

Abstract
The ubiquitin-proteasome system is implicated in cell apoptosis that is frequently dysregulated in human cancers. POH1/rpn11/PSMD14, as a part of the 19S proteasomal subunit, contributes to the progression of malignancy, but its role in apoptosis remains unclear. Here, we showed that POH1 expression was increased and associated with poor outcomes in three independent cohorts of patients with hepatocellular carcinoma (HCC), esophageal cancer (EC), and colorectal cancer (CRC). The knockdown of POH1 significantly inhibited tumor cell proliferation and induced apoptosis mediated by the mitochondrial pathway in vitro. Intratumoral injection of POH1 small interfering RNA (siRNA) significantly reduced the progression of tumor growth and induced apoptosis in vivo. Furthermore, p53 or Bim siRNA markedly attenuated the apoptosis induced by POH1 depletion. POH1 depletion resulted in cell apoptosis by increasing the stability of p53 and Bim and inhibiting their ubiquitination. Overall, POH1 knockdown induced cell apoptosis through increased expression of p53 and Bim via enhanced protein stability and attenuated degradation. Thus, POH1 may serve as a potential prognostic marker and therapeutic target in human cancers.
Abbreviations: Bcl-2, B-cell lymphoma-2; HCC, hepatocellular carcinoma; EC, esophageal cancer; CRC, colorectal cancer; POH1, Pad1 homologue, proteasome 26S subunit, non-ATPase 14; IHC, Immunohistochemistry; qRT-PCR, quantitative reverse transcription polymerase chain reaction; TMA, tissue microarray; SDS-PAGE, sodium dodecyl sulfate polyacrylamide gel electrophoresis; PARP1, poly (ADP-ribose) polymerase 1; TUNEL, terminal deoxynucleotidyl transferase (TdT) dUTP nick-end labeling; siRNA, small interfering RNA
Introduction
Apoptosis is a programmed cell death, which is a highly regulated and controlled process essential for organisms to remove the damaged, dysfunctional, or excess cells [1]. Dysregulation in apoptosis may lead to a variety of diseases such as cancer. In general, there are two apoptotic pathways: intrinsic and extrinsic pathway. The intrinsic pathway of apoptosis is controlled by the B-cell lymphoma-2 (Bcl-2) family of anti-apoptotic (such as Bcl-2 and Bcl-xL) and pro-apoptotic proteins (such as Bad, Bid, Bax and Bim) [2]. When the cells are suffering severe cellular damage or stress, the Bcl-2 family of proteins regulates apoptosis by controlling mitochondrial permeability and the release of cytochrome c. Released cytochrome c binds Apaf1 and forms an activation complex with caspase 9, which gives rise to the formation of apoptosome, which is responsible for induction of mitochondrial apoptosis [3], [4], [5]. Bim is a member of Bcl-2 family proapoptotic proteins, which translocates to mitochondria in response to death stimuli, including survival factor withdrawal [6], [7]. In addition, p53 is involved in the regulation of mitochondrial apoptotic pathway, mainly through the transcriptional regulation of the mitochondrial membrane Bcl-2 family proteins [8], [9]. Upon exposure of cells to certain stimuli (DNA damage, oxidative stress, etc.), the mitochondrial membrane permeability increases, resulting in the release of cytochrome c and the subsequent induction of mitochondrial apoptosis [10], [11], [12]. Cell apoptosis is frequently dysregulated in human cancers, and emerging evidence indicates that cancer cells adopt various strategies to override apoptosis [13], [14].
The proteasome is an abundant multienzyme complex that provides the main pathway for the degradation of intracellular proteins in eukaryotic cells. The 26S proteasome consists of one 20S core complex for proteolysis and two 19S regulatory complexes for protein degradation [15], [16], [17]. Accumulating evidence indicates that the loss of control over the ubiquitin proteasome system may induce cell apoptosis [18], [19]. POH1, a deubiquitinating enzyme within the 19S proteasomal subunit, is responsible for substrate deubiquitination during proteasomal degradation [20], [21]. POH1 functions in various biological processes, including protein stability [22], [23], aggresome clearance and disassembly [24], cellular proliferation [25] double-strand DNA break responses [26], and embryonic stem cell differentiation [27]. In normal cells, POH1 small interfering RNA (siRNA) may induce reduction in cell proliferation [28]. POH1 is also known to play an important role in the progression of tumors. For instance, siRNA-mediated knockdown of POH1 had a considerable impact on cell viability and induced cell arrest in the G0-G1 phase, ultimately leading to senescence [28]. Wang et al. [29] proposed that the aberrant upregulation of nuclear POH1-mediated E2F1 stabilization promotes tumor formation in hepatocellular carcinoma (HCC). It is suggested that targeting POH1 may overcome proteasome inhibitor (such as bortezomib) resistance in multiple myeloma by inducing cell apoptosis [30]. Whether POH1 deregulation contributes to the intrinsic pathway of apoptosis in cancer is questionable.
In this study, we detected the expression of POH1 at both mRNA and protein levels in HCC, esophageal carcinoma (EC), and colorectal cancer (CRC) tissues and determined the relationship between POH1 and clinicopathological features of patients with these cancers. Furthermore, we observed that POH1 silencing induced cell apoptosis through an increase in the expression of p53 and Bim mediated by enhanced protein stability. Our study, therefore, describes a previously unknown mechanism that p53 and Bim expression is regulated by POH1 and its implication in apoptosis.
Materials and Methods
Patients, Tissue Specimens, and Follow-Up
A total of 461 paraffin-embedded HCC specimens, 216 paraffin-embedded EC specimens and 314 paraffin-embedded CRC specimens were obtained from the archives of the Department of Pathology of the Sun Yat-sen University Cancer Center (SYSUCC) between January 2000 and December 2015. Fifty-nine cases of paired fresh HCC and adjacent nontumorous liver tissues, 12 cases of paired fresh EC and adjacent nontumorous esophageal tissues, and 20 cases of paired fresh CRC tissues and adjacent nontumorous colon tissues were collected from patients at the time of surgical resection for the determination of POH1 mRNA and protein expression. None of the patients received any chemotherapy or radiotherapy before the surgery. The follow-up period was defined as the interval from the date of surgery to the date of death or the last follow-up. This study was approved by the Institutional Review Board and Human Ethics Committee of SYSUCC.
Tissue Microarray (TMA) Construction and Immunohistochemistry (IHC)
Using a tissue array instrument (Minicore Excilone, Minicore, UK), a tissue core (0.6 mm in diameter) was punched from the marked areas and re-embedded. All specimens were fixed with 4% paraformaldehyde in 0.1 M phosphate buffer for 24 hours and embedded in paraffin wax. The paraffin-embedded tissues sections were sliced into 4-μm sections and mounted onto glass slides. After dewaxing, the slides were treated with 3% hydrogen peroxide in methanol and blocked with a biotin-blocking kit (DAKO, Germany). After blocking, the slides were overnight incubated with POH1 monoclonal antibody (1:50, Abcam, US), p53 monoclonal antibody (1:50, Santa Cruz, US), and Bim monoclonal antibody (1:50, CST, US) in a moist chamber at 4°C. After washing thrice in phosphate-buffered saline (PBS), the slides were incubated with biotinylated goat anti-rabbit antibodies for 1 hour. The slides were stained with DAKO liquid 3,3′-diaminobenzidine tetrahydrochloride (DAB), followed by their counterstaining with Mayer’s hematoxylin and observation under a microscope.
The protein level of POH1 was determined by semiquantitative IHC detection. The positively stained samples were scored as follows: 1, ≤25% of positively stained cells; 2, >25%-≤50% of positively stained cells; 3, >50%-≤75% of positively stained cells; 4, >75% of positively stained cells. The intensity of staining was scored according to the following standard: 0, negative staining; 1, weak staining; 2, moderate staining; and 3, strong staining. The final score was calculated by multiplying the percentage score by the staining intensity score. The scores were independently determined by two pathologists (Dr. Jing-Ping Yun and Dr. Yong Li). The median IHC score was chosen as the cutoff value for defining high and low expression.
Chemicals and Reagents
Dulbecco’s modified Eagle’s medium (DMEM) was obtained from Gibco (Gibco, Gaithersburg, MD). The radioimmunoprecipitation assay (RIPA) buffer supplemented with a protease inhibitor cocktail (P8340) and cycloheximide (CHX) was purchased from Sigma-Aldrich (MA, USA), and MG132 was from Millipore. Phosphatase inhibitors were supplied by Roche Diagnostics (Shanghai, China), and monoclonal antibodies against POH1 were purchased from Abcam (USA). Monoclonal antibodies against Bim, Bad, Bid, Bak, Bax, puma, Noxa, p21, Bcl-xl, caspase-3, and caspase-9 were purchased from Cell Signaling Technology (USA). Monoclonal antibodies against β-actin, p53, Mcl-1, hemagglutinin (HA), PARP1, ubiquitin (Ub), a nd horseradish peroxidase (HRP)–conjugated anti-rabbit and anti-mouse antibodies were obtained from Santa Cruz Biotechnology Inc (CA, USA). Cytochrome c was purchased from Affinity Biosciences (USA), and Annexin V-FLOUS double staining kit was from Roche (Basel., Switzerland). Other routine laboratory reagents obtained were of Commercial Sources Of Analytical (Guangzhou, China).
Cell Lines and Cell Culture
The human esophageal carcinoma cell line EC109 and human hepatic carcinoma cell line QGY-7701 were obtained from the Chinese Academy of Sciences (Shanghai, China). Human colorectal carcinoma cell line HCT116 was acquired from the American Type Culture Collection (ATCC, Manassas, VA), while HCT116 p53−/− cells were a generous gift from Professor Tiebang Kang at SYSUCC. All of EC109, QGY-7701, and HCT116 are p53 wild-type cell lines. These cells were cultured in DMEM supplemented with 10% heat-inactivated fetal bovine serum (FBS, HyClone, Logan, UT) at 37°C in a humidified atmosphere of 5% CO2.
Small Interfering RNA (siRNA)
siRNA duplexes targeting POH1 (siRNA#1: 5′-GAACAAGUCUAUAUCUCUUUU-3′; siRNA#2: 5′-GGCAUUAAUUCAΜGGACUAUU-3′), p53 (siRNA#1: 5′-CCACUΜGAΜGGAGAGUAUU-3′; siRNA#2: 5′-TGCGTGTGGAGTATTTGGATG-3′), and Bim (siRNA#1: 5′-CCCAΜGAGUΜGΜGACAAAUTT-3′; siRNA#2: 5′-GACAGAGCCACAAGGGUAAUTT-3′) as well as a negative control (NC) siRNA duplex (forward: 5′-UUCUCCGAACGUGUCACGUTT-3′; reverse: 5′-ACGUGACACGUUCGGAGAATT-3′) were chemically synthesized by Shanghai GenePharma Co. Ltd. (Shanghai, China). Transfection was performed using Lipofectamine RNAiMAX (Invitrogen; Carlsbad, CA) according to the manufacturer’s instructions.
Quantitative Real-Time Reverse Transcription Polymerase Chain Reaction (qRT-PCR)
Total RNA was extracted from clinical samples and cultured cells using Trizol reagent (Invitrogen, Carlsbad, CA) following manufacturer’s instruction. Reverse transcription was performed with random primers using M-MLV reverse transcriptase (Promega Inc., USA) according to the manufacturer's instructions. SYBR green–based real-time PCR was carried out to measure the expression of POH1, p53, Bim, and 18S using CFX Connect Real-time PCR system (Bio-Rad, USA). Primers designed were as follows: human POH1, forward: 5′-TTGCTATGCCACAGTCAGGA-3′ and reverse: 5′-AACAACCATCTCCGGCCTTC-3′; human p53, forward: 5′-CAGCACATGACGGAGGTTGT-3′ and reverse: 5′-TCATCCAAATACTCCACACGC-3′; human Bim, forward: 5′-TAAGTTCTGAGTGTGACCGAGA-3′ and reverse: 5′- GCTCTGTCTGTAGGGAGGTAGG-3′; 18S, forward: 5′-TGAGAAACGGCTACCACATCC-3′ and reverse: 5′-ACCAGACTTGCCCTCCAATG-3′. qRT-PCR conditions were as follows: initial denaturation at 95°C for 10 minutes and 40 cycles of 95°C for 30 seconds, 60°C for 30 seconds, 72°C for 30 seconds, and a final extension of 10 minutes. SDS 2.3 software (Applied Biosystems) was used to quantify and analyze the relative mRNA levels. Relative quantification of POH1 mRNA was performed using the 2−ΔCt method. The experiments were performed independently at least thrice, and all samples were in triplicates.
Proliferation and Colony Formation Assays
After transfection with POH1 siRNAs or Flag (3 × Flag)-tagged POH1 plasmid and scramble siRNA or vector plasmid, the cells were seeded in each well of 96-well plates (3 × 104 cells/ml) in 100 μl medium and cultured for 5 days. The proliferation assay was performed by treating cells with 20 μl of 3-(4,5-dimethylthiazol-2-yl)-2,5-diphenyltetrazolium bromide (MTT) reagent (5 mg/ml, AMRESCO, Solon, OH) for 4 h at 37°C. The formazan crystals formed were dissolved in dimethyl sulfoxide (150 μl/well). The absorbance of each sample was measured using a multilabel plate reader (PerkinElmer) at 490 nm wavelength. For the colony formation assay, 500 cells were seeded into six-well plates and incubated at 37°C in a humidified atmosphere containing 5% CO2 for 10-14 days. Colonies were fixed with methanol, stained with 0.1% crystal violet, and counted.
Analysis of Protein Ubiquitination
We transfected QGY-7701, EC109, and HCT116 cells (107 cells) with UB-HA and POH1 siRNAs (or scramble siRNA), and 48 hours after transfection, cells were treated with 10 μM MG132 or DMSO for 9 hours. The total protein was extracted, and precleared cell lysates were incubated with protein A/G-agarose (Santa Cruz) and 8 μg Ub antibody overnight at 4°C. Precipitated proteins were dissolved in sodium dodecyl sulfate (SDS) loading buffer and fractionated by SDS polyacrylamide gel electrophoresis (SDS-PAGE). The proteins were visualized by immunoblotting, and ubiquitination was detected with p53 or Bim antibody.
Apoptosis Assays
To detect the apoptosis induced by POH1, we first transfected QGY-7701, EC109, and HCT116 cells with POH1 or scrambled siRNA for 48 hours. The cells were collected by centrifugation and treated with Annexin V-FITC and propidium iodide (Annexin V-FLUOS Staining kit from Roche) for 15 minutes at 26°C in the dark according to the manufacturer’s protocol. The cell suspension was immediately analyzed by flow cytometry (ACEA Novocyte, ACEA Biosciences, Inc, CA) to determine cell apoptosis. At least 2 × 104 events were collected for each sample at a flow rate of 80–100 cells per second.
For mitochondrial staining, each sample was incubated with JC-1 (Sigma-Aldrich, MO) at 37°C in the dark for 15 minutes followed by washing thrice with PBS. The samples were immediately analyzed using flow cytometry. At least 1 × 104 events were collected for each sample at a flow rate of 80 to 100 cells per second. The interest cell population was gated on the forward and side scatter to exclude debris and aggregates. For JC-1 fluorescence analysis, FL1 and FL2 channels were used for the detection of green fluorescence (488 nm) and red fluorescence (580 nm), respectively. The mitochondrial transmembrane potential (ψm) was characterized by the ratio of red/green fluorescence.
For Hoechst 33342 staining, cells transfected for 48 hours were washed twice with PBS and fixed with 4% paraformaldehyde containing 0.25% Triton X-100 for 10 minutes. Cells were treated with Hoechst 33342 for 15 minutes at room temperature in the dark. The excess of Hoechst 3342 was discarded. Cells were washed twice with PBS, and their morphology was observed under a fluorescence microscope.
In Vivo Tumor Growth
Male athymic nude mice (BALB/c-nu, 4 weeks, 18-22 g) were bred at the animal facility of the Center of Experimental Animals, Sun Yat-sen University (China). In brief, each nude mouse was implanted subcutaneously under the right armpit with 5 × 106 relative cells (QGY-7701, EC109, or HCT116). Each cell line included three groups, and each group contained six or seven mice. Ten days later, the mice with each cell line xenograft were injected with the following reagents: (1) negative control (NC) siRNA duplex, (2) POH1 siRNA-1, and (3) POH1 siRNA-2. The injection was twice a week, and the tumor size was measured with digital calipers before injection. Two weeks later, the mice were sacrificed, and tumors were collected for further measurement. Five monitoring points for tumor volumes of mice were collected. The mean of tumor volumes was calculated using the following formula: volume = length × width2 × 0.5. All animal studies were conducted with the approval of the Medical Experimental Animal Care Commission of Sun Yat-sen University Cancer Center.
Statistical Analysis
Statistical analysis was performed using SPSS software (version 19.0, Chicago, IL). Data from at least three experiments were depicted as mean values ± standard deviation (SD). Data for POH1 expression were analyzed using the Student’s t test. Survival curves were evaluated with the Kaplan-Meier method (log-rank test). A multivariate Cox proportional-hazard regression model was constructed to evaluate the independent influence of POH1 on prognosis. Differences were considered significant for P value less than .05.
Results
POH1 Expression Is Increased and Associated with Poor Outcomes in HCC, EC, and CRC
The expression of POH1 was determined in fresh HCC tissues by Western blot and qRT-PCR analyses. The results showed that POH1 protein level was upregulated in six pairs of fresh HCC, EC, and CRC tissues (Figure 1A1–A3). A significant increase in POH1 mRNA expression level was observed in HCC (n=59), EC (n=12), and CRC (n=20) fresh samples as compared with the adjacent nontumorous tissues (Figure 1B1–B3).
Figure 1.

POH1 gene and protein expression levels are increased in HCC, EC, and CRC and correlated with poor prognosis. (A) Expression of POH1 protein in several paired tumor (T) tissues and adjacent non-tumorous (N) tissues examined by Western blot (A1, HCC; A2, EC; A3, CRC). (B) The mRNA levels of POH1 in tumor (T) and corresponding adjacent nontumorous (N) tissues were determined with qRT-PCR (B1, HCC; B2, EC; B3, CRC). (C) POH1 was detected in the cytoplasm and nucleus within tumor and non-tumorous cells by IHC. The micrographs show strong staining in tumor (T) and weak staining in nontumorous (N) tissues (C1, HCC; C2, EC; C3, CRC) (top panel). Reproducibility of the measurement in all patients was calculated using the Wilcoxon matched paired test (bottom panel). (D) Kaplan-Meier survival analyses were conducted to evaluate the significance of POH1 in overall survival based on the protein expression in our cohort (D1, HCC; D2, EC; D3, CRC). Data are mean ± SD (all *P<.05 and **P<.01).
To further determine the upregulation of POH1 in HCC, EC, and CRC, three independent cohorts of patients with these cancers were recruited. TMA-based IHC analysis showed that the immunoreactivity of POH1 was mainly observed in the nucleus and cytoplasm of most cancer cells. POH1 expression was markedly increased in tumor samples as compared with the adjacent nontumorous tissues (Figure 1C1–C3, top panel). The alteration in POH1 expression was statistically significant (Figure 1C1–C3, bottom panel, P < .001, Wilcoxon matched-paired test).
Kaplan-Meier survival analysis was conducted to determine the prognostic impact of POH1 in HCC, EC, and CRC patients. HCC cases (n=461) with high POH1 expression were often associated with a worse prognosis in terms of overall survival (log-rank test; P = .012; Figure 1D1). In addition, CRC cases (n=314) with high POH1 expression were also associated with a worse prognosis in terms of overall survival (log-rank test; P = .013; Figure 1D3). However, we failed to detect similar results in EC patients (Figure 1D2). According to the mRNA levels of POH1 in 182 EC patients of TGCA dataset, high expression of POH1 suggests a trend of poor prognosis in terms of overall survival (log-rank test; P = .030; Supplementary Figure 1). Multivariate analysis indicated POH1 as an independent prognostic factor of overall survival in HCC (hazard ratio = 1.229, 95% confidence interval: 1.013-1.491, P = .036) (Table 1) and CRC (hazard ratio = 1.848, 95% confidence interval: 1.031-3.310, P =0.039) (Table 2).
Supplementary Figure 1.

Kaplan-Meier survival analyses were conducted to evaluate the significance of POH1 in overall survival, based on the mRNA level in EC TCGA cohort (n=182).
Table 1.
Univariate and Multivariate Analyses of Clinicopathological and POH1 Expression for Overall Survival in HCC Patients (n=461).
| Variables | Univariate Analysis |
Multivariate Analysis |
||
|---|---|---|---|---|
| HR (95% CI) | P Value | HR (95% CI) | P Value | |
| Age (<49 vs. ≥49 years) | 0.884 (0.736-1.062) | .188 | ||
| Gender (female vs. male) | 1.082 (0.797-1.469) | .613 | ||
| HBV (positive vs. negative) | 1.186 (0.927-1.519) | .175 | ||
| Tumor size (<5 vs. ≥5 cm) | 1.562 (1.248-1.955) | .000 | 1.251 (0.975-1.605) | .078 |
| Tumor multiplicity (single vs. multiple) | 1.334 (1.105-1.611) | .003 | 0.996 (0.796-1.247) | .971 |
| Tumor capsule (absent vs. present) | 0.707 (0.585-0.855) | .000 | 0.890 (0.728-1.089) | .258 |
| Liver cirrhosis (yes vs. no) | 0.674 (0.530-0.858) | .001 | 0.754 (0.586-0.970) | .028 |
| AFP (<20 vs. ≥20 ng/ml) | 1.514 (1.217-1.885) | .000 | 1.317 (1.050-1.652) | .017 |
| Vascular invasion (yes vs. no) | 2.109 (1.612-2.529) | .000 | 1.431 (1.115-1.837) | .005 |
| Tumor differentiation | 1.377 (1.145-1.657) | .001 | 1.211 (1.002-1.464) | .048 |
| TNM (I-II vs. III-IV) | 1.836 (1.523-2.213) | .000 | 1.380 (1.067-1.784) | .014 |
| POH1 expression (low vs. high) | 1.277 (1.055-1.545) | .012 | 1.229 (1.013-1.491) | .036 |
AFP, a-fetoprotein; HBsAg, hepatitis B surface antigen; HR, hazard ratio; CI, confidence interval.
Table 2.
Univariate and Multivariate Analyses of Clinicopathological and POH1 Expression for Overall Survival in CRC Patients (n=315)
| Variables | Univariate Analysis |
Multivariate Analysis |
||
|---|---|---|---|---|
| HR (95% CI) | P Value | HR (95% CI) | P Value | |
| Age (<59 vs. ≥59 years) | 1.047 (0.732-1.497) | .803 | ||
| Gender (female vs. male) | 0.954 (0.661-1.376) | .800 | ||
| BMI (<24 vs. ≥24) | 1.546 (1.072-2.231) | .020 | 1.593 (1.092-2.325) | .016 |
| Hypertension (yes vs. no) | 1.373 (0.923-2.042) | .118 | ||
| Diabetes (yes vs. no) | 2.084 (1.311-3.312) | .002 | 2.025 (1.253-3.274) | .004 |
| T stage(I-II vs. III-IV) | 2.995 (1.647-5.445) | .000 | 1.941 (0.898-4.195) | .092 |
| Metastasis (yes vs. no) | 4.015 (2.725-5.917) | .000 | 0.477 (0.241-0.945) | .034 |
| TNM (I-II vs. III-IV) | 5.212 (3.438 -7.900) | .000 | 5.931 (2.692-13.07) | .000 |
| LNM (yes vs. no) | 2.182 (1.497-3.181) | .000 | 2.182 (1.361-3.497) | .001 |
| Tumor differentiation | 1.496 (0.963-2.325) | .073 | ||
| R0 resection (yes vs. no) | 0.466 (0.301-0.719) | .001 | 0.509 (0.321-0.807) | .004 |
| CEA (<5 vs. ≥5 μg/l) | 2.345 (1.590-3.458) | .000 | 1.277 (0.828-1.968) | .268 |
| CA19-9 (<37 vs. ≥37 U/ml) | 3.149 (2.182-4.545) | .000 | 1.251 (1.761-3.981) | .000 |
| POH1 expression (low vs. high) | 1.983 (1.135-3.465) | .016 | 1.848 (1.031-3.310) | .039 |
BMI, body mass index; LNM, lymph node metastasis; CEA, carcinoembryonic antigen; CA19-9, carbohydrate antigen 19-9; HR, hazard ratio; CI, confidence interval.
POH1 Knockdown Inhibits Cancer Cell Proliferation
To investigate the potential role of POH1 in cancer progression, QGY-7701, EC109, and HCT116 cells were transfected with two POH1 siRNAs. The knockdown of POH1 expression in three cell lines was confirmed by Western blot analysis (Figure 2A). MTT assay revealed that the knockdown of POH1 resulted in a significant inhibition of cell viability (Figure 2B). The colony formation assay confirmed that POH1 silencing reduced cell proliferation in HCC, EC, and CRC, as the number of colonies formed by POH1-silenced cells was much lower than that in the control groups (Figure 2C). The overexpression of POH1 protein in these three cell lines was confirmed by Western blot analysis (Figure 2D). MTT and colony formation assays revealed that the overexpression of POH1 failed to promote cell proliferation (Figure 2E–F).
Figure 2.

POH1 knockdown inhibits cell proliferation. (A) The effect of POH1 siRNA was confirmed by Western blot analysis. (B) POH1 knockdown impaired QGY-7701, EC109, and HCT116 cell viability. Cells transfected with POH1 siRNA (siPOH1-1 or siPOH1-2) or scrambled siRNA were cultured in a 96-well plate for 4 days. Cell viability was determined using the MTT assay. (C) POH1 depletion weakened the colony formation ability of the above cell lines. The number of colonies is shown. (D) The effect of POH1 overexpression was confirmed by Western blot analysis. (E) POH1 overexpression failed to affect the viability of QGY-7701, EC109, and HCT116. Cells transfected with POH1 or vector were cultured in a 96-well plate for 4 days. Cell viability was determined using MTT assay. (F) POH1 overexpression failed to affect monoclonal formation ability of above cell lines. The number of colonies is shown. All *P<.05 and **P<.01.
Intratumoral Injection of POH1 siRNA Decreases Tumor Growth and Induces Apoptosis In Vivo
To assess the effect of POH1 knockdown on tumor growth in vivo, each nude mouse was implanted subcutaneously under the right armpit with 5 × 106 relative cells (QGY-7701, EC109, or HCT116). Ten days later, the mice with each cell line xenograft were injected directly into the tumor with POH1 siRNA-liposome complex or scrambled siRNA-liposome twice a week, and the tumor size was measured with digital calipers before injection. Two weeks later, the mice were sacrificed, and tumors were collected for further measurement. Intratumoral injection of POH1 siRNA significantly inhibited tumor growth as compared with the scrambled siRNA injection (Figure 3A1, B1, & C1). Furthermore, the tumor weight was lighter in POH1 siRNA groups as compared with the control groups (Figure 3A2, B2, & C2). The tumor volumes were significantly smaller in POH1 siRNA groups (Figure 3A3, B3, & C3). The analyses of terminal deoxynucleotidyl transferase (TdT) dUTP nick-end labeling (TUNEL) assay revealed more apoptotic cells in POH1 siRNA groups as compared to the control groups (Figure 3D), and the differences observed were statistically significant (Figure 3E). Analyses of IHC staining results of the xenograft confirmed that the expression of POH1 was lower in POH1 siRNA groups than the control groups, while the expression of p53, Bim, caspase-3, and caspase-9 was higher in POH1 siRNA groups as compared with the control groups (Supplementary Figure 2). These data suggest that the intratumoral injection of POH1 siRNA decreased tumor growth and induced apoptosis in vivo.
Figure 3.

POH1 siRNA intratumoral injection inhibits tumor proliferation. Each nude mouse was implanted with relative cells (QGY-7701, EC109, or HCT116), and 10 days later, mice with each cell line xenograft were injected with POH1 siRNA (30 μg) or scrambled siRNA twice a week. The tumor size was measured with digital calipers before the injection. (A) QGY-7701 cells were used for xenograft. The tumor volume (A1) and weight (A2) were measured after the mice were sacrificed. Representative images of the xenograft are shown (A3). (B) EC109 cells were used for xenografts. The tumor volume (B1) and weight (B2) were measured after the mice were sacrificed. Representative images of the xenograft are shown (B3). (C) HCT116 cells were used for xenograft. The tumor volume (C1) and weight (C2) were measured after the mice were sacrificed. Representative images of the xenograft are shown (C3). (D) TUNEL assay for cell apoptosis in POH1 siRNAs or scrambled siRNA groups. (E) Apoptotic index was counted according to the apoptotic cell number under three randomly selected fields. All *P<.05 and **P<.01.
Supplementary Figure 2.

Paraffin-embedded tumor tissue sections were stained with anti-POH1, anti-p53, anti-Bim, anti-caspase-3, and anti-caspase-9 antibodies and counterstained with hematoxylin. Representative images of the staining are shown (400× magnification).
POH1 Knockdown Induces Apoptosis of Cancer Cells via Mitochondrial Pathway
To determine the mechanism underlying apoptosis mediated by POH1 knockdown, we examined whether the intrinsic apoptosis was involved in this process. Following POH1 siRNA treatment for 48 hours, PARP1, caspase-9, and caspase-3 were activated, and the level of cytochrome c was increased in the cytoplasm (Figure 4A). Furthermore, knockdown of POH1 upregulated the protein levels of p53 and its targets such as p21, Bax, and Puma but not Noxa. As Bcl-2 family proteins play important roles in mitochondria- and caspase-mediated apoptosis, we determined the expression profile of Bcl-2 family members. The proapoptotic protein Bim was notably upregulated in cells upon POH1 knockdown, but the expression of other apoptosis-related proteins, including Bak, Bcl-xl, and Bid, remained unchanged (Figure 4B). In addition, knockdown of POH1 upregulated the protein levels of Mcl-1, but the overexpression of POH1 failed to affect the expression of MCl-1 (Supplementary Figure 6).
Figure 4.

POH1 knockdown induces cell apoptosis. Cells were treated with POH1 siRNA (siPOH1-1 or siPOH1-2) or scrambled siRNA for 48 hours. (A) Expressions of PARP1, caspase-3, caspase-9, and cytochrome c were examined by Western blot analysis. Cytochrome c was extracted from cell cytoplasm. (B) Expressions of p53, p21, and Bcl-2 family of proteins, including Bak, Bid, Bcl-xl, and Bim, were examined by Western blot analysis. (C) Hoechst 33342 staining of QGY-7701, EC109, and HCT116 cells detected by fluorescent microscopy. Highly condensed or fragmented nuclei represent apoptotic cells. Intact nuclei represent viable cells (×100) (left panel). The percentage of apoptotic cells was recorded (right panel). (D) POH1 silencing induced apoptosis in QGY-7701, EC109, and HCT116 cells. Cells were transfected with POH1 siRNA (siPOH1-1 or siPOH1-2) or scrambled siRNA for 48 hours and then stained with Annexin V and propidium iodide (PI) (left panel). The percentage of apoptotic cells was recorded (right panel). (E) POH1 siRNA induced a decrease in the mitochondrial membrane potential. Cells were transfected with POH1 siRNA (siPOH1-1 or siPOH1-2) or scrambled siRNA for 48 hours and stained with JC-1 for 15 minutes. The mitochondrial transmembrane potential (ψm) was determined by FACS (left panel). Graph depicts mean ± SD of three independent experiments (right panel). All *P<.05 and **P<.01.
Supplementary Figure 6.

The effects of knockdown (A) and overexpression (B) of POH1 on Mcl-1 levels by western blot analysis.
We investigated the effects of POH1 depletion on cell morphology by staining QGY-7701, EC109, and HCT116 cells with Hoechst 33342, followed by their observation under a fluorescence microscope. Treatment of cells with POH1 siRNAs for 48 hours resulted in an increase in nuclear fragmentation and condensation (Figure 4C). POH1 knockdown induced HCC cell apoptosis. The average percentage of apoptotic cells was 47.1% and 49.4% in QGY-7701 cells transfected with two POH1 siRNAs versus 7% in control cells; 28.1% and 33% in EC109 cells transfected with two POH1 siRNAs versus 6% in the control group; and 49.6% and 44% in HCT116 cells transfected with two POH1 siRNAs versus 8% in the control groups (Figure 4D). JC-1 staining showed that POH1 knockdown increased the red-to-green fluorescence intensity ratio, indicative of the damage of the mitochondrial transmembrane potential—one of the signatures of apoptosis mediated by the mitochondrial pathway (Figure 4E). Thus, POH1 may regulate apoptosis via modulation of mitochondrial proteins.
POH1 Knockdown Attenuates Degradation of p53 and Bim
In this study, both p53 and Bim were upregulated after POH1 knockdown. As POH1 is a deubiquitinating enzyme responsible for substrate deubiquitination during proteasomal degradation, we determined whether POH1 binds to p53 or Bim and induces ubiquitination and proteasomal degradation. Both knockdown and overexpression of POH1 had no effect on the mRNA level of p53 and Bim, indicative of the posttranslational regulation of p53 and Bim by POH1 (Supplementary Figure 3). We studied the interaction between POH1 and p53 (or Bim) in QGY-7701 cells by co-immunoprecipitation (Supplementary Figure 4) and examined if POH1 affected the half-life of p53 and Bim using CHX (protein synthesis inhibitor). We observed that p53 and Bim levels rapidly decreased in control cells following treatment with 20 μg/ml CHX for 9 hours. Knockdown of POH1 dramatically attenuated the degradation of p53 and Bim in QGY-7701 (Figure 5A), EC109 (Figure 5B), and HCT116 (Figure 5C) cells. POH1 knockdown decreased p53 and Bim ubiquitination in QGY-7701, EC109, and HCT116 cells (Figure 5D & E). These data confirmed that POH1 depletion attenuates the proteasome-mediated degradation of p53 and Bim.
Supplementary Figure 3.

Neither overexpression nor knockdown of POH1 influences the mRNA level of p53 and Bim. Fig S3A is overexpression of POH1 and S3B-D is siRNA of POH1. The mRNA level of siRNA control (NC) was used as a control in every experiment. All **P<0.01.
Supplementary Figure 4.

POH1 interacts with p53 or Bim. (A, B) The interaction between endogenous POH1 and p53 or Bim. QGY-7701 cells were collected for co-immunoprecipitation (Co-IP) assays using antibodies as indicated. IgG was used as a control.
Figure 5.

POH1 knockdown attenuates the degradation of p53 and Bim. POH1 contributed to the stability of p53 and Bim proteins. (A) QGY-7701 cells with POH1 siRNAs were treated with 20 μg/ml CHX for indicated periods. The expression of POH1, p53, and Bim was detected (A1). The decrease in p53 (A2) and Bim (A3) proteins was normalized and shown. (B) EC109 cells with POH1 siRNAs were treated with 20 μg/ml CHX for indicated periods. The expression of POH1, p53, and Bim was detected (B1). The decrease in p53 (B2) and Bim (B3) proteins was normalized and shown. (C) HCT116 cells with POH1 siRNAs were treated with 20 μg/ml CHX for indicated periods. The expression of POH1, p53, and Bim was detected (C1). The decrease in p53 (C2) and Bim (C3) protein was normalized and shown. β-Actin was used as a loading control. (D) POH1 knockdown attenuated p53 ubiquitination. Cells were transfected with combinations of plasmids encoding HA-Ub, POH1 siRNA, or scramble siRNA and treated with MG132 9 hours before the ubiquitination assay. (E) POH1 knockdown attenuated Bim ubiquitination. Cells were transfected with combinations of plasmids encoding HA-Ub, POH1 siRNA, or scramble siRNA and treated with MG132 9 hours before the ubiquitination assay. Bound and input proteins were detected by immunoblotting using antibodies as indicated. At least three experiments were performed. Data are mean ± SD (All *P<.05 and **P<.01).
Knockdown of p53 or Bim Decreased Apoptosis Mediated by POH1 Depletion
We assessed if p53 and Bim were involved in apoptosis mediated by POH1 depletion. Cells treated with POH1 siRNA and p53 or Bim siRNA for 48 hours showed attenuation in the cleaved form of caspase-9 and caspase-3 as compared with those treated with only POH1 siRNA (Figure 6A). p53 or Bim siRNA markedly decreased the apoptosis driven by POH1 depletion. The average percentage of apoptotic cells was 35.2% and 30.5% in QGY-7701 cells co-transfected with POH1 siRNA and p53 or Bim siRNA, respectively, versus 48.2% in POH1-knockdown cells (Figure 6B); 18.5% and 14.9% in EC109 cells co-transfected with POH1 siRNA and p53 or Bim siRNA, respectively, versus 26.2% in POH1-knockdown cells (Figure 6C); and 31.2% and 29.5% in HCT116 cells co-transfected with POH1 siRNA and p53 or Bim siRNA, respectively, versus 45.1% in POH1-knockdown cells (Figure 6D). These data suggest the critical role of p53 and Bim in POH1-mediated apoptosis.
Figure 6.

p53 and Bim siRNA attenuated the apoptosis driven by POH1 depletion. POH1 and p53 or Bim siRNAs were co-transfected into QGY-7701, EC109, and HCT116 cells for 48 hours. (A) Expressions of p53, Bim, PARP1, caspase-3, and caspase-9 were examined by Western blot analysis. (B) Apoptosis was determined by flow cytometry (left panel). Graph depicts mean ± SEM of three independent experiments (right panel). (C) POH1 and p53 or Bim siRNAs were co-transfected into QGY-7701 cells for 48 hours. Apoptosis was determined by flow cytometry (left panel). Graph depicts mean ± SEM of three independent experiments (right panel). (D) POH1 and p53 or Bim siRNAs were co-transfected into HCT116 cells for 48 hours. Apoptosis was determined by flow cytometry (left panel). Graph depicts mean ± SD of three independent experiments (right panel). All *P<.05 using paired Student’s t test.
Discussion
As a deubiquitinating enzyme responsible for substrate deubiquitination during proteasomal degradation, POH1 is known to be upregulated in several cancer types [29], [30], [31]. However, the prognostic implication of POH1 is unclear. Here, we studied large cohorts of 461 patients with HCC and 314 patients with CRC and found that POH1 expression was markedly increased and closely correlated with poor prognosis. In addition, a large cohort of 216 patients with EC showed marked increase in POH1 expression, which had no closed correlation with poor prognosis; however, the high POH1 mRNA expression level in 182 EC patients of TCGA dataset suggests a trend of poor prognosis in terms of overall survival. Two recent studies revealed the correlation between high POH1 expression and poor overall survival in multiple myeloma and breast cancer [30], [31]. Our data are consistent with these studies, suggesting the potential role of POH1 as a clinically significant biomarker of poor prognosis in patients with HCC, EC, and CRC.
POH1, a 19 S proteasomal subunit, has been shown to be involved in many biological functions, such as cellular viability [25], cell cycle [31], and double-strand DNA break responses [26]. The failure of DNA damage repair usually induces cell apoptosis, suggestive of the role of POH1 in apoptosis. We studied the oncogenic function of POH1 in solid tumors; knockdown of POH1 expression in QGY-7701, EC109, and HCT116 cells decreased cell viability and induced apoptosis. In addition, the intratumoral injection of POH1 siRNAs decreased tumor growth and induced apoptosis in vivo. Furthermore, POH1 silencing resulted in the activation of PARP1, caspase-9, caspase-3, and cytochrome c and upregulation of p53, p21, Bax, Puma, Noxa, and Bim. In addition, our results suggest that MCl-1 is upregulated after POH1 silenced, and several studies have shown that inhibitor of the 26S proteasome induced the expression of Mcl-1 [32], [33]. Taken together, we hypothesize that silence of POH1 causes dysfunction of proteasome and increases the expression of MCl-1. JC-1 staining showed that POH1 knockdown decreased mitochondrial transmembrane potential. Recent studies have suggested that knockdown of POH1 may induce cancer cell apoptosis [30], [31]. Taken together, POH1 knockdown may induce apoptosis mediated via mitochondrial pathway.
Our data show that p53 and Bim were upregulated in POH1-knockdown cells. We, therefore, hypothesized that POH1 is an important regulator of p53 and Bim, which were involved in apoptosis. The regulation of p53 [34] and Bim [35] may affect cell apoptosis. We observed that p53 and Bim siRNA markedly decreased the apoptosis driven by POH1 depletion. As p53 siRNA and Bim siRNA failed to completely inhibit the apoptosis driven by POH1 depletion, the effect of POH1 knockdown may, at least in part, be dependent on p53 and Bim. In addition, our data showed POH1 knockdown induced higher apoptosis in HCT116 p53+/+ cells as compared with HCT116 p53−/− cells (Supplementary Figure 5). Thus, we demonstrate that POH1 regulates cell apoptosis partially through the increased expression of p53 and Bim.
Supplementary Figure 5.
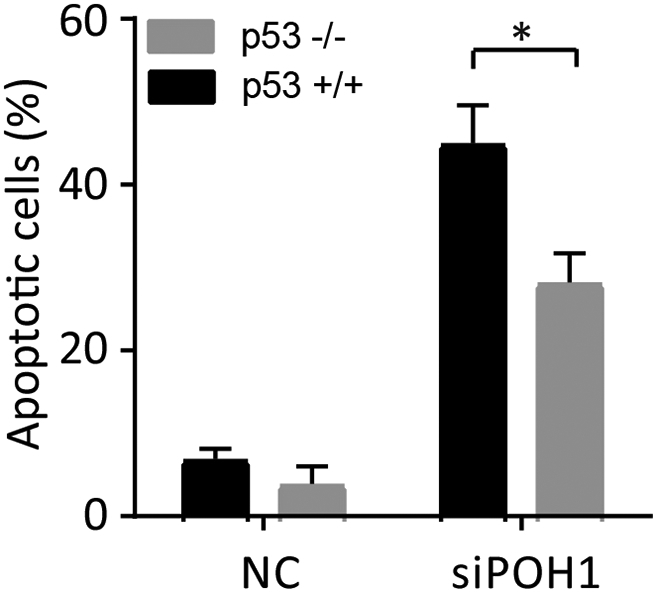
The effect of POH1 knockdown on the apoptosis of HCT116 p53+/+ and HCT116 p53−/− cells. HCT116 p53+/+ and HCT116 p53−/− cells were transfected with POH1 or scramble siRNA and harvested 48 h post-transfection for flow cytometry analysis. The percentage of apoptotic cells was recorded. All *P<0.05.
There are no reports on the regulation between POH1 and p53 or Bim. Given that p53 and Bim were upregulated in POH1-knockdown cells, we evaluated the mechanism underlying p53 and Bim regulation mediated via POH1 depletion. As a key subunit of 19S proteasome, POH1 can regulate the stability of several proteins. We found that POH1 knockdown increased the protein level of p53 and Bim but failed to affect their mRNA levels. Furthermore, the half-life of both p53 and Bim was longer in POH1-knockdown cells, and the levels of ubiquitinated p53 and Bim decreased in POH1-knockdown cells. Although it may seem a bit contradictory, we demonstrate that knockdown of POH1 (a deubiquitining enzyme) could diminish the ubiquitin modification of p53 and Bim, and we speculated there were some reasons. First, POH1 may affect some intermediate proteins that regulate p53 or Bim, such as JNK and PKM2, which could regulate the stability of p53 or Bim [35], [36], [37]. Similarly, as a deubiquitinating enzyme, USP22 silenced leads to increased FBP1 ubiquitination and decreased FBP1 protein occupancy at the p21 gene [38]. Unfortunately, knockdown of POH1 had no effect on the mRNA and protein level of JNK and PKM2 in our study (Data not shown). So we hope to detect other effective proteins involved in the regulation of p53 and bim in POH1-knockdown cells. In addition, some studies show that deubiquitination of substrates before association with the proteasome can rescue proteins from irreversible degradation, whereas deubiquitination of proteasome-associated substrates can either promote or prevent degradation, depending on the extent and timing of deubiquitination in relation to substrate release or proteolysis [20], [39], [40]. Collectively, we proposed that knockdown of POH1 would increase stability and inhibit ubiquitination of p53 and Bim.
These results suggest that POH1 may interact with p53 or Bim. We determined the interaction between POH1 and p53 (or Bim) in QGY-7701 cells by co-IP (Supplementary Figure 4). However, the specific interaction between POH1 and p53 (or Bim) needs to be further validated. Several studies suggest the involvement of the regulation p53 and Bim stability in apoptosis. For instance, Bim is essential for initiating the intrinsic apoptotic pathway, and its stability is shown to be regulated by protein kinase A [41], pyruvate kinase M2 [35], and differentiation-related gene-1 [42]. Furthermore, Zhang et al. [43] reported that TRAF6 not only inhibits p53 localization into mitochondria but also links p53 to K-63 ubiquitin chain, thereby accelerating p53 degradation and subsequently inhibiting cell apoptosis. Another study showed that knockdown of RNF2 induces apoptosis by enhancing p53 stability [44]. Together with these observations, we demonstrated that POH1 is very important for the stability and degradation of p53 and Bim and therefore suggest a new approach for the regulation of p53 and Bim in apoptosis of human cancers.
In summary, our data show that the expression of POH1 is remarkably increased in patients with HCC, EC, and CRC and is associated with unfavorable clinical outcomes. POH1 depletion, on the other hand, induces cell apoptosis by increasing stability and inhibiting ubiquitination of p53 and Bim. The intratumoral injection of POH1 siRNAs decreased tumor growth and induced apoptosis in vivo. This study suggests the potential role of POH1 as a prognostic marker and therapeutic target in human cancers.
The following are the supplementary data related to this article.
Acknowledgements
We are grateful to Yongzhong Liu for providing us with the POH1 plasmid and Prof. Tiebang Kang for the HCT116 p53−/− cell line. The authenticity of this article has been validated by uploading the key raw data onto the Research Data Deposit public platform (www.researchdata.org.cn), with the approval RDD number as RDDB2018000284.
Footnotes
Authors’ contributions: Conceived and designed the experiments: W.-C.H., L.-L.L., L.-Y.; performed the experiments: W.-C.H., Y.-X., H.-Y.F.; analyzed the data: W.-C.H., L.-L.L.; contributed reagents/materials/analysis tools: L.-S.X., C.-S.L., C.-S.H., W.-H.; and wrote the paper: W.-C.H., L.-Y., Z.-C.Z., Y.-J.P. All authors read and approved the final manuscript.
Consent for publication: Not applicable.
Ethics approval and consent to participate: This study has been approved by the Institutional Review Board and Human Ethics Committee of SYSUCC. Written consent for using the samples for research purposes was obtained from all patients prior to surgery. The project has been examined by Animal Ethical and Welfare Committee of SYSU and is in compliance with animal protection, animal welfare, and ethical principles and the relevant provisions of the National Laboratory Animal Welfare Ethics.
Competing interests: The authors declare that they have no competing interests.
The study was supported by grants from the National Key R&D Program of China (2017YFC1309000), the National Natural Science Foundation of China (No. 81702755, 81602135), and Science and Technology Program of Guangzhou (No. 201707020038).
Contributor Information
Chun-Hua Wang, Email: wangchunh@sysucc.org.cn.
Shi-Xun Lu, Email: lushix@sysucc.org.cn.
Li-Li Liu, Email: liulil@sysucc.org.cn.
Yong Li, Email: liyong@sysucc.org.cn.
Xia Yang, Email: yangxia1@sysucc.org.cn.
Yang-Fan He, Email: heyangf@sysucc.org.cn.
Shi-Lu Chen, Email: chenshil@sysucc.org.cn.
Shao-Hang Cai, Email: caishaoh@sysucc.org.cn.
Hong Wang, Email: wanghong@sysucc.org.cn.
Jing-Ping Yun, Email: yun_lab@hotmail.com.
References
- 1.Jacobson MD, Weil M, Raff MC. Programmed cell death in animal development. Cell. 1997;88:347–354. doi: 10.1016/s0092-8674(00)81873-5. [DOI] [PubMed] [Google Scholar]
- 2.Johnstone RW, Ruefli AA, Lowe SW. Apoptosis: a link between cancer genetics and chemotherapy. Cell. 2002;108:153–164. doi: 10.1016/s0092-8674(02)00625-6. [DOI] [PubMed] [Google Scholar]
- 3.Wang X. The expanding role of mitochondria in apoptosis. Genes Dev. 2001;15:2922–2933. [PubMed] [Google Scholar]
- 4.Ashkenazi A. Targeting the extrinsic apoptotic pathway in cancer: lessons learned and future directions. J Clin Invest. 2015;125:487–489. doi: 10.1172/JCI80420. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Russo A, Terrasi M, Agnese V, Santini D, Bazan V. Apoptosis: a relevant tool for anticancer therapy. Ann Oncol. 2006;17(Suppl. 7):vii115–vii123. doi: 10.1093/annonc/mdl963. [DOI] [PubMed] [Google Scholar]
- 6.Bouillet P, Huang DC, O'Reilly LA, Puthalakath H, O'Connor L, Cory S, Adams JM, Strasser A. The role of the pro-apoptotic Bcl-2 family member bim in physiological cell death. Ann N Y Acad Sci. 2000;926:83–89. doi: 10.1111/j.1749-6632.2000.tb05601.x. [DOI] [PubMed] [Google Scholar]
- 7.O'Connor L, Strasser A, O'Reilly LA, Hausmann G, Adams JM, Cory S, Huang DC. Bim: a novel member of the Bcl-2 family that promotes apoptosis. EMBO J. 1998;17:384–395. doi: 10.1093/emboj/17.2.384. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Follis AV, Chipuk JE, Fisher JC, Yun MK, Grace CR, Nourse A, Baran K, Ou L, Min L, White SW. PUMA binding induces partial unfolding within BCL-xL to disrupt p53 binding and promote apoptosis. Nat Chem Biol. 2013;9:163–168. doi: 10.1038/nchembio.1166. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Wolff S, Erster S, Palacios G, Moll UM. p53's mitochondrial translocation and MOMP action is independent of Puma and Bax and severely disrupts mitochondrial membrane integrity. Cell Res. 2008;18:733–744. doi: 10.1038/cr.2008.62. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Czabotar PE, Westphal D, Dewson G, Ma S, Hockings C, Fairlie WD, Lee EF, Yao S, Robin AY, Smith BJ. Bax crystal structures reveal how BH3 domains activate Bax and nucleate its oligomerization to induce apoptosis. Cell. 2013;152:519–531. doi: 10.1016/j.cell.2012.12.031. [DOI] [PubMed] [Google Scholar]
- 11.Letai A, Bassik MC, Walensky LD, Sorcinelli MD, Weiler S, Korsmeyer SJ. Distinct BH3 domains either sensitize or activate mitochondrial apoptosis, serving as prototype cancer therapeutics. Cancer Cell. 2002;2:183–192. doi: 10.1016/s1535-6108(02)00127-7. [DOI] [PubMed] [Google Scholar]
- 12.Leshchiner ES, Braun CR, Bird GH, Walensky LD. Direct activation of full-length proapoptotic BAK. Proc Natl Acad Sci U S A. 2013;110:E986–E995. doi: 10.1073/pnas.1214313110. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Fernald K, Kurokawa M. Evading apoptosis in cancer. Trends Cell Biol. 2013;23:620–633. doi: 10.1016/j.tcb.2013.07.006. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Vucic D, Dixit VM, Wertz IE. Ubiquitylation in apoptosis: a post-translational modification at the edge of life and death. Nat Rev Mol Cell Biol. 2011;12:439–452. doi: 10.1038/nrm3143. [DOI] [PubMed] [Google Scholar]
- 15.Finley D. Recognition and processing of ubiquitin-protein conjugates by the proteasome. Annu Rev Biochem. 2009;78:477–513. doi: 10.1146/annurev.biochem.78.081507.101607. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Matyskiela ME, Martin A. Design principles of a universal protein degradation machine. J Mol Biol. 2013;425:199–213. doi: 10.1016/j.jmb.2012.11.001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Jung T, Catalgol B, Grune T. The proteasomal system. Mol Asp Med. 2009;30:191–296. doi: 10.1016/j.mam.2009.04.001. [DOI] [PubMed] [Google Scholar]
- 18.Zhang X, Schulz R, Edmunds S, Kruger E, Markert E, Gaedcke J, Cormet-Boyaka E, Ghadimi M, Beissbarth T, Levine AJ. MicroRNA-101 suppresses tumor cell proliferation by acting as an endogenous proteasome inhibitor via targeting the proteasome assembly factor POMP. Mol Cell. 2015;59:243–257. doi: 10.1016/j.molcel.2015.05.036. [DOI] [PubMed] [Google Scholar]
- 19.Powers MV, Clarke PA, Workman P. Dual targeting of HSC70 and HSP72 inhibits HSP90 function and induces tumor-specific apoptosis. Cancer Cell. 2008;14:250–262. doi: 10.1016/j.ccr.2008.08.002. [DOI] [PubMed] [Google Scholar]
- 20.Verma R, Aravind L, Oania R, McDonald WH, Yates JR, III, Koonin EV, Deshaies RJ. Role of Rpn11 metalloprotease in deubiquitination and degradation by the 26S proteasome. Science. 2002;298:611–615. doi: 10.1126/science.1075898. [DOI] [PubMed] [Google Scholar]
- 21.Glickman MH, Adir N. The proteasome and the delicate balance between destruction and rescue. PLoS Biol. 2004;2 doi: 10.1371/journal.pbio.0020013. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Nabhan JF, Ribeiro P. The 19 S proteasomal subunit POH1 contributes to the regulation of c-Jun ubiquitination, stability, and subcellular localization. J Biol Chem. 2006;281:16099–16107. doi: 10.1074/jbc.M512086200. [DOI] [PubMed] [Google Scholar]
- 23.Schwarz T, Sohn C, Kaiser B, Jensen ED, Mansky KC. The 19S proteasomal lid subunit POH1 enhances the transcriptional activation by Mitf in osteoclasts. J Cell Biochem. 2010;109:967–974. doi: 10.1002/jcb.22475. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Hao R, Nanduri P, Rao Y, Panichelli RS, Ito A, Yoshida M, Yao TP. Proteasomes activate aggresome disassembly and clearance by producing unanchored ubiquitin chains. Mol Cell. 2013;51:819–828. doi: 10.1016/j.molcel.2013.08.016. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Gallery M, Blank JL, Lin Y, Gutierrez JA, Pulido JC, Rappoli D, Badola S, Rolfe M, Macbeth KJ. The JAMM motif of human deubiquitinase Poh1 is essential for cell viability. Mol Cancer Ther. 2007;6:262–268. doi: 10.1158/1535-7163.MCT-06-0542. [DOI] [PubMed] [Google Scholar]
- 26.Butler LR, Densham RM, Jia J, Garvin AJ, Stone HR, Shah V, Weekes D, Festy F, Beesley J, Morris JR. The proteasomal de-ubiquitinating enzyme POH1 promotes the double-strand DNA break response. EMBO J. 2012;31:3918–3934. doi: 10.1038/emboj.2012.232. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Buckley SM, Aranda-Orgilles B, Strikoudis A, Apostolou E, Loizou E, Moran-Crusio K, Farnsworth CL, Koller AA, Dasgupta R, Silva JC. Regulation of pluripotency and cellular reprogramming by the ubiquitin-proteasome system. Cell Stem Cell. 2012;11:783–798. doi: 10.1016/j.stem.2012.09.011. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Byrne A, McLaren RP, Mason P, Chai L, Dufault MR, Huang Y, Liang B, Gans JD, Zhang M, Carter K. Knockdown of human deubiquitinase PSMD14 induces cell cycle arrest and senescence. Exp Cell Res. 2010;316:258–271. doi: 10.1016/j.yexcr.2009.08.018. [DOI] [PubMed] [Google Scholar]
- 29.Wang B, Ma A, Zhang L, Jin WL, Qian Y, Xu G, Qiu B, Yang Z, Liu Y, Xia Q. POH1 deubiquitylates and stabilizes E2F1 to promote tumour formation. Nat Commun. 2015;6:8704. doi: 10.1038/ncomms9704. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Song Y, Li S, Ray A, Das DS, Qi J, Samur MK, Tai YT, Munshi N, Carrasco RD, Chauhan D. Blockade of deubiquitylating enzyme Rpn11 triggers apoptosis in multiple myeloma cells and overcomes bortezomib resistance. Oncogene. 2017;36:5631–5638. doi: 10.1038/onc.2017.172. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Luo G, Hu N, Xia X, Zhou J, Ye C. RPN11 deubiquitinase promotes proliferation and migration of breast cancer cells. Mol Med Rep. 2017;16:331–338. doi: 10.3892/mmr.2017.6587. [DOI] [PubMed] [Google Scholar]
- 32.Hu J, Dang N, Menu E, De Bruyne E, Xu D, Van Camp B, Van Valckenborgh E, Vanderkerken K. Activation of ATF4 mediates unwanted Mcl-1 accumulation by proteasome inhibition. Blood. 2012;119:826–837. doi: 10.1182/blood-2011-07-366492. [DOI] [PubMed] [Google Scholar]
- 33.Combaret V, Boyault S, Iacono I, Brejon S, Rousseau R, Puisieux A. Effect of bortezomib on human neuroblastoma: analysis of molecular mechanisms involved in cytotoxicity. Mol Cancer. 2008;7:50. doi: 10.1186/1476-4598-7-50. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Tsuchiya A, Kanno T, Shimizu T, Nakao S, Tanaka A, Tabata C, Nakano T, Nishizaki T. A novel PP2A enhancer induces caspase-independent apoptosis of MKN28 gastric cancer cells with high MEK activity. Cancer Lett. 2014;347:123–128. doi: 10.1016/j.canlet.2014.01.034. [DOI] [PubMed] [Google Scholar]
- 35.Hu W, Lu SX, Li M, Zhang C, Liu LL, Fu J, Jin JT, Luo RZ, Zhang CZ, Yun JP. Pyruvate kinase M2 prevents apoptosis via modulating Bim stability and associates with poor outcome in hepatocellular carcinoma. Oncotarget. 2015;6:6570–6583. doi: 10.18632/oncotarget.3262. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Topisirovic I, Gutierrez GJ, Chen M, Appella E, Borden KL, Ronai ZA. Control of p53 multimerization by Ubc13 is JNK-regulated. Proc Natl Acad Sci U S A. 2009;106:12676–12681. doi: 10.1073/pnas.0900596106. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Ha Thi HT, Lim HS, Kim J, Kim YM, Kim HY, Hong S. Transcriptional and post-translational regulation of Bim is essential for TGF-beta and TNF-alpha-induced apoptosis of gastric cancer cell. Biochim Biophys Acta. 2013;1830:3584–3592. doi: 10.1016/j.bbagen.2013.03.006. [DOI] [PubMed] [Google Scholar]
- 38.Atanassov BS, Dent SY. USP22 regulates cell proliferation by deubiquitinating the transcriptional regulator FBP1. EMBO Rep. 2011;12:924–930. doi: 10.1038/embor.2011.140. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Guterman A, Glickman MH. Complementary roles for Rpn11 and Ubp6 in deubiquitination and proteolysis by the proteasome. J Biol Chem. 2004;279:1729–1738. doi: 10.1074/jbc.M307050200. [DOI] [PubMed] [Google Scholar]
- 40.Yao T, Cohen RE. A cryptic protease couples deubiquitination and degradation by the proteasome. Nature. 2002;419:403–407. doi: 10.1038/nature01071. [DOI] [PubMed] [Google Scholar]
- 41.Moujalled D, Weston R, Anderton H, Ninnis R, Goel P, Coley A, Huang DC, Wu L, Strasser A, Puthalakath H. Cyclic-AMP-dependent protein kinase A regulates apoptosis by stabilizing the BH3-only protein Bim. EMBO Rep. 2011;12:77–83. doi: 10.1038/embor.2010.190. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42.Ambrosini G, Seelman SL, Schwartz GK. Differentiation-related gene-1 decreases Bim stability by proteasome-mediated degradation. Cancer Res. 2009;69:6115–6121. doi: 10.1158/0008-5472.CAN-08-3024. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43.Zhang X, Li CF, Zhang L, Wu CY, Han L, Jin G, Rezaeian AH, Han F, Liu C, Xu C. TRAF6 Restricts p53 Mitochondrial Translocation, Apoptosis, and Tumor Suppression. Mol Cell. 2016;64:803–814. doi: 10.1016/j.molcel.2016.10.002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44.Wen W, Peng C, Kim MO, Ho Jeong C, Zhu F, Yao K, Zykova T, Ma W, Carper A, Langfald A. Knockdown of RNF2 induces apoptosis by regulating MDM2 and p53 stability. Oncogene. 2014;33:421–428. doi: 10.1038/onc.2012.605. [DOI] [PMC free article] [PubMed] [Google Scholar]