

Abstract
Mouse models of lupus have shown that multiple immune cell types contribute to autoimmune disease. This study sought to investigate the involvements of B cells and dendritic cells (DCs) in supporting the expansion of inflammatory and regulatory CD4+ T cells that are critical for lupus pathogenesis. We used the lupus-prone B6.NZM2410.Sle1.Sle2.Sle3 (TC) and congenic B6 control mice to investigate how the genetic predisposition of these two cell types controls the activity of normal B6 T cells. Using an allogenic in vitro assay, we showed that TC B1-a and B2 cells expanded Th17 cells significantly more than their B6 counterparts. This expansion was dependent on CD86 and IL-6 expression, and mapped to the Sle1 lupus-susceptibility locus. In vivo, TC as compared to B6 B cells promoted the differentiation of CD4+ T cells into Th1 and follicular helper T (Tfh) cells, but limited the expansion of Treg cells as compared to B6 B cells. Finally, when normal B6 CD4+ T cells were introduced into Rag1−/− mice, TC myeloid/stromal cells caused their heightened activation, decreased Treg differentiation and increased renal infiltration of Th1 and Th17 cells, as compared with B6 myeloid/stromal cells. The current results show that B cells from lupus mice amplify inflammatory CD4+ T cells in a non-redundant manner with myeloid/stromal cells.
Keywords: Lupus, B cells, dendritic cells, CD4+ T cells, Tfh, Th17, autoimmunity
Introduction
B cells participate in a variety of effector functions. In addition to being precursors of antibody-secreting plasmablasts and plasma cells, they can be particularly efficient APCs that provide co-stimulatory factors as well as cytokines that can be either pro-inflammatory (IL-6, TFNα) or anti-inflammatory (IL-10). It has been suggested that these antibody-independent B cell functions modulate autoimmune processes including those that lead to systemic lupus erythematosus (SLE) (1). Although there have been numerous studies dissecting the mechanisms by which CD4+ T cells contribute to the production of autoantibodies by lupus B cells, less is known concerning how B cells regulate effector functions of CD4+ T cells in lupus (2–4).
Conventional dendritic cells (DCs) are professional antigen presenting cells (APCs) that modulate the function of cognate naïve CD4+ T cells through the production of cytokines. Multiple studies in humans and mouse models have established the involvement of DCs in lupus, in which both intrinsic and extrinsic pathways lead to the accumulation of activated DCs that secrete inflammatory cytokines (5). Depletion of DCs in lupus-prone MRL/lpr mice reduced autoimmune pathology as well as T cell expansion (6). DC-depleted MRL/lpr mice had reduced levels of autoAbs, but their total IgG did not differ from DC-sufficient controls. Surprisingly, however, depletion of DCs had very little impact on spontaneous T cell activation, inferring that B cells rather than DCs were responsible for the activation of T cells by autoAg. Evidence supporting a specific contribution of B cells was revealed by the analysis of B-cell vs. DC-specific MyD88-deficient MRL/lpr mice (7), a model in which TLR7 and TLR9 are required for the production of anti-RNA and anti-DNA autoAbs, respectively, signaling through the MyD88 adaptor (8). AutoAbs were eliminated in mice with MyD88-deficient B cells but only modestly reduced in mice with MyD88-deficient DCs. Since these autoAbs are T-dependent, the results demonstrated a critical role for TLR-activated B cells in stimulating autoreactive T cells. Accordingly, only CD4+ T cells from B-cell deficient MRL/lpr mice or MRL/lpr mice with MyD88-deficient B cells showed a significant reduction in spontaneous CD4+ T cell activation. This suggests that B cells activate T cells in a qualitatively different manner than DCs, which may be more favorable to autoreactive T cells. Interestingly, both DC-and B-cell-specific MyD88 deficiencies caused reductions in Tfh cells in MRL/lpr mice. However, in autoimmune B6.Sle1b mice, B cells secreting IFN-γ as well as signaling through IFN-γR and STAT1 were required for a full induction of spontaneously arising Tfh cells and autoAbs (9). Overall, these results suggest that B cells may play a more critical role in the activation of autoreactive T cells in lupus as compared with non-autoimmune mice, at least partly due to their chronic TLR activation by nucleic acids.
B cell subsets representing different stages of development have overlapping but distinct functions (10). There is evidence for skewed distributions of these B cell subsets in lupus mice (11) and patients (12) that could impinge on their ability to cause T cell activation. Among these subsets, innate-like B1-a cells are expanded in lupus mice (13), and lupus patients (14). B1-a cells are generally excluded from T-dependent immune responses (15) but their enhanced APC function as compared to conventional B cells (B2) was recognized over 20 years ago (16). Peritoneal B-1a (pB1a) cells promote the expansion of IL-10, IFNγ and IL-4 producing CD4+ T cells in an Ag-dependent manner, while splenic B-1a cells more efficiently promoted the expansion of Th17 cells as compared to conventional B cells in vitro (17). In vivo, Ag-pulsed pB1a cells induced a greater CD4+ T cell proliferation (17). A stronger Ag-dependent expansion of inflammatory CD4+ T cells secreting IFNγ, IL-17 and TNFα was induced in vitro by allogeneic pB1a cells, while B2 cells in the same conditions expanded Foxp3 regulatory CD4+ (Treg) T cells (18). In addition to Ag presentation, CD44 and CD86 expression were required for the pB1a cells to expand inflammatory T cells (19). Conversely, IL-17A expanded pulmonary B1-a cells during a viral infection by inducing Blimp-1 and NF-kB, which are key transcription factors for B1-a cell differentiation (20). This suggests a mutual amplification of B1-a cells and Th17 cells may play a protective role against pathogens.
We have used the B6.NZM2410.Sle1.Sle2.Sle3 (TC) mouse model of lupus model and related single congenic strains to characterize interactions among immune cells that were essential to disease development (21). These strains share at least 95% of their genetic background with non-autoimmune C57BL/6J (B6) mice, including the MHC, the immunoglobulin and T cell receptor genes. Using this model, we showed that autoreactive CD4+ T cells driven by the expression of the Sle1 and Sle3 loci are essential to the production of autoAbs (22; 23). DCs from TC mice reduce Treg expansion and functions (24), and they activate B cell proliferation and Ab production (25; 26). In the current study, we examine the role of B cells from TC mice in activating and inducing the production of inflammatory cytokines by CD4+ T cells. We show by both in vitro and in vivo assays that B cells from TC mice caused B6 CD4+ T cells to expand in both the spleen and kidneys with a skewing towards more activated inflammatory phenotypes, and that IL-6 plays a major role in this process. We also show that non-lymphoid cells from TC mice induced overlapping but distinct phenotypes in CD4+ T cells. We have previously identified an intrinsic hyperactivation of CD4+ T cells and B cells in this model of lupus (27; 28). Here we show that DCs from TC mice exhibit an intrinsically activated phenotype in the absence of lymphocytes. Overall, our results demonstrate the activation of CD4+ T cells that drives autoimmune pathogenesis in TC mice results from interactions with both B cells and DCs that amplify cell-intrinsic defects imparted by the expression of lupus susceptibility genes.
Materials and Methods
Mice
The TC, B6.Sle1 and B6.Sle2 strains have been previously described (29; 30). B6, B6.C-H2bm12/KhEg (bm12), and B6.Rag1−/− (B6.Rag) mice were originally purchased from the Jackson Laboratory (Bar Harbor, ME, USA). TC.Rag1−/− (TC.Rag) mice were produced by breeding the Rag1−/− allele to the Sle1, Sle2, and Sle3 loci as previously described for other alleles (31). B6.Il21VFP mice were produced by the insertion of an IRES-VFP (Venus-fluorescent protein) cassette in a non-coding exon on the Il21 gene, resulting in the tagging of IL-21 expressing cells with VFP (32). Only female mice were used in this study, and they were housed by strain of origin. B cell donors were isolated from at least 5 months of age and age-matched within experiments. CD4+ T cell donors were isolated from 2 to 6 months of age. B6.Rag and TC.Rag recipients were used between 2 and 4 month old. All experimental groups within an experiment were tested simultaneously to avoid environmental variations. All experiments were conducted according to protocols approved by the University of Florida IACUC.
In vitro T cell polarization
Splenic CD4+ T cells and CD43− B cells (sB2) were isolated by negative selection with magnetic beads (Miltenyi Biotec, Auburn, CA, USA) yielding sB2 and CD4+ T cell population with a purity >95%. Peritoneal B1-a cells (pB1a) were isolated as previously described (33). Briefly, non-adherent peritoneal cells were first negatively selected with biotinylated anti-Thy1.2 (53-2.1) and anti-CD3ε (145-2C11) Abs, and then positively selected with biotinylated anti-CD5 (53-7.3) Ab. This protocol led to a B220int CD5+ cell population with an average of 80% purity. CD4+ T cells (2 × 105) from bm12 mice were co-cultured with pB1a or sB2 from the congenic NZM strains or B6 mice (1 × 105) for 5 d in T cell polarizing media without anti-CD3 or anti-CD28 Abs. Th17 polarizing media contained TGFβ (3 ng/ml), IL-6 (50 ng/ml), 6-Formylindolo (3,2-b) carbazole (FICZ, 300 nM; Enzo Life Sciences, Farmingdale, NY, USA), anti-IL-4 Ab and anti-IFNγ Ab (10 ug/ml each). Th1 polarizing media contained IL-12 (10 ng/ml) and anti-IL-4 Ab (10 ug/ml), and Treg polarizing media contained TGFβ (20 ng/ml) and IL-2 (100 U). All cytokines were purchased from Peprotech (Rocky Hill, NJ, USA). In some experiments, blocking Abs to CD86 (GL1, 10 ug/ml), CD44 (either IM7 or KM114 clones, 20 ug/ml) or IL-6 (1 ug/ml, Peprotech) were added to the co-cultures.
Flow Cytometry
Single-cell suspensions were prepared from spleens using standard procedures. After red blood cell lysis, cells were blocked with anti-CD16/32 Ab (2.4G2), and stained in FACS staining buffer (2.5% FBS, 0.05% sodium azide in PBS). Fluorochrome-conjugated Abs used were to detect BCL6 (K112-91), BTK (M4G3LN), CD4 (RAM4-5), CD11c (HL3), CD19 (1D3), CD44 (IM7), CD45 (30-F11), CD69 (H1.2F3), CD80 (16-10A1), CD86 (GL1), Foxp3 (FJK-16s), I-Ab (AF6-120.1), IFN-γ (XMG1.2), IL-6 (MP5-20F3), IL-17A (TC11-18H10.1), IL-21 (FFA21), PD-1 (RMP1-30), PGSL-1 (CD162, clone 2PH1). These antibodies were purchased from BD Biosciences (San Jose, CA, USA), eBioscience (San Diego, CA, USA), or BioLegend (San Diego, CA, USA). CXCR5 was stained in a 3-step process first with the purified antibody (2G8) followed by biotinylated anti-rat IgG (Jackson ImmunoResearch Lab, West Grove, PA, USA) then PerCP-labeled streptavidin. Dead cells were excluded with fixable viability dye (eFluor780, eBioscience). Data were collected on LSRFortessa (BD Biosciences) and analyzed using FCS Express (DeNovo, Glendale, CA, USA) and FlowJo (LLC, Ashland, OR, USA). To analyze intracellular cytokine production, cells were treated with the leukocyte activation cocktail (LKA, BD Biosciences) for 5 h and fixed with the Fixation/Permeabilization kit (eBiosciences).
Cytokine measurement
Marginal zone (MZ) and follicular (FO) B cells were sorted from purified splenic CD43− B cells as IgM+CD21+CD23− for MZ B cells and IgM+CD21−CD23+ for FO B cells, and stimulated for 3 d with 1 ug/ml CpG (Invivogen). IL-6 was measured in the supernatant by ELISA (OptEIA kit, BD Biosciences). Purified sB2 cells were stimulated with 10 ug/ml LPS or 1 ug/ml CpG for 24 h and IL-6 was measured by ELISA. Cells were further stimulated with LKA cocktail for IL-6 intracellular FACS analysis. IL-6 was also measured in CD11c+ splenic DCs from B6.Rag and TC.Rag mice by FACS after 24 h stimulation of total splenocytes, as described for sB2 cells. Bone-marrow (BM)-derived DCs were differentiated with GM-CSF and IL-4 as previously described (26). Cytokines were measured before and after 6 h or 24 h stimulation with 1 ug/ml LPS by qRT-PCR and ELISA, as previously described (34).
Adoptive transfers
Five millions splenic CD4+ T cells isolated from B6.Il21VFP mice were co-transferred intravenously with an equal number of B6 or TC sB2 cells into B6.Rag mice. Recipient mice were sacrificed 1 week later. To evaluate the role of IL-6 in the process, pairs of B6.Rag mice were injected with B6, TC or Sle1 sB2 cells from the same mouse along with B6.Il21VFP CD4+ T cells, and one member of the pair was treated with 100 ug of anti-IL-6 neutralizing Ab or isotype control one day after cell transfer. The immunophenotypes of the donor and recipient CD4+ T cells were analyzed in the spleen and kidneys. Kidney lymphocytes were obtained after collagenase digestion as previously described (35).
Statistical Analysis
Statistical analyses were performed using the GraphPad Prism 6.0 software. Unless indicated, data was normally distributed, and graphs show means and standard deviations of the mean (SEM) for each group. Results were compared with 2-tailed t tests with a minimal level of significance set at P < 0.05. Bonferroni corrections were applied for multiple comparisons.
Results
TC B cells expand Th17 cells in vitro
To compare the effect of B cells from lupus-prone and control B6 mice on T cell polarization, we used an allogeneic assay in which bm12 CD4+ T cells were co-cultured with pB1a or sB2 cells from the congenic NZM strains or B6 mice for 5 d in polarizing media. The polarization of bm12 CD4+ T cells in the presence of MHC Aβ-mismatched pB1a and sB2 B cells confirmed results obtained with fully allogenic B and T cells (36): B6 pB1a cells promoted the expansion of Th17 cells more effectively than B6 sB2 cells (Fig. 1A), and conversely B6 sB2 cells promoted the expansion of Treg cells more than B6 pB1a cells (Fig. 1B). A similar relative expansion of Th17 cells by pB1a cells and Treg cells by sB2 cells, respectively, was observed for B cells of TC origin. Importantly, both TC pB1a and sB2 cells expanded Th17 cells significantly more than their B6 counterparts (Fig. 1A). The presence of TC as compared to B6 B cells also resulted in significantly higher absolute numbers of Th17 cells (pB1a: 5 ± 0.53 × 104 vs. 0.08 ± 0.23 × 104, p < 0.001; sB2: 9.19 ± 1.14 × 104 vs. 1.84 ± 0.24 × 104, p < 0.001). However, no difference between strains was observed for Treg cells, which were expanded by sB2 cells from either strain over 10 fold more than by pB1a cells (Fig. 1B). There was also no difference between strains for Th1 cells, which were expanded more by pB1a than sB2 cells (Fig. 1C). These results not only confirmed in a different model that pB1a cells preferentially expand Th17 cells and sB2 cells preferentially expand Treg cells. More importantly, both pB1a and sB2 cells from the lupus-prone TC mice enhanced Th17 polarization relative to cells from their B6 counterparts in the context of in vitro allogeneic stimulation.
Figure 1. TC B cells expand Th17 T cells significantly more than B6 B cells in vitro.
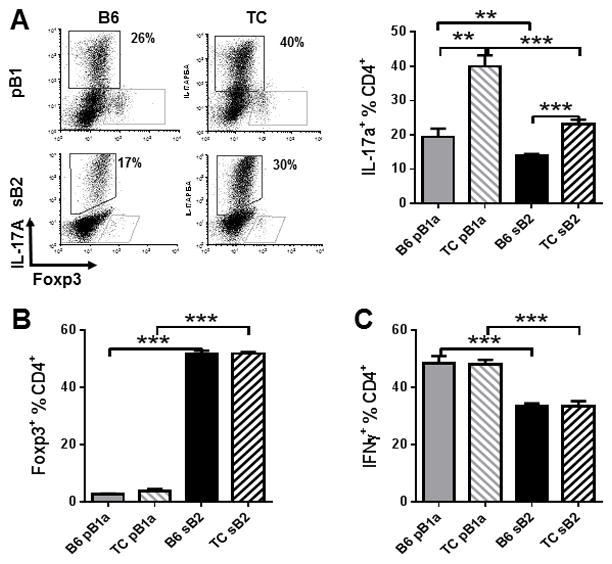
Phenotypes of bm12 CD4+ T cells cultured in polarizing conditions in the presence of B6 or TC pBa1and sB2 cells for 5 d. A. Th17 polarization: representative FACS plots of CD4+-gated cells stained with IL-17A and Foxp3, and frequency of IL-17A+ CD4+ T cells. B. Treg polarization: Frequency of Foxp3+ CD4+ T cells. C. Th1 polarization: frequency of IFN-γ+ CD4+ T cells. Data represents n = 5–10 mice, expressed as means ± SEM. ** P < 0.01, *** P < 0.001.
Expansion of Th17 cells by TC B cells is CD86 and IL-6 dependent
To understand the mechanisms responsible for the enhanced Th17 polarization by TC B cells, we evaluated the known contributors to Th17 polarization in non-autoimmune mice. Th17 cell expansion by pB1a cells depends on CD86 and CD44 expression (19), but not on specific B1-a cell markers, including PD-L2 (37). As determined by mean fluorescence intensity (MFI), TC sB2 cells expressed a significantly higher level of both CD44 and CD86 than B6 sB2 cells (Fig. 2A). The reverse, however, was observed for pB1a cells (Fig. 2A), which we have confirmed by gene expression profiling (Morel unpublished). This indicated that the capacity of TC B cells to enhance Th17 polarization was not simply due to an over-expression of CD44 or CD86. There was no strain difference for the expression of either CD80 or phosphorylated BTK, a marker of elevated BCR signaling characteristic of B-1a cells (Fig. 2A). We directly tested the requirement of CD44 and CD86 expression in the ability of sB2 TC cells to expand Th17 cells by using blocking mAbs. Polarization in the presence of the IM7 anti-CD44 Ab further expanded Th17 cells stimulated by either B6 or TC sB2 cells (Fig. 2B and C). However the KM114 anti-CD44 Ab, which blocks CD44 binding to hyaluronic acid (38), completely inhibited Th17 induction (Fig. 2C). Overall, these results showed that CD44 binding is involved in Th17 induction, and suggests that its increased expression by TC sB2 cells may contribute to their increased ability to expand effector T cells. The mechanism by which CD44 expressed on TC sB2 cells expand bm12 Th17 differentiation may be different from that of BALB/c B1-a cells expanding B6 Th17 differentiation, with the functional binding of CD44 being to HA in the former and to osteopontin in the latter. Blocking CD86 access dramatically reduced the frequency and number (data not shown) of Th17 and Th1 cells with either B6 or TC sB2 cells (Fig. 2B and D), while cell viability was not affected (data not shown).
Figure 2. Th17 expansion by TC B cells depends on CD86 and IL-6.

A. Expression levels of CD44, CD86, CD80 and pBTK in B6 or TC pB1a and sB2 cells. B. Representative FACS plots of bm12 CD4+ gated cells stained with IL-17A and IFNγ that have been cultured with TC sB2 cells alone (-), or with anti-CD44 or anti-CD86 blocking Abs. C. Frequency of IL-17A+ bm12 CD4+ T cells cultured in Th17 polarizing conditions in the presence of B6 or TC sB2 cells with or without IM7 or blocking KM114 anti-CD44 antibody. D. Frequency of IL-17A+ (left) and IFN-γ+ (right) bm12 CD4+ T cells cultured in Th17 or Th1 polarizing conditions in the presence of B6 or TC sB2 cells with or without anti-CD86 Ab. E. IL-6 production by sorted B6 and TC FO and MZ B cells stimulated with CpG. F. Frequency of IL-17A+ bm12 CD4+ T cells cultured in Th17 polarizing conditions in the presence B6 or TC sB2 cells with or without anti-IL-6 Ab or in the absence of IL-6 in the Th17 polarizing media. Data represents n = 9 mice (A), 6 mice (C and D), 3 mice (E and F), expressed as means ± SEM. * P < 0.05, ** P < 0.01, *** P < 0.001.
Th17 polarization requires IL-6 and we have shown (26) that TC FO and MZ B cells stimulated by CpG or apoptotic cells (Fig. 2E) produce a higher amount of IL-6 than B6 B cells. We confirmed this results with total sB2 cells stimulated with LPS (Fig. 3D). However, only background levels of Th17 induction were detected in the absence of IL-6 with either B6 or TC sB2 cells (Fig. 2F), indicating that the excess IL-6 produced by TC sB2 cells was not sufficient to induce Th17 differentiation. Blocking anti-IL-6 normalized Th17 cell polarization by TC sB2 cells to a level similar to those for B6 sB2 cells (Fig. 2F). This suggested that excess IL-6 contributed to the skewing observed with TC sB2 cells. Overall, these results showed that CD44, CD86 and IL-6 signaling contribute to the expansion of Th17 by sB2 cells and suggested that the high levels of CD44 and CD86 expression as well as IL-6 secretion by TC sB2 cells may contribute to their enhanced Th17 and Th1 polarizing activity.
Figure 3. Sle1 contributes to Th17 expansion by B cells.

A–B. Frequency and absolute numbers of IL-17A-producing bm12 CD4+ T cells in Th17 polarizing media co-cultured with sB2 cells from B6, B6.Sle1, B6.Sle2, or TC mice (A) with representative FACS plots shown in (B). C. CD86 and CD44 expression on B6, B6.Sle1, B6.Sle2, and TC sB2 cells. D. IL-6 measured in the supernatants of sB2 cells stimulated with LPS. E. Intracellular IL-6 in sB2 cells stimulated with LPS. Representative FACS plots are shown on the right. Data represents n = 4 – 5 mice, expressed as means ± SEM. Comparisons were made to B6 values, * P < 0.05, ** P < 0.01, *** P < 0.001.
Sle1 contributes to the ability of TC B cells to expand Th17 cells
Among the three susceptibility loci present in TC mice, expression of both Sle1 and Sle2 (27; 28), but not Sle3 (39), results in intrinsic B cell activation. We therefore compared the expansion of allogenic Th17 cells by TC sB2 cells to either Sle1 or Sle2 sB2 cells. Sle1 sB2 cells expanded allogenic Th17 cells significantly more than B6 sB2 cells, while there was no difference between Sle2 and B6 sB2 cells (Fig. 3A and B). This occurred in spite of similar levels of CD86 expression by Sle1 and B6 sB2 cells (Fig. 3C), suggesting that CD86 is required but its overexpression is not necessary for the enhanced Th17 expansion. IL-6 levels, however, correlated with Th17 cell expansion, with Sle1 but not Sle2 sB2 cells stimulated with LPS producing significantly more IL-6 than B6 sB2 cells, to a level equivalent or higher than TC sB2 cells (Fig. 3D and E). Similar results were obtained with sB2 cells stimulated with CpG (data not shown). This suggests that Sle1 is the main contributor to the ability of TC sB2 cells to promote Th17 expansion through the production of IL-6.
TC CD4+ T cells have an inflammatory cytokine profile
As we have previously reported (40; 41), TC mice with active disease have increased numbers of CD4+ T cells in the spleen and kidneys (Fig. 4A). Among these T cells, the frequencies of IFNγ+ T cells were increased in the spleen (Fig. 4B). In the kidneys, there was no difference in the frequency of IFNγ+ T cells, but an increased absolute number of TC IFNγ+ CD4+ T cells (data not shown). The frequency of splenic IL-17A+ CD4+ T cells was very low and similar between TC and B6 mice (Fig. 4C). Similar results were obtained in the kidneys, when considering either CD4+ T cells or CD4−CD8− T cells (data not shown). The absolute numbers of IL-17A+ CD4+ T cells were higher in TC spleens (data not shown), and TC kidneys (Fig. 4C). These results are consistent with the higher numbers of total CD4+ T cells in TC spleens and kidneys (Fig. 4A). There was a trend for an increased frequency of IL-21-producing CD4+ T cells in TC spleens (Fig. 4D), but the intracellular detection of this cytokine is notoriously poor. However, quantitation of splenic CD4+PD-1hiCXCR5hiBcl6+Foxp3− Tfh cells (Fig. 4E) and renal CD45+CD4+CD44hiPD-1+PGSL-1lo Tfh cells (Fig. 4F) revealed a higher frequency and number (data not shown) of Tfh cells in TC mice. These results confirm that TC mice exhibited a relatively expanded frequency of IFNγ+ CD4+ T cells, and of Tfh cells in both spleen and kidneys. They also showed increased numbers of IFNγ+ CD4+ T cells, Tfh and IL-17+ CD4+ T cells in the kidneys of TC mice, strongly suggesting that these effector cells play a role in pathogenesis.
Figure 4. Expanded effector CD4+ T cell populations in TC mice.

A. Absolute numbers of CD4+ T cells in the spleen and kidney of B6 and TC mice. B. Frequencies of splenic IFN-γ+ CD4+ T cells. C. Frequencies of IL-17A+ CD4+ T cells in the spleen and absolute numbers in the kidneys. D. Frequencies of splenic IL-21+ CD4+ T cells. Frequencies of splenic (E, CD4+CXCR5hiPD-1hiBCL6+FOXP3−) and renal (F, CD45+CD4+CD44hiPSGL-1loPD-1+) Tfh cells with gating strategy shown on the right. Mice were 6–7 months old. Data represents n = 4–7 mice, expressed as means ± SEM. * P < 0.05, ** P < 0.01.
TC sB2 cells expand Tfh and IFNγ-producing CD4+ T cells in vivo
To compare the effects on TC and B6 sB2 cells on T cell development in vivo, we used an adoptive transfer model of CD4+ T cells obtained from B6.Il21VFP mice. These mice express an Il-21-VFP reporter construct (Sup. Fig. 1A) in a Tfh cell-specific manner (32), which we have validated in the CD4+ T cells that we used for adoptive transfers (Sup. Fig. 1B). Total CD4+ T cells from B6.Il21VFP mice were injected in B6.Rag mice along with TC or B6 sB2 cells. After one week, a significantly higher number of CD4+ T cells was recovered from spleens and kidneys of mice that received TC sB2 cells (Fig. 5A). The frequency of IL-21-VFP+ CD4+ T cells was expanded over 10-fold in spleens of all B6.Rag recipients in the presence of either B6 or TC sB2 cells, ranging from about 4% of the donor CD4+ T cells expressing IL-21-VFP to over 40% (Fig. 5B–D). However, the frequencies and numbers of IL-21-VFP+ CD4+ T cells were higher in the presence of TC sB2 cells in both spleens (Fig. 5C) and kidneys (Fig. 5D). Similar results were obtained in the blood and mesenteric lymph node (data not shown). In addition, the frequencies of splenic Treg cells were reduced (Fig. 5E) and the frequencies and numbers of splenic IFNγ+ CD4+ T cells were expanded (Fig. 5F) in the presence of TC sB2 cells. No change was observed, however, for these T cell subsets in the kidneys (data not shown). The frequency of IL-17A+ CD4+ T cells was also lower in the spleen (Fig. 5G) and the kidneys (data not shown) of recipients of TC sB2 cells. To evaluate the potential role of IL-6 on T cell phenotypes induced by TC sB2 cells in vivo, B6.Rag mice were treated with anti-IL-6 neutralizing Ab or isotype control one day after cell transfer (Fig. 5H). Blocking IL-6 decreased the TC sB2 cell induced-expansion of CD4+ T cells as well as the level of expression of IL-21 on Tfh cells. This is consistent with studies showing that IL-6 induces IL-21 expression in murine (42; 43) and human (44) CD4+ T cells. Blocking IL-6 had no effect on the frequency of IL-21+ CD4+ T cells or Tfh cells, although as a consequence of the reduced number of total CD4+ T cells, it decreased the numbers of these cells (data not shown). IL-6 inhibition had also no effect on the frequency of Treg and Th1 cells induced by TC B cells, but it increased the frequency of IFNγ+ producing cells in the presence of B6 s2B cells. In this cohort, contrary to the previous cohort (Fig. 5G), TC B cells enhanced Th17 expansion, and this was reduced by IL-6 inhibition. The difference between the two cohorts performed over one year apart may be due to drifts in environmental conditions, to which Th17 cells are exquisitely sensitive. Overall, these results showed that TC sB2 cells promote CD4+ T cell expansion in vivo with a skewing away from Treg and towards Th1 and Tfh cell polarization. Further, IL-6 produced or induced by TC sB2 cells contributes to some of these phenotypes, specifically the expansion of total CD4+ T cells and the amount of IL-21 produced by Tfh cells, which have both been associated with lupus pathogenesis.
Figure 5. TC sB2 expand the frequencies of IL-21 and IFN-γ-producing CD4+ T cells in vivo.

B6 or TC SB2 cells were co-transferred with CD4+ T cells from B6.Il21VFP mice into B6.Rag mice and analysis of recipient mice was conducted one week later. A. Numbers of total CD4+ T cells in the spleen (left) and kidneys (right). B. Representative FACS plots showing IL-21-VFP staining in CD4+ gated cells in spleens and kidneys. Frequencies and numbers of IL-21-VFP+ CD4+ T cells in spleens (C) and kidneys (D). E. Frequencies of splenic Treg cells. F. Frequencies and numbers of splenic IFN-γ+ CD4+ T cells. G. Frequencies of splenic IL-17A+ CD4+ T cells. H. B6.Il21VFP CD4+ T cell phenotypes in B6.Rag transferred with B6, TC or Sle1 sB2 cells and treated with anti-Il-6 neutralizing Ab (+) or isotype control (-). Data represents n = 7 (A–G) mice per group, and 3 for B6, 4 for TC and 1 Sle1 sB2 per pair of B6.Rag recipients (H), expressed as means ± SEM. * P < 0.05, ** P < 0.01, *** P < 0.001.
Since Sle1 sB2 cells also produce high levels of IL-6 (Fig. 3D–E), we evaluated their effect on CD4+ T cells in the same transfer model (Fig. 5G). Sle1 sB2 cells also expanded CD4+ T cells relative to B6 sB2 B cells, but it was not dependent on IL-6. IL-6 inhibition had also no effect on the modest increase in IL-21 expression conferred by Sle1 sB2 cells, or the frequency of Treg or Tfh cell expansion. Interestingly, Sle1 sB2 cells induce a large Th1 expansion, which was, as for B6 sB2 cells, enhanced by the IL-6 blocking Ab. These results show that although Sle1 B cells produce high levels of Il-6, they do not recapitulate the phenotypes induced by TC sB2 cells, implying that TC B cells impact CD4+ T cell fate through other mechanism(s) in addition to IL-6 production.
TC myeloid/stromal cells expand Tfh cells in vivo
To assess the contribution of non-B cells to T cell expansion and polarization, we transferred non-autoimmune B6 CD4+ T cells into either TC.Rag or B6.Rag mice. The recipient mice differed by the expression of the three Sle loci in their myeloid cells, including DCs, as well as stromal cells that can secrete immunomodulatory cytokines. The number of B6 CD4+ T cells recovered from either the spleens or kidneys was higher in TC.Rag than in B6.Rag mice (Fig. 6A), and these cells were also more activated, as shown by the expression of CD69 (Fig. 6B). Conversely, the frequencies of Treg cells was decreased in TC.Rag recipients (Fig. 6C), but the frequencies and numbers of Tfh cells were increased, leading to about three-fold more Tfh cells in TC.Rag as compared to B6.Rag mice (Fig. 6D). Th1 and Th17 polarizations were similar between the two recipient strains, even though the absolute numbers of Th1 and Th17 cells were higher in TC.Rag spleens due to their higher numbers of total CD4+ T cells (Fig. 6E and G). The kidneys of TC.Rag recipients were infiltrated with higher frequencies of IFNγ+ CD4+ T cells but lower frequencies of IL-17A+ CD4+ T cells as compared to kidneys of B6.Rag recipients (Fig. 6F and H). However, as in the spleen, TC.Rag kidneys contained higher numbers of both IFNγ+ and IL-17A+ CD4+ T cells as compared to B6.Rag kidneys due to the higher numbers of total CD4+ T cells. These results showed that, similar to B cells, TC myeloid/stromal cells expand CD4+ T cells and polarize them away from Treg and toward Tfh phenotypes. The expansion of IFNγ+ CD4+ T cells by TC myeloid/stromal cells occurred in the kidneys but not in the spleen. Overall, as with TC sB2 cells, the numbers of IFNγ+ and IL-17A+ CD4+ T cells were higher in the spleens and kidneys of TC.Rag than B6.Rag mice that received the same non-autoimmune T cells.
Figure 6. TC myeloid/stromal cells expand Tfh and IFNγ-producing T cells in vivo.

B6 CD4+ T cells were transferred into B6.Rag or TC.Rag mice and analysis was conducted one week later in recipient mice. A. Numbers of total CD4+ T cells in the spleen and kidneys. Frequencies and numbers of splenic CD69+CD4+ T cells (B), Treg (C, CD4+FOXP3+) and Tfh (D, CD4+CXCR5+PD-1+FOXP3−) cells. Frequencies and numbers of IFNγ+ (E–F) and IL-17A+ (G–F) CD4+ T cells in the spleen and kidneys. Data represents n = 5 mice, expressed as means ± SEM. * P < 0.05, ** P < 0.01, *** P < 0.001.
TC DCs exhibit an intrinsic inflammatory phenotype
Among myeloid and stromal cells capable of differentially affecting the phenotypes of CD4+ T cells in TC.Rag mice, we examined DCs, since these cells have a well-established immunoregulatory effect on T cells. DCs in TC mice have an inflammatory phenotype, including high production of IL-6 (24–26) and a type 1 interferon signature (45). To address whether these phenotypes are secondary to B and T cell activation, including through stimulation by autoAb immune complexes, we compared DC phenotypes between TC.Rag, B6.Rag and TC mice. The numbers and frequencies of DCs were higher in TC.Rag than in B6.Rag spleens (Fig. 7A). As expected, the frequency of DCs was higher in TC.Rag than TC spleens due to the absence of lymphocytes in the former. However, the total number of DCs was lower in TC.Rag mice that do not develop splenomegaly as seen in TC mice (data not shown). The frequencies of splenic DCs producing IL-6 were equivalent between the two Rag-deficient strains, but were significantly higher than in TC mice (Fig. 7B). The absolute numbers of IL-6+ DCs were also higher in TC.Rag than in B6.Rag spleens. Not only TC.Rag spleens contain more IL-6+ DCs, but a greater percentage of TC.Rag splenic DCs expressed IL-6 in response to LPS or CpG stimulation in vitro (Fig. 7C). To better define the cytokine profile of DCs from TC mice, we compared gene expression and cytokine secretion by BMDCs from Rag-deficient and wild-type TC and B6 mice. As we have previously shown (24; 26), TC BMDCs expressed and secreted more IL-6 than B6 BMDCs (Fig. 7D). This was also the case for BMDCs of TC.Rag as compared to B6.Rag mice. TC and TC.Rag BMDCs also expressed and secreted more IL-1β and IL-33 than their B6 counterparts (Fig. 7E and F). It should be noted that the differential gene expression of these pro-inflammatory cytokines was obtained in unstimulated BMDCs, and it was amplified by LPS stimulation Consistent with the expansion of IFNγ+ CD4+ T cells in TC mice, TC BMDCs secreted more IL-12 than B6 BMDCs after LPS stimulation, although there was only a trend at the message level (Fig. 7G). Finally, TC.Rag BMDCs expressed higher levels of Tnfa and lower levels of Il10 transcripts than those from B6.Rag mice at 6 h after stimulation with LPS (Fig. 7H). Overall, these results demonstrate that the expression of the Sle loci In TC mice resulted in an expansion of peripheral DCs as well as their skewing toward expression of multiple inflammatory cytokines.
Figure 7. TC DCs present an intrinsic inflammatory phenotype.

Frequencies and numbers of total splenic DCs (A) and IL6+ DCs (B) in B6.Rag and TC.Rag mice, as compared to age-matched TC mice. C. IL-6 intracellular staining in splenic DCs from B6.Rag (black) and TC.Rag (white) 24 h after LPS or CpG stimulation. Cytokine gene expression in unstimulated, and secretion in supernatant by BMDCs from the four indicated strain before and after stimulation with LPS: IL-6 (D); IL-1b (E), and IL-33 (F). G. Il12b gene expression and protein expression in supernatants of BMDCs stimulated with LPS for 6 h (qRT-PCR) and 24 h (ELISA). H. Tnfa, and Il10 gene expression in BMDCs, without stimulation for Tnfa, and 6 h after LPS stimulation for Il10. Gene expression was normalized to the B6 mean values. # p < 0.05 one-tailed t test; * p < 0.05, ** p < 0.01, *** p < 0.001. n = 4–6 per strain.
Overlapping but distinct CD4+ T cell activation by TC B cells and myeloid cells
Our experimental design for adoptive transfers cannot evaluate the effect of B cells in the absence of myeloid and stromal cells, but we can compare the effects of TC or B6 B cells added to normal B6 myeloid/stromal cells to the effects of B6 or TC myeloid/stromal cells alone (Fig. 8). The addition of B cells of either strain resulted in a greater expansion of total CD4+ T cells both in the spleen and the kidney (Fig. 8A), and Treg cells (Fig. 8B), with the lowest frequency of Treg cells being observed in TC.Rag recipients (i.e. TC myeloid cells no B cells). This latter result is an agreement with our in vitro assay showing that TC B2 cells promote Treg cell polarization (Fig. 1). We have previously shown in vitro that both TC B cells and DCs contributed to TC Treg cell dysfunction linked to their IL-6 production (24; 25). However, the side-by-side comparison of these in vivo results suggests that DCs from TC mice play a greater role than B cells in inducing Treg dysfunction. The frequency of splenic IL-21+ Tfh cells was reduced by the presence of B6 but not TC B cells (Fig. 8C). However, B cells from either strain resulted in a higher number of Tfh cells in the spleen and kidneys, and a higher frequency of Tfh cells in the kidneys (Fig. 8C). Since both lupus B cells and DCs produce IL-6, the differential effect may reside in other cytokines such as IFNγ as suggested in Sle1b mice (9), and/or specific co-stimulatory pathways that need to be characterized. As for Tfh cells, the frequency of Th1 cells was reduced by the presence of B6 but not TC B cells in the spleen and by B cells from either strain in the kidneys (Fig. 8D). We have shown that DCs from TC mice produce a similar amount of IL-12 to B6 DCs, which is still higher than the amount of IL-12 produced by B cells, possibly explaining our results. The number of Th1 cells, however, was enhanced by B cells as for the other CD4+ T cell subsets. Finally, myeloid/stromal cells expanded the frequency of Th17 cells significantly more than B cells in both strains, and the numbers of Th17 cells in the kidneys were significantly higher in the absence of B cells (Fig. 8E). Interestingly, the highest numbers of Th17 cells (which are quite low as compared to other T cell subsets) was observed in the kidneys of TC.Rag recipients. Overall, these results showed that both TC B cells and DCs promoted inflammatory phenotypes in normal CD4+ T cells, but these two cell types showed notable differences, with TC B cells promoting greater T cell numbers and Tfh cell expansion, while TC DCs enhanced Th1 and Th17 cell polarization.
Figure 8. Comparison of the effects of B cells and DCs of CD4+ T cells phenotypes in vivo.

CD4+ T cells from B6.Il21VFP mice into B6.Rag or TC.Rag mice with or without B6 or TC B cells. For simplicity, the strain background of the recipient mice is indicated by the strain of origin of the recipient DCs. A. Numbers of total CD4+ T cells in spleens and kidneys. B. Frequencies and numbers of splenic Treg cells. Frequencies and numbers of splenic and kidney IL-21+ CD4+ T cells (C), IFNγ+ CD4+ T cells (D) and IL-17A+ CD4+ T cells (E). Data represents n = 5 mice, expressed as means ± SEM. * P < 0.05, ** P < 0.01, *** P < 0.001.
Discussion
In this study, we used the TC congenic mouse model of lupus to dissect the roles of specific cell types, here B cells and myeloid/stromal cells with a focus on DCs, on the activation of CD4+ T cells. We used non-autoimmune T cells and showed that both B cells and myeloid/stromal cells contribute independently to their expansion, activation and polarization toward inflammatory phenotypes that are associated with lupus pathogenesis. To examine the ability of B cells to support CD4+ T cell polarization in vitro, we used an MHC-class II mismatch as the source of CD4+ T cell stimulation in lieu of traditional anti-CD3/CD28 stimulation. With this allogenic assay, we confirmed results previously described by the Rothstein group in a fully allogeneic assay, in which pB1a cells polarized CD4+ T cells toward Th17 and Th1 phenotypes and sB2 cells polarized CD4+ T cells toward a Treg phenotype (18). B-1a cells are expanded in several mouse models of lupus (13) and SLE patients (14). In lupus-prone mice, B-1a cells are found not only in their expected location in the peritoneal and pleural cavities, but also in inflamed tissues, such as the kidneys and the thymus (46), in a CXCL13/CXCR5-dependent manner (47). Moreover, there is evidence that CD4+ T cells migrate to the peritoneal cavity where they encounter B-1a cells (17). In addition, B-1a cells from the lupus-prone NZM2410 mouse express high levels of co-stimulatory molecules and have high APC functions (48). Finally, B-1a cells from lupus mice have a different receptor repertoire (49), and they are enriched for a sub-population expressing PD-L2 that is more autoreactive (50), but whose function is currently unknown (51). Overall, given their ability to expand pro-inflammatory T cells, their number, location in inflamed tissues, autoreactive repertoire and high co-stimulatory activity, it is most likely that lupus B-1a cells contribute to the activation of autoreactive T cells, therefore participating in the multicellular feedforward activation loop that sustains lupus pathogenesis (52). In support of this hypothesis, we found that the expression of the Sle2c1 locus, which is responsible for the B-1a cell expansion in the NZM2410 model, increases Th17 cell polarization and promotes nephritis in Fas-deficient mice (35). Conversely, the expanded B1-a cells in SLE patients induced a greater in vitro proliferation of allogeneic CD4+ T cells than B1-a cells from healthy controls (14).
Polarization to Treg or Th1 cells was not impacted by the strain of origin of either pB1a or sB2 cells. However, both pB1a cells and sB2 cells from TC mice expanded Th17 cells to a greater extent than their B6 counterparts. Furthermore, this phenotype mapped to the Sle1 locus as does the ability of B cells to produce IL-6. Th17 cell expansion by TC B cells also involved CD44 and CD86, as was previously shown for pB1a cells in an allogenic assay (19). In our assay, CD44 expression on CD4+ T cells was not a variable since all T cells were from a normal B6 source. However, CD44 expression is increased on lupus CD4+ T cells, including in TC mice (41; 53), and CD44 expression on T cells also contributes to Th17 cell expansion (19), providing an amplification loop through B-T cell interactions. However, it needs mentioned that Th17 cells represent a very small subset of CD4+ T cells in TC mice in comparison to the robust numbers and frequencies of Th1 and Tfh cells that are found in lupus mice and patients (54–57). We cannot exclude the possibility that this minor subset contributes to autoimmune disease in our mouse model. Using a transfer system of normal and lupus B cells along with normal T cells in lymphopenic mice, we showed that lupus B cells favors the homeostatic expansion of total CD4+ T cells, with a skewing toward Tfh and Th1 cells and a relative decrease of Treg cells. The CD4+ T cell phenotypes induced by TC sB2 cells were largely abrogated by IL-6 blockade. Our results confirm the recent identification of IL-6 production as a critical mechanism by which self-reactive B cells induce GC formation and Tfh responses using another model of systemic autoimmunity (58). Our results demonstrate that lupus B cells play a significant role in the pathological skewing of lupus CD4+ T cells, in part through the production of IL-6.
Using the same model of homeostatic expansion, we showed that the non-lymphoid TC cells also confer an inflammatory phenotype to normal CD4+ T cells. Within these non-lymphoid cells, we focused on DCs, which are known to be major regulators of T cell effector functions. Our study cannot rule out that other TC BM-derived cells also regulate T cells. Stromal cells from TC.Rag mice could also in principle modulate T cells through the production of chemokines and cytokines. Indeed, hematopoietic cells other than professional APCs as well as non-hematopoietic cells can modulate CD4+ T cells responses not only through the production of cytokines and chemokines, but also through non-classical antigen presentation (59). Our previous work with individual Sle loci has shown, however, that their immune phenotypes were entirely supported by BM-derived cells (27; 39; 60–63), making it unlikely that stromal cells play a major role in modulating the T cell inflammatory phenotypes in TC mice. Here we showed that in TC mice, DCs exhibited an intrinsic inflammatory phenotype in the absence of lymphocytes, and importantly, in the absence of autoantibodies. DCs in this lupus model, and most likely in other related models, present an interferon signature that precedes autoimmune manifestations (45). Our results go one step further by showing that autoantibody immune complexes are not required for TC DCs to produce inflammatory cytokines. We have previously shown that Sle3 expression affects B and T cells functions through myeloid cells (39). It is therefore possible that Sle3 is at least partially responsible for the phenotypes that we have observed in CD4+ T cells transferred into TC.Rag mice.
Overall our findings show that cell intrinsic defects in B cells and DCs contribute independently to the polarization of CD4+ T cells toward inflammatory phenotypes in the TC model of lupus. Although it is well documented that both DCs and B cells support Tfh cell development and maintenance (64), our results suggest that lupus B contribute disproportionally to their pathological expansion. A distinctive cytokine response to stimulation of the same TLR ligands has been shown for normal B cells and DCs (65), and it is likely that such differences are consequential in lupus. This disorder is historically viewed as a B cell-dependent humoral autoimmune disease, but it has been appreciated that non-antibody functions of B cells may play a critical role (66). Our results document the ability of lupus B cells to drive the expansion of inflammatory T cells that drive the expansion of B cells through which pathogenic autoAbs are ultimately produced.
Supplementary Material
Acknowledgments
We thank Leilani Zeumer and Nathalie Kanda for outstanding technical help.
Abbreviations
- Ab
antibody
- Ag
antigen
- Bm12
B6.C-H2bm12/KhEg
- BM
bone-marrow
- B6
C57BL/6J
- FO
follicular
- MZ
marginal zone
- pB1a
peritoneal B1-a cells
- sB2
splenic conventional B cells
- SLE
systemic lupus erythematosus
- TC
B6.NZM2410.Sle1.Sle2.Sle3
- Tfh
follicular helper T cells
- Treg
Foxp3 regulatory CD4+ T cells
- VFP
Venus-fluorescent protein
Footnotes
Author Contributions
Author contribution: ZX, SCC, HY and LM designed experiments; ZX, SCC, WL, and HY carried out experiments; ZX, SCC, HY and LM analyzed data; DR and HCM produced the B6.IL-21-VFP mice; and SCC, DR, HCM and LM wrote the paper.
Conflict of Interest
The authors declare no conflict of interest.
References
- 1.Mariño E, Grey ST. B cells as effectors and regulators of autoimmunity. Autoimmunity. 2012;45:377–387. doi: 10.3109/08916934.2012.665527. [DOI] [PubMed] [Google Scholar]
- 2.Vinuesa CG, Sanz I, Cook MC. Dysregulation of germinal centres in autoimmune disease. Nat Rev Immunol. 2009;9:845–857. doi: 10.1038/nri2637. [DOI] [PubMed] [Google Scholar]
- 3.Craft JE. Follicular helper T cells in immunity and systemic autoimmunity. Nat Rev Rheumatol. 2012;8:337–347. doi: 10.1038/nrrheum.2012.58. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Blanco P, Ueno H, Schmitt N. T follicular helper (Tfh) cells in lupus: Activation and involvement in SLE pathogenesis. Eur J Immunol. 2016;46:281–290. doi: 10.1002/eji.201545760. [DOI] [PubMed] [Google Scholar]
- 5.Sozzani S, Del Prete A, Bosisio D. Dendritic cell recruitment and activation in autoimmunity. J Autoimmun. 2017;85:126–140. doi: 10.1016/j.jaut.2017.07.012. [DOI] [PubMed] [Google Scholar]
- 6.Teichmann LL, Ols ML, Kashgarian M, Reizis B, Kaplan DH, Shlomchik MJ. Dendritic cells in lupus are not required for activation of T and B cells but promote their expansion, resulting in tissue damage. Immunity. 2010;33:967–978. doi: 10.1016/j.immuni.2010.11.025. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Teichmann LL, Schenten D, Medzhitov R, Kashgarian M, Shlomchik MJ. Signals via the adaptor MyD88 in B cells and DCs make distinct and synergistic contributions to immune activation and tissue damage in lupus. Immunity. 2013;38:528–540. doi: 10.1016/j.immuni.2012.11.017. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Christensen SR, Shupe J, Nickerson K, Kashgarian M, Flavell RA, Shlomchik MJ. Toll-like receptor 7 and TLR9 dictate autoantibody specificity and have opposing inflammatory and regulatory roles in a murine model of lupus. Immunity. 2006;25:417–428. doi: 10.1016/j.immuni.2006.07.013. [DOI] [PubMed] [Google Scholar]
- 9.Domeier PP, Chodisetti SB, Soni C, Schell SL, Elias MJ, Wong EB, Cooper TK, Kitamura D, Rahman ZSM. IFN-γ receptor and STAT1 signaling in B cells are central to spontaneous germinal center formation and autoimmunity. J Exp Med. 2016;213:715–732. doi: 10.1084/jem.20151722. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Allman D, Pillai S. Peripheral B cell subsets. Curr Opin Immunol. 2008;20:149–157. doi: 10.1016/j.coi.2008.03.014. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Sang A, Zheng YY, Morel L. Contributions of B cells to lupus pathogenesis. Mol Immunol. 2013;62:329. doi: 10.1016/j.molimm.2013.11.013. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Anolik JH. B cell biology: implications for treatment of systemic lupus erythematosus. Lupus. 2013;22:342–349. doi: 10.1177/0961203312471576. [DOI] [PubMed] [Google Scholar]
- 13.Duan B, Morel L. Role of B-1a cells in autoimmunity. Autoimmun Rev. 2006;5:403–408. doi: 10.1016/j.autrev.2005.10.007. [DOI] [PubMed] [Google Scholar]
- 14.Griffin DO, Rothstein TL. A small CD11b(+) human B1 cell subpopulation stimulates T cells and is expanded in lupus. J Exp Med. 2011;208:2591–2598. doi: 10.1084/jem.20110978. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Berland R, Wortis HH. Origins and functions of B-1 cells with notes on the role of CD5. Ann Rev Immunol. 2002;20:253–300. doi: 10.1146/annurev.immunol.20.100301.064833. [DOI] [PubMed] [Google Scholar]
- 16.Zimecki M, Kapp JA. Presentation of antigen by B cell subsets. II. The role of CD5 B cells in the presentation of antigen to antigen-specific T cells. Arch Immunol Ther Exp (Warsz) 1994;42:349–353. [PubMed] [Google Scholar]
- 17.Margry B, Wieland WH, van Kooten PJ, van Eden W, Broere F. Peritoneal cavity B-1a cells promote peripheral CD4+ T-cell activation. Eur J Immunol. 2013;43:2317–2326. doi: 10.1002/eji.201343418. [DOI] [PubMed] [Google Scholar]
- 18.Zhong X, Gao W, Degauque N, Bai C, Lu Y, Kenny J, Oukka M, Strom TB, Rothstein TL. Reciprocal generation of Th1/Th17 and T(reg) cells by B1 and B2 B cells. Eur J Immunol. 2007;37:2400–2404. doi: 10.1002/eji.200737296. [DOI] [PubMed] [Google Scholar]
- 19.Wang Y, Rothstein TL. Induction of Th17 cell differentiation by B-1 cells. Front Immunol. 2012;3:281. doi: 10.3389/fimmu.2012.00281. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Wang X, Ma K, Chen M, Ko KH, Zheng BJ, Lu L. IL-17A promotes pulmonary B-1a cell differentiation via induction of blimp-1 expression during influenza virus infection. PLoS Pathog. 2016;12:e1005367. doi: 10.1371/journal.ppat.1005367. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Morel L. Mapping lupus susceptibility genes in the NZM2410 mouse model. Adv Immunol. 2012;115:113–139. doi: 10.1016/B978-0-12-394299-9.00004-7. [DOI] [PubMed] [Google Scholar]
- 22.Mohan C, Yu Y, Morel L, Yang P, Wakeland EK. Genetic dissection of Sle pathogenesis: Sle3 on murine chromosome 7 impacts T cell activation, differentiation, and cell death. J Immunol. 1999;162:6492–6502. [PubMed] [Google Scholar]
- 23.Chen Y, Cuda C, Morel L. Genetic determination of T cell help in loss of tolerance to nuclear antigens. J Immunol. 2005;174:7692–7702. doi: 10.4049/jimmunol.174.12.7692. [DOI] [PubMed] [Google Scholar]
- 24.Wan S, Xia C, Morel L. IL-6 produced by dendritic cells from lupus-prone mice inhibits CD4+CD25+ T cell regulatory functions. J Immunol. 2007;178:271–279. doi: 10.4049/jimmunol.178.1.271. [DOI] [PubMed] [Google Scholar]
- 25.Wan S, Zhou Z, Duan B, Morel L. Dendritic cells from lupus prone mice directly interact with B cells to increase their effector functions. Arthritis Rheum. 2008;58:1741–1750. doi: 10.1002/art.23515. [DOI] [PubMed] [Google Scholar]
- 26.Sang A, Zheng YY, Yin Y, Dozmorov I, Li H, Hsu HC, Mountz JD, Morel L. Dysregulated cytokine production by dendritic cells modulates B cell responses in the NZM2410 mouse model of lupus. PLoS One. 2014;9:e102151. doi: 10.1371/journal.pone.0102151. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Sobel ES, Satoh M, Chen WF, Wakeland EK, Morel L. The major murine systemic lupus erythematosus susceptibility locus Sle1 results in abnormal functions of both B and T cells. J Immunol. 2002;169:2694–2700. doi: 10.4049/jimmunol.169.5.2694. [DOI] [PubMed] [Google Scholar]
- 28.Xu Z, Butfiloski EJ, Sobel ES, Morel L. Mechanisms of peritoneal B-1a cells accumulation induced by murine lupus susceptibility locus Sle2. J Immunol. 2004;173:6050–6058. doi: 10.4049/jimmunol.173.10.6050. [DOI] [PubMed] [Google Scholar]
- 29.Morel L, Mohan C, Yu Y, Croker BP, Tian N, Deng A, Wakeland EK. Functional dissection of systemic lupus erythematosus using congenic mouse strains. J Immunol. 1997;158:6019–6028. [PubMed] [Google Scholar]
- 30.Morel L, Croker BP, Blenman KR, Mohan C, Huang G, Gilkeson G, Wakeland EK. Genetic reconstitution of systemic lupus erythematosus immunopathology with polycongenic murine strains. Proc Natl Acad Sci USA. 2000;97:6670–6675. doi: 10.1073/pnas.97.12.6670. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Sang A, Niu H, Cullen J, Choi SC, Zheng YY, Wang H, Shlomchik MJ, Morel L. Activation of rheumatoid factor-specific B cells is antigen dependent and occurs preferentially outside of germinal centers in the lupus-prone NZM2410 mouse model. J Immunol. 2014;193:1609–1621. doi: 10.4049/jimmunol.1303000. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Marnik EA, Wang X, Sproule TJ, Park G, Christianson GJ, Lane-Reticker SK, Jain S, Duffy T, Wang H, Carter GW, Morse HC, 3rd, Roopenian DC. Precocious interleukin 21 expression in naive mice Identifies a natural helper cell population in autoimmune disease. Cell Rep. 2017;21:208–221. doi: 10.1016/j.celrep.2017.09.036. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Xu Z, Potula HH, Vallurupalli A, Perry D, Baker H, Croker BP, Dozmorov I, Morel L. Cyclin-dependent kinase inhibitor Cdkn2c regulates B cell homeostasis and function in the NZM2410-derived murine lupus susceptibility locus Sle2c1. J Immunol. 2011;186:6673–6682. doi: 10.4049/jimmunol.1002544. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Lu S, Zeumer L, Sorensen H, Yang H, Ng Y, Yu F, Riva A, Croker B, Wallet S, Morel L. The murine Pbx1-d lupus susceptibility allele accelerates mesenchymal stem cell differentiation and impairs their immunosuppressive function. J Immunol. 2015;194:43–55. doi: 10.4049/jimmunol.1401851. [DOI] [PubMed] [Google Scholar]
- 35.Xu Z, Cuda CM, Croker BP, Morel L. The NZM2410-derived lupus susceptibility locus Sle2c1 increases TH17 polarization and induces nephritis in Fas-deficient mice. Arthritis Rheum. 2011;63:764–774. doi: 10.1002/art.30146. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Zhong X, Gao W, Degauque N, Bai C, Lu Y, Kenny J, Oukka M, Strom TB, Rothstein TL. Reciprocal generation of Th1/Th17 and T(reg) cells by B1 and B2 B cells. Eur J Immunol. 2007;37:2400–2404. doi: 10.1002/eji.200737296. [DOI] [PubMed] [Google Scholar]
- 37.Zhong X, Lau S, Bai C, Degauque N, Holodick NE, Steven SJ, Tumang J, Gao W, Rothstein TL. A novel subpopulation of B-1 cells is enriched with autoreactivity in normal and lupus-prone mice. Arthritis Rheum. 2009;60:3734–3743. doi: 10.1002/art.25015. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Greyner HJ, Wiraszka T, Zhang LS, Petroll WM, Mummert ME. Inducible macropinocytosis of hyaluronan in B16-F10 melanoma cells. Matrix Biol. 2010;29:503–510. doi: 10.1016/j.matbio.2010.06.004. [DOI] [PubMed] [Google Scholar]
- 39.Sobel ES, Morel L, Baert R, Mohan C, Schiffenbauer J, Wakeland EK. Genetic dissection of systemic lupus erythematosus pathogenesis: Evidence for functional expression of Sle3/5 by non-T cells. J Immunol. 2002;169:4025–4032. doi: 10.4049/jimmunol.169.7.4025. [DOI] [PubMed] [Google Scholar]
- 40.Xu Z, Duan B, Croker BP, Morel L. STAT4 deficiency reduces autoantibody production and glomerulonephritis in a mouse model of lupus. Clin Immunol. 2006;120:189–198. doi: 10.1016/j.clim.2006.03.009. [DOI] [PubMed] [Google Scholar]
- 41.Yin Y, Choi SC, Xu Z, Perry DJ, Seay H, Croker BP, Sobel ES, Brusko TM, Morel L. Normalization of CD4+ T cell metabolism reverses lupus. Sci Transl Med. 2015;7:274ra218. doi: 10.1126/scitranslmed.aaa0835. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42.Dienz O, Eaton SM, Bond JP, Neveu W, Moquin D, Noubade R, Briso EM, Charland C, Leonard WJ, Ciliberto G, Teuscher C, Haynes L, Rincon M. The induction of antibody production by IL-6 is indirectly mediated by IL-21 produced by CD4+ T cells. J Exp Med. 2009;206:69–78. doi: 10.1084/jem.20081571. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43.Eddahri F, Denanglaire S, Bureau F, Spolski R, Leonard WJ, Leo O, Andris F. Interleukin-6/STAT3 signaling regulates the ability of naive T cells to acquire B-cell help capacities. Blood. 2009;113:2426–2433. doi: 10.1182/blood-2008-04-154682. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44.Diehl SA, Schmidlin H, Nagasawa M, Blom B, Spits H. IL-6 triggers IL-21 production by human CD4+ T cells to drive STAT3-dependent plasma cell differentiation in B cells. Immunol Cell Biol. 2012;90:802–811. doi: 10.1038/icb.2012.17. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45.Sriram U, Varghese L, Bennett HL, Jog NR, Shivers DK, Ning Y, Behrens EM, Caricchio R, Gallucci S. Myeloid dendritic cells from B6.NZM Sle1/Sle2/Sle3 lupus-prone mice express an IFN signature that precedes disease onset. J Immunol. 2012;189:80–91. doi: 10.4049/jimmunol.1101686. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46.Morshed SRM, Mannoor K, Halder RC, Kawamura H, Bannai M, Sekikawa H, Watanabe H, Abo T. Tissue-specific expansion of NKT and CD5+B cells at the onset of autoimmune disease in (NZB×NZW)F1 mice. Eur J Immunol. 2002;32:2551–2561. doi: 10.1002/1521-4141(200209)32:9<2551::AID-IMMU2551>3.0.CO;2-C. [DOI] [PubMed] [Google Scholar]
- 47.Ishikawa S, Matsushima K. Aberrant B1 cell trafficking in a murine model for lupus. Front Biosci. 2007;12:1790–1803. doi: 10.2741/2188. [DOI] [PubMed] [Google Scholar]
- 48.Mohan C, Morel L, Yang P, Wakeland EK. Accumulation of splenic B1a cells with potent antigen-presenting capability in NZM2410 lupus-prone mice. Arthritis Rheum. 1998;41:1652–1662. doi: 10.1002/1529-0131(199809)41:9<1652::AID-ART17>3.0.CO;2-W. [DOI] [PubMed] [Google Scholar]
- 49.Holodick NE, Zeumer L, Rothstein TL, Morel L. Expansion of B-1a cells with germline heavy chain sequence in lupus mice. Front Immunol. 2016;7:108. doi: 10.3389/fimmu.2016.00108. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 50.Zhong X, Lau S, Bai C, Degauque N, Holodick NE, Steven SJ, Tumang J, Gao W, Rothstein TL. A novel subpopulation of B-1 cells is enriched with autoreactivity in normal and lupus-prone mice. Arthritis Rheum. 2009;60:3734–3743. doi: 10.1002/art.25015. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 51.Lee RA, Mao C, Vo H, Gao W, Zhong X. Fluorescence tagging and inducible depletion of PD-L2-expressing B-1 B cells in vivo. Ann N Y Acad Sci. 2015;1362:77–85. doi: 10.1111/nyas.12865. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 52.Liu Z, Davidson A. Taming lupus-a new understanding of pathogenesis is leading to clinical advances. Nat Med. 2012;18:871–882. doi: 10.1038/nm.2752. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 53.Niu Y, Sengupta M, Titov AA, Choi SC, Morel L. The PBX1 lupus susceptibility gene regulates CD44 expression. Molecular Immunology. 2017;85:148–154. doi: 10.1016/j.molimm.2017.02.016. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 54.Pollard KM, Cauvi DM, Toomey CB, Morris KV, Kono DH. Interferon-gamma and systemic autoimmunity. Discov Med. 2013;16:123–131. [PMC free article] [PubMed] [Google Scholar]
- 55.Munroe ME, Lu R, Zhao YD, Fife DA, Robertson JM, Guthridge JM, Niewold TB, Tsokos GC, Keith MP, Harley JB, James JA. Altered type II interferon precedes autoantibody accrual and elevated type I interferon activity prior to systemic lupus erythematosus classification. Ann Rheum Dis. 2016;75:2014–2021. doi: 10.1136/annrheumdis-2015-208140. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 56.Choi JY, Ho JH, Pasoto SG, Bunin V, Kim ST, Carrasco S, Borba EF, Goncalves CR, Costa PR, Kallas EG, Bonfa E, Craft J. Circulating follicular helper-like T cells in systemic lupus erythematosus: association with disease activity. Arthritis Rheumatol. 2015;67:988–999. doi: 10.1002/art.39020. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 57.Choi JY, Seth A, Kashgarian M, Terrillon S, Fung E, Huang L, Wang LC, Craft J. Disruption of pathogenic cellular networks by IL-21 blockade leads to disease amelioration in murine lupus. J Immunol. 2017;198:2578–2588. doi: 10.4049/jimmunol.1601687. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 58.Arkatkar T, Du SW, Jacobs HM, Dam EM, Hou B, Buckner JH, Rawlings DJ, Jackson SW. B cell-derived IL-6 initiates spontaneous germinal center formation during systemic autoimmunity. J Exp Med. 2017;214:3207–3217. doi: 10.1084/jem.20170580. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 59.Kambayashi T, Laufer TM. Atypical MHC class II-expressing antigen-presenting cells: can anything replace a dendritic cell? Nat Rev Immunol. 2014;14:719–730. doi: 10.1038/nri3754. [DOI] [PubMed] [Google Scholar]
- 60.Sobel ES, Mohan C, Morel L, Schiffenbauer J, Wakeland EK. Genetic dissection of SLE pathogenesis: Adoptive transfer of Sle1 mediates the loss of tolerance by bone marrow-derived B cells. J Immunol. 1999;162:2415–2421. [PubMed] [Google Scholar]
- 61.Cuda CM, Wan S, Sobel ES, Croker BP, Morel L. Murine lupus susceptibility locus Sle1a controls regulatory T cell number and function through multiple mechanisms. J Immunol. 2007;179:7439–7447. doi: 10.4049/jimmunol.179.11.7439. [DOI] [PubMed] [Google Scholar]
- 62.Perry DJ, Yin Y, Telarico T, Baker HV, Dozmorov I, Perl A, Morel L. Murine lupus susceptibility locus Sle1c2 mediates CD4+ T cell activation and maps to estrogen-related receptor gamma. J Immunol. 2012;189:793–803. doi: 10.4049/jimmunol.1200411. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 63.Choi SC, Hutchinson TE, Titov AA, Seay HR, Li S, Brusko TM, Croker BP, Salek-Ardakani S, Morel L. The lupus susceptibility gene Pbx1 regulates the balance between follicular helper T cell and regulatory T cell differentiation. J Immunol. 2016;197:458–469. doi: 10.4049/jimmunol.1502283. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 64.Crotty S. T follicular helper cell differentiation, function, and roles in disease. Immunity. 2014;41:529–542. doi: 10.1016/j.immuni.2014.10.004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 65.Barr TA, Brown S, Ryan G, Zhao J, Gray D. TLR-mediated stimulation of APC: Distinct cytokine responses of B cells and dendritic cells. Eur J Immunol. 2007;37:3040–3053. doi: 10.1002/eji.200636483. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 66.Pisetsky DS, Grammer AC, Ning TC, Lipsky PE. Are autoantibodies the targets of B-cell-directed therapy? Nat Rev Rheumatol. 2011;7:551–556. doi: 10.1038/nrrheum.2011.108. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.