild type unoperated showed a decrease in distance traveled between the two tests, likely representing an effect of repeated testing. In order to quantify the relative effect of CLP survival versus repeated testing across groups, the percent change in distance traveled from baseline to 14 days was calculated for each mouse. Total distance traveled demonstrated a greater decrease in wild-type mice after CLP compared to the effect of repeated testing alone (Fig. 8C, ANOVA p < 0.0001, post-hoc p < 0.01 for wild-type CLP compared to wild-type unoperated). Relative change in distance traveled was also greater after CLP in S100A9−/− mice compared to wild-type mice (Fig. 8C, p = 0.002). Anxiety-like behavior as measured by time spent in the center of the open field also revealed increased anxiety-like behavior after CLP in S100A9−/− mice. There was an overall effect of time (Fig. 8B, 2-way repeated measures ANOVA, p = 0.002) and an interaction between test session and group (p<0.0001). Post-hoc tests showed a significant decrease in the amount of time spent in the center of the open field in S100A9 knockout mice after CLP (p<0.0001), but not in unoperated wild-type or wild-type after CLP. Relative change in time in center was greater for S100A9−/− compared to wild-type mice after CLP (Fig. 8D, p = 0.008), but there was not a significant decrease after CLP in wild-type compared to unoperated mice (p = 0.052).
Fig. 8.
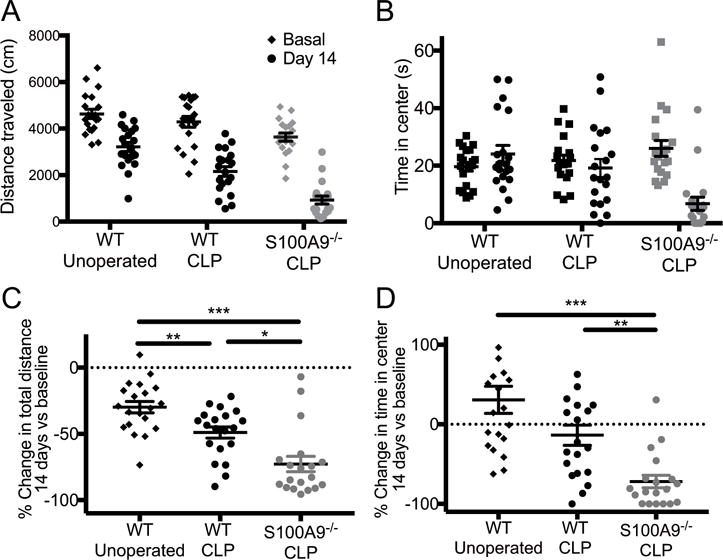
S100A9 knockout increases anxiety-like behavior after CLP. Basal and 14-day repeated open field tests show decreased total distance traveled in wild-type unoperated mice (N = 10 male, 10 female), wild type mice before and after CLP (N = 10 male, 10 female), and S100A9−/− mice before and after CLP (N = 7 male, 12 female) (A, 2-way ANOVA, effect of group p < 0.001, effect of time p < 0.001, interaction p = 0.0013). Time in the center of the open field was significantly decreased only in S100A9−/− mice after CLP (B, 2-way ANOVA, effect of time p = 0.0015 and time/group interaction p < 0.001). In order to isolate the effect of CLP from the effect of repeated measures and baseline effect of genotype, we calculated the relative change in both distance and time measures for each individual mouse over the experiment. Wild-type CLP mice demonstrated a greater decrease in total distance moved than did unoperated mice (p < 0.01) and S100A9−/− mice demonstrated a greater effect of CLP than did wild-type controls (p = 0.0026) (C). The change in time in center of the arena did not differ significantly among wild-type unoperated controls and sepsis survivors (p = 0.052) but S100A9−/− sepsis survivors spent significantly less time in the center after CLP compared to wild-type sepsis survivors (D, p < 0.008). *** p < 0.001, ** p < 0.01, * p < 0.05
Discussion
Using a combination of post-mortem human specimens and a mouse model, we have shown that the DAMP S100A8/A9 is expressed by multiple cell types in the brain during and after sepsis. Using a mouse model of sepsis, we have found that S100A8/A9 expression contributes to persistent neuroinflammation after sepsis through several pathways. As in other disease models, S100A8/A9 serves as important signal for neutrophil chemotaxis. S100A8/A9 expression also alters microglial/macrophage function. Although we found that brain resident myeloid cells are not constitutively activated after sepsis, they are primed in sepsis survivor mice to produce ROS and TNFα upon secondary stimulation, and this priming effect is S100A8/A9 dependent. Surprisingly, we found that despite the CNS effects of S100A8/A9, S100A9 deficiency led to worsened anxiety-life behavior in sepsis survivor mice.
S100A8/A9 both promotes and suppresses inflammation in both humans and murine models of disease. S100A8/A9 is known to play a role in the innate immune response as an endogenous ligand of the Toll-like receptor 4, amplifying the production of TNFα in response to endotoxemia (47). S100A8/A9 contributes to bacterial dissemination and death in early E. coli peritonitis, but is protective in both Gram-negative and Gram-positive pneumonia (45, 46, 52). In part, S100A8/A9 appears to protect against excessive activation of the immune system in infection by modulating neutrophil chemotaxis and function (45, 52, 53). In chronic inflammation associated with autoimmunity and malignancy, S100A8/A9 has been found to exert other anti-inflammatory effects, including maintenance of myeloid derived suppressor cell populations (54, 55).
Neuroinflammation, especially attributed to microglial activation, has been associated with brain dysfunction in both sterile and infectious models of sepsis survival (18, 19, 22). Multiple cytokines and chemokines act directly on neurons and disturb normal brain function, apart from actions in immunity (56–58). Here, however, we found that S100A8/A9 deficiency results in worsened anxiety-like behavior in sepsis survivor mice, despite the role of S100A8/A9 in promoting neutrophilia, ROS production, and TNFα secretion. Although this result was unexpected, it demonstrates that S100A8/A9 expression is not simply an epiphenomenon of inflammation in sepsis, but that S100A8/A9 plays an important role in the brain function of sepsis survivors.
S100A8/A9 deficiency may alter the brain response to sepsis at via several different mechanisms. First, the absence of S100A8/A9 could significantly alter the early systemic immune response and overall severity of sepsis. Although a recent report noted complete protection of S100A9−/− mice from CLP-induced mortality, we did not we did not find any difference in mortality or weight loss and regain among S100A9−/− and wild-type mice (44). Therefore, it is unlikely that the differences we observed were due to survivor bias or severity of early illness. We did, however, find that S100A8/A9 is required to clear the intraperitoneal infection associated with CLP. While this chronic infection did not lead to death, weight loss or worsening disseminated infection, it could lead to ongoing sickness behaviors. Of note, however, dissemination of aerobic bacteria to the brain was not significantly increased in S100A9 deficient mice, though this does not exclude differences in brain exposure to anaerobic or uncultured organisms. Second, the S100A8/A9 dependent properties we identified in the CNS response – neutrophilia and priming of ROS and cytokine production – may not contribute to tissue damage but rather tissue repair and suppression of ongoing deleterious inflammation. S100A8/A9 and other cytokines contribute to recruitment and expansion of myeloid derived suppressor cells in a variety of diseases, including sepsis (44, 59–61). Furthermore, in sterile forms of brain injury, such as stroke, some forms of inflammation lead to functional recovery (62).
Third, it is likely that pro-inflammatory pathways serve different functions as the response to sepsis evolves, and that signals that are important for pathogen clearance early in the response lead to undesirable tissue damage in survivors. This hypothesis mirrors the clinical observations common to clinical trials of immunomodulatory treatments of sepsis – therapies that reduce harmful inflammation in animal models had no net benefit in patients, likely due to the competing beneficial effects of the innate immune response on the underling infection (63, 64). Further studies that employ temporal control of S100A8/A9 action, either by conditional control of gene expression or pharmacologic inhibition of S100A8/A9 or its receptors, will be needed to disentangle the roles of this pathway in pathogen clearance and brain injury. These approaches are beyond the limitations of the current investigation, however.
In addition to raising the question of when S100A8/A9 signaling is important to brain injury in sepsis, this study also highlights the multiple sources from which S100A8/A9 may be produced. S100A8/A9 is highly expressed by bone-marrow derived cells in the periphery during sepsis or endotoxemia (46, 47). Sepsis and other states of severe systemic inflammation are characterized by disruption of the blood brain barrier, and thus it is possible that S100A8/A9 and other DAMPS enter the brain from the circulation. Indeed, the discordance between induction of S100A8/A9 protein in the brain and lack of upregulation of S100A8 mRNA that we observed in sham-operated mice 5 days after operation suggests that S100A8/A9 enters the brain from the periphery even in states of mild systemic inflammation. However, we have found both through immunohistochemical studies of postmortem human brains and cell-specific analysis of gene expression in sepsis survivor mice that S100A8/A9 is expressed by cells within the brain parenchyma, including astrocytes, microglia, and infiltrating myeloid cells. Although S100A9 is known to be an early component of amyloid plaque formation in Alzheimer’s disease, we did not observe plaque-like deposits of S100A8 in postmortem human tissue (27). However, given that many, but not all patients who died of sepsis also had Alzheimer’s disease, and the small size of the cohort, the possible confounding contribution of chronic neurodegenerative disease to S100A8/A9 expression is a limitation of this study.
Our study is limited by the lack of immunostaining for S100A8/A9 in murine tissues. S100A8 is expressed in both CCR2 expressing monocytes and CX3CR1 expressing microglia/macrophages. CX3CR1 expressing cells may be yolk-sac origin microglia or brain associated, bone-marrow derived macrophages (65). Infiltrating monocytes may play a specific role in brain dysfunction related to neuroinflammation (58, 66). The increased number of CX3CR1-GFPlowCCR2-RFP+ cells in the brains of sepsis survivors suggests that circulating monocytes are entering the brain parenchyma and assuming a resident cell phenotype. Importantly, although we did not differentiate between these two subpopulations, the cells identified as microglia/macrophages, either CX3CR1-GFP expressing or CD45mid/CD11b+/CD64+, exclude circulating myeloid cells and reflect inflammation and immune response within the brain parenchyma. In addition to expression by myeloid cells, immunostaining in human autopsy revealed S100A8 expression in astrocytes. In situ hybridization revealed that astrocytes also express S100A8 in sepsis survivor mice. This technique, however, does not allow us to determine if astrocyte specific expression is changed in sepsis survivor mice, nor are we able to quantify the relative contribution of microglia/macrophages, monocytes, and astrocytes to total brain expression of S100A8/A9 expression. Expression of S100A8/A9 by cells within the brain parenchyma, regardless of the specific cellular source, suggests that the neuroinflammatory response after sepsis may be both compartmentalized and localized, not just a diffuse response to systemic inflammation and injury. Future studies will require cell-specific knockout of S100A9 to determine the importance of S100A8/A9 expression from specific cellular sources.
In addition to its effect as a chemokine, S100A8/A9 also modulates the action of microglia. It is well known that severe inflammation, as in sepsis and endotoxemia, typically induces tolerance to further stimulation in immune cells (67, 68). This effect is most often observed over the course of 24-72 hours after the initial stimulus, and depending on the nature of the second insult may limit deleterious inflammation or contribute to mortality through inappropriate immunoparalysis (69). S100A8/A9 expression is associated with induction of endotoxin tolerance in peripheral blood mononuclear cells, and pretreatment with S100A8/A9 for 24 hours decreases the subsequent cytokine response to TLR4 ligands both in vivo and in vitro (70, 71). Microglia also demonstrate cell-autonomous tolerance to LPS, although data from co-culture models suggest that LPS tolerance in vivo proceeds from a mixture of cell-autonomous and network mechanisms (72, 73).
Here, however, we found that S100A8/A9 plays an important role in priming of microglia rather than tolerance to subsequent stimulation. Priming of splenocytes after CLP has been observed for several weeks following sepsis, and has been shown to depend on the DAMP HMGB1 (74). In mice that have recovered from systemic infection with Salmonella typhimurium, intracerebral injection of Salmonella derived LPS resulted in increased expression of immunohistochemical markers of microglial activation, including CD11c and MHCII in both parenchymal microglia and perivascular recruited cells (75). We have shown that after sepsis, microglial are also functionally primed to respond to LPS, and that this priming depends on S100A9. Tolerance to LPS depends on epigenetic regulation of pro-inflammatory gene expression, and future studies will determine whether S100A8/A9 or S100A9 contributes to priming via epigenetic mechanisms or via persistent signaling via TLR4 and RAGE.
In addition to priming of cytokine production, CLP also results in priming of microglial ROS production. Although persistent cytokine stimulation can induce microglia to produce ROS, we found that microglia are minimally responsive to PMA in their naïve state (76). Five days after CLP, however, microglia are primed to inducibly produce ROS in response to PMA. ROS production in response to bacterial infection and systemic inflammation may typically be considered in the context of bactericidal activity. However, ROS also serve as a second messenger, and are necessary for S100A8/A9 induced cytokine production in blood mononuclear cells (77).
Our results indicate that S100A8/A9 is an important mediator of persistent neuroinflammation after sepsis. S100A8/A9 is expressed by cells within the brain parenchyma and leads to functional priming of microglial responses and recruitment of neutrophils to the CNS after sepsis. Future studies will focus on whether S100A8/A9 signaling outside the period of acute infection is neuroprotective or deleterious, and whether sustained S100A8/A9 expression alters microglial priming via ongoing signal transduction or epigenetic reprogramming.
Supplementary Material
Acknowledgments
We thank the Michigan Interdisciplinary Critical Care Research Workgroup for comments on this manuscript.
Grant support: Funding was provided by the National Institutes for Health: T32HL00774921, K08NS101054 (to B.H.S.), RO1MH104261 (to H.A.) and R01HL123515 (to T.J.S.). Additional support was provided by the University of Michigan Depression Center and the Michigan Institute for Clinical and Health Research (CTSA UL1TR000433) (to B.H.S). The University of Michigan Brain Bank is supported by National Institute of Aging P30 AG 053760 as well as the University of Michigan Protein Folding Disorders Initiative.
Abbreviations
- DAMP
damage associated molecular pattern
- CLP
cecal ligation and puncture
- ROS
reactive oxygen species
- LPS
lipopolysaccharide
References
- 1.Heron M. Deaths: Leading Causes for 2014. Natl Vital Stat Rep. 2016;65:1–96. [PubMed] [Google Scholar]
- 2.Hopkins RO, Jackson JC. Long-term neurocognitive function after critical illness. Chest. 2006;130:869–878. doi: 10.1378/chest.130.3.869. [DOI] [PubMed] [Google Scholar]
- 3.Stevenson EK, Rubenstein AR, Radin GT, Wiener RS, Walkey AJ. Two decades of mortality trends among patients with severe sepsis: a comparative meta-analysis*. Crit Care Med. 2014;42:625–31. doi: 10.1097/CCM.0000000000000026. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Kaukonen KM, Bailey M, Suzuki S, Pilcher D, Bellomo R. Mortality related to severe sepsis and septic shock among critically ill patients in Australia and New Zealand, 2000–2012. JAMA. 2014;311:1308–16. doi: 10.1001/jama.2014.2637. [DOI] [PubMed] [Google Scholar]
- 5.Elixhauser A, Friedman B, Stranges E. Septicemia in US Hospitals, 2009: HCUP Statistical Brief #122. 2011 [PubMed] [Google Scholar]
- 6.Sutton JP, Friedman B. Trends in Septicemia Hospitalizations and Readmissions in Selected HCUP States, 2005 and 2010: Statistical Brief #161. 2006 [PubMed] [Google Scholar]
- 7.Bienvenu OJ, Gellar J, Althouse BM, Colantuoni E, Sricharoenchai T, Mendez-Tellez PA, Shanholtz C, Dennison CR, Pronovost PJ, Needham DM. Post-traumatic stress disorder symptoms after acute lung injury: a 2-year prospective longitudinal study. Psychol Med. 2013;43:2657–71. doi: 10.1017/S0033291713000214. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Bienvenu OJ, Colantuoni E, Mendez-Tellez PA, Shanholtz C, Dennison-Himmelfarb CR, Pronovost PJ, Needham DM. Cooccurrence of and remission from general anxiety, depression, and posttraumatic stress disorder symptoms after acute lung injury: a 2-year longitudinal study. Crit Care Med. 2015;43:642–53. doi: 10.1097/CCM.0000000000000752. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Rabiee A, Nikayin S, Hashem MD, Huang M, Dinglas VD, Bienvenu OJ, Turnbull AE, Needham DM. Depressive Symptoms After Critical Illness: A Systematic Review and Meta-Analysis. Crit Care Med. 2016;44:1744–1753. doi: 10.1097/CCM.0000000000001811. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Parker AM, Sricharoenchai T, Raparla S, Schneck KW, Bienvenu OJ, Needham DM. Posttraumatic stress disorder in critical illness survivors: a metaanalysis. Crit Care Med. 2015;43:1121–9. doi: 10.1097/CCM.0000000000000882. [DOI] [PubMed] [Google Scholar]
- 11.Jackson JC, Hart RP, Gordon SM, Shintani A, Truman B, May L, Ely EW. Six-month neuropsychological outcome of medical intensive care unit patients. Crit Care Med. 2003;31:1226–1234. doi: 10.1097/01.CCM.0000059996.30263.94. [DOI] [PubMed] [Google Scholar]
- 12.Pandharipande PP, Girard TD, Jackson JC, Morandi A, Thompson JL, Pun BT, Brummel NE, Hughes CG, Vasilevskis EE, Shintani AK, Moons KG, Geevarghese SK, Canonico A, Hopkins RO, Bernard GR, Dittus RS, Ely EW. Long-term cognitive impairment after critical illness. N Engl J Med. 2013;369:1306–1316. doi: 10.1056/NEJMoa1301372. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Annane D, Sharshar T. Cognitive decline after sepsis. Lancet Respir Med. 2015;3:61–69. doi: 10.1016/S2213-2600(14)70246-2. [DOI] [PubMed] [Google Scholar]
- 14.Warford J, Lamport AC, Kennedy B, Easton AS. Human Brain Chemokine and Cytokine Expression in Sepsis: A Report of Three Cases. Can J Neurol Sci / J Can des Sci Neurol. 2017;44:96–104. doi: 10.1017/cjn.2016.310. [DOI] [PubMed] [Google Scholar]
- 15.Sharshar T, Annane D, de la Grandmaison GL, Brouland JP, Hopkinson NS, Francoise G. The neuropathology of septic shock. Brain Pathol. 2004;14:21–33. doi: 10.1111/j.1750-3639.2004.tb00494.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Lemstra AW, Groen in’t Woud JCM, Hoozemans JJM, van Haastert ES, Rozemuller AJM, Eikelenboom P, van Gool WA. Microglia activation in sepsis: A case-control study. J Neuroinflammation. 2007;4:4. doi: 10.1186/1742-2094-4-4. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Singer BH, Newstead MW, Zeng X, Cooke CL, Thompson RC, Singer K, Ghantasala R, Parent JM, Murphy GG, Iwashyna TJ, Standiford TJ. Cecal Ligation and Puncture Results in Long-Term Central Nervous System Myeloid Inflammation. PLoS One. 2016;11:e0149136. doi: 10.1371/journal.pone.0149136. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Weberpals M, Hermes M, Hermann S, Kummer MP, Terwel D, Semmler A, Berger M, Schafers M, Heneka MT. NOS2 gene deficiency protects from sepsis-induced long-term cognitive deficits. J Neurosci. 2009;29:14177–14184. doi: 10.1523/JNEUROSCI.3238-09.2009. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Chavan SS, Huerta PT, Robbiati S, Valdes-Ferrer SI, Ochani M, Dancho M, Frankfurt M, Volpe BT, Tracey KJ, Diamond B. HMGB1 Mediates Cognitive Impairment in Sepsis Survivors. Mol Med. 2012;18:930–937. doi: 10.2119/molmed.2012.00195. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Ma KC, Schenck EJ, Pabon MA, Choi AMK. The Role of Danger Signals in the Pathogenesis and Perpetuation of Critical Illness. Am J Respir Crit Care Med. 2018;197:300–309. doi: 10.1164/rccm.201612-2460PP. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Sankowski R, Mader S, Valdés-Ferrer SI. Systemic inflammation and the brain: novel roles of genetic, molecular, and environmental cues as drivers of neurodegeneration. Front Cell Neurosci. 2015;9:28. doi: 10.3389/fncel.2015.00028. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Gasparotto J, Girardi CS, Somensi N, Ribeiro CT, Moreira JCF, Michels M, Sonai B, Rocha M, Steckert AV, Barichello T, Quevedo J, Dal-Pizzol F, Gelain DP. Receptor for advanced glycation endproducts mediates sepsis-triggered amyloid-β accumulation, tau phosphorylation, and cognitive impairment. J Biol Chem. 2017 doi: 10.1074/jbc.M117.786756. jbc M117.786756. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Schiopu A, Cotoi OS. S100A8 and S100A9: DAMPs at the crossroads between innate immunity, traditional risk factors, and cardiovascular disease. Mediators Inflamm. 2013;2013:828354. doi: 10.1155/2013/828354. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Wache C, Klein M, Ostergaard C, Angele B, Häcker H, Pfister HW, Pruenster M, Sperandio M, Leanderson T, Roth J, Vogl T, Koedel U. Myeloid-Related Protein 14 Promotes Inflammation and Injury in Meningitis. J Infect Dis. 2015;212:247–57. doi: 10.1093/infdis/jiv028. [DOI] [PubMed] [Google Scholar]
- 25.Lu SM, Yu CJ, Liu YH, Dong HQ, Zhang X, Zhang SS, Hu LQ, Zhang F, Qian YN, Gui B. S100A8 contributes to postoperative cognitive dysfunction in mice undergoing tibial fracture surgery by activating the TLR4/MyD88 pathway. Brain Behav Immun. 2015;44:221–34. doi: 10.1016/j.bbi.2014.10.011. [DOI] [PubMed] [Google Scholar]
- 26.Kamphuis W, Kooijman L, Schetters S, Orre M, Hol EM. Transcriptional profiling of CD11c-positive microglia accumulating around amyloid plaques in a mouse model for Alzheimer’s disease. Biochim Biophys Acta - Mol Basis Dis. 2016;1862:1847–1860. doi: 10.1016/j.bbadis.2016.07.007. [DOI] [PubMed] [Google Scholar]
- 27.Wang C, Klechikov AG, Gharibyan AL, Wärmländer SKTS, Jarvet J, Zhao L, Jia X, Shankar SK, Olofsson A, Brännström T, Mu Y, Gräslund A, Morozova-Roche LA. The role of pro-inflammatory S100A9 in Alzheimer’s disease amyloid-neuroinflammatory cascade. Acta Neuropathol. 2014;127:507–522. doi: 10.1007/s00401-013-1208-4. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Kim HJ, Chang KA, Ha TY, Kim J, Ha S, Shin KY, Moon C, Nacken W, Kim HS, Suh YH. S100A9 knockout decreases the memory impairment and neuropathology in crossbreed mice of Tg2576 and S100A9 knockout mice model. PLoS One. 2014;9:e88924. doi: 10.1371/journal.pone.0088924. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Gruden MA, Davydova TV, Wang C, Narkevich VB, Fomina VG, Kudrin VS, Morozova-Roche LA, Sewell RDE. The misfolded pro-inflammatory protein S100A9 disrupts memory via neurochemical remodelling instigating an Alzheimer’s disease-like cognitive deficit. Behav Brain Res. 2016;306:106–116. doi: 10.1016/j.bbr.2016.03.016. [DOI] [PubMed] [Google Scholar]
- 30.Lodeiro M, Puerta E, Ismail MAM, Rodriguez-Rodriguez P, Rönnbäck A, Codita A, Parrado-Fernandez C, Maioli S, Gil-Bea F, Merino-Serrais P, Cedazo-Minguez A. Aggregation of the Inflammatory S100A8 Precedes Aβ Plaque Formation in Transgenic APP Mice: Positive Feedback for S100A8 and Aβ Productions. J Gerontol A Biol Sci Med Sci. 2017;72:319–328. doi: 10.1093/gerona/glw073. [DOI] [PubMed] [Google Scholar]
- 31.Manitz MP, Horst B, Seeliger S, Strey A, Skryabin BV, Gunzer M, Frings W, Schonlau F, Roth J, Sorg C, Nacken W. Loss of S100A9 (MRP14) Results in Reduced Interleukin-8-Induced CD11b Surface Expression, a Polarized Microfilament System, and Diminished Responsiveness to Chemoattractants In Vitro. Mol Cell Biol. 2003;23:1034–1043. doi: 10.1128/MCB.23.3.1034-1043.2003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Pasparakis M, Alexopoulou L, Episkopou V, Kollias G. Immune and inflammatory responses in TNF alpha-deficient mice: a critical requirement for TNF alpha in the formation of primary B cell follicles, follicular dendritic cell networks and germinal centers, and in the maturation of the humoral immune response. J Exp Med. 1996;184:1397–1411. doi: 10.1084/jem.184.4.1397. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Cardona AE, Pioro EP, Sasse ME, Kostenko V, Cardona SM, Dijkstra IM, Huang D, Kidd G, Dombrowski S, Dutta R, Lee JC, Cook DN, Jung S, Lira SA, Littman DR, Ransohoff RM. Control of microglial neurotoxicity by the fractalkine receptor. Nat Neurosci. 2006;9:917–924. doi: 10.1038/nn1715. [DOI] [PubMed] [Google Scholar]
- 34.Yamasaki R, Lu H, Butovsky O, Ohno N, Rietsch AM, Cialic R, Wu PM, Doykan CE, Lin J, Cotleur AC, Kidd G, Zorlu MM, Sun N, Hu W, Liu L, Lee JC, Taylor SE, Uehlein L, Dixon D, Gu J, Floruta CM, Zhu M, Charo IF, Weiner HL, Ransohoff RM. Differential roles of microglia and monocytes in the inflamed central nervous system. J Exp Med. 2014;211:1533–1549. doi: 10.1084/jem.20132477. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Saederup N, Cardona AE, Croft K, Mizutani M, Cotleur AC, Tsou CL, Ransohoff RM, Charo IF. Selective chemokine receptor usage by central nervous system myeloid cells in CCR2-red fluorescent protein knock-in mice. PLoS One. 2010;5:e13693. doi: 10.1371/journal.pone.0013693. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Schindelin J, Arganda-Carreras I, Frise E, Kaynig V, Longair M, Pietzsch T, Preibisch S, Rueden C, Saalfeld S, Schmid B, Tinevez JY, White DJ, Hartenstein V, Eliceiri K, Tomancak P, Cardona A. Fiji: an open-source platform for biological-image analysis. Nat Methods. 2012;9:676–82. doi: 10.1038/nmeth.2019. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Walley KR, Lukacs NW, Standiford TJ, Strieter RM, Kunkel SL. Balance of inflammatory cytokines related to severity and mortality of murine sepsis. Infect Immun. 1996;64:4733–4738. doi: 10.1128/iai.64.11.4733-4738.1996. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Frank MG, Wieseler-Frank JL, Watkins LR, Maier SF. Rapid isolation of highly enriched and quiescent microglia from adult rat hippocampus: immunophenotypic and functional characteristics. J Neurosci Methods. 2006;151:121–130. doi: 10.1016/j.jneumeth.2005.06.026. [DOI] [PubMed] [Google Scholar]
- 39.Ballinger MN, Newstead MW, Zeng X, Bhan U, Horowitz JC, Moore BB, Pinsky DJ, Flavell RA, Standiford TJ, Theodore J. TLR Signaling Prevents Hyperoxia-Induced Lung Injury by Protecting the Alveolar Epithelium from Oxidant-Mediated Death. J Immunol. 2012;189:356–364. doi: 10.4049/jimmunol.1103124. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Chen SH, Lo CZ, Su SY, Kuo BH, Hsiung CA, Lin CY. UPS 2.0: unique probe selector for probe design and oligonucleotide microarrays at the pangenomic/genomic level. BMC Genomics. 2010;11:S6. doi: 10.1186/1471-2164-11-S4-S6. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.Banati RB, Rothe G, Valet G, Kreutzberg GW. Respiratory burst activity in brain macrophages: a flow cytometric study on cultured rat microglia. Neuropathol Appl Neurobiol. 1991;17:223–230. doi: 10.1111/j.1365-2990.1991.tb00718.x. [DOI] [PubMed] [Google Scholar]
- 42.Gao S, Yang Y, Fu Y, Guo W, Liu G. Diagnostic and prognostic value of myeloid-related protein complex 8/14 for sepsis. Am J Emerg Med. 2015;33:1278–82. doi: 10.1016/j.ajem.2015.06.025. [DOI] [PubMed] [Google Scholar]
- 43.Ulas T, Pirr S, Fehlhaber B, Bickes MS, Loof TG, Vogl T, Mellinger L, Heinemann AS, Burgmann J, Schöning J, Schreek S, Pfeifer S, Reuner F, Völlger L, Stanulla M, von Köckritz-Blickwede M, Glander S, Barczyk-Kahlert K, von Kaisenberg CS, Friesenhagen J, Fischer-Riepe L, Zenker S, Schultze JL, Roth J, Viemann D. S100-alarmin-induced innate immune programming protects newborn infants from sepsis. Nat Immunol. 2017;18:622–632. doi: 10.1038/ni.3745. [DOI] [PubMed] [Google Scholar]
- 44.Dai J, Kumbhare A, Youssef D, McCall CE, El Gazzar M. Intracellular S100A9 Promotes Myeloid-Derived Suppressor Cells during Late Sepsis. Front Immunol. 2017;8:1565. doi: 10.3389/fimmu.2017.01565. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45.Achouiti A, Vogl T, Van Der Meer AJ, Stroo I, Florquin S, De Boer OJ, Roth J, Zeerleder S, Van’T Veer C, De Vos AF, Van Der Poll T. Myeloid-related protein-14 deficiency promotes inflammation in staphylococcal pneumonia. Eur Respir J. 2015;46:464–473. doi: 10.1183/09031936.00183814. [DOI] [PubMed] [Google Scholar]
- 46.van Zoelen MAD, Vogl T, Foell D, Van Veen SQ, van Till JWO, Florquin S, Tanck MW, Wittebole X, Laterre PF, Boermeester MA, Roth J, van der Poll T. Expression and Role of Myeloid-related Protein-14 in Clinical and Experimental Sepsis. Am J Respir Crit Care Med. 2009;180:1098–1106. doi: 10.1164/rccm.200810-1552OC. [DOI] [PubMed] [Google Scholar]
- 47.Vogl T, Tenbrock K, Ludwig S, Leukert N, Ehrhardt C, van Zoelen MAD, Nacken W, Foell D, van der Poll T, Sorg C, Roth J. Mrp8 and Mrp14 are endogenous activators of Toll-like receptor 4, promoting lethal, endotoxin-induced shock. Nat Med. 2007;13:1042–1049. doi: 10.1038/nm1638. [DOI] [PubMed] [Google Scholar]
- 48.Valdés-Ferrer SI, Rosas-Ballina M, Olofsson PS, Lu B, Dancho ME, Ochani M, Li JH, Scheinerman JA, Katz DA, Levine YA, Hudson LK, Yang H, Pavlov VA, Roth J, Blanc L, Antoine DJ, Chavan SS, Andersson U, Diamond B, Tracey KJ. HMGB1 mediates splenomegaly and expansion of splenic CD11b+ Ly-6C(high) inflammatory monocytes in murine sepsis survivors. J Intern Med. 2013;274:381–90. doi: 10.1111/joim.12104. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49.Stein VM, Czub M, Hansen R, Leibold W, Moore PF, Zurbriggen A, Tipold A. Characterization of canine microglial cells isolated ex vivo. Vet Immunol Immunopathol. 2004;99:73–85. doi: 10.1016/j.vetimm.2004.01.002. [DOI] [PubMed] [Google Scholar]
- 50.Richardson M, Ayliffe M, Helbert M, Davies E. A simple flow cytometry assay using dihydrorhodamine for the measurement of the neutrophil respiratory burst in whole blood: comparison with the quantitative nitrobluetetrazolium test. J Immunol Methods. 1998;219:187–193. doi: 10.1016/s0022-1759(98)00136-7. [DOI] [PubMed] [Google Scholar]
- 51.Bitzinger DI, Schlachetzki F, Lindner R, Trabold B, Dittmar MS. Flow-cytometric measurement of respiratory burst in rat polymorphonuclear granulocytes: Comparison of four cell preparation procedures, and concentration–response evaluation of soluble stimulants. Cytom Part A. 2008;73A:643–650. doi: 10.1002/cyto.a.20539. [DOI] [PubMed] [Google Scholar]
- 52.Achouiti A, Vogl T, Urban CF, Röhm M, Hommes TJ, van Zoelen MAD, Florquin S, Roth J, van ’t Veer C, de Vos AF, van der Poll T. Myeloid-related protein-14 contributes to protective immunity in gram-negative pneumonia derived sepsis. PLoS Pathog. 2012;8:e1002987. doi: 10.1371/journal.ppat.1002987. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 53.De Filippo K, Neill DR, Mathies M, Bangert M, McNeill E, Kadioglu A, Hogg N. A new protective role for S100A9 in regulation of neutrophil recruitment during invasive pneumococcal pneumonia. FASEB J. 2014;28:3600–3608. doi: 10.1096/fj.13-247460. [DOI] [PubMed] [Google Scholar]
- 54.Sade-Feldman M, Kanterman J, Ish-Shalom E, Elnekave M, Horwitz E, Baniyash M. Tumor necrosis factor-α blocks differentiation and enhances suppressive activity of immature myeloid cells during chronic inflammation. Immunity. 2013;38:541–54. doi: 10.1016/j.immuni.2013.02.007. [DOI] [PubMed] [Google Scholar]
- 55.Zheng R, Chen S, Chen S. Correlation between myeloid-derived suppressor cells and S100A8/A9 in tumor and autoimmune diseases. Int Immunopharmacol. 2015;29:919–925. doi: 10.1016/j.intimp.2015.10.014. [DOI] [PubMed] [Google Scholar]
- 56.Liu X, Quan N. Microglia and CNS Interleukin-1: Beyond Immunological Concepts. Front Neurol. 2018;9:8. doi: 10.3389/fneur.2018.00008. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 57.Cazareth J, Guyon A, Heurteaux C, Chabry J, Petit-Paitel A. Molecular and cellular neuroinflammatory status of mouse brain after systemic lipopolysaccharide challenge: importance of CCR2/CCL2 signaling. J Neuroinflammation. 2014;11:132. doi: 10.1186/1742-2094-11-132. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 58.Garré JM, Silva HM, Lafaille JJ, Yang G. CX3CR1+ monocytes modulate learning and learning-dependent dendritic spine remodeling via TNF-α. Nat Med. 2017;23:714–722. doi: 10.1038/nm.4340. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 59.Sinha P, Okoro C, Foell D, Freeze HH, Ostrand-Rosenberg S, Srikrishna G. Proinflammatory S100 proteins regulate the accumulation of myeloid-derived suppressor cells. J Immunol. 2008;181:4666–75. doi: 10.4049/jimmunol.181.7.4666. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 60.Choksawangkarn W, Graham LM, Burke M, Lee SB, Ostrand-Rosenberg S, Fenselau C, Edwards NJ. Peptide-based systems analysis of inflammation induced myeloid-derived suppressor cells reveals diverse signaling pathways. Proteomics. 2016;16:1881–1888. doi: 10.1002/pmic.201500102. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 61.Uhel F, Azzaoui I, Grégoire M, Pangault C, Dulong J, Tadié JM, Gacouin A, Camus C, Cynober L, Fest T, Le Tulzo Y, Roussel M, Tarte K. Early expansion of circulating granulocytic myeloid-derived suppressor cells predicts development of nosocomial infections in patients with sepsis. Am J Respir Crit Care Med. 2017;196:315–327. doi: 10.1164/rccm.201606-1143OC. [DOI] [PubMed] [Google Scholar]
- 62.Wattananit S, Tornero D, Graubardt N, Memanishvili T, Monni E, Tatarishvili J, Miskinyte G, Ge R, Ahlenius H, Lindvall O, Schwartz M, Kokaia Z. Monocyte-Derived Macrophages Contribute to Spontaneous Long-Term Functional Recovery after Stroke in Mice. J Neurosci. 2016;36:4182–95. doi: 10.1523/JNEUROSCI.4317-15.2016. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 63.Opal SM, Laterre PF, Francois B, LaRosa SP, Angus DC, Mira JP, Wittebole X, Dugernier T, Perrotin D, Tidswell M, Jauregui L, Krell K, Pachl J, Takahashi T, Peckelsen C, Cordasco E, Chang CS, Oeyen S, Aikawa N, Maruyama T, Schein R, Kalil AC, Van Nuffelen M, Lynn M, Rossignol DP, Gogate J, Roberts MB, Wheeler JL, Vincent JL. Effect of eritoran, an antagonist of MD2-TLR4, on mortality in patients with severe sepsis: the ACCESS randomized trial. JAMA. 2013;309:1154–62. doi: 10.1001/jama.2013.2194. [DOI] [PubMed] [Google Scholar]
- 64.Rice TW, Wheeler AP, Bernard GR, Vincent JL, Angus DC, Aikawa N, Demeyer I, Sainati S, Amlot N, Cao C, Ii M, Matsuda H, Mouri K, Cohen J. A randomized, double-blind, placebo-controlled trial of TAK-242 for the treatment of severe sepsis. Crit Care Med. 2010;38:1685–94. doi: 10.1097/CCM.0b013e3181e7c5c9. [DOI] [PubMed] [Google Scholar]
- 65.Yona S, Kim KW, Wolf Y, Mildner A, Varol D, Breker M, Strauss-Ayali D, Viukov S, Guilliams M, Misharin A, Hume DA, Perlman H, Malissen B, Zelzer E, Jung S. Fate mapping reveals origins and dynamics of monocytes and tissue macrophages under homeostasis. Immunity. 2013;38:79–91. doi: 10.1016/j.immuni.2012.12.001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 66.Wohleb ES, Powell ND, Godbout JP, Sheridan JF. Stress-induced recruitment of bone marrow-derived monocytes to the brain promotes anxiety-like behavior. J Neurosci. 2013;33:13820–13833. doi: 10.1523/JNEUROSCI.1671-13.2013. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 67.Foster SL, Hargreaves DC, Medzhitov R. Gene-specific control of inflammation by TLR-induced chromatin modifications. Nature. 2007;447:972. doi: 10.1038/nature05836. [DOI] [PubMed] [Google Scholar]
- 68.Wheeler DS, Lahni PM, Denenberg AG, Poynter SE, Wong HR, Cook JA, Zingarelli B. Induction of endotoxin tolerance enhances bacterial clearance and survival in murine polymicrobial sepsis. Shock. 2008;30:267–73. doi: 10.1097/shk.0b013e318162c190. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 69.Deng JC, Cheng G, Newstead MW, Zeng X, Kobayashi K, Flavell RA, Standiford TJ. Sepsis-induced suppression of lung innate immunity is mediated by IRAK-M. J Clin Invest. 2006;116:2532–42. doi: 10.1172/JCI28054. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 70.Austermann J, Friesenhagen J, Fassl S, Ortkras T, Burgmann J, Barczyk-Kahlert K, Faist E, Zedler S, Pirr S, Rohde C, Müller-Tidow C, von Köckritz-Blickwede M, von Kaisenberg CS, Flohé SB, Ulas T, Schultze JL, Roth J, Vogl T, Viemann D. Alarmins MRP8 and MRP14 Induce Stress Tolerance in Phagocytes under Sterile Inflammatory Conditions. Cell Rep. 2014;9:2112–2123. doi: 10.1016/j.celrep.2014.11.020. [DOI] [PubMed] [Google Scholar]
- 71.Fontaine M, Planel S, Peronnet E, Turrel-Davin F, Piriou V, Pachot A, Monneret G, Lepape A, Venet F. S100A8/A9 mRNA induction in an ex vivo model of endotoxin tolerance: roles of IL-10 and IFNγ. PLoS One. 2014;9:e100909. doi: 10.1371/journal.pone.0100909. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 72.Chu CH, Wang S, Li CL, Chen SH, Hu CF, Chung YL, Chen SL, Wang Q, Lu RB, Gao HM, Hong JS. Neurons and astroglia govern microglial endotoxin tolerance through macrophage colony-stimulating factor receptor-mediated ERK1/2 signals. Brain Behav Immun. 2016;55:260–272. doi: 10.1016/j.bbi.2016.04.015. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 73.Schaafsma W, Zhang X, van Zomeren KC, Jacobs S, Georgieva PB, Wolf SA, Kettenmann H, Janova H, Saiepour N, Hanisch UK, Meerlo P, van den Elsen PJ, Brouwer N, Boddeke HWGM, Eggen BJL. Long-lasting pro-inflammatory suppression of microglia by LPS-preconditioning is mediated by RelB-dependent epigenetic silencing. Brain Behav Immun. 2015;48:205–221. doi: 10.1016/j.bbi.2015.03.013. [DOI] [PubMed] [Google Scholar]
- 74.Valdés-Ferrer SI, Rosas-Ballina M, Olofsson PS, Lu B, Dancho ME, Li J, Yang H, Pavlov VA, Chavan SS, Tracey KJ. High-mobility group box 1 mediates persistent splenocyte priming in sepsis survivors: evidence from a murine model. Shock. 2013;40:492–5. doi: 10.1097/SHK.0000000000000050. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 75.Püntener U, Booth SG, Perry VH, Teeling JL. Long-term impact of systemic bacterial infection on the cerebral vasculature and microglia. J Neuroinflammation. 2012;9:146. doi: 10.1186/1742-2094-9-146. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 76.Siddiqui TA, Lively S, Schlichter LC. Complex molecular and functional outcomes of single versus sequential cytokine stimulation of rat microglia. J Neuroinflammation. 2016;13:66. doi: 10.1186/s12974-016-0531-9. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 77.Simard JC, Cesaro A, Chapeton-Montes J, Tardif M, Antoine F, Girard D, Tessier PA. S100A8 and S100A9 induce cytokine expression and regulate the NLRP3 inflammasome via ROS-dependent activation of NF-κB(1.) PLoS One. 2013;8:e72138. doi: 10.1371/journal.pone.0072138. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.