

Abstract
HDM2 and HDMX are two homologs essential for controlling p53 tumor suppressor activity under normal conditions. Both proteins bind different sites on the p53 N‐terminus, and while HDM2 has E3 ubiquitin ligase activity towards p53, HDMX does not. Nevertheless, HDMX is required for p53 polyubiquitination and degradation, but the underlying molecular mechanism remains unclear. Alone, HDMX and HDM2 interact via their respective C‐terminal RING domains but here we show that the presence of p53 induces an N‐terminal interface under normal cellular conditions. This results in an increase in HDM2‐mediated p53 polyubiquitination and degradation. The HDM2 inhibitor Nutlin‐3 binds the N‐terminal p53 binding pocket and is sufficient to induce the HDM2‐HDMX interaction, suggesting that the mechanism depends on allosteric changes that control the multiprotein complex formation. These results demonstrate an allosteric interchange between three different proteins (HDMX‐HDM2‐p53) and help to explain the molecular mechanisms of HDM2‐inhibitory drugs.
Keywords: HDM2, HDMX, p53, allosteric interactions, E3 ubiquitin ligase substrate specificity
Introduction
Many different stress pathways converge on the tumor suppressor p53 transcription factor to activate a specific cellular response, such as cell cycle arrest in G1 or G2, DNA repair, or irreversible events such as apoptosis or senescence.1, 2, 3 Under normal cellular conditions p53 is tightly down regulated by the two nonredundant paralogous proteins, HDM2 and HDMX (MDM2 and MDMX in mice).4, 5 HDM2 is an E3 ubiquitin ligase carrying a C‐terminal RING (Really Interesting New Gene) domain, whereas HDMX lacks this activity despite sharing 75% overall similarity in this region. The C‐terminal HDM2 and HDMX RING‐RING interaction is essential to ensure low levels of p53 under normal conditions, consequently stabilizing HDM2 and providing an optimal scaffold for E2 ubiquitin conjugate interaction.6, 7, 8 Mice harboring MDMX lacking the RING‐like domain show an embryonic lethal phenotype, similar to MDMX‐null mice, which can be rescued by the concomitant deletion of p53.9 Both proteins bind p53 through their respective N‐terminal regions,10, 11 and even though the overall structure of the HDM2‐p53 interaction also is preserved in the HDMX‐p53 interface, the hydrophobic pocket of HDMX to which p53 binds is slightly different in size and shape.12, 13 This explains why some antagonists of the HDM2‐p53 interaction, such as the Nutlin‐3, are ineffective for inhibiting the HDMX‐p53 association. An additional site for the interaction between the HDM2 acidic domain and the p53 DNA‐binding core domain is promoted by ligands that bind the N‐terminal hydrophobic pocket of HDM2 and promote p53 ubiquitination.14, 15, 16, 17 More recently, a secondary allosteric interaction site involving the acidic and C‐terminal domains of HDMX with the p53 DNA binding domain has been described. This interaction was found to repress p53 DNA binding capacity.18 In addition to the cooperative interactions of p53‐HDM2 and p53‐HDMX that control p53 activity, allosteric interactions between HDM2 and HDMX also play important roles in controlling p53 activity. Under conditions of DNA damage, ATM‐mediated phosphorylation of HDMX and HDM2 at serine residues 403 and 395, respectively, promote HDM2 and HDMX heterodimer formation and induce their interaction with the p53 mRNA, which creates an interface between the HDMX and HDM2 N‐termini. This interface switches HDM2‐mediated ubiquitination away from p53 and instead towards HDMX and HDM2, allowing p53 activation.19, 20, 21, 22 Hence, the previous model, where HDM2 is responsible for p53 polyubiquitination and HDMX for preventing p53 transactivation, is now complemented by a more dynamic model, where allosteric regulation involving the different domains of the p53‐HDM2‐HDMX multiprotein complex regulate the E3 ubiquitin ligase activity of HDM2 and stability of p53 under different conditions.6, 23
Here we show the role of p53 in enhancing the interaction between HDM2 and HMDX under normal conditions. In the absence of p53, the C‐termini of HDMX and HDM2 are crucial for the interaction. In the presence of p53, the HDM2‐RING domain is not necessary for proper recognition of HDMX, and the N‐terminus is a site of a primary interaction. This highlights the importance of all domains of the protein in proper protein–protein recognition.
Results
p53 promotes the HDM2‐HDMX interaction in vitro and in cells
Emerging models show that HDMX assists HDM2‐mediated p53 ubiquitination, and we have previously shown that allosteric changes in HDMX and HDM2 following DNA damage create a new interface between the two proteins, which redirects HDM2 E3 ligase activity away from p53 and towards itself and HDMX.21 In accordance to previous reports we observed that HDMX facilitates HDM2 E3 ligase activity towards p53 under normal cellular conditions [Fig. 1(A)]; however, the interaction between HDMX and HDM2 is weaker under normal conditions compared to DNA damage21, 24 [Fig. 1(B)]. This observation prompted us to investigate the mechanism of HDM2‐HDMX heterodimerization under normal conditions in presence of p53. By using recombinant purified proteins [Fig. 1(C)], we firstly tested the affinity of HDMX versus HDM2 in the absence or presence of p53. In Figure 1(D) we describe the ELISA assay we used to analyze the HDMX and HDM2 interaction. ELISA plates were coated with 10 ng/μL of HDMX, to which an increasing amount of HDM2 (0–10 ng/μL) was added, with or without the addition of a fixed amount of p53 (10 ng/μL). The p53‐dependent regulation of HDMX‐HDM2 affinity was estimated using a 2A10 anti‐HDM2 antibody [Fig. 1(D)]. This revealed that the HDMX‐HDM2 interaction increased in the presence of p53 from a K diss of 131 nM in absence of p53 to 39 nM in the presence of the p53 protein [Fig. 2(A)]. As a positive control we performed an ELISA of the interaction between HDM2 and p53 with a K diss of 8.5 nM, HSP90 protein was used as a negative control of the interaction with HDM2 and both assays were developed with 2A10 anti‐HDM2 antibody [Fig. 2(B)]. The positive effect of p53 on HDM2‐HDMX binding was also observed by a coimmunoprecipitation assay using the H1299 cell line, which is p53 null [Fig. 2(C)]. In comparison with HDM2‐mediated p53 ubiquitination, establishment of the p53‐HDMX‐HDM2 triple interaction resulted in the polyubiquitination of p53 [Fig. 1(A)]. To confirm this effect in cells we performed a proximity ligase assay (PLA), which gives a specific signal if two different epitopes are located within 40 nm of each other.25 We expressed the HDM2 in the H1299 cell line that in addition to be p53 null, has a double knock out on HDMX and HDM2 (H1299DKO) to avoid endogenous background. Cells were transfected with HDM2 and HDMX in either the presence or absence of p53. As expected, we observed the same p53‐dependent induction of the interaction between HDMX and the HDM2, it is worth to note that the interaction is mostly cytoplasmic [Fig. 3(A,B)].
Figure 1.
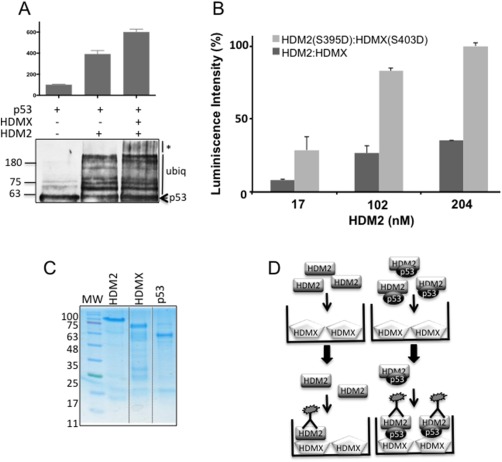
(A) In vitro ubiquitination of recombinant p53 with HDM2 in the presence or absence of HDMX. The presence of HDMX increased the size of the p53‐ubiquitinated forms. The assay was developed with CM1 anti‐p53. (B) ELISA assay showing the interaction between HDMX and HDM2 versus HDMX(S403D) and HDM2(S395D) phosphomimetic proteins. The plates were covered with a fixed amount of HDMX (204 nM) and increasing amounts of HDM2 (0 to 10 ng/μL). The interaction of the two phosphomimetic proteins is around three times bigger in comparison with the two wild‐types proteins. (C) Recombinant purified HDMX, HDM2 and p53 proteins. (D) Scheme representing the ELISA assay performed in absence or presence of p53. The plates were covered with HDMX, HDM2, or HDM2 plus p53 and developed with an antibody YB25 against HDM2.
Figure 2.
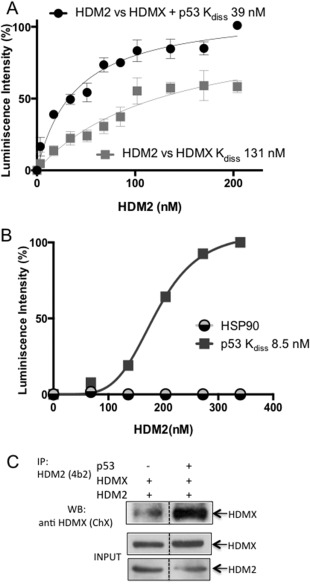
p53 enhances the interaction between HDMX and HDM2. (A) ELISA assay showing the effect of p53 on the HDMX‐HDM2 interaction. The plates were covered with a fixed amount of HDMX (10 ng/μL) and increasing amounts of HDM2 (0–10 ng/μL) in the presence or absence of a fixed amount of recombinant purified p53 (10 ng/μL). The interaction between HDMX and HDM2 increases in the presence of p53. Anti‐HDM2 YB25 antibody was used to develop the assay. The average of five independent experiments is shown. (B) ELISA assay showing that HDM2 interacts with p53 in a specific manner; HSP90 was used as a negative control of the interaction. The plates were covered with a fixed amount of p53 or HSP90 (20 ng/μL) and increasing amounts of HDM2 (0–20 ng/μL) and developed with 2A10 anti‐HDM2 antibody. (C) Coimmunoprecipitation assay performed in the H1299 cell line. Cells were transfected with HDMX and HDM2, with or without p53. The 4B2 anti‐HDM2 antibody was used to pull down HDM2, and the ChX chicken anti‐HDMX antibody was used to develop the western blot.
Figure 3.
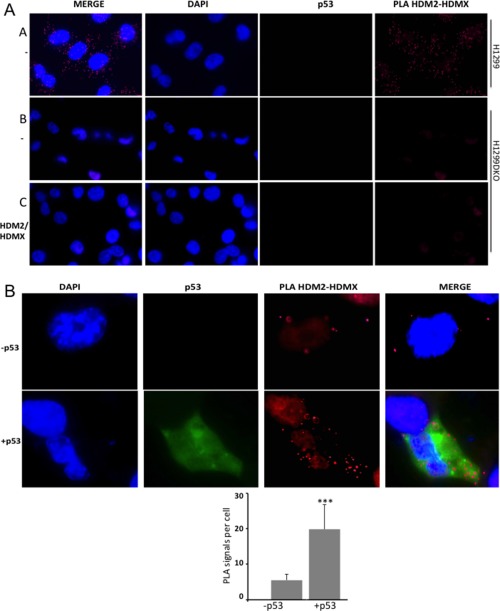
p53 enhances the interaction between HDMX and HDM2 in cells. (A) Controls of the proximity ligase assay (PLA) experiments. Panel A shows nontransfected cells H1299 that were analyzed for the HDMX‐HDM2 interaction. Dots representing the interaction of the endogens proteins can be appreciated. A double knockout H1299 HDM2/HDMX (H1299DKO) cells was done in order to observe the interaction of the transfected constructs avoiding the interferences of the endogenous proteins. Panel B shows PLA of the interaction of the HDMX‐HDM2 using nontransfected double knockout cells H1299DKO. Panel C shows transfected cells H1299DKO with HDMX and HDM2, the PLA was made using just one primary antiHDMX 5.1 antibody as a control. (B) The PLA for the HDMX and HDM2 interaction in the presence or absence of p53. H1299DKO cells were transfected with HDMX and HDM2 (upper panel) or with HDMX, HDM2 and p53 (lower panel). Anti‐HDM2 rabbit and anti‐HDMX mouse 5.1 were used. For IF of p53 anti‐p53 chicken antibody was used. Cell nuclei is visualized in blue with DAPI, p53 in green and the interaction in red. At least 30 cells were analyzed for each experiment. The quantification of the interaction was done with Image J software (*P < 0.05, **P < 0.01, ***P < 0.001).
Both HDMX and HDM2 contain a p53‐binding site in their respective N‐terminus domains and form heterodimers via their C‐terminal RING domains, thus, there are several possible scenarios for p53‐induced HDMX‐HDM2 interaction. One possibility is an increase in RING‐RING affinity or by p53 forming a “sandwich” between HDM2 and HDMX as have been previously suggested.8 Other possibilities include allosteric effects related to p53 binding HDM2 and/or HDMX.
The RING‐RING interaction is not required for the p53‐dependent induction of HDM2‐HDMX interaction
To evaluate these possible models, we first created a deletion of HDM2 C‐terminal domain to investigate their role in the RING‐RING interaction. The construct lacked the C‐terminal RING domain of HDM2(1–432) [Fig. 4(A)]. The construct was well expressed and purified using a recombinant expression system [Fig. 4(B)]. It was tested for their interaction with the full‐length HDMX protein in the presence or absence of p53 by ELISA assays. We observed that deletion of the RING domain of HDM2(1–432) abolished the interaction with HDMX, which is in line with previous studies that have shown that the RING domain is essential for the heterodimerization. Interestingly, the stimulatory effect of p53 on the HDM2(1–432)‐HMDX interaction remained [Fig. 4(C)]. In order to confirm that this result was not restricted to in vitro, we also carried out a PLA. We expressed the HDM2(1–432) deletion in the H1299DKO, in either the presence or absence of p53. This resulted in the same p53‐dependent induction of the interaction between HDMX and the HDM2(1–432) and we also observed that the interaction remains cytoplasmic (Fig. 5). These data show that p53 stimulates the interaction between HDMX and HDM2 via an interface that is independent of the RING domain, then may be located in the N‐terminal region of HDM2 or HDMX.
Figure 4.
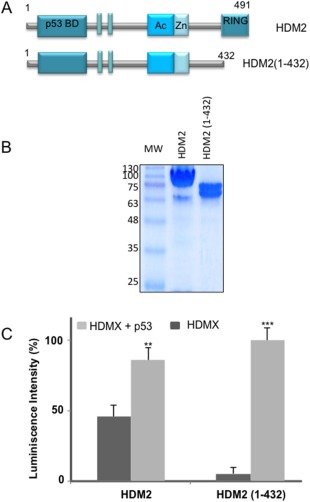
p53 increases the HDMX‐HDM2 interaction in a RING‐RING independent manner. (A) Cartoon illustrating HDM2 and its construct lacking a portion of the C‐terminal. HDM2 comprises 491 amino acid organized in a most N‐terminal domain that regulates the hydrophobic pocket containing the p53 binding site, the nuclear localization signal and nuclear export signal. An acidic domain, the zinc‐finger domain, and in the very C‐terminus contains the Really Interesting New Gene (RING) domain. The construct used in this work HDM2(1–432) (amino acids 1–432) lacking the RING C‐terminal domain. (B) Recombinants purified HDM2 and the HDM2 (1–432) construct. (C) ELISA assay using a fixed amount of HDMX (10 ng/μL) and a fixed amount of HDM2(1–432) (10 ng/μL) in the presence or absence of p53 (10 ng/μL). Anti‐HDM2 2A10 antibody was used to develop the assay (*P < 0.05, **P < 0.01, ***P < 0.001).
Figure 5.
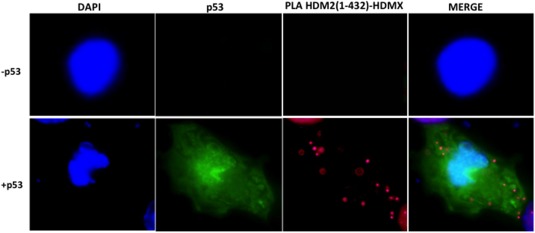
Proximity ligase assay for the HDMX and HDM2 (1–432) interaction, with or without p53 protein. H1299DKO cells were transfected with HDMX and HDM2(1–432) (upper panel) or with HDMX, HDM2(1–432), and p53 (lower panel). Anti HDM2 rabbit and anti‐HDMX‐mouse 5.1 were used, for IF of p53 anti‐p53 chicken antibody was used. Cell nuclei is visualized in blue with DAPI, p53 in green and the interaction in red.
The role of the N‐termini of HDMX and HMD2 on the p53 effect
Having shown that the RING‐RING interaction between HDMX and HDM2 is not required for p53‐dependent induction of HDM2‐HDMX heterodimers, we turned our attention to the N‐terminus of HDMX and HDM2 that both bind p53. We tested the affinity of HDM2 to a recombinant HDMX protein that lacks the N‐terminus p53‐binding domain, corresponding to the alternative translated HDMXp60 isoform initiated within the seventh AUG codon at position 12826 in the presence or absence of p53 [Fig. 6(A)]. The addition of p53 led to a slightly increased interaction at low concentrations but at higher concentrations the effect was lost. On the other hand, by using a construct of HDM2(241–491) that lacks the N‐terminal region, the presence of p53 in this case marginally decreased the interaction between HDMX and the HDM2(241–491) construct [Fig. 6(B)].
Figure 6.
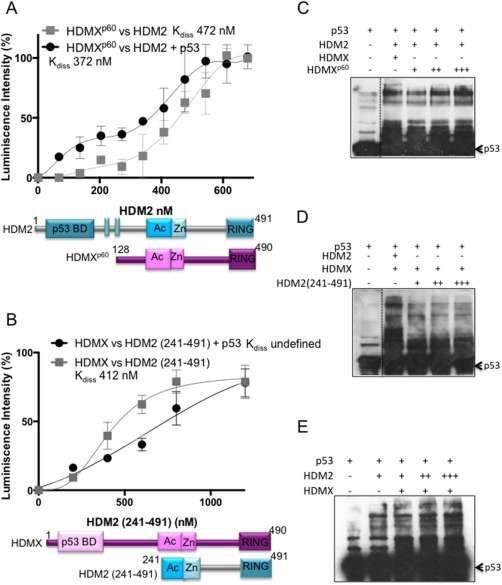
Effect of the N‐terminal domain of HDMX and HMD2 on the heterodimer formation. (A) ELISA assay using a fixed amount of recombinant purified HDMXp60 (10 ng/μL) and increasing amounts of HDM2 (0–40 ng/μL) in the presence or absence of p53 (10 ng/μL). The average of five independent experiments and SD are shown. (B) ELISA assay using a fixed (10 ng/μL) amount of recombinant purified HDMX and increasing amounts of HDM2(241–491) (0–60 ng/μL) in the presence or absence of p53 (10 ng/μL). The average of five independent experiments and SD are shown. (C) In vitro ubiquitination of recombinant p53 with HDM2 in the presence of HDMX or increasing concentrations of HDMXp60. The presence of higher concentrations of HDMXp60 increased the p53‐ubiquitination (D). In vitro ubiquitination of recombinant p53 with increasing concentrations of HDM2(241–492) in the presence of HDMX. The presence of higher concentrations of HDM2(241–492) impairs the p53‐ubiquitination, showing the importance of the N‐terminal of HDM2 protein in the effect of p53. (E) In vitro ubiquitination of recombinant p53 with increasing amounts of HDM2 in the presence of HDMX. The presence of higher concentrations of HDM2 increased the p53‐ubiquitination as expected. The assays of ubiquitination were developed with CM1 anti‐p53.
We then performed in vitro p53 ubiquitination assays using the HDM2 full‐length and increasing concentrations of the alternative translated isoform HDMXp60. Our aim was to determine if the absence of the N‐terminal part of HDMX mimicry the p53 effect on the heterodimer formation. That could explain the lack of effect with this construct but the resulted heterodimer could support p53 poly‐ubiquitination. The reaction with HDM2 and HDMXp60 resulted dose dependent and in the highest abundance of HDMXp60 highest amount of ubiquitinated p53, as we expected [Fig. 6(C)]. We performed a similar experiment but using increasing concentration of the HDM2(241–491). In this case we observed less amount of ubiquitinated p53 [Fig. 6(D)]. Likewise, when we increased HDM2 as a control, we observed a higher abundance of ubiquitinated p53 [Fig. 6(E)]. Together these experiments provide evidence that the binding of p53 with the N‐termini of both proteins is critical for the p53 positive effect in the HDM2‐HDMX heterodimer formation.
The observation that using the isoforms HDMXp60 or HDM2(241–491), that lack of the N‐termini of the proteins, the p53 effect in the binding disappear, together with the fact that the ubiquitination of HDM2(241–491), that contains the RING E3‐activity, decrease the p53 ubiquitination, strongly suggest that the binding of p53 on HDM2 N‐termini promotes a conformation that increased the formation of a functional heterodimer. This implicates an allosteric change on the proteins induced by p53 binding. To probe our hypothesis we performed an ELISA, but instead of p53 we used Nutlin‐3, which binds the same hydrophobic pocket in HDM2 as p53. Increasing concentrations of Nutlin‐3 (0–700 μM) resulted in similar induction of the HDM2‐HDMX interaction [Fig. 7(A)], indicating that ligands that bind the hydrophobic pocket of HDM2 induce a conformation change that affects the ability of HDM2 to regulate HDM2 and HDMX heterodimer formation; the effect is observed since 300 μM of Nutlin‐3. Similarly, the interaction between HDMX and the C‐terminal‐deletion HDM2(1–432) construct is increased in the presence of Nutlin‐3 [Fig. 7(B)], in this case the effect is stronger since it is observed at 100 μM of the compound. These results suggest that in the presence of Nutlin‐3 the interaction between HDM2 and HDMX increases, then the ubiquitination of HDMX by HDM2 also would increase. To test this hypothesis, we performed in vitro and in cells ubiquitination experiments. In vitro ubiquitination shows increased bands of HDMX ubiquitinated in the presence of Nutlin‐3 [Fig. 7(C)]. In cells however, the degradation of HDMX in the presence of Nutlin‐3 was so strong that we were not able to see the ubiquitination bands, but the disappearance of HDMX protein, even in the presence of MG132 proteasome inhibitor [Fig. 7(D)]. Both experiments confirm that in the presence of Nutlin‐3 the interaction between the HDMX and HDM2 increases, as well as the HDMX ubiquitination.
Figure 7.
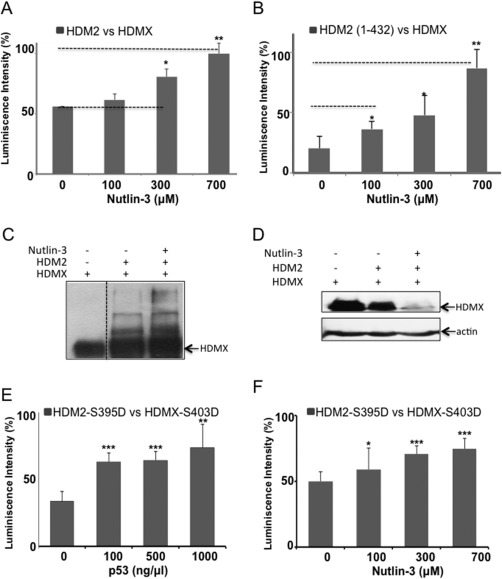
Effect of Nutlin‐3 on the HDMX‐HDM2 interaction. (A) ELISA assay using a fixed amount of HDMX (10 ng/μL) and a fixed amount of HDM2 (10 ng/μL) in the presence of different concentrations of Nutlin‐3 (0–700 μM). As with p53 protein, the presence of Nutlin‐3 increased the interaction between HDMX and HDM2. (B) ELISA assay as in (A) but using HDM2 (1–432) construct that lacks the C‐terminal domain (C) In vitro ubiquitination of recombinant HDMX with HDM2 in presence Nutlin‐3. The compound increased HMDX ubiquitination mediated by HDM2. (D) In cells ubiquitination of HDMX with HDM2 in presence of Nutlin‐3. The compound degraded HMDX even with MG132 proteasome inhibitor. The assays of ubiquitination were developed with 4.1 anti‐HDMX (E) ELISA assay using a fixed amount of HDMX(S403D) (10 ng/μL) and a fixed amount of HDM2(S395D) (10 ng/μL) in the presence of different concentrations of p53 (0–20 ng/μL). p53 increase the binding between the two phosphomimetic proteins. (F) ELISA assay using a fixed amount of HDMX(S403D) (10 ng/μL) and a fixed amount of HDM2(S395D) (10 ng/μL) in the presence of different concentrations of Nutlin‐3 (0–700 μM). As with p53 protein, the Nutlin‐3 increased the interaction between HDMX and HDM2. The assays were developed with YB25 anti‐HDM2 (*P < 0.05, **P < 0.01 and ***p < 0.001).
Finally, we tested if the effects of p53 and Nutlin‐3 were also observed with the phosphomimetic HDMX(S403D) and HDM2(S395D) proteins. We perform ELISA assays between these two proteins with increasing concentrations of p53 or Nutlin‐3. An augment in the interaction between the two proteins was shown in the presence either of p53 protein or Nutlin‐3 [Fig. 7(E, F)]. These experiments confirm our previous results showing that the phosphomimetic HDM2(S395D) binds p53 protein, and only the presence of p53 mRNA and its binding to the RING domain of HDM2(S395D) avoids the interaction with the p53 protein.21
Taken together, the allosteric changes in HDM2 conformation induced by Nutlin‐3 and how this regulates the interaction with HDMX resembles the interaction cascade taking place between p53 and HDM2 following Nutlin‐3 treatment.14, 27, 28
Discussion
HDM2 is able to inhibit p53 activity through its E3 ubiquitin ligase activity and p53 polyubiquitination. Despite not having its own E3 ligase activity, HDMX assists HMD2‐mediated polyubiquitination of p53 by stabilizing HDM2 to obtain the proper conformation to engage with its E2 ligase.24 Studies on the heterodimerization of HDM2 and HMDX have suggested that the absence of the HDM2 RING domain inhibits its interaction with HDMX.6, 7, 17, 19, 29, 30, 31 However, the results presented here show that under normal conditions the RING domain of HDM2 is crucial for the interaction, but only in the absence of p53. The presence of p53 enhances the interaction between HDMX and HDM2, even without the RING domain of HDM2, increasing the p53 polyubiquitination. In the lack of the N‐terminal p53‐binding region of HDM2 or HDMX, the presence of p53 did not stimulate the interaction between HDMX‐HDM2. This could be interpreted as a conformational change induced by the binding of p53. The effect of p53 on promoting the HDM2‐HDMX interaction is mimicked by Nutlin‐3, which binds the p53 hydrophobic pocket of HDM2 but not HDMX promoting HDMX ubiquitination (Fig. 8). This interaction takes place in the cytoplasm of the cells (Figs. 3 and 5), in concordance with the previous report of Wang et al., 2011, where they showed that the p53 ubiquitination by MDM2 and MDMX is mainly cytoplasmic.24
Figure 8.
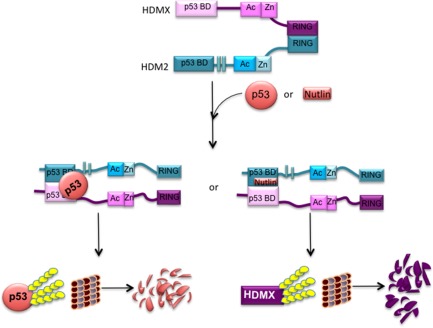
Model of the mechanism underlying the formation of the ternary complex between p53, HDMX, and HDM2.
Previous reports have shown that HDM2 and p53 interaction involves more than one domain14, 15, 16; for instance, the HDMX‐p53 interaction takes place within a second cooperative interacting site between the p53 DNA binding domain with the acidic and RING domains of HDMX.18 In addition, the interaction between the N‐terminal of both HDMX and HDM2 under DNA damage conditions is assisted by the p53 mRNA.21 All those cases exemplify a more dynamic model where the entire structure of the proteins becomes necessary for the proper recognition and function. Our results support a model in which a ternary complex between HDM2, HDMX, and p53 involves the interaction between the N‐terminal domains of the three proteins promoting p53 poly‐ubiquitination
Using deleting‐domains constructs, we were able to observe that even if a specific activity belongs to a specific domain, the rest of the protein serves as a scaffold to ensure proper recognition and function. Taken together, we observed that the plasticity of multidomain proteins facilitate their interaction with many different partners in order to perform multiple functions. While each domain has a specific function, the whole protein plays a crucial role in providing an additional level of regulation.
Materials and Methods
Plasmids and antibodies
The cDNA for HDM2(1–432), HDM2(241–491), and HDMXp60 were PCR amplified and cloned in pET28a, and/or pcDNA. Proteins were detected with 4B2, CM1, 2A10, 4.1, and 5.1 from RECAMO and ChX‐anti‐HDMX and YB25‐anti HDM2 that are chicken anti‐HDMX and rabbit anti‐HDM2 homemade antibodies; mouse anti‐HIS (Invitrogen). IP's were performed using protein G‐Sepharose beads (Sigma) and developed with ECL mix (GE healthcare).
Purification of recombinant protein
Histidine tagged recombinant proteins HDM2, HDMX, HDMXp60, HDM2(1–432) and HDM2(241–491), were produced in E. coli BL21 (DE3) cells cultured at 37°C and purified using HiTrap Nickel columns (GE healthcare). Lysis and buffer A (50 mM Tris, 200 mM NaCl, 40 mM Imidazol, 10 μM ZnSO4, 10% glycerol, were complemented with 10 mg of phenylmethanesulfonyl fluoride (PMSF) (SIGMA). The pellet was resuspended in 10 mL of Urea buffer (Urea 8M, 50 mM Tris pH 8.5, 150 mM NaCl, 10 mM Imidazole) and incubated for 2 h at room temperature. After centrifugation the supernatant was loaded into the nickel column and washed with the following buffers; 25 mL of urea buffer, 25 mL of urea buffer plus 0.2% triton and 25 mL of buffer A plus 10 mM of β‐mercaptoethanol. The refolding of the proteins takes place into the column with the last washed step, and we took out the urea. Recombinant proteins already folded were then eluted from Nickel columns with buffer B (50 mM Tris pH 8.5, 200 mM NaCl, 10 µM ZnSO4, 300 mM imidazole). Protein were dialyzed against phosphate buffer with 10 μM ZnSO and concentrated in centrifugal filter units Amicon, (Merck Millipore). p53 was induced at 15°C ON and purified on a HiTrap Nickel column from the soluble fraction. Proteins were quantified and analyzed in SDS–PAGE.
ELISA
Ninety‐six well plates (Thermo scientific) were coated with 10 ng/μL of protein in 0.1M NaHCO3 (200 μL/well) overnight at 4°C. After incubation, plates were washed 6 × 200 μL with 0.1% PBS‐Tween and blocked for 1 h at room temperature with 0.1% PBS‐Tween containing BSA 3% and then washed 6 × 200 μL with 0.1% PBS‐Tween. The second bound protein was diluted (from 0 to 10 or 20 μg/μL) and added to the plate. For ternary complexes, p53 protein (10 ng/μL) was mixed with the second bound protein and incubated 1 h at 4°C, prior to loading in the plates. Plates were washed 6 × 200 μL with 0.1% PBS‐Tween and incubated with the primary specific antibody (1:1000) for 1 h at room temperature. After washing 6 × 200 μL with 0.1% PBS‐Tween, secondary antibody was incubated for 1 h at room temperature. Plates were washed and incubated with 100 μL/well of ECL mix (GE healthcare) and luminescence was measured in a Cary eclipse fluorescence spectrophotometer.
In vitro ubiquitination assays
Ubiquitination reactions contained 25 mM HEPES (pH 8.0); 10 mM MgCl2; 4 mM ATP; 0.5 mM DTT; 0.05% (v/v) Triton X‐100; 0.25 mM benzamidine; 10 mM creatine phosphate; 3.5 units/mL creatine kinase; ubiquitin or His‐tagged ubiquitin (2 μg); and E1 (100 nM), E2 (1 μM), and the proteins of interest; p53 at 5 μg, HDMX and HDMXp60 at 10 μg. Reactions were started with the addition of HDM2 or HDM2(241–491), incubated for 15 min at 30°C, and analyzed with 5–10% SDS–PAGE followed by immunoblotting.
HDM2/HDMX knockout in H1299 (H1299DKO)
HDM2 and HDMX knockouts in H1299 cell line were generated using CRISPR Cas9 expressed in lentiviral expression system. We used lentiCRISPR‐V2 vector to target coding region of human HDM2 at p53 binding domain, sequence: 5′‐ATCGTTTAGTCATAATATACTGG‐3′ (PAM sequence underlined). Annealed oligos 5′CACCGATCGTTTAGTCATAATATAC3′ and 5′AAACGTATATTATGACTAAACGATC3′ were cloned into lentiCRISPR‐V2 vector using protocol described by Shalem et al. (https://www.ncbi.nlm.nih.gov/pubmed/24336571). HDMX knockout was also designed to target coding sequence of HDMX gene at p53 binding domain, sequence 5′‐ATCTTCAAAAGCGGCAGTTTTGG‐3′, using primers 5′‐CACCGATCTTCAAAAGCGGCAGTTT‐3′ and 5′‐AAACAAACTGCCGCTTTTGAAGATC‐3′. HDMX sg RNA was cloned into lentiCRISPR‐V2‐Blast in which the original puromycin resistance gene was replaced by blasticidin to enable double selection.
The lentivirus was produced in HEK293 cells using standard protocol (https://www.ncbi.nlm.nih.gov/pubmed/24336571). The double knockouts of HDM2 and HDMX in H1299 cell line were performed stepwise. The cells transduced by HDM2 CRISPR were selected by puromycin (5 μg/mL). The cell clone containing HDM2 knockout was then subjected to transduction with HDMX CRISPR and selected by blasticidin (10 μg/mL). The selected cells were cloned from single cell. The original lentiCRISPR v2 was a gift from Feng Zhang (Addgene plasmid # 52961).
PLA assay
Proximal Ligation Assay (PLA) method was performed according to the manufacturer's instructions. Briefly, H1299 cells double knockout HDMX and HDM2 (H1299DKO) were grown on coverslips and transfected with the indicated constructs, then fixed in 4% paraformaldehyde for 10 min before permeabilization and blocked in PBS containing 0.1% saponine PBS 1× and BSA3% for 30 min. Primary antibodies were incubated anti‐HDM2‐rabbit and anti‐HDMX‐mouse 5.1 and for IF of p53 anti‐p53 chicken antibody were incubated 1 h at 37°C each. After washing the cells, PLA probes were added, followed by hybridization, ligation 30 min 37°C and amplification 100 min at 37°C. DNA, p53, and the interaction were visualized after incubation with the detection solution in blue, green, and in red, respectively. The slides were analyzed by fluorescence microscopy and the ImageJ software. We counted dots in 30 cells randomly selected during the imaging. Nontransfected cells were excluded from the statistical analyses.
Conflict of Interest
The authors claim no conflict of interest.
Author Contributions
Conceived and designed experiment: VO‐I and IM‐M, analyzed data: VO‐I and RF; performed the experiments: MM‐S, IM‐M, PM and JH‐M; wrote of manuscript: VOI.
Acknowledgments
We thank to Yolanda Rebolloso‐Gómez and Celina González‐Gallegos for technical support.
References
- 1. Olivares‐Illana V, Fåhraeus R (2010) p53 isoforms gain functions. Oncogene 29:5113–5119. [DOI] [PubMed] [Google Scholar]
- 2. Vogelstein B, Lane D, Levine AJ (2000) Surfing the p53 network. Nature 408:307–310. [DOI] [PubMed] [Google Scholar]
- 3. Levine AJ, Oren M (2009) The first 30 years of p53: growing ever more complex. Nat Rev Cancer 9:749–758. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4. Parant J, Chavez‐Reyes A, Little NA, Yan W, Reinke V, Jochemsen AG, Lozano G (2001) Rescue of embryonic lethality in Mdm4‐null mice by loss of Trp53 suggests a nonoverlapping pathway with MDM2 to regulate p53. Nat Genet 29:92–95. [DOI] [PubMed] [Google Scholar]
- 5. Montes de Oca Luna R, Wagner DS, Lozano G (1995) Rescue of early embryonic lethality in mdm2‐deficient mice by deletion of p53. Nature 378:203–206. [DOI] [PubMed] [Google Scholar]
- 6. Gu J, Kawai H, Nie L, Kitao H, Wiederschain D, Jochemsen AG, Parant J, Lozano G, Yuan Z‐M (2002) Mutual dependence of MDM2 and MDMX in their functional inactivation of p53. J Biol Chem 277:19251–19254. [DOI] [PubMed] [Google Scholar]
- 7. Linke K, Mace PD, Smith CA, Vaux DL, Silke J, Day CL (2008) Structure of the MDM2/MDMX RING domain heterodimer reveals dimerization is required for their ubiquitylation in trans. Cell Death Differ 15:841–848. [DOI] [PubMed] [Google Scholar]
- 8. Wade M, Wahl GM (2009) Targeting Mdm2 and Mdmx in cancer therapy: better living through medicinal chemistry? Mol Cancer Res 7:1–11. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9. Pant V, Xiong S, Iwakuma T, Quintas‐Cardama A, Lozano G (2011) Heterodimerization of Mdm2 and Mdm4 is critical for regulating p53 activity during embryogenesis but dispensable for p53 and Mdm2 stability. Proc Natl Acad Sci U S A 108:11995–12000. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10. Shvarts A, Steegenga WT, Riteco N, van Laar T, Dekker P, Bazuine M, van Ham RCA, van der Houven van Oordt W, Hateboer G, van der Eb AJ, Jochemsen AG (1996) MDMX: a novel p53‐binding protein with some functional properties of MDM2. EMBO J 15:5349–5357. [PMC free article] [PubMed] [Google Scholar]
- 11. Picksley SM, Vojtesek B, Sparks A, Lane DP (1994) Immunochemical analysis of the interaction of p53 with MDM2–fine mapping of the MDM2 binding site on p53 using synthetic peptides. Oncogene 9:2523–2529. [PubMed] [Google Scholar]
- 12. Kussie PH, Gorina S, Marechal V, Elenbaas B, Moreau J, Levine AJ, Pavletich NP (1996) Structure of the MDM2 oncoprotein bound to the p53 tumor suppressor transactivation domain. Science 274:948–953. [DOI] [PubMed] [Google Scholar]
- 13. Popowicz GM, Czarna A, Holak TA (2008) Structure of the human Mdmx protein bound to the p53 tumor suppressor transactivation domain. Cell Cycle 7:2441–2443. [DOI] [PubMed] [Google Scholar]
- 14. Wallace M, Worrall E, Pettersson S, Hupp TR, Ball KL (2006) Dual‐site regulation of MDM2 E3‐ubiquitin ligase activity. Mol Cell 23:251–263. [DOI] [PubMed] [Google Scholar]
- 15. Shimizu H, Burch LR, Smith AJ, Dornan D, Wallace M, Ball KL, Hupp TR (2002) The conformationally flexible S9‐S10 linker region in the core domain of p53 contains a novel MDM2 binding site whose mutation increases ubiquitination of p53 in vivo. J Biol Chem 277:28446–28458. [DOI] [PubMed] [Google Scholar]
- 16. Yu GW, Rudiger S, Veprintsev D, Freund S, Fernandez‐Fernandez MR, Fersht AR (2006) The central region of HDM2 provides a second binding site for p53. Proc Natl Acad Sci U S A 103:1227–1232. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17. Kawai H, Wiederschain D, Yuan ZM (2003) Critical contribution of the MDM2 acidic domain to p53 ubiquitination. Mol Cell Biol 23:4939–4947. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18. Wei X, Wu S, Song T, Chen L, Gao M, Borcherds W, Daughdrill GW, Chen J (2016) Secondary interaction between MDMX and p53 core domain inhibits p53 DNA binding. Proc Natl Acad Sci U S A 113:E2558–E2563. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19. Tanimura S, Ohtsuka S, Mitsui K, Shirouzu K, Yoshimura A, Ohtsubo M (1999) MDM2 interacts with MDMX through their RING finger domains. FEBS Lett 447:5–9. [DOI] [PubMed] [Google Scholar]
- 20. Leslie PL, Ke H, Zhang Y (2015) The MDM2 RING domain and central acidic domain play distinct roles in MDM2 protein homodimerization and MDM2‐MDMX protein heterodimerization. J Biol Chem 290:12941–12950. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21. Medina‐Medina I, García‐Beltrán P, de la Mora‐de la Mora I, Oria‐Hernández J, Millot G, Fahraeus R, Reyes‐Vivas H, Sampedro JG, Olivares‐Illana V (2016) Allosteric interactions by p53 mRNA governs HDM2 E3 ubiquitin ligase specificity under different conditions. Mol Cell Biol 36:2195–2205. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22. Malbert‐Colas L, Ponnuswamy A, Olivares‐Illana V, Tournillon A‐S, Naski N, Fåhraeus R (2014) HDMX folds the nascent p53 mRNA following activation by the ATM kinase. Mol Cell 54:500–511. [DOI] [PubMed] [Google Scholar]
- 23. Badciong JC, Haas AL (2002) MdmX is a RING finger ubiquitin ligase capable of synergistically enhancing Mdm2 ubiquitination. J Biol Chem 277:49668–49675. [DOI] [PubMed] [Google Scholar]
- 24. Wang X, Wang J, Jiang X (2011) MdmX protein is essential for Mdm2 protein‐mediated p53 polyubiquitination. J Biol Chem 286:23725–23734. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25. Weibrecht I, Leuchowius K‐J, Clausson C‐M, Conze T, Jarvius M, Howell WM, Kamali‐Moghaddam M, Söderberg O (2010) Proximity ligation assays: a recent addition to the proteomics toolbox. Expert Rev Proteomics 7:401–409. [DOI] [PubMed] [Google Scholar]
- 26. Tournillon A‐S, López I, Malbert‐Colas L, Naski N, Olivares‐Illana V, Fåhraeus R (2015) The alternative translated MDMX(p60) isoform regulates MDM2 activity. Cell Cycle 14:449–458. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27. Worrall EG, Wawrzynow B, Worrall L, Walkinshaw M, Ball KL, Hupp TR (2009) Regulation of the E3 ubiquitin ligase activity of MDM2 by an N‐terminal pseudo‐substrate motif. J Chem Biol 2:113–129. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28. Hernychova L, Man P, Verma C, Nicholson J, Sharma CA, Ruckova E, Teo JY, Ball K, Vojtesek B, Hupp TR (2013) Identification of a second Nutlin‐3 responsive interaction site in the N‐terminal domain of MDM2 using hydrogen/deuterium exchange mass spectrometry. Proteomics 13:2512–2525. [DOI] [PubMed] [Google Scholar]
- 29. Waning DL, Lehman JA, Batuello CN, Mayo LD (2011) c‐Abl phosphorylation of Mdm2 facilitates Mdm2‐Mdmx complex formation. J Biol Chem 286:216–222. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30. Wang X (2011) p53 regulation: teamwork between RING domains of Mdm2 and MdmX. Cell Cycle 10:4225–4229. [DOI] [PubMed] [Google Scholar]
- 31. Kostic M, Matt T, Martinez‐Yamout MA, Dyson HJ, Wright PE (2006) Solution structure of the Hdm2 C2H2C4 RING, a domain critical for ubiquitination of p53. J Mol Biol 363:433–450. [DOI] [PubMed] [Google Scholar]