

Abstract
Aims
c-jun N-terminal kinase (JNK) is a critical stress response kinase that activates in a wide range of physiological and pathological cellular processes. We recently discovered a pivotal role of JNK in the development of atrial arrhythmias in the aged heart, while cardiac CaMKIIδ, another pro-arrhythmic molecule, was also known to enhance atrial arrhythmogenicity. Here, we aimed to reveal a regulatory role of the stress kinase JNK2 isoform on CaMKIIδ expression.
Methods and results
Activated JNK2 leads to increased CaMKIIδ protein expression in aged human and mouse atria, evidenced from the reversal of CaMKIIδ up-regulation in JNK2 inhibitor treated wild-type aged mice. This JNK2 action in CaMKIIδ expression was further confirmed in HL-1 myocytes co-infected with AdMKK7D-JNK2, but not when co-infected with AdMKK7D-JNK1. JNK2-specific inhibition (either by a JNK2 inhibitor or overexpression of inactivated dominant-negative JNK2 (JNK2dn) completely attenuated JNK activator anisomycin-induced CaMKIIδ up-regulation in HL-1 myocytes, whereas overexpression of JNK1dn did not. Moreover, up-regulated CaMKIIδ mRNA along with substantially increased phosphorylation of JNK downstream transcription factor c-jun [but not activating transcription factor2 (ATF2)] were exhibited in both aged atria (humans and mice) and transiently JNK activated HL-1 myocytes. Cross-linked chromatin-immunoprecipitation assays (XChIP) revealed that both c-jun and ATF2 were bound to the CaMKIIδ promoter, but significantly increased binding of c-jun only occurred in the presence of anisomycin and JNK inhibition alleviated this anisomycin-elevated c-jun binding. Mutated CaMKII consensus c-jun binding sites impaired its promoter activity. Enhanced transcriptional activity of CaMKIIδ by anisomycin was also completely reversed to the baseline by either JNK2 siRNA or c-jun siRNA knockdown.
Conclusion
JNK2 activation up-regulates CaMKIIδ expression in the aged atrium. This JNK2 regulation in CaMKIIδ expression occurs at the transcription level through the JNK downstream transcription factor c-jun. The discovery of this novel molecular mechanism of JNK2-regulated CaMKII expression sheds new light on possible anti-arrhythmia drug development.
Keywords: c-Jun N-terminal kinase, Calcium/camodulin kinase II, c-Jun transcription factor, Atrial fibrillation
1. Introduction
The c-jun N-terminal kinases (JNKs) are serine/threonine protein kinases and belong to an important family of mitogen-activated protein kinases.1 JNK is activated in response to stress challenges, and is critical in the development of cancers, diabetes, and cardiovascular diseases (CVDs; e.g. myocardial infarction, atherosclerosis, heart failure).1–5 Our laboratory recently reported for the first time6,7 that activated JNK plays an important role in the development of atrial fibrillation (AF), the most common arrhythmia in the aging population.8–10 Although aging is a major risk factor for AF development, there are no clear age-related cell signalling pathways known to promote the association between age and AF.
Accumulating studies suggest a crucial role of CaMKII in calcium (Ca2+) mishandling triggered AF.11–14 CaMKIIδ is a well-known cardiac pro-arrhythmic molecule that phosphorylates Ca2+ handling proteins including phospholamban, Ca2+ releasing ryanodine receptors (RyRs), inositol 1, 4, 5-trisphosphate receptors (IP3R), and L-type Ca2+ channels, thereby playing a crucial role in the excitation-contraction coupling in the normal heart and enhanced arrhythmogenicity in the pathologically remodelled heart.15–19 Thus, CaMKIIδ inhibition has been considered as a potential anti-arrhythmic intervention.20–22 However the outcomes have not been as optimal as expected due to some off-target effects.23 Understanding how CaMKIIδ expression is regulated will greatly aid in developing novel anti-arrhythmic drugs to inhibit CaMKIIδ.
Here we discovered that JNK activation is concurrently linked to significantly increased CaMKIIδ expression in the human atrium with increasing age. We further demonstrated that this JNK enhanced CaMKIIδ expression is a JNK2 isoform-specific action. By using silico analysis and a unique cross-linked chromatin-immunoprecipitation (XChIP) assay, we revealed for the first time that JNK downstream transcription factors c-jun and activating transcription factor2 (ATF2) are bound to the CaMKIIδ proximal promoter region. We further discovered that increased binding of c-jun to the CaMKII promoter leads to elevated promoter activity by using either JNK ablation or c-jun knockdown. Our findings reveal a previously unrecognized molecular mechanism of JNK2 regulated CaMKIIδ expression that underlies the pathogenesis of atrial arrhythmias in the elderly. Our results provide an important basis for modulating JNK2 activity as a potential novel therapeutic approach for AF.
2. Methods
2.1 Human atrial specimens
Human atrial tissues were obtained from 11 human donor hearts provided by Illinois Gift of Hope Organ and Tissue Donor Network (GOH). These hearts were not used for heart transplantation but had no history of AF and/or major CVDs. Table 1 shows de-identified data (age, gender, race, etc.) from the donors. The studies were approved by the Human Study Committees of Loyola University Chicago, Rush University Medical Center, and Illinois GOH. Consent was obtained by GOH from the donors’ families for use of donor hearts for research. These human heart studies were performed to conform to the Declaration of Helsinki.
Table 1.
De-identified donor information
| Age | Gdr | Eth | HW/ BW | Afib | VD | CAD | HLD | DM | MI | HF | HTN | Meds |
|---|---|---|---|---|---|---|---|---|---|---|---|---|
| 26 | M | Cau | 4.76 | − | − | − | − | − | − | − | − | − |
| 26 | F | Cau | 5.11 | − | − | − | − | − | − | − | − | − |
| 29 | M | Cau | 3.63 | − | − | − | − | − | − | − | − | − |
| 47 | F | Cau | 5.16 | − | − | − | − | − | − | − | − | − |
| 52 | F | AA | 3.73 | − | − | − | − | − | − | − | − | − |
| 52 | F | Cau | 5.41 | − | − | + | − | − | − | − | + | − |
| 58 | F | Cau | 4.44 | − | − | − | − | − | − | − | + | − |
| 59 | F | Cau | 9.30 | − | − | − | − | − | − | − | − | − |
| 61 | M | Cau | 5.22 | − | − | − | − | − | − | − | − | − |
| 68 | M | Cau | 4.34 | − | − | − | − | − | − | − | − | − |
| 78 | F | Hisp | 8.53 | − | − | − | − | − | − | − | + | +a |
Gdr, gender; Eth, ethnicity; F, Female; M, Male; Cau, Caucasian; AA, African-American; Hisp, Hispanic; HW/BW, heart weight (g) to body weight (kg) ratio; Afib, Atrial Fibrillation; VD, Valve Disease; CAD, coronary artery disease; HLD, hyperlipidaemia; MI, myocardial infarction; HTN, hypertension; DM, Diabetes Mellitus; HF, heart failure; Meds, outpatient medications
Atenolol, hydrochlorothiazide, lisinopril.
2.2 Animal preparations
Wild-type (WT) C57B/6j mice (Jackson Laboratory, ME) at 26 months (aged) and 2–2.5 months (young) were studied. To assess the functional action of JNK on CaMKIIδ expression, we used 2-month old cardiac-specific inducible MKK7D (αMHC-cre) young transgenic mice (a generous gift from Dr. Yibin Wang, UCLA) that express cardiac-specific MKK7D to robustly activate JNK with tamoxifen treatment (50 mg/kg I.P. for 5 constitutive days.7,24 Mice were studied five days after the last dose of tamoxifen treatment. Mice were sedated via isoflurane inhalation (2% isoflurane delivered in 100% oxygen) and cardiac function was assessed using echocardiography. Left ventricular (LV) dimensions (from both short axis view and long axis four-chamber view) of mouse hearts were assessed using 2D echocardiography (vevo2100) as described previously in.6,15 Ejection fraction (EF) and fractional shortening (FS) were analysed as previously described.6,15 In addition, aged WT mice were treated in vivo with a JNK2 inhibitor JNK2I-IX25 (10 mg/kg, I.P. every other day for a total of 10 days). Mice were sacrificed under a surgical plane of anaesthesia induced with a mixture of 100 mg/kg ketamine and 4.5 mg/kg Xylazine (I.P.) for terminal studies. All animal studies followed the Guide for the Care and Use of Laboratory Animals (NIH Publication, 8th Edition, 2011) and were approved by the Institutional Animal Care and Use Committees of Loyola University Chicago.
2.3 Reagents and antibodies
JNK activator anisomycin and JNK-specific inhibitors SP600125 and JNK2I-IX were used as previously described.6,25,26 Antibodies recognizing JNK1, JNK2, phosphorylated total JNK (JNK-P) (Cell Signalling) and CaMKIIδ (Abcam) were used for immunoblotting assays. The antibodies against c-jun or ATF2 used for XChIP were from Cell Signalling and Abcam, respectively.
2.4 Plasmid construction
The sequence ∼1.2 kb upstream of the mouse CaMKIIδ transcriptional start codon was amplified by using PCR from a mouse genomic clone with primers harbouring KpnI and BglII sites and inserted into the luciferase reporter vector pGL3 at the corresponding restriction sites. The inserted sequences were verified by sequencing.
2.5 Culture and treatment of HL-1 myocytes
Mouse HL-1 myocytes were kindly provided by Dr William Claycomb (Louisiana State University) and cells were strictly maintained and passaged as previously described.6,27 On the fourth day after cell plating, cells were treated with and without JNK activator anisomycin6 (50 ng/ml; EMD) for 24 h and cells were harvested on day five. To activate JNK, cells were also co-infected with adenoviral (Ad) MKK7D (constitutively activated JNK upstream activator; from Dr Yibin Wang, UCLA) and JNK2 or JNK1 [Seven Hills Bioreagents (SHB)] for 48 h. To inhibit JNK, anisomycin-treated HL-1 atrial myocytes were pre-treated with JNK specific inhibitors SP600125 (2 µmol/l; SP, EMD, Millipore, USA)6 or JNK2 inhibitor (170 nmol/l; JNK2I-IX, EMD),25 or co-infected with AdMKK7D and AdJNK2dn (inactivated dominant negative JNK2, SHB) or AdMKK7D and AdJNK1dn (inactivated dominant negative JNK1, SHB).
2.6 Knockdown of JNK2 and c-jun with siRNAs
HEK293 cells were transfected with scrambled siRNA (a negative control), JNK2- or c-jun-specific siRNA at 20 nmol/L with Lipofectamine 2000 transfection reagent (Invitrogen). Six hours after the transfection, cells were treated with anismomycin for 24 h. The sequences of siRNAs were:
Scrambled siRNA, sense: CGUUAAUCGCGUAUAAUACGCGUAT
Anti-sense: AUACGCGUAUUAUACGCGAUUAACGAC
JNK2 siRNA, sense: ACAUUGUUGUGAAAUCAGACUGCAC
Anti-sense: GUGCAGUCUGAUUUCACAACAAUGUUG
c-jun siRNA, sense: GCAAUAGAGACUGUAGAUUGCUUCT
anti-sense: AGAAGCAAUCUACAGUCUCUAUUGCAG
2.7 Western blotting
The heart tissues from humans and mice were homogenized in RIPA buffer. Mouse atrial HL-1 cells were also lysed in RIPA. After clearing by centrifugation, lysates (20 µg) were subjected to Western blotting using specific antibodies. The proteins were visualized with ECL reagent (Thermo Scientific) and scanned with a Bio-Rad gel imaging system. The protein levels were quantified with the gel imager software and normalized with internal loading control glyceraldehyde 3-phosphate dehydrogenase (GAPDH).
2.8 RNA preparation and quantitative real time PCR
Total RNA was extracted with Trizol reagent (Thermo Fisher Scientific). The quantity and quality of the prepared RNA were determined by UV spectrometry. An aliquot of RNA (1 µg) was reverse-transcripted to cDNA. The amounts of mRNA were then detected by qPCR with specific primers and GoTaq SYBR Master Mix (Promega) using the CFX96 real-time PCR detection system (Bio-Rad), and normalized with GAPDH. The sequences of CaMKIIδ primers used for real time PCR were: Forward: GGCAGACTTCGGCTTAGCCATAG; Reverse: TCATCCCAGAAGGGTGGGTACC
2.9 Cross-link chromatin immunoprecipitation
The experiment was started 24 h after treatment of HL-1 cells with anisomycin as previously described in.6,7 In brief, cells were cross-linked with 1% formaldehyde for 10 min at room temperature, followed by 5 min incubation with 0.125 mol/l glycine. The cells were lysed in a SDS lysis buffer containing protease inhibitors (Sigma-Aldrich) followed by sonication. The supernatant of the cell lysates was incubated with 0.5 mg anti-c-jun antibody (Cell Signalling), 1 µg/ml anti-ATF2 antibody (Abcam) or rabbit IgG, respectively, and immunoprecipitated (IP-ed) with 20 µl protein A/G conjugated magnetic beads (EMD). The elutes containing bound DNAs from the c-jun or ATF2 immunoprecipitates were concentrated to 200 mmol/l with NaCl and incubated at 62°C for 2 h, then subjected to 95°C for 10 min. After purification, the IP-ed DNA was quantified by real-time PCR using the primers flanking the potential AP-1 binding site in the mouse CaMKIIδ proximal promoter region (sequences of primers, forward: CAGGCTAGGCACGCTCACGTGAC, reverse: GGGCGGTAGG AAGGTTGGCT) and the SYBR master mix (Promega). The quantitative amounts of DNA were normalized to the total input and presented as enrichment.
2.10 Promoter activity assay
Plasmid pGL3-CaMKIIδ-WT or CaMKIIδ-mu that contain either an inserted WT or mutated (replaced -716 T/-715 G to A/C) mouse CaMKIIδ promoter sequence upstream from the encoded Firefly luciferase were transfected into HEK293 cells together with a reference plasmid pTK-RL, which constitutively expresses Firefly luciferase. The following day, cells were incubated with anisomycin at 50 ng/ml for 24 h. Luciferase activity assay was measured by Pherastar (BMG LABTECH) using the Dual-Luciferase Reporter System (Promega) following the manufacturer’s instructions. The results were normalized with Renilla luciferase activity to correct the variation in transfection efficiency.
2.11 In vitro JNK activity assay
Mouse heart tissue was homogenized in a lysis buffer (mM: TrisHCl 40, NaCl 150, ß-glycerophosphate 5, NaF 10, Na3VO4 0.2, and protease inhibitor cocktail, pH 7.4). Lysates were incubated with 1 µg/ml anti-JNK1 or JNK2 antibody (Abcam) and 7.5 µl of protein G beads followed by kinase activity assay. In brief, the resultant IP was resuspended in the kinase reaction buffer in the presence of 20 µM ATP and 1 µg c-jun, and incubated at room temperature for 45 min. At the end of the reaction, the kinase activity detection was conducted using the ADP-Glo Kinase Assay Kit (Promega) following the manufacturer’s instructions. The results are presented as relative light units.
2.12 Statistical analysis
All data are presented as means ± SEM. Differences between two groups were evaluated using One-Way ANOVA with Tukey post hoc test and P < 0.05 was considered to be significant.
3. Results
3.1 JNK activation is linked to a significantly increased CaMKIIδ protein level in human atria with increasing age
We assessed protein expression of CaMKIIδ in human atria that were obtained from donors with a normal cardiac function and no history of AF or major CVDs (Table 1). Quantitative immunoblotting assays showed that increased CaMKIIδ protein was positively correlated with elevated JNK-P (activated JNK) in the human atrium with increasing age (Figure 1A and B). In contrast, levels of JNK1 and JNK2 proteins were not different between the human atrial samples with increasing age (Figure 1C).
Figure 1.

Concordantly activated JNK and up-regulated CaMKIIδ protein in the human atrium with increasing age. (A,B) Representative immunoblotting images and quantitative data showing elevated JNK-P and increased CaMKIIδ protein in the aged human atrium compared with that of young. GAPDH serves as a loading control. (C) Representative immunoblotting images and summarized data of quantified levels of JNK1, JNK2 protein expression in human atria with increasing age.
This concurrent increase of JNK-P and CaMKIIδ in the aged human atria led us to investigate the functional role of JNK activation on CaMKIIδ expression. To measure the JNK specific action in CaMKIIδ expression without interfering with other potential confounding factors in the aged heart, cardiac specific tamoxifen-inducible MKK7D mice were studied.24 Induction of constitutively activated MKK7, an upstream activator of JNK, led to significant activation of JNK (Figure 2A). Previous studies have suggested that long-term MKK7 activation (2-months post-tamoxifen induction) leads to an impaired cardiac function.24 However, MKK7D Tg mice (MKK7D+-Tx) with a short-term post-tamoxifen induction (5 days) showed significantly activated JNK but a comparable ventricular EF and FS to that of tamoxifen-treated WT littermates (MKK7D−-Tx; Figure 2B). As shown in Figure 2C, CaMKIIδ was markedly increased in JNK activated MKK7D+ mice, suggesting JNK activation contributes to enhanced CaMKIIδ expression.
Figure 2.

Activated JNK in cardiac-specific inducible MKK7D young mice leads to up-regulated CaMKIIδ. Immunoblotting images and summarized data showing markedly activated JNK (A) but preserved cardiac function (EF and FS); (B) in MKK7D mice (MKK7D+-Tx) with tamoxifen-induced expression of the constitutively active upstream JNK activator MKK7D compared with MKK7D negative littermates treated with tamoxifen (MKK7D--Tx). (C) Immunoblotting images and pooled data indicating up-regulated CaMKIIδ protein expression in tamoxifen-treated MKK7D mice compared with that of controls.
3.2 JNK2 isoform specific action in CaMKIIδ protein expression in aged mice
JNK1 and JNK2 are the two major isoforms expressed in myocytes and the two isoforms were found to have distinctive functions.2,28,29 Therefore, we assessed JNK isoform-specific action in CaMKIIδ expression. JNK1 and JNK2 proteins in the aged mouse heart were first IP-ed using JNK isoform specific antibodies. Using an ADP-Glo enzyme activity assay, we found that the activity of the JNK2 isoform was significantly increased, while JNK1 was unchanged in aged WT mice compared with that of young WT controls (Figure 3A and B). Next, we discovered that CaMKIIδ protein was remarkably elevated (by 60%) in aged WT mice compared with that of young mice (Figure 3C). JNK2 inhibition in aged WT mice reversed this age-associated up-regulation of CaMKIIδ proteins (Figure 3C, far right). Taken together, the results from both aged humans and aged mice strongly suggest that JNK2 is required for upregulated CaMKIIδ protein expression in the aged heart.
Figure 3.
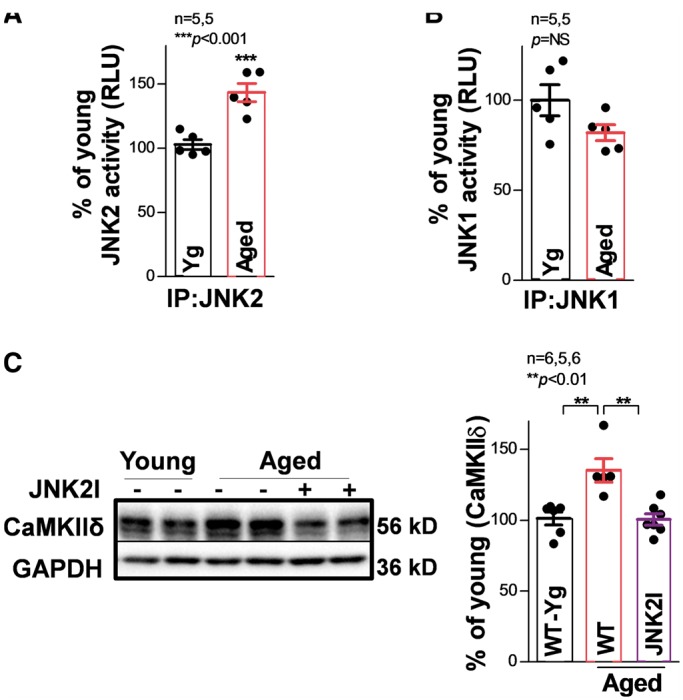
JNK2 contributes to increased CaMKIIδ protein expression in the aged mouse heart. In vitro kinase activity assay shows that immunoprecipitated JNK2 (IP: JNK2; A) but not JNK1 (IP: JNK1; (B) activity is significantly elevated in the hearts of aged WT mice. (C) Example immunoblotting images and summarized data suggesting significantly increased CaMKIIδ protein expression along with unchanged JNK2 total protein in aged WT mice, while JNK2 inhibitor in vivo treatment effectively reversed CaMKIIδ proteins to a level comparable to young controls.
3.3 Activation of JNK2 leads to increased CaMKIIδ protein expression in cultured HL-1 myocytes
To further measure the action of activated JNK2 on CaMKIIδ expression in myocytes, we took advantage of a well-characterized mouse atrial HL-1 myocyte line. HL-1 myocytes have been shown to express cardiac genes and proteins, including ion channels and mature isoforms of sarcomeric contractile proteins normally found in adult myocytes.6,27,30–32 Co-expression of the JNK activator MKK7D and JNK2 in HL-1 myocytes led to increased CaMKIIδ expression (by 40%), whereas co-expression of MKK7D and JNK1 did not (Figure 4A). Similarly, stimulation of cells with anisomycin (a JNK activator)6 for 24 h led to a marked increase in the CaMKIIδ protein level (by ∼60% compared with the control, Figure 4B). We acknowledge that anisomycin may not be a JNK specific activator. To exclude potential off-target anisomycin actions, we have included the use of specific JNK2 inhibition either by a JNK2 inhibitor or overexpressed inactivated JNK2 dominant negative proteins to help delineate the contribution of JNK2 in anisomycin-induced CaMKII expression in myocytes. When cultured HL-1 myocytes were pre-treated with a JNK2-specific inhibitor JNK2I-IX, this anisomycin-induced up-regulation of CaMKIIδ was completely abolished (Figure 4B, far right lanes/bar). Moreover, we infected HL-1 myocytes with adenovirus expressing either inactivated dominant negative JNK1 (JNK1dn) or dominant negative JNK2 (JNK2dn) followed by anisomycin treatment for 24 h. We found that inactive JNK2dn expression attenuated this anisomycin-induced upregulation of CaMKIIδ, while overexpression of JNK1dn had no such preventive effect on CaMKIIδ proteins (Figure 4C and D). Collectively, these results demonstrate a JNK2 isoform-specific action in enhanced CaMKIIδ protein expression.
Figure 4.

Activation of JNK2 but not JNK1 contributes to the increase in CaMKIIδ protein expression in cultured HL-1 myocytes. (A) Co-infection of constitutively activated JNK upstream regulator MKK7D and JNK2, but not JNK1, increases the level of CaMKIIδ. (B) Treatment of HL-1 myocytes with anisomycin (JNK activator) for 24 h increases CaMKIIδ protein, which is prevented in the presence of JNK2-specific inhibitor JNK2I-IX. The lower panel shows phospho-JNK is increased by anisomycin. immunoblotting images and summarized data showing that overexpression of inactivated dominant negative JNK2 (JNK2dn) completely reverses anisomycin-induced upregulation of CaMKIIδ (C). As control, adenovirus expressing LacZ has no effect on anisomycin action. In contrast, overexpressing dominant negative JNK1 (JNK1dn) does not affect the expression of CaMKIIδ (D).
3.4 Up-regulated CaMKIIδ mRNA in the aged human atrium and JNK-activated cultured HL-1 myocytes
Using quantitative real-time PCR, we found that CaMKIIδ mRNA was significantly increased in the human atrium, and it was positively correlated with age (Figure 5A). This was consistent with the results we observed for CaMKIIδ protein expression in the human atrium (Figure 1B). Similarly, in HL-1 myocytes, CaMKIIδ mRNA was remarkably elevated by both JNK activator anisomycin (about 100% increase, Figure 5B) and the upstream activating kinase MKK7D (about 70% increase, Figure 5C). Altogether, these results indicate that JNK up-regulates CaMKIIδ mRNA expression.
Figure 5.
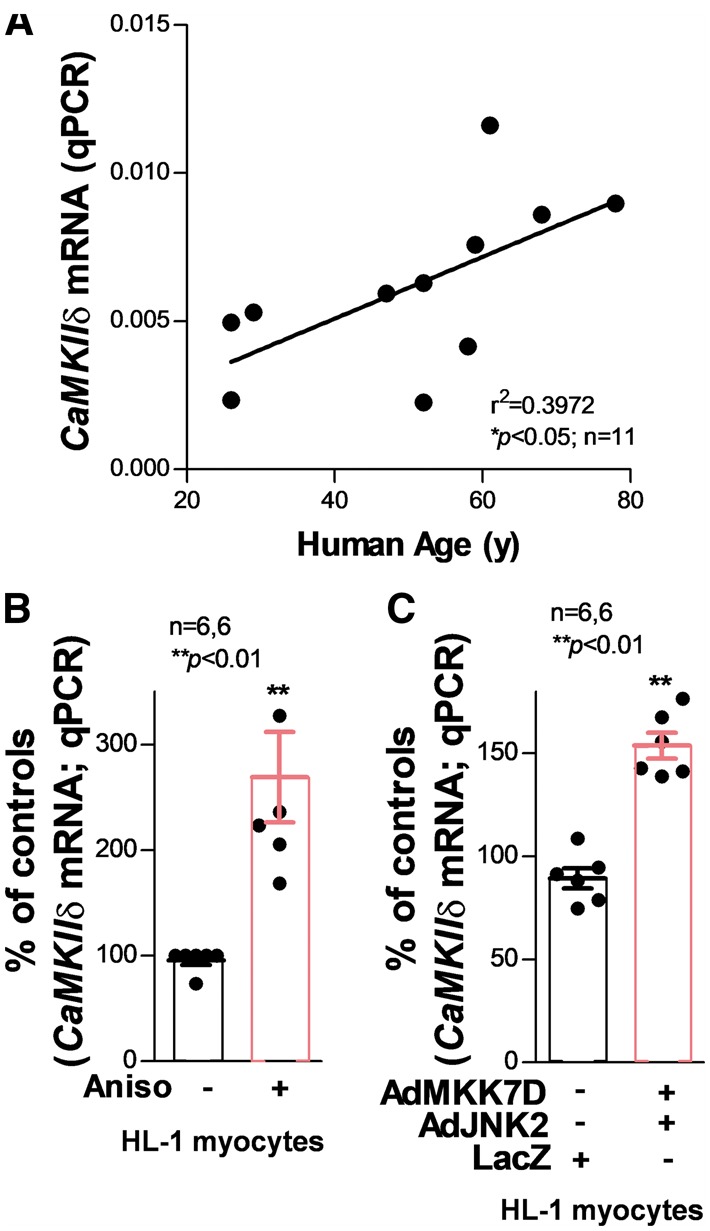
Dramatically increased CaMKIIδ mRNA expression in aged human atrium and JNK-activated myocytes. (A) Regression analysis shows a positive correlation between CaMKIIδ mRNA level and age in the healthy human atrium, indicating CaMKIIδ transcription activity increases with human age. (B) Co-infection of constitutively activated JNK upstream regulator MKK7D and JNK2 dramatically increases the transcription of CaMKIIδ. (C) JNK activator anisomycin-treated myocytes also shows a significantly increased level of CaMKIIδ mRNA.
3.5 JNK enhances the binding of JNK downstream transcription factor c-jun to the CaMKIIδ gene promoter
JNKs mediate multiple cellular processes through phosphorylation of their downstream targets, particularly the transcription factor activator protein-1 (AP-1) complex.33–35 Thus, we analysed the phosphorylation status of JNK downstream AP-1 transcription factors, c-jun and ATF2, using phospho-specific antibodies. As shown in Figure 6A and B, phosphorylated c-jun was significantly increased in aged mouse hearts, while phosphorylated ATF2 remained unchanged compared with that of young controls. Similarly, phospho-c-jun was also increased in anisomycin-treated HL-1 myocytes compared with sham controls, while fractionation of HL-1 myocytes showed that anisomycin remarkably increased c-jun-P proteins in the nuclei (Figure 6C). It is conceivable that activated c-jun triggers CaMKIIδ transcription. However, which transcription factors regulate CaMKIIδ gene expression has not been defined to date. Whether or not c-jun and ATF2 bind to and regulate the CaMKII gene promoter remains completely unknown.
Figure 6.

JNK increases in situ binding of c-jun but not ATF2 to the CaMKIIδ gene. (A,B) Summarized immunoblotting data suggest a markedly increased activated c-jun (c-jun-P) recognized by a phospo-specific antibody, while ATF2-P was unchanged; JNK2 ablation in the aged JNK2KO mouse heart completely attenuated this age-related activation of c-jun. (C) JNK activator anisomycin increased c-jun phosphorylation in total cell lysates as well as nuclear extracts in the HL-1 myocytes treated with anisomycin for 24 h. (D). Example images and summarized data of in situ XChIP assays suggesting that JNK activation significantly increases the binding of c-jun to the CaMKIIδ gene but not ATF2 in anisomycin-treated HL-1 myocytes, while JNK specific inhibition using JNK inhibitor SP600125 reverses the anisomycin-induced effect on the c-jun/CaMKIIδ interaction.
To test this hypothesis, we first performed silico analysis of CaMKIIδ gene with the JASPAR programme (http://jaspar.genereg.net) and identified a highly matched c-jun and ATF2 binding consensus 1210 bp upstream of the mouse CaMKIIδ transcription start site. Importantly, this consensus is also conserved in the human CaMKIIδ gene. To examine the binding of c-jun and ATF2 to the CaMKIIδ gene, we employed a unique XChIP assay using c-jun or ATF2 protein immunoprecipitation and quantitative real-time qPCR with primers encompassing the identified putative AP-1 binding site of the CaMKIIδ promoter. We discovered that both c-jun and ATF2 bind to the CaMKIIδ gene. Interestingly, we found that the binding of c-jun was markedly enhanced when myocytes were treated with JNK activator anisomycin for 24 h (Figure 6D), while ATF2 was unchanged. Moreover, JNK specific inhibitor SP600125 significantly reduced binding of c-jun proteins to the CaMKIIδ promoter (Figure 6D, far right). These results indicate that activation of JNK increases phosphorylation of the downstream target c-jun and thereby preferentially promotes the binding of c-jun to the CaMKIIδ gene promoter.
3.6 Specific action of JNK2 in CaMKIIδ up-regulation via c-jun-enhanced CaMKIIδ promoter activity
To address whether enhanced binding of c-jun to the CaMKIIδ gene leads to an increase in CaMKIIδ transcription, JNK action on CaMKIIδ promoter activity was assessed. We amplified a 1.2 kb-long fragment from the mouse CaMKIIδ gene containing our newly identified binding sites as described above and inserted it into a luciferase reporter vector (Figure 7A). The activity of firefly luciferase downstream of the CaMKIIδ promoter was significantly augmented by anisomycin treatment for 24 h (Figure 7B, left bar). Further, we mutated the putative AP-1 binding consensus (by replacing -716 T/-715 G to A/C) on the CaMKIIδ promoter. This AP-1 binding consensus mutation not only eliminated anisomycin-augmented activity of the CaMKIIδ promoter, but also markedly reduced the baseline CaMKIIδ promoter activity (Figure 7B). These results suggest a critical regulatory role of AP-1 in CaMKIIδ expression.
Figure 7.

Specific action of JNK2 in up-regulated CaMKIIδ transcription via c-jun enhanced CaMKIIδ promoter activity. (A) schematic diagram of CaMKIIδ promoter vector containing a Firefly luciferase reporter. (B) Summarized data of in vitro promoter reporter assays showing that anisomycin (24 h) significantly increases CaMKIIδ-WT (WT) promoter activity. AP-1 binding consensus mutation of the CaMKIIδ promoter [CaMKIIδ-mu (mu)] suppressed both baseline activity and anisomycin-induced promoter activity. Data were normalized with co-transfected plasmid expressing Renilla luciferase as an internal control of the assay. (C) Immunoblotting images confirmed the successful siRNA knockdown effect on JNK2 proteins ompared with that of scrambled control siRNA. Summarized data of in vitro promoter reporter assays showing that JNK2 siRNA knockdown significantly suppressed anisomycin-induced (24 h) CaMKIIδ promoter activity compared with that of scrambled control siRNA in HEK293 cells transfected with Firefly luciferase reporter tagged CaMKIIδ promoter vectors. (D) Summarized data of in vitro promoter reporter assays showing that c-jun siRNA knockdown also significantly prevented anisomycin-induced elevation of CaMKIIδ promoter activity. (E) Schematic diagram of proposed underlying molecular mechanism of JNK2 up-regulated CaMKIIδ expression in the aged atrium.
Just as we have demonstrated that JNK2 is crucial in the up-regulation of CaMKIIδ expression, we have also assessed the effect of JNK2 siRNA knockdown on CaMKIIδ promoter activity. We found that JNK2 depletion attenuated anisomycin-induced enhancement of CaMKIIδ promoter activity (Figure 7C). Moreover, knockdown of the JNK downstream transcription factor c-jun with c-jun specific siRNA also dramatically suppressed CaMKIIδ promoter activity (Figure 7D). Taken together, these results revealed for the first time that the binding of c-jun to the CaMKIIδ gene is functional and the JNK2 isoform directly up-regulates the transcriptional activity of CaMKIIδ via a JNK-enhanced c-jun binding to the CaMKIIδ promoter (Figure 7E).
4. Discussion
We have discovered a previously unrecognized molecular mechanism of JNK2 isoform-specific action in up-regulating the expression of a pro-arrhythmic molecule CaMKIIδ in the aged atrium. We have also revealed a novel underlying molecular mechanism where activated JNK2 increases CaMKIIδ transcription activity via direct binding of the JNK downstream target c-jun to the CaMKIIδ promoter.
4.1 Role of JNK in CaMKIIδ up-regulation and enhanced AF propensity in aging
JNK, originally identified as a stress protein kinase, is activated in response to a variety of extrinsic and intrinsic stresses.2,36 During the aging process, cardiomyocytes undergo various cellular changes such as enhanced oxidative stress as well as accumulating other chemical or biophysical stresses in the heart.37,38 Although JNK may be activated with increasing age as the stresses accumulate,6,39 our current and previous results suggest that activated atrial JNK is a consistent feature of aged atrium among different species (humans, rabbits, and mice).6,7 Aging is an important risk factor for AF, the most common arrhythmia, affecting millions worldwide.8–10 Our laboratory recently discovered a causal link between activated JNK and AF development in the aged heart.6,7 Although the underlying mechanisms of age-associated AF propensity remain incompletely understood, our current studies reveal that stress kinase JNK2 plays a pivotal role in regulating the expression of CaMKIIδ, a pro-arrhythmic molecule.15–19
The JNK contribution to the CaMKII/RyR remodelling in fibrillating atria warrants further investigation. But, this investigation may be confounded by the complexity of AF causes, duration of AF progression, age and many other factors. This makes it difficult to distinguish whether CaMKII-dependent Ca2+ mishandling in the fibrillating atrium is a pre-existing arrthythmogenic remodelling or the consequence of AF-begets-AF. Our current results from aged human and animal atria, in hearts with no history of AF or CVD history, provide evidence of age-related CaMKIIδ up-regulation and enhanced atrial arrhythmogenicity by activated JNK, predisposing the heart to the onset of AF.
Recent studies indicate that alterations of CaMKIIδ-dependent phosphorylation of Ca2+ triggered Ca2+ release RyR channels are present in the atria of chronic AF patients. Moreover, results in several animal models show that altered SR Ca2+ handling proteins contribute to enhanced diastolic SR Ca2+ leak and AF development.13,14,40 Many studies have shown LV CaMKII expression and activity are significantly increased in patients with advanced heart failure as well as in animal models of cardiac hypertrophy and heart failure.41–43 We and others have previously demonstrated that enhanced activation of ventricular CaMKIIδ is critically involved in phosphorylation of Ca2+ handling proteins, which results in sensitization of RyR channels and consequent triggered activities and arrhythmia.15,43–49 Thus, development of drugs to target CaMKIIδ activity has been proposed as a potential anti-arrhythmic intervention.20,21
Although a variety of CaMKII inhibitors are currently available for research purposes, their off-target effects hinder their use for clinical applications.23 Thus, additional upstream or downstream components of the CaMKII signalling cascades are being considered as new therapeutic targets. With the findings of increased CaMKIIδ protein expression in the aged atria (our current finding) and in failing hearts,15,17,46 modulating CaMKIIδ expression could be an alternative approach to targeting the CaMKIIδ molecule. Our discovery of the isoform-specific action of JNK2 in CaMKIIδ expression has shed new light on the possibility of inhibiting isoform specific JNK2 activity as a novel therapeutic intervention to prevent or treat cardiac arrhythmias.
4.2 Novel findings of isoform specific action of JNK2 in CaMKIIδ expression
CaMKIIδ is critical in regulating a large number of cellular substrates including ion channels, pumps, transporters and transcription factors.22 Yet, how the expression of CaMKIIδ itself is controlled remains surprisingly understudied. Here, we uncovered the underlying molecular mechanism to explain how JNK2 up-regulates CaMKIIδ gene expression.
JNK1 and JNK2 are the two predominant isoforms expressed in the heart.2,6 JNK1 has been shown to be critical in preserving cardiac function, preventing nerve degeneration, and promoting apoptosis in hearts with in vitro ischemia-reperfusion.28 Conversely, the functions of JNK2 in the heart have not been well defined to date. Here, we have identified JNK2 as a key factor in up-regulated CaMKIIδ expression. While the concordantly activated JNK and upregulated CaMKIIδ were found in both aged human atria and animal models (with either JNK naturally activated by aging in WT mice or activated genetically in MKK7D young mice), the rescue effect of JNK2 ablation on JNK-induced CaMKII upregulation from JNK2 inhibitor-treated aged mice and JNK activator anisomycin-treated myocytes, demonstrate a key regulatory role of the JNK2 isoform in CaMKIIδ protein expression.
It is known that JNK regulates its downstream gene expression through active or inactive AP-1, which is composed of JNK-regulated transcription factors (TFs) including c-jun and ATF2.7,33 The AP-1 complex regulates its target gene expression by binding the AP-1 consensus site(s) in the promoter region of target genes, or by dissociating from the promoter region to activate or suppress the specific gene expression.7,33 To date, transcription factors that regulate CaMKIIδ gene expression remain completely unknown. We discovered, for the first time, that JNK downstream transcription factor c-jun and ATF2 both bind to the CaMKIIδ gene and play a critical role in regulating CaMKII expression. This is supported by our results of suppressed CaMKII promoter activity when the AP-1 binding consensus sites were mutated. Another novel finding here is that JNK increases the binding of c-jun, but not ATF2, to the CaMKIIδ promoter. Further, JNK inhibition alleviated this enhanced c-jun binding, and JNK2 or c-jun specific siRNA knockdown rescued JNK-driven c-jun action on CaMKIIδ promoter activity. These intriguing rescue results of JNK2 inhibition in CaMKIIδ expression suggest that manipulating JNK2 activity could be an appealing anti-arrhythmia therapeutic approach.
4.3 Conclusions and potential implications of the results
Age-associated AF vulnerability is clinically significant. Our focus was to begin defining the role of JNK in CaMKII expression in normal aged atria that have no history of AF or CVD. We have demonstrated a causative action of activated JNK2 in up-regulating the pro-arrhythmic molecule CaMKIIδ in the aged atrium. Given that our current studies have revealed JNK2 to be a key regulator of the pro-arrhythmic CaMKIIδ, our new findings are clinically important and shed light on the possibility of modulating isoform specific activity of JNK as a novel therapeutic intervention approach to prevent or treat AF, with even broader applications for other cardiac arrhythmias in the elderly and heart failure patients.
Clearly, JNK-driven CaMKII expression in aged and failing ventricles as well as possible atria-ventricle chamber differences deserve further studies. Also, CaMKII was reported to contribute to cardiac remodelling.50 Although the absence of atrial structural remodelling in normal aging hearts has not been observed,6,51 the relationship between JNK, CaMKII, and structural remodelling also warrants further investigation in future studies.
Acknowledgements
We graciously thank Dr Yibin Wang for his generous gift of MKK7D Tg mice and Sarah Burris for providing her service through the Departmental Small Animal Core facilities at Loyola University Chicago.
Conflict of interest: none declared.
Funding
This work was supported by Natural Science Foundation of China [81400257 to J.Z.]; American Heart Association [10GRNT3770030 to X.A.]; and National Institutes of Health [HL113640 and AA024769 to X.A.].
References
- 1. Davis RJ. Signal transduction by the JNK group of MAP kinases. Cell 2000;103:239–252. [DOI] [PubMed] [Google Scholar]
- 2. Rose BA, Force T, Wang Y.. Mitogen-activated protein kinase signaling in the heart: angels versus demons in a heart-breaking tale. Physiol Rev 2010;90:1507–1546. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3. Karin M, Gallagher E.. From JNK to pay dirt: jun kinases, their biochemistry, physiology and clinical importance. IUBMB Life 2005;57:283–295. [DOI] [PubMed] [Google Scholar]
- 4. Sun A, Zou Y, Wang P, Xu D, Gong H, Wang S, Qin Y, Zhang P, Chen Y, Harada M, Isse T, Kawamoto T, Fan H, Yang P, Akazawa H, Nagai T, Takano H, Ping P, Komuro I, Ge J.. Mitochondrial aldehyde dehydrogenase 2 plays protective roles in heart failure after myocardial infarction via suppression of the cytosolic JNK/p53 pathway in mice. J Am Heart Assoc 2014;3:e000779.. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5. Liu Y, Wang J, Qi SY, Ru LS, Ding C, Wang HJ, Zhao JS, Li JJ, Li AY, Wang DM.. Reduced endoplasmic reticulum stress might alter the course of heart failure via caspase-12 and JNK pathways. Can J Cardiol 2014;30:368–375. [DOI] [PubMed] [Google Scholar]
- 6. Yan J, Kong W, Zhang Q, Beyer EC, Walcott G, Fast VG, Ai X.. c-Jun N-terminal kinase activation contributes to reduced connexin43 and development of atrial arrhythmias. Cardiovasc Res 2013;97:589–597. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7. Yan J, Thomson JK, Zhao W, Wu X, Gao X, DeMarco D, Kong W, Tong M, Sun J, Bakhos M, Fast VG, Liang Q, Prabhu SD, Ai X.. The stress kinase JNK regulates gap junction Cx43 gene expression and promotes atrial fibrillation in the aged heart. J Mol Cell Cardiol 2018;114:105–115. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8. Go AS, Hylek EM, Phillips KA, Chang Y, Henault LE, Selby JV, Singer DE.. Prevalence of diagnosed atrial fibrillation in adults: national implications for rhythm management and stroke prevention: the AnTicoagulation and Risk Factors in Atrial Fibrillation (ATRIA) Study. J Amer Med Assoc 2001;285:2370–2375. [DOI] [PubMed] [Google Scholar]
- 9. Benjamin EJ, Levy D, Vaziri SM, D'Agostino RB, Belanger AJ, Wolf PA.. Independent risk factors for atrial fibrillation in a population-based cohort. The Framingham Heart Study. J Amer Med Assoc 1994;271:840–844. [PubMed] [Google Scholar]
- 10. Rich MW. Epidemiology of atrial fibrillation. J Interv Card Electrophysiol 2009;25:3–8. [DOI] [PubMed] [Google Scholar]
- 11. Yan J, Zhao W, Thomson JK, Gao X, DeMarco DM, Carrillo E, Chen B, Wu X, Ginsburg KS, Bakhos M, Bers DM, Anderson ME, Song LS, Fill M, Ai X.. Stress signaling JNK2 crosstalk with CaMKII underlies enhanced atrial arrhythmogenesis. Circ Res 2018;doi: 10.1161/CIRCRESAHA.117.312536. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12. Ai X. SR calcium handling dysfunction, stress-response signaling pathways, and atrial fibrillation. Front Physiol 2015;6:46.. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13. Chelu MG, Sarma S, Sood S, Wang S, van Oort RJ, Skapura DG, Li N, Santonastasi M, Muller FU, Schmitz W, Schotten U, Anderson ME, Valderrabano M, Dobrev D, Wehrens XH.. Calmodulin kinase II-mediated sarcoplasmic reticulum Ca2+ leak promotes atrial fibrillation in mice. J Clin Invest 2009;119:1940–1951. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14. Neef S, Dybkova N, Sossalla S, Ort KR, Fluschnik N, Neumann K, Seipelt R, Schondube FA, Hasenfuss G, Maier LS.. CaMKII-dependent diastolic SR Ca2+ leak and elevated diastolic Ca2+ levels in right atrial myocardium of patients with atrial fibrillation. Circ Res 2010;106:1134–1144. [DOI] [PubMed] [Google Scholar]
- 15. Ai X, Curran JW, Shannon TR, Bers DM, Pogwizd SM.. Ca2+/calmodulin-dependent protein kinase modulates cardiac ryanodine receptor phosphorylation and sarcoplasmic reticulum Ca2+ leak in heart failure. Circ Res 2005;97:1314–1322. [DOI] [PubMed] [Google Scholar]
- 16. Anderson ME, Brown JH, Bers DM.. CaMKII in myocardial hypertrophy and heart failure. J Mol Cell Cardiol 2011;51:468–473. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17. Zhang R, Khoo MS, Wu Y, Yang Y, Grueter CE, Ni G, Price EE Jr., Thiel W, Guatimosim S, Song LS, Madu EC, Shah AN, Vishnivetskaya TA, Atkinson JB, Gurevich VV, Salama G, Lederer WJ, Colbran RJ, Anderson ME.. Calmodulin kinase II inhibition protects against structural heart disease. Nat Med 2005;11:409–417. [DOI] [PubMed] [Google Scholar]
- 18. Maxwell JT, Natesan S, Mignery GA.. Modulation of inositol 1, 4, 5-trisphosphate receptor type 2 channel activity by Ca2+/calmodulin-dependent protein kinase II (CaMKII)-mediated phosphorylation. J Biol Chem 2012;287:39419–39428. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19. Zhang T, Guo T, Mishra S, Dalton ND, Kranias EG, Peterson KL, Bers DM, Brown JH.. Phospholamban ablation rescues sarcoplasmic reticulum Ca(2+) handling but exacerbates cardiac dysfunction in CaMKIIdelta(C) transgenic mice. Circ Res 2010;106:354–362. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20. Anderson ME. Calmodulin kinase signaling in heart: an intriguing candidate target for therapy of myocardial dysfunction and arrhythmias. Pharmacol Ther 2005;106:39–55. [DOI] [PubMed] [Google Scholar]
- 21. Anderson ME. Pathways for CaMKII activation in disease. Heart Rhythm 2011;8:1501–1503. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22. Rokita AG, Anderson ME.. New therapeutic targets in cardiology: arrhythmias and Ca2+/calmodulin-dependent kinase II (CaMKII). Circulation 2012;126:2125–2139. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23. Hund TJ, Mohler PJ.. Role of CaMKII in cardiac arrhythmias. Trends Cardiovasc Med 2015;25:392–397. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24. Petrich BG, Molkentin JD, Wang Y.. Temporal activation of c-Jun N-terminal kinase in adult transgenic heart via cre-loxP-mediated DNA recombination. Faseb J 2003;17:749–751. [DOI] [PubMed] [Google Scholar]
- 25. Petrich BG, Eloff BC, Lerner DL, Kovacs A, Saffitz JE, Rosenbaum DS, Wang Y.. Targeted activation of c-Jun N-terminal kinase in vivo induces restrictive cardiomyopathy and conduction defects. J Biol Chem 2004;279:15330–15338. [DOI] [PubMed] [Google Scholar]
- 26. Angell RM, Atkinson FL, Brown MJ, Chuang TT, Christopher JA, Cichy-Knight M, Dunn AK, Hightower KE, Malkakorpi S, Musgrave JR, Neu M, Rowland P, Shea RL, Smith JL, Somers DO, Thomas SA, Thompson G, Wang R.. N-(3-Cyano-4, 5, 6, 7-tetrahydro-1-benzothien-2-yl)amides as potent, selective, inhibitors of JNK2 and JNK3. Bioorg Med Chem Lett 2007;17:1296–1301. [DOI] [PubMed] [Google Scholar]
- 27. Yan J, Thomson JK, Zhao W, Fast VG, Ye T, Ai X.. Voltage and calcium dual channel optical mapping of cultured HL-1 atrial myocyte monolayer. J Vis Exp 2015;97:e52542. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28. Bogoyevitch MA. The isoform-specific functions of the c-Jun N-terminal Kinases (JNKs): differences revealed by gene targeting. Bioessays 2006;28:923–934. [DOI] [PubMed] [Google Scholar]
- 29. Sabapathy K, Hochedlinger K, Nam SY, Bauer A, Karin M, Wagner EF.. Distinct roles for JNK1 and JNK2 in regulating JNK activity and c-Jun-dependent cell proliferation. Mol Cell 2004;15:713–725. [DOI] [PubMed] [Google Scholar]
- 30. Xia M, Salata JJ, Figueroa DJ, Lawlor AM, Liang HA, Liu Y, Connolly TM.. Functional expression of L- and T-type Ca2+ channels in murine HL-1 cells. J Mol Cell Cardiol 2004;36:111–119. [DOI] [PubMed] [Google Scholar]
- 31. White SM, Constantin PE, Claycomb WC.. Cardiac physiology at the cellular level: use of cultured HL-1 cardiomyocytes for studies of cardiac muscle cell structure and function. Am J Physiol Heart Circ Physiol 2004;286:H823–H829. [DOI] [PubMed] [Google Scholar]
- 32. Claycomb WC, Lanson NA Jr., Stallworth BS, Egeland DB, Delcarpio JB, Bahinski A, Izzo NJ. Jr.,. HL-1 cells: a cardiac muscle cell line that contracts and retains phenotypic characteristics of the adult cardiomyocyte. Proc Natl Acad Sci U S A 1998;95:2979–2984. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33. Gius D, Botero A, Shah S, Curry HA.. Intracellular oxidation/reduction status in the regulation of transcription factors NF-kappaB and AP-1. Toxicol Lett 1999;106:93–106. [DOI] [PubMed] [Google Scholar]
- 34. Teunissen BE, Jansen AT, van Amersfoorth SC, O'Brien TX, Jongsma HJ, Bierhuizen MF.. Analysis of the rat connexin 43 proximal promoter in neonatal cardiomyocytes. Gene 2003;322:123–136. [DOI] [PubMed] [Google Scholar]
- 35. Zhang Y, Wu S, Ma J, Xia Y, Ai X, Sun J.. Bacterial protein AvrA stabilizes intestinal epithelial tight junctions via blockage of the C-Jun N-terminal kinase pathway. Tissue Barriers 2015;3:e972849.. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36. Ai X, Yan J, Carrillo E, Ding W.. The stress-response MAP kinase signaling in cardiac arrhythmias. Rev Physiol Biochem Pharmacol 2016;172:77–100. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37. Yang Z, Shen W, Rottman JN, Wikswo JP, Murray KT.. Rapid stimulation causes electrical remodeling in cultured atrial myocytes. J Mol Cell Cardiol 2005;38:299–308. [DOI] [PubMed] [Google Scholar]
- 38. Juhaszova M, Rabuel C, Zorov DB, Lakatta EG, Sollott SJ.. Protection in the aged heart: preventing the heart-break of old age? Cardiovasc Res 2005;66:233–244. [DOI] [PubMed] [Google Scholar]
- 39. Peart JN, Gross ER, Headrick JP, Gross GJ.. Impaired p38 MAPK/HSP27 signaling underlies aging-related failure in opioid-mediated cardioprotection. J Mol Cell Cardiol 2007;42:972–980. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40. Chiang DY, Kongchan N, Beavers DL, Alsina KM, Voigt N, Neilson JR, Jakob H, Martin JF, Dobrev D, Wehrens XH, Li N.. Loss of microRNA-106b-25 cluster promotes atrial fibrillation by enhancing ryanodine receptor type-2 expression and calcium release. Circ Arrhythm Electrophysiol 2014;7:1214–1222. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41. Maier LS. CaMKIIdelta overexpression in hypertrophy and heart failure: cellular consequences for excitation-contraction coupling. Braz J Med Biol Res 2005;38:1293–1302. [DOI] [PubMed] [Google Scholar]
- 42. Anderson ME. CaMKII and a failing strategy for growth in heart. J Clin Invest 2009;119:1082–1085. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43. Hoch B, Meyer R, Hetzer R, Krause EG, Karczewski P.. Identification and expression of delta-isoforms of the multifunctional Ca2+/calmodulin-dependent protein kinase in failing and nonfailing human myocardium. Circ Res 1999;84:713–721. [DOI] [PubMed] [Google Scholar]
- 44. Yeh YH, Wakili R, Qi XY, Chartier D, Boknik P, Kaab S, Ravens U, Coutu P, Dobrev D, Nattel S.. Calcium-handling abnormalities underlying atrial arrhythmogenesis and contractile dysfunction in dogs with congestive heart failure. Circ Arrhythm Electrophysiol 2008;1:93–102. [DOI] [PubMed] [Google Scholar]
- 45. Greiser M, Neuberger HR, Harks E, El-Armouche A, Boknik P, de Haan S, Verheyen F, Verheule S, Schmitz W, Ravens U, Nattel S, Allessie MA, Dobrev D, Schotten U.. Distinct contractile and molecular differences between two goat models of atrial dysfunction: aV block-induced atrial dilatation and atrial fibrillation. J Mol Cell Cardiol 2009;46:385–394. [DOI] [PubMed] [Google Scholar]
- 46. Zhang T, Maier LS, Dalton ND, Miyamoto S, Ross J Jr., Bers DM, Brown JH.. The deltaC isoform of CaMKII is activated in cardiac hypertrophy and induces dilated cardiomyopathy and heart failure. Circ Res 2003;92:912–919. [DOI] [PubMed] [Google Scholar]
- 47. Respress JL, van Oort RJ, Li N, Rolim N, Dixit SS, deAlmeida A, Voigt N, Lawrence WS, Skapura DG, Skardal K, Wisloff U, Wieland T, Ai X, Pogwizd SM, Dobrev D, Wehrens XH.. Role of RyR2 phosphorylation at S2814 during heart failure progression. Circ Res 2012;110:1474–1483. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48. Maier LS, Zhang T, Chen L, DeSantiago J, Brown JH, Bers DM.. Transgenic CaMKIIdeltaC overexpression uniquely alters cardiac myocyte Ca2+ handling: reduced SR Ca2+ load and activated SR Ca2+ release. Circ Res 2003;92:904–911. [DOI] [PubMed] [Google Scholar]
- 49. Sossalla S, Fluschnik N, Schotola H, Ort KR, Neef S, Schulte T, Wittkopper K, Renner A, Schmitto JD, Gummert J, El-Armouche A, Hasenfuss G, Maier LS.. Inhibition of elevated Ca2+/calmodulin-dependent protein kinase II improves contractility in human failing myocardium. Circ Res 2010;107:1150–1161. [DOI] [PubMed] [Google Scholar]
- 50. Kreusser MM, Lehmann LH, Keranov S, Hoting MO, Oehl U, Kohlhaas M, Reil JC, Neumann K, Schneider MD, Hill JA, Dobrev D, Maack C, Maier LS, Grone HJ, Katus HA, Olson EN, Backs J.. Cardiac CaM Kinase II genes delta and gamma contribute to adverse remodeling but redundantly inhibit calcineurin-induced myocardial hypertrophy. Circulation 2014;130:1262–1273. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 51. Platonov PG, Mitrofanova LB, Orshanskaya V, Ho SY.. Structural abnormalities in atrial walls are associated with presence and persistency of atrial fibrillation but not with age. J Am Coll Cardiol 2011;58:2225–2232. [DOI] [PubMed] [Google Scholar]