

Abstract
Transglutaminase from Streptomyces mobaraensis (MTG) has become a powerful tool to covalently and highly specifically link functional amines to glutamine donor sites of therapeutic proteins. However, details regarding the mechanism of substrate recognition and interaction of the enzyme with proteinaceous substrates still remain mostly elusive. We have determined the crystal structure of the Streptomyces papain inhibitory protein (SPIp), a substrate of MTG, to study the influence of various substrate amino acids on positioning glutamine to the active site of MTG. SPIp exhibits a rigid, thermo‐resistant double‐psi‐beta‐barrel fold that is stabilized by two cysteine bridges. Incorporation of biotin cadaverine identified Gln‐6 as the only amine acceptor site on SPIp accessible for MTG. Substitution of Lys‐7 demonstrated that small and hydrophobic residues in close proximity to Gln‐6 favor MTG‐mediated modification and are likely to facilitate introduction of the substrate into the front vestibule of MTG. Moreover, exchange of various surface residues of SPIp for arginine and glutamate/aspartate outside the glutamine donor region influences the efficiency of modification by MTG. These results suggest the occurrence of charged contact areas between MTG and the acyl donor substrates beyond the front vestibule, and pave the way for protein engineering approaches to improve the properties of artificial MTG‐substrates used in biomedical applications.
Keywords: Streptomyces papain inhibitor, crystal structure, transglutaminase, glutamine donor, Streptomyces mobaraensis
Short abstract
PDB Code(s): http://firstglance.jmol.org/fg.htm?mol=5NTB
Abbreviations
- aa
amino acids
- DAIP
dispase autolysis inducing protein
- MBC
(mono)biotin cadaverine
- MTG
microbial transglutaminase from Streptomyces mobaraensis
- SPI
Streptomyces papain inhibitor
Introduction
Microbial transglutaminase from streptomycetes (MTG, EC 2.3.2.13, P81453) has been originally produced to crosslink glutamine and lysine residues of food proteins or to alter protein charge by endo‐glutamine hydrolysis.1 Primary amines are remarkable acyl acceptor molecules and may replace lysine donor proteins in a competitive manner. The efficient and selective incorporation of functional amines into proteins may be one reason for the increasing usage of MTG in biotechnological applications. For instance, therapeutic proteins such as human growth hormone, apomyoglobulin, or human interferon were successfully coupled to polyethylene glycol amine to improve the pharmacokinetic properties.2, 3, 4 The benefit of MTG was even recognized in the preparation of antibody drug conjugates.5, 6, 7 Biologicals contain a large number of glutamines that are potential acyl donor sites for MTG. However, human interferon and the PEGylated amine were linked by MTG at only one, human growth factor at two glutamine positions.2, 4 Immunoglobulin G even needs the complete removal of the carbohydrate moiety to render a single glutamine for MTG accessible.8 These examples demonstrate the high specificity of MTG for distinct glutamines although interactions between the enzyme and the substrate proteins are poorly understood. As alternative strategy to overcome absence of favorable glutamine‐sites, suitable MTG‐tags were attached to terminal peptides of therapeutic antibodies in order to create a highly specific glutamine donor substrate.5, 7
Transglutaminase from Streptomyces mobaraensis (formerly Streptoverticillium mobaraense) has a distinct compact, disk‐shaped structure (PDB 1IU4), unrelated to the structure of other proteins.9 The amino acids of the catalytic triad (Cys‐64, Asp‐255, and His‐274) reside at the bottom of a 16 Å deep cleft in such a way that Cys‐64 and His‐274 are more directed to the front and rear vestibules, respectively. Asp‐255 is arranged in close proximity to the cysteine and is regarded to be the proton acceptor. Comparison with blood coagulation factor XIII‐like transglutaminases and sequence patterns of MTG substrate peptides suggest, that the front vestibule of MTG is the binding site for glutamine donor proteins (acyl donors).9 Such interaction between MTG and the glutamine donor substrate protein allows the immediate attack of the Cys‐64 thiolate onto the γ‐carboxamide function of a protruding glutamine residue and the formation of the acyl‐enzyme intermediate. Acidic amino acids such as Glu‐249, Glu‐300, and Asp‐304 line the rear vestibule of the active site cleft, which led to the assumption that these attract lysine donor proteins or primary amines (acyl acceptors) to complete the conjugation reaction.9 Unfortunately, crystal structures of MTG exhibiting suitable glutamine peptides or inhibitors attached to MTG are not available, which would provide a deeper insight into the binding features of acyl donor substrates. In their early report, Kashiwagi et al. already pointed out the importance of large conformational flexibility and small residues upstream of the accessible glutamine/acyl donor site.9 Mutagenesis experiments then revealed MTG variants with increased activity towards the small model peptide benzyloxycarbonylglutaminylglycine (ZQG) if amino acids such as Asp‐3, Trp‐59, Val‐65, Gln‐74, Tyr‐75, and Thr‐77, all near the active site at the acyl donor vestibule, are replaced for smaller and more hydrophobic residues.10 Moreover, sequence requirements around the glutamines of numerous peptide substrates were extensively examined, even using large peptide and mRNA libraries.11, 12, 13, 14 From the obtained sequence information, it remained largely elusive how acyl donor proteins are recognized and attached to the crosslinking enzyme. In an additional approach, the short tag sequence LLQG was incorporated into ∼90 surface accessible regions of an anti‐EGFR antibody.5 Beside terminally modified variants, nine proteins carrying sequence‐internal LLQG insertions were labeled by the MTG‐mediated incorporation of a fluorescent amine. However, proofs for structural integrity were not provided, and only C‐terminally linked antibody drug conjugates were tested in biological studies. Thus, it still remains elusive whether glutamine donor proteins require small or large contact areas to position the proper glutamine to the front vestibule of microbial transglutaminase.
Recently, we determined the first structure of a physiological transglutaminase substrate from S. mobaraensis, the dispase autolysis inducing protein (DAIP) that adopts a seven‐bladed beta‐propeller fold, where four out of the five present glutamines are clustered on the DAIP‐surface.15 The three most reactive glutamines (Gln‐39, Gln‐298, and Gln‐345) remarkably differ from less reactive glutamines (Gln‐65 and Gln‐144) in sequence environment, position, and secondary structure. The sequence around Gln‐39 only contains uncharged amino acids, mostly serines and threonines, and presumably forms a highly flexible loop. Gln‐298 is flanked by aromatic residues and is located in a rigid β‐turn resulting in an exposed, protruding carboxamide side‐chain. Ultimately, Gln‐345 is located in the flexible carboxy terminus and is flanked by charged amino acids. Incorporation of a biotinylated amine revealed Gln‐39 as the prime labeling position of MTG, thus supporting the early predictions by Kashiwagi et al. that reactive glutamines must occur in a flexible protein structure element with small amino acids.9 However, the study could not answer the question how MTG distinguishes between several adjacent γ‐carboxamide side‐chains of the clustered glutamines of DAIP.
Following the determination of the DAIP structure, in this report, we describe the structure of the Streptomyces papain inhibitor protein (SPIp). SPIp was previously determined as a substrate of transglutaminase to illuminate enzyme function in the life cycle of S. mobaraensis.16 The 12 kDa SPIp protein folds into a rigid double‐psi beta barrel domain and contains three glutamines, but only one glutamine (Gln‐6) located at the rigid amino terminus is a substrate of MTG for incorporation of a biotinylated amine, albeit with a slower rate as compared to DAIP. Apart from structure determination, we used extensive mutagenesis of surface exposed residues on SPIp, on the one hand to analyze determinants of enzymatic labeling rate and efficiency, and on the other hand to provide new insights into the recognition and interaction of MTG with the acyl donor substrate protein.
Results
Primary structure of the transglutaminase substrate
Streptomyces mobaraensis secretes a transglutaminase (MTG) substrate of 12 kDa in size that was previously purified from heated culture broths by two ion exchange chromatographies.16 The inhibitory activity for papain‐like cysteine proteases caused us to designate the protein as Streptomyces papain inhibitor (SPI). Recent investigations now revealed that SPI consists of two components, the 12 kDa protein (SPIp) and a small molecule (SPIac) possessing the inhibitory activity (unpublished results). Presently, we are unaware of the interaction between SPIp and SPIac since accumulation of the small inhibitor with sufficient purity is extremely difficult. In any case, SPIp, recombinantly produced in Escherichia coli for this study (Fig. 1), is a glutamine and lysine donor substrate of MTG but has no papain inhibitory activity. Its structural integrity was verified by measuring melting points of the thermo‐resistant protein.
Figure 1.
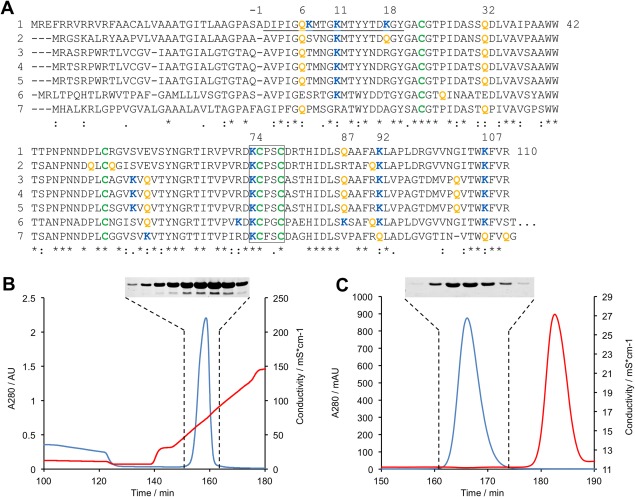
Sequence alignment and protein purification of SPIp. (A) Sequence of the transglutaminase substrate (SPIp) from Streptomyces mobaraensis and comparison with putative Streptomyces proteins. (1) P86242, S. mobaraensis; (2) Q9X5U4, S. lavendulae; (3) A0A0L8NMC1, S. griseoflavus; (4) A0A0M8QYC6, S. rimosus subsp. rimosus; (5) A0A0L8PS48, S. aureofaciens; (6) K4QWL3, S. davawensis; (7) F8JME3, S. cattleya. Multiple sequence alignment was carried out by Clustal Omega. Cysteine, glutamine, and lysine residues of the mature protein are depicted in green, orange, and blue. The N‐terminal peptide determined by Edman degradation and the inversely configured thioredoxin motif are underlined or framed by a box, respectively. (B, C) Purification of rSPIp. Supernatant proteins of lysed E. coli BL21 (DE3) pLysS were precipitated by pH‐shift from 8.0 to 5.0 and separated by Fractogel EMD SO3¯ chromatography at pH 5.0 (B) and Superdex 75 chromatography at pH 8.0 (C). Blue, A280; red, conductivity. Inserts: Protein patterns of Coomassie‐stained 15% SDS polyacrylamide gels.
The primary structure of SPIp consists of a 33‐amino acid (aa) signal peptide and the 110‐aa mature protein [Fig. 1(A)]. Orthologs are available in the genomes of various streptomycetes, though no transglutaminase producer is found among them. The biological function of SPIp is still unknown but there is structural, but not necessarily functional relationship to expansins (F1DTC5, Meloidogyne javanica), rare lipoproteins (A0A0M8QYC6, S. rimosus subsp. rimosus), speract/scavenger receptor domain‐containing proteins (D3BU04, Polysphondylium pallidum), pherophorin‐dz1 proteins (C5P8S7, Coccidioides posadasii), proline‐and threonine‐rich proteins (Q3HYB9, Coccidioides posadasii), barwin‐like endoglucanases (S3E833, Glarea lozoyensis), or allergens such as Asp f 7 (H0ENE8, Glarea lozoyensis). All mentioned proteins have four cysteines in common that are highly conserved (alignment not shown). The four cysteines most likely contribute to the heat‐resistance of SPIp.16 The cysteine motif KCPXC (X may be Gly, Ser, and Thr) in the posterior third of the protein is particularly striking and resembles the inversely configured, N‐terminal active site motif, CGPCK, of the oxidoreductase thioredoxin (Trx1, P0AA25, E. coli).
Molecular structure of wtSPIp
We used X‐ray crystallography to gain insights into the molecular structure of SPIp. SPIp crystals were obtained from purified protein derived from recombinant rSPIp as well as from wtSPIp isolated from S. mobaraensis heated culture broth.16 While crystals were obtained from both proteins, wtSPIp crystallized in space group P212121 with two monomers per asymmetric unit resulting in diffraction data to 1.5 Å resolution, whereas rSPIp crystallized in space group P1 with four monomers per asymmetric unit and diffraction data to 2.4 Å resolution. Structures were solved using the D1 domain of expansin from Clavibacter michiganensis (PDB 4JCW) as a molecular replacement model. The structures of wtSPIp and rSPIp are largely identical and overlay with an overall R.M.S.D of 0.30–0.38 Å (Fig. S1, Supporting Information); we thus only report on the high‐resolution wtSPIp structure, which we refer to as the SPIp structure hereafter. A complete model of SPIp with all residues (1–110) has been built and refined to R work/R free of 16.3/18.4 (Table SI, Supporting Information). The structure of SPIp comprises a single domain with eight β‐strands, one α‐helix and four 310‐helices, and two disulfide bonds formed by Cys‐23/Cys‐53 and Cys‐75/Cys‐78 [Fig. 2(A)]. SPIp and the D1 domain of expansin share 30% sequence identity, and both structures superpose well with an RMSD of 0.75 Å [Fig. 2(B, C)]. Expansin D1 and SPIp both adopt a double‐psi‐beta‐barrel (DPBB) fold, which was first described for the structure of barwin, a plant defense protein, and the endoglucanase V from Humicola insolens.17, 18 The DPBB domain is also found in the conserved N‐terminal domain of rare lipoprotein A (RlpA), a lytic transglycosylase involved in the cell division process of Pseudomonas aeruginosa.19 Beside enzymatic functionality, DPBBs were found to be involved in cofactor binding as well as ligand binding.20
Figure 2.
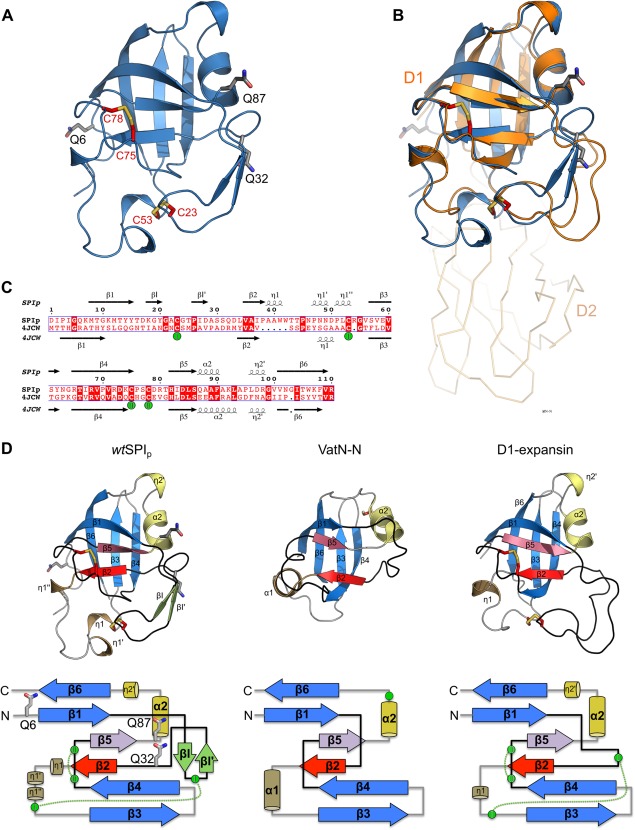
Crystal structure of the transglutaminase substrate (SPIp) from Streptomyces mobaraensis and tertiary structure comparison with DPBB‐containing proteins. (A) Cartoon representation of SPIp glutamines (carbons gray) and cysteines (carbons red) shown in stick representation. (B) Superposition of SPIp with the D1 N‐terminal domain of expansin (PDB 4JCW). SPIp is colored blue and expansin orange. The C‐terminal domain D2 of expansin is shown as ribbon representation. (C) Sequence alignment of SPIp and expansin D1 (PDB 4JCW) with secondary structure assigned by the DSSP algorithm (η indicates a 310‐helix). Disulfide‐bridges are numbered and cysteines involved are marked with green spheres. (D) Protein fold (cartoon representation) and topology comparison of SPIp (left), VatN‐N (middle, PDB 1CZ4), and the D1 domain of C. michiganensis’ expansin (right, PDB 4JCW). Each double psi‐beta‐barrel is composed of six β‐strands. β2 and β5, which are the central strands of each psi–loop (black), are colored red and violet, respectively. Remaining β‐strands that are part of the double‐psi‐beta barrel are highlighted in blue. For SPIp, two short β‐strands (βI and βI′) are inserted after β1 and are colored light green. Right‐handed α‐helices are colored sand (α1) and yellow (α2). 310‐helices (e.g., η1 or η2) are colored the same way. Cysteines in structures are shown as sticks with carbon atoms in red. Cysteines in the topology drawing are shown as green circles. Disulfide‐bridges are indicated by green dotted lines. Glutamine positions are shown as sticks with carbon atoms in gray for SPIp only.
The DPBB belongs to a subclass of six‐stranded β‐barrels. The core barrel is assembled by two interlocking psi‐motifs,20 where each motif comprises three β‐strands that resemble the Greek letter Ψ in top view. The first canonical psi‐motif, exemplified by the structure of the N‐terminal substrate recognition domain of the AAA ATPase VatN (VatN‐N; PDB 1CZ4), is formed by β1 and β2 and its connecting “psi”‐loop, crossing β5.21, 22 Equivalently, the second motif is formed by β4 and β5 with the second psi‐loop crossing β2 [Fig. 2(D)]. The lateral strands β3 and β6 complete the six‐stranded β‐barrel. Despite the structurally conserved central DPBB, connecting loops between the β‐strands show high variability in secondary structure composition and length, as seen for SPIp, expansin, and Vat‐N‐N [Fig. 2(D)]. And while the first psi‐loop in VatN‐N comprises only 10 aa, the loop in SPIp and expansin is extended to 18 aa and 22 aa, respectively. These longer loops are further stabilized by a disulfide‐bridge (SPIp: Cys‐23/Cys‐53, expansin: Cys‐23/Cys‐48), which is not found in VatN‐N since it only contains one cysteine.
Determination of the only glutamine donor site of SPIp
The mature MTG substrate SPIp contains the three glutamines Gln‐6, Gln‐32, and Gln‐87 that are located in solvent accessible regions, as well as six lysines (Lys‐7, Lys‐11, Lys‐18, Lys‐74, Lys‐92, and Lys‐107), scattered over the surface of SPIp (Fig. S2, Supporting Information). These positions are highly conserved in SPIp‐like Streptomyces proteins, with exception of Lys‐7 and Lys‐18 [Fig. 1(A)]. Importantly, both proteins, the rSPIp produced in E. coli and wtSPI from S. mobaraensis, showed no structural differences and exhibited approximately the same melting point, suggesting similar, high stability in solution (Table 1, Fig. 2). We thus used rSPIp and Δ2Q‐deficient variants (rSPIp‐Q1 variants) to determine potential glutamine donor sites for MTG. The Q1 variants were obtained by substitution of different glutamine pairs for asparagines through site directed mutagenesis, leaving only one of the three surface‐exposed glutamine residues on SPIp in place (Table 1). The immediate effect of the amino acid exchange was a significant drop in the melting points by 9–16°C, indicative of a stabilizing role of the glutamines on SPIp structure. This conclusion was further underlined by the Δ3Q‐deficient variant (SPIp‐Q0), where all three glutamines were replaced by asparagines, resulting in a strong reduction in melting temperature by ∼20°C to below 65°C.
Table 1.
Melting Points and Biotinylation Grade of Various rSPIp Variants at 20 h of Incubation
| Variant | Amino acid exchange | Glutamine flanking amino acids | Melting point (°C) | Rel. biotinylation | |
|---|---|---|---|---|---|
| SPIp(CEX) | Q3 | none | 84.8 | 1.89 | |
| wtSPIp(CEX + SEC) | Q3 | none | 85.0 | 1.16 | |
| rSPIp | Q3 | none | 84.2 | 1.00 | |
| Q6 | Q1 | Q32N, Q87N | D IPIG QK MTG K | 68.6 | 0.83 |
| Q32 | Q1 | Q6N, Q87N | I D ASS QD LVAI | 70.4 | 0.04 |
| Q87 | Q1 | Q6N, Q32N | H I D LS Q AAFA K | 75.4 | 0.05 |
| N6 | Q2 | Q6N | D IPIG NK MTG K | 81.0 | 0.07 |
| Q0 | Q0 | Q6N, Q32N, Q87N | 64.2 | 0.05 | |
| G7 | Q3 | K7G | D IPIG QG MTG K | 82.3 | 13.66 |
| A7 | Q3 | K7A | D IPIG QA MTG K | 85.9 | 2.44 |
| D7 | Q3 | K7D | D IPIG QD MTG K | 83.8 | 0.55 |
| rDAIP | Q5 | none | 48.7 | 18.96 | |
| rDAIP‐Q39 | Q1 | Q65N, Q144N, | 51.9 | 14.76 | |
| Uncharged → Charged | Q298N, Q345N | ||||
| R9 | Q3 | T9R | D IPIG QK M R G K | 83.3 | 0.55 |
| D9 | Q3 | T9D | D IPIG QK M D G K | 81.6 | 2.13 |
| E9 | Q3 | T9E | D IPIG QK M E G K | 83.4 | 1.76 |
| R40 | Q3 | A40R | 78.5 | 0.78 | |
| E40 | Q3 | A40E | 82.6 | 1.08 | |
| R49 | Q3 | N49R | 81.9 | 1.16 | |
| R55 | Q3 | G55R | 77.6 | 2.05 | |
| E55 | Q3 | G55E | 81.4 | 1.25 | |
| R61 | Q3 | S61R | 79.7 | 2.65 | |
| E61 | Q3 | S61E | 84.7 | 0.83 | |
| R105 | Q3 | T105R | 79.3 | 3.96 | |
| R109 | Q3 | V109R | 75.8 | 4.25 | |
| E109 | Q3 | V109E | 78.0 | 1.46 | |
| Charge inversion | |||||
| E65 | Q3 | R65E | 78.5 | 1.27 | |
| E68 | Q3 | R68E | 77.9 | 2.85 | |
| E92 | Q3 | R92E | 74.3 | 1.96 | |
| R97 | Q3 | D97R | 82.0 | 1.21 | |
| E107 | Q3 | K107E | 82.2 | 1.46 | |
| E110 | Q3 | R110E | 81.1 | 1.71 |
CEX, cationic exchange chromatography; SEC, size exclusion chromatography.
The destabilizing effect of glutamine mutations can be explained with the SPIp structure. The side chains of Gln‐32 and Gln‐87 coordinate several water molecules and form intra‐molecular interactions. The Gln‐32 side‐chain oxygen interacts with the backbone amide hydrogen of Asp‐28 introducing a stabilizing H‐bond within the first psi‐loop. The Gln‐87 side‐chain carboxamide‐NH2 interacts with the side‐chain oxygen and the backbone carbonyl oxygen of Ser‐30 forming two additional H‐bonds between loop (β4‐β5) and the first psi‐loop (Fig. S3, Supporting Information). Disturbing or eliminating those interactions is likely to cause or contribute to the observed drop in melting temperature for Q 1/Q 0 variants, as seen for variants containing the Gln‐87‐Asn substitution that results in the most significant drop in melting temperature. Furthermore, the observed intra‐molecular contacts of Gln‐32 and Gln‐87 would restrict access of MTG to the carboxamide group, whereas the carboxamide side‐chain of Gln‐6 is solvent exposed and shows no direct interaction (up to 3.5Å) with surrounding residues or water molecules.
In line with that observation, replacement of Gln‐6 by asparagine only resulted in a minor drop in melting temperature by ∼3°C (Table 1). Furthermore, Gln‐6 was the only MTG conjugation site for the covalent incorporation of the biotinylated amine and replacement by asparagine completely abolished biotin labeling by MTG (Table 1, Fig. 3). Interestingly, compared to rSPIp, the overall biotinylation rate of wtSPI produced in Streptomyces mobaraensis was twice as large after 20 h. However, similar labeling efficiency was achieved if wtSPI was subjected to additional purification procedures such as size exclusion chromatography or dialysis [Fig. 3(B)]. We assume that, when wtSPI is purified from Streptomyces mobaraensis, the access of MTG to the wtSPI glutamine is enhanced by small molecules, either the papain inhibitory molecule (SPIac) or amphiphilic compounds from culture broth as was earlier reported for detergents such as N‐lauroyl sarcosine and N‐lauroylamido‐3‐N′,N′‐dimethyl propylamine.23, 24 We therefore used a final purification step by size exclusion chromatography for all proteins used in this work, unless stated otherwise. The difference to the first characterized MTG substrate, the dispase autolysis inducing protein (DAIP), was particularly striking. MTG‐mediated biotinylation of rDAIP or the Δ4Q‐deficient variant rDAIP‐Q39 (DAIP‐Q1) was more than 10‐fold faster than for wtSPI (Fig. 3, Table 1).15 The likely explanation is that, in contrast to the major DAIP glutamine donor site Gln‐39,15 all sequences flanking the three glutamines in SPIp contain charged amino acids within ± five residues (Fig. 1, Table 1), which are likely to disfavor modification by MTG.15 It must be emphasized, however, that, in contrast to DAIP, rSPIp was selectively modified at a single glutamine. The finding allowed usage of thermo‐resistant rSPIp‐Q3, the control protein hereinafter, and the study of single amino acid substitutions influencing MTG catalysis, thus expanding our knowledge on MTG selectivity.
Figure 3.
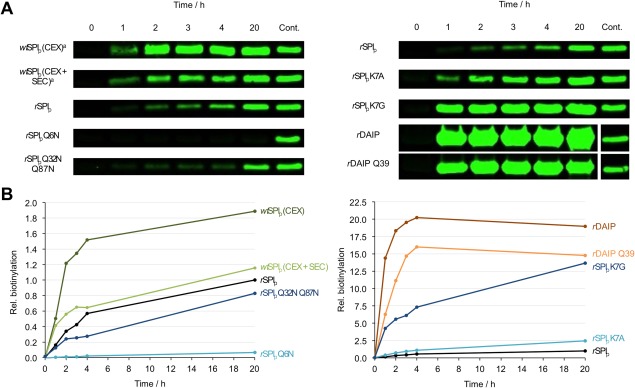
Determination of SPIp glutamine crosslinking sites and hot spots by transglutaminase‐mediated biotinylation. Various glutamine donor proteins (15 µM), biotin cadaverine (200 µM) and MTG (0.3 µM) were incubated at 37°C in 50 mM Tris pH 8.0 containing 100 mM NaCl up to 20 h. Upon electrophoresis and blotting, the biotinylated proteins were visualized by the addition of IRDye 800 CW streptavidin conjugates. (A) Examples of proteins biotinylated upon the indicated times. rSPIp was the control after 20 h incubation; (B) biotinylation grade of indicated proteins expressed as multiple fluorescence of rSPIp upon the indicated times.
Influence of donor site environment on labeling efficiency
As shown above, the only MTG‐substrate glutamine in SPIp is Gln‐6, which is located in the N‐terminal loop of SPIp. With the exception of Asp‐1, only hydrophobic and small amino acids (1DIPIGQ6) are located upstream of Gln‐6. The bottom area of the front vestibule of MTG, representing the putative glutamine donor binding site, is mainly hydrophobic with patches of weak positive charge (Tyr‐62, Val‐65, Trp‐69, Tyr‐75, Pro‐76, Thr‐77, and Phe‐254) whereas one side of the front vestibule is negatively charged (formed by Asp‐1, Asp‐3, Asp‐4, Glu‐54, Glu‐55, and Glu‐58) and the opposite side is positively charged (Arg‐208, Arg‐215, and Arg‐238). The hydrophobic residues in proximity to Gln‐6 in SPIp might thus accommodate close to the catalytic MTG‐Cys‐64 at the rather uncharged bottom of the glutamine donor binding site, and thus be favorable for MTG‐mediated modification, as we reported before.15 But despite the presence of small and hydrophobic residues, the efficiency of SPIp labeling is reduced compared to DAIP Gln‐39, which we reasoned to be potentially caused by the presence of the downstream situated basic amino acid Lys‐7.10
To test this hypothesis and further analyze the influence of residues flanking Gln‐6, we decided to replace Lys‐7 by uncharged/small/hydrophobic (Gly‐7 and Ala‐7) and acidic (Asp‐7) residues and to evaluate the variants by enzymatic biotinylation (Table 1, Fig. 3). It should be noticed that the modified rSPIp proteins exhibited all three, structure‐stabilizing glutamines and that the melting point was only reduced by less than 2°C by Lys‐7 substitutions (Table 1). While replacement of Lys‐7 by aspartate resulted in a reduction of labeling efficiency, replacement with the small amino acid glycine (Lys‐7‐Gly) considerably increased the MTG‐mediated amine incorporation (>13‐fold), thus supporting previous observations by us, Kashiwagi et al. and Yokoyama et al.9, 10, 15 Even alanine in Lys‐7‐Ala increased biotinylation by MTG. However, the Lys‐7‐Gly variant exhibited a 5‐ to 6‐fold higher labeling efficiency compared to Lys‐7‐Ala, most likely due to the methyl group at the alanine‐Cα. In line with previous observations, the occurrence of small and hydrophobic amino acids around appropriate glutamine donor sites thus seems to be a necessary prerequisite for the interaction and efficient modification by MTG, at least for rigid acyl donor proteins with limited accessibility such as SPIp, whereas incorporation of a charged amino acid, such as Lys‐7 in wt or Lys‐7‐Asp, impairs MTG‐mediated labeling (Table 1).
Effect of amino acids outside the glutamine donor region of rSPIp
MTG substrate peptides are increasingly used in biotechnological applications for a highly specific modification of therapeutic proteins. In order to better understand the influence of regions outside the glutamine donor region, we analyzed the impact of changing the surface charge distribution by mutagenesis of surface exposed residues on SPIp. Apart from forming charged, complementary patches promoting tight, short‐range interactions between two molecules, charged residues on the surface also influence the interaction at longer distance by a process termed “electrostatic steering”, that contributes to the association speed in the initial binding event between two molecules. Thus, a detailed understanding of the influence of charged surface residues on the interactions between MTG and its physiological substrate SPIP might contribute towards improving the properties of artificial MTG‐substrates for biomedical applications by protein engineering.
In this regard, an MTG‐SPI complex structure would greatly contribute to our understanding of these short‐ and long‐range interactions regulating MTG‐substrate interaction. But despite extensive efforts, crystallization of the binary MTG‐SPIp complex was not successful, potentially also due to instability of the thioester intermediate generated by the MTG‐Cys‐64 thiolate attack onto the SPIp‐Gln‐6 γ‐carboxamide. Simulation of the protein–protein interactions by docking experiments still provided unreliable results due to the conformational flexibility of proteins and their special potency of mutual structural adaptation. Thus, to uncover the influence of surface residues outside of the region surrounding Gln‐6 on directing the only glutamine substrate residue into the MTG active site, several water‐exposed, uncharged amino acids at the surface of SPIp were replaced by arginines and glutamates/aspartates. The selection was based on the wtSPIp structure to provide an even distribution across the whole molecule (Fig. 4, Table 1). All variants contained the stabilizing glutamines and consistently maintained the high SPIp melting points, suggesting no major destabilizing effect of the mutations. Seven residues were mutated to Arg and five to Glu (and Asp for Thr‐9) (Table 1). Of these, arginine variants R49 (Asn‐49‐Arg), R55 (Gly‐55‐Arg), R61 (Ser‐61‐Arg), R105 (Thr‐105‐Arg), and R109 (Val‐109‐Arg) displayed, despite the presence of Lys‐7, a considerably higher (∼1.2‐ to 4.2‐fold) amine incorporation rate than rSPIp. R9 (Thr‐9‐Arg) instead resulted in a reduction of amine incorporation by ∼45% as compared to rSPIp. However, contrary to our expectations, only for E61 (Ser‐61‐Glu) and E9/D9 (Thr‐9‐Glu/Asp) introduction of the opposite charge resulted in the inverse effect seen for the Arg‐substitutions, namely a slight reduction in amine incorporation for E61 (0.83) and an increased labeling efficiency for E9 and D9 (1.76 and 2.13, respectively). Instead, variants E55 (Gly‐55‐Glu) and E109 (Val‐109‐Glu) both resulted in an increased amine incorporation efficiency, albeit to a lower extent (1.25–1.46) as compared to the respective R55 and R109 variants (2.05–4.25). In contrast, mutation of Ala‐40 to Arg only slightly reduced the labeling efficiency (Ala‐40‐Arg) or had no major influence when mutated to Glu (Ala‐40‐Glu). Thus, only positions Thr‐9 and Ser‐61 exhibit the expected, opposite effect on amine incorporation upon mutation to a basic or acidic residue, suggesting that surface patches close these positions are involved in short‐range, charged interaction with MTG. However, for the other three positions (Gly‐55, Ser‐61, and Val‐109), interaction of MTG and rSPIp does not appear to be governed by compatible charges on both proteins, but rather by other charge‐mediated effects, that we cannot explain based on our current knowledge of the structures and properties of MTG and rSPIp (Fig. S4, Supporting Information). Apart from the aforementioned variants, we additionally inverted present charges on the surface of rSPIp by substituting Arg/Lys with Glu or by substituting Asp with Arg. Interestingly, inverting the charge resulted in an increased amine incorporation (∼1.2–2.9) in all analyzed substitutions, comprising Arg‐65‐Glu, Arg‐68‐Glu, Arg‐92‐Glu, Asp‐97‐Arg, Lys‐107‐Glu, and Arg‐110‐Glu.
Figure 4.
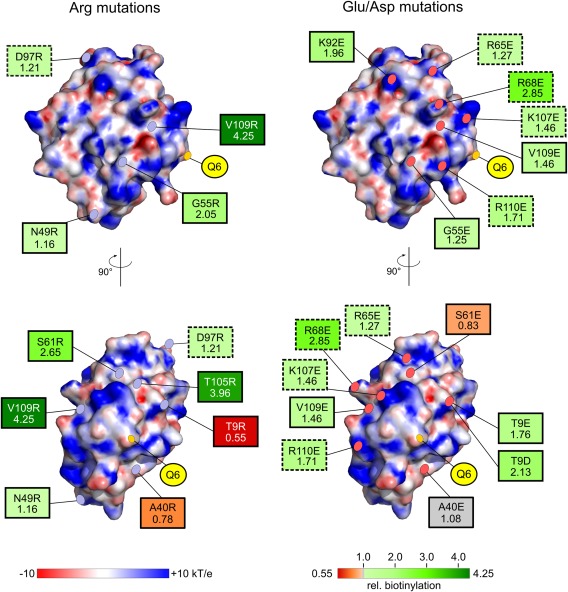
Summary of the mutational studies of SPIp surface residues to influence MTG‐mediated biotinylation. Positions of mutations (with rel. biotinylation rate, see also Table II) are marked and labeled on the electrostatic surface of SPIp (–10 to +10 kT/e). Arg‐substitutions are shown on the left‐hand side and Glu/Asp mutations on the right‐hand side. Effect on the relative biotinylation rate of SPIp by MTG is indicated by a red to green color gradient with the variant resulting in the lowest rate colored in dark red (A40R = 0.55) and the variant with the highest rate colored in dark green (V109R = 4.25). Relative biotinylation rate of wtSPIp was set to 1.00 and is indicated in gray, as well as the A40E mutant with a rel. biotinylation rate of 1.08. Charge inversions (e.g., Arg to Asp) are marked with dashed borders and uncharged to charged substitutions are indicated by solid borders.
Discussion
The interactions between enzymes and substrate proteins that do not form stable intermediates are poorly understood, in particular the mode of attraction, the proper orientation, the conformational adaptation and the protein dissociation. Transglutaminases catalyze the crosslinking of proteins via acyl transfer between glutamine and lysine donor proteins, thereby establishing high‐energy thioester transition states that decompose in the presence of primary amines (e.g., lysine residues). Most recently, we have reported on microbial transglutaminase (MTG) that modifies the dispase autolysis inducing protein (DAIP), a seven‐bladed beta‐propeller, at a distinct protein edge clustering four out of five glutamine residues.15 The glutamine in DAIP position 39 (Gln‐39) proved to be the most preferred biotinylation site. MTG mediated labeling of Gln‐298 and Gln‐345 was comparably low. In contrast, the Streptomyces papain inhibitor protein (SPIp), subject of this study, has only a single MTG‐accessible glutamine (Gln‐6) site at the rigid, wedge‐shaped N terminus of a double‐psi‐beta‐barrel fold. Like DAIP Gln‐39, the microenvironment surrounding SPIp Gln‐6 is largely composed of hydrophobic amino acids, promoting the interaction with the front vestibule of MTG.9 However, SPIp even exhibits a disruptive (possibly regulatory) element, Lys‐7, that obviously reduces labeling efficiency by MTG. Noticeably, Lys‐7 is replaced by serine, threonine or proline in the sequence of SPIp‐like proteins from other streptomycetes lacking a gene for transglutaminase. Substitution of Lys‐7 by glycine (Lys‐7‐Gly variant) considerably increased amine incorporation (13‐ to 14‐fold compared to rSPIp), thus achieving labeling efficiency of DAIP Q‐39. The result was in line with former observations that small and hydrophobic residues in close vicinity of the substrate glutamine favor protein crosslinking.9, 15 Although the Lys‐7‐Gly variant of rSPIp achieved a similar biotinylation grade as DAIP Gln‐39 upon 20 h, the velocity of MTG mediated conjugation was considerably slower. It thus seems obvious that the overall affinity of MTG for DAIP should be higher than for SPIp. In water, long‐range forces of soluble, that is largely hydrophilic polymers are mainly determined by charged functions.25 The surface of the disk‐shaped MTG for glutamine donor substrates (front surface) is characterized at least by four extended patches comprising two acidic (left side of Cys‐64) and two basic areas (right side), respectively. Hence, higher biotinylation rate by MTG might be explained by stronger attractive forces of DAIP compared to SPIp. Unfortunately, protein–protein docking failed to reveal SPIp charged areas complementary to the MTG front surface. We therefore decided to replace distinct solvent‐exposed SPIp residues against arginine or glutamate/aspartate. All other amino acids, especially the three glutamines and Lys‐7, were left unchanged to avoid conformational changes and distortion of the results. Interestingly, of the five residues that were replaced by both, a basic Arg and an acidic Asp/Glu, only positions Thr‐9 and Ser‐61 showed inverse effects on labeling efficiency (with Ala‐40 only showing mild inverse effect). For the other two, Gly‐55 and Val‐109, substitution with Arg or Glu, in both cases resulted in an increased labeling efficiency, which however was overall more prominent for Arg‐substitutions. This suggests that, in contrast to Gly‐55 and Val‐109, only Thr‐9 and Ser‐61 are likely to be located in charged patches involved in mediating charge‐complementary, close‐contact interactions between rSPIp and MTG. Especially substitution of amino acids Thr‐105 and Val‐109 with Arg resulted in an ∼4‐fold increased labeling efficiency by MTG, and these residues are located near the SPIp C terminus, mainly characterized by the positively charged Lys‐107 and Arg‐110. We thus assume that arginine in position 105 and position 109, close to the C‐terminal end, might strengthen long‐range attractive forces between MTG and SPIp. However, due to the lack of a SPIp–MTG complex structure, it remains to be determined whether these basic SPIp residues indeed address acidic MTG areas comprising Asp‐1, Asp‐3 and Asp‐4 or Glu‐54, Glu‐55, and Glu‐58. And since we cannot currently explain the molecular basis of the charge‐mediated influence on amine incorporation, further structural and functional investigations of additional substrates from S. mobaraensis are in progress, which may help to unravel similarities in the interaction between MTG and various acyl donor proteins and to further improve our understanding of the interaction between MTG and its substrates.
The observation, that the majority of amino acid substitutions results in an increased labeling efficiency, further raises two interesting questions, (i) why is SPIp significantly less efficiently labeled by MTG as compared to DAIP and (ii) why did no mutations accumulate during evolution that turn SPIP into a better MTG‐substrate? In other words, are distinct surface functions of SPIp required to enable self‐assembly with other MTG substrate proteins prior to crosslinking? If so, in vivo Gln‐6 and Lys‐7 are likely to protrude from the SPIp accommodating protein layer to be linked by MTG to adjacent lysine and glutamine donor functions in a zipper‐like mechanism. In vitro however, Lys‐7‐Gly/Ala variants would lead to the removal of the positive charge, which in turn would facilitate access of MTG to Gln‐6 and result in the observed increase in amine incorporation. Even though not utilized by evolution, the mutagenesis experiments presented here demonstrate that in vitro bioengineering represents a promising approach to increase overall labeling efficiency and to optimize MTG substrates for biotechnological and medicinal applications.
Conclusions
In this work, we have determined the crystal structure of the MTG‐substrate SPIp, which adopts a thermo‐resistant double‐psi‐beta‐barrel fold. We further identified Gln‐6 as the only amine acceptor site on SPIp accessible for MTG. In line with previous observations, substitution of Lys‐7 demonstrated that small and hydrophobic residues in close proximity to Gln‐6 favor MTG‐mediated modification and are likely to facilitate introduction of the substrate into the front vestibule of MTG. Apart from studying the influence of amino acids in close proximity to the substrate Gln‐6, we analyzed the influence of various SPIp surface amino acids on labeling efficiency by MTG. Those mutagenesis experiments demonstrate that overall labeling efficiency of MTG substrates can be improved by surface modification. New approaches are in progress, on the one hand to further define the interaction between microbial transglutaminase and glutamine donor substrates, and on the other hand to adapt surface properties allowing for the optimization of artificial MTG‐substrates for biomedical applications and potentially the genetic adaptation of any protein (polymer) to become a MTG glutamine substrate.
Materials and Methods
Materials
Microbial transglutaminase (MTG) and wtSPI were produced by Streptomyces mobaraensis, strain 40847 (German Collection of Microorganisms and Cell Cultures, Braunschweig, Germany), as described.16, 26 The production strain for rSPIp and variants derived from was E. coli BL21 (DE3) pLysS ([F–ompT gal dcm lon hsdSB(rB‐ mB‐) λ(DE3 [lacI lacUV5‐T7 gene 1 ind1 sam7 nin5])] from Merck‐Millipore (Darmstadt, Germany). (Mono)biotin cadaverine (5‐N‐biotinamido pentyl amine, MBC) and IRDye 800CW streptavidin conjugates were purchased from Zedira (Darmstadt, Germany) and LI‐COR Biotechnology (Bad Homburg, Germany), respectively.
Protein production of rSPIp
The gene encoding SPIp was synthesized by GenScript (NJ) and inserted into pET‐22b(+) using standard procedures. Upon heat‐shock transformation, E. coli BL21 (DE3) pLysS was cultured in LB medium (10 g/L peptone, 5 g/L yeast extract, and 5 g/L NaCl) at 37°C. Protein production was started at OD600 of 0.6–0.8 by the addition of 1 mM IPTG at 28°C. A preceding pelB signal peptide allowed secretion into the periplasm. Supernatant proteins of the sonicated cells were precipitated by reducing the pH from 8.0 (50 mM Tris/HCl) to 5.0 (50 mM acetate) and separated by Fractogel EMD SO3¯ chromatography at pH 5.0 as described16 and Superdex75 chromatography (GE Healthcare, Frankfurt, Germany) using 100 mM NaCl in 50 mM Tris/HCl pH 8.0. The replacement of amino acids was performed by site directed mutagenesis (SDM) or size overlap extension (SOE) PCR. In brief, appropriate primers including the corresponding nucleotide substitutions were used in SDM to amplify spi opt in pET‐22b(+) by Pfu DNA polymerase (Thermo Scientific, Darmstadt, Germany). Hydrolysis of parental DNA by DpnI for 1 h at 37°C and E. coli XL 1 blue selection resulted in pET‐22b(+) containing the modified gene for rSPIp variants. To perform SOE PCR in some cases, the mutated primers along with an additional primer pair, attaching up‐ and downstream of spi opt, yielded two PCR products that were combined by a second PCR. The replacement of several amino acids occurred by successive SDM or SOE as required.
Determination of protein melting points
Highly purified proteins were dissolved in or dialyzed against 100 mM NaCl in 50 mM Tris/HCl pH 8.0 to obtain concentrations of 350 µg/mL. While increasing temperature by 1°C/min the intrinsic fluorescence was monitored at 330 and 350 nm using Prometheus NT. 48 (Nanotemper Technologies, Munich, Germany). Protein unfolding was expressed by rfu350/330 ratios.
Evaluation of rSPIp variants by MTG mediated biotinylation
The enzymatic incorporation of biotin cadaverine, separation by electrophoresis and blotting on PVDF membranes were performed as described.15 The biotinylated and blotted proteins were then used to determine the degree of labeling as increase in fluorescence per min (rfu/min) of equal areas upon addition of IRDye 800CW streptavidin conjugates. The null glutamine variant, Q0 (Gln‐6‐Asn, Gln‐32‐Asn, and Gln‐87‐Asn), and N6 (Gln‐6‐Asn) were the controls. The biotinylation grade is expressed as the fluorescence ratio of rSPIp variants versus rSPIp (rfu rSPIp variant/rfu rSPIp) at a given time.
Crystallization and data processing
Protein for crystallization was purified as described above, lyophilized and stored at 4°C as described above. wtSPIp crystals grew in 2.2M ammonium sulfate and 0.2M lithium chloride from drops with equal volumes of precipitant and protein (15 mg/mL). Crystals started to grow after three weeks and reached their full size (∼200 × 250 × 20 µm) after seven weeks at 20°C. A data set with 1.5 Å resolution was collected with λ = 0 1.007 Å on a PILATUS 6M detector at ID30B (ESRF, Grenoble). Data reduction and scaling was done in space group P212121 using the XDS package.27 Since no homolog structures for SPI were available, MRage from the Phenix suite28 was used for homology fold search and automated molecular replacement (MR) with the structure of expansin from Clavibacter michiganensis (PDB 4JCW). The initial SPIp model was manually completed in COOT32 and used as an MR model for the P212121 crystal form. Iterative building and refinement cycles were carried out using Coot and Phenix.refine,29 respectively. Electrostatic surfaces of SPIp were calculated with the Adaptive Poisson‐Boltzmann Solver.30
Data Deposition
The atomic coordinates and structure factors (code: 5NTB) have been deposited in the Protein Data Bank (http://www.pdb.org/).
Conflicts of Interest
The authors declare that they have no conflicts of interest with the contents of this article.
Supporting information
Supporting Information
Acknowledgments
We are grateful to the staff of beamline at ID30B at ESRF, Grenoble, for assistance with data collection and processing.
Contributor Information
Hans‐Lothar Fuchsbauer, Email: hans-lothar.fuchsbauer@h-da.de.
Andrea Scrima, Email: andrea.scrima@helmholtz-hzi.de.
References
- 1. Ando H, Adachi M, Umeda K, Matsuura A, Nonaka M, Uchio R, Tanaka H, Motoki M (1989) Purification and characterization of a novel transglutaminase derived from micro‐organisms. Agric Biol Chem 53:2613–2617. [Google Scholar]
- 2. Fontana A, Spolaore B, Mero A, Veronese FM (2008) Site‐specific modification and PEGylation of pharmaceutical proteins mediated by transglutaminase. Adv Drug Deliv Rev 60:13–28. [DOI] [PubMed] [Google Scholar]
- 3. Zhao X, Shaw AC, Wang J, Chang CC, Deng J, Su J (2010) A novel high‐throughput screening method for microbial transglutaminases with high specificity toward Gln141 of human growth hormone. J Biomol Screen 15:206–212. [DOI] [PubMed] [Google Scholar]
- 4. Spolaore B, Raboni S, Satwekar AA, Grigoletto A, Mero A, Montagner IM, Rosato A, Pasut G, Fontana A (2016) Site‐specific transglutaminase‐mediated conjugation of interferon α‐2b at glutamine or lysine residues. Bioconjugate Chem 27:2695–2706. [DOI] [PubMed] [Google Scholar]
- 5. Strop P, Liu S‐H, Dorywalska M, Delaria K, Dushin RG, Tran T‐T, Ho W‐H, Farias S, Casas MG, Abdiche Y, Zhou D, Chandrasekaran R, Samain C, Loo C, Rossi A, Rickert M, Krimm S, Wong T, Chin SM, Yu J, Dilley J, Chaparro‐Riggers J, Filzen GF, O'Donnell CJ, Wang F, Myers JS, Pons J, Shelton DL, Rajpal A (2013) Location matters: site of conjugation modulates stability and pharmacokinetics of antibody drug conjugates. Chem Biol 20:161–167. [DOI] [PubMed] [Google Scholar]
- 6. Dennler P, Chiotellis A, Fischer E, Brégeon D, Belmant C, Gauthier L, Lhospice F, Romagne F, Schibli R (2014) Transglutaminase‐based chemo‐enzymatic conjugation approach yields homogeneous antibody‐drug conjugates. Bioconjug Chem 25:569–578. [DOI] [PubMed] [Google Scholar]
- 7. Siegmund V, Schmelz S, Dickgiesser S, Beck J, Ebenig A, Fittler H, Frauendorf H, Piater B, Betz UAK, Avrutina O, Scrima A, Fuchsbauer HL, Kolmar H (2015) Locked by design: a conformationally constrained transglutaminase tag enables efficient site‐specific conjugation. Angew Chem Int Ed 54:13420–13424. [DOI] [PubMed] [Google Scholar]
- 8. Jeger S, Zimmermann K, Blanc A, Grünberg J, Honer M, Hunziker P, Struthers H, Schibli R (2010) Site‐specific and stoichiometric modification of antibodies by bacterial transglutaminase. Angew Chem Int Ed 49:9995–9997. [DOI] [PubMed] [Google Scholar]
- 9. Kashiwagi T, Yokoyama K, Ishikawa K, Ono K, Ejima D, Matsui H, Suzuki E (2002) Crystal structure of microbial transglutaminase from Streptoverticillium mobaraense . J Biol Chem 277:44252–44260. [DOI] [PubMed] [Google Scholar]
- 10. Yokoyama K, Utsumi H, Nakamura T, Ogaya D, Shimba N, Suzuki E, Taguchi S (2010) Screening for improved activity of a transglutaminase from Streptomyces mobaraensis created by a rational mutagenesis and random mutagenesis. Appl Microbiol Biotechnol 87:2087–2096. [DOI] [PubMed] [Google Scholar]
- 11. Ohtsuka T, Ota M, Nio N, Motoki M (2000) Comparison of substrate specificities of transglutaminases using synthetic peptides as acyl donors. Biosci Biotechnol Biochem 64:2608–2613. [DOI] [PubMed] [Google Scholar]
- 12. Sugimura Y, Yokoyama K, Nio N, Maki M, Hitomi K (2008) Identification of preferred substrate sequences of microbial transglutaminase from Streptomyces mobaraensis using phage‐displayed peptide library. Arch Biochem Biophys 477:379–383. [DOI] [PubMed] [Google Scholar]
- 13. Lee JH, Song C, Kim DH, Park IH, Lee SG, Lee YS, Kim BG (2013) Glutamine (Q)‐peptide screening for transglutaminase reaction using mRNA display. Biotechnol Bioeng 110:353–362. [DOI] [PubMed] [Google Scholar]
- 14. Malesevic M, Migge A, Hertel TC, Pietzsch M (2015) A fluorescence‐based array screen for transglutaminase substrates. ChemBioChem 16:1169–1174. [DOI] [PubMed] [Google Scholar]
- 15. Fiebig D, Schmelz S, Zindel S, Ehret V, Beck J, Ebenig A, Ehret M, Fröls S, Pfeifer F, Kolmar H, Fuchsbauer HL, Scrima A (2016) Structure of the dispase autolysis‐inducing protein from Streptomyces mobaraensis and glutamine cross‐linking sites for transglutaminase. J Biol Chem 291:20417–20426. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16. Sarafeddinov A, Arif A, Peters A, Fuchsbauer HL (2011) A novel transglutaminase substrate from Streptomyces mobaraensis inhibiting papain‐like cysteine proteases. J Microbiol Biotechnol 21:617–626. [PubMed] [Google Scholar]
- 17. Ludvigsen S, Poulsen FM (1992) Three‐dimensional structure in solution of barwin, a protein from barley seed. Biochemistry 31:8783–8789. [DOI] [PubMed] [Google Scholar]
- 18. Davies GJ, Dodson GG, Hubbard RE, Tolley SP, Dauter Z, Wilson KS, Hjort C, Mikkelsen JM, Rasmussen G, Schülein M (1993) Structure and function of endoglucanase V. Nature 365:362–364. [DOI] [PubMed] [Google Scholar]
- 19. Jorgenson MA, Chen Y, Yahashiri A, Popham DL, Weiss DS (2014) The bacterial septal ring protein RlpA is a lytic transglycosylase that contributes to rod shape and daughter cell separation in Pseudomonas aeruginosa . Mol Microbiol 93:113–128. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20. Castillo RM, Mizuguchi K, Dhanaraj V, Albert A, Blundell TL, Murzin AG (1999) A six‐stranded double‐psi beta barrel is shared by several protein superfamilies. Structure 7:227–236. [DOI] [PubMed] [Google Scholar]
- 21. Coles M, Diercks T, Liermann J, Gröger A, Rockel B, Baumeister W, Koretke KK, Lupas A, Peters J, Kessler H (1999) The solution structure of VAT‐N reveals a ‘missing link’ in the evolution of complex enzymes from a simple betaalphabetabeta element. Curr Biol 9:1158–1168. [DOI] [PubMed] [Google Scholar]
- 22. Coles M, Hulko M, Djuranovic S, Truffault V, Koretke K, Martin J, Lupas AN (2006) Common evolutionary origin of swapped‐hairpin and double‐psi beta barrels. Structure 14:1489–1498. [DOI] [PubMed] [Google Scholar]
- 23. Schmidt S, Adolf F, Fuchsbauer HL (2008) The transglutaminase‐activating metalloprotease inhibitor from Streptomyces mobaraensis is a glutamine and lysine donor substrate of the intrinsic transglutaminase. FEBS Lett 582:3132–3138. [DOI] [PubMed] [Google Scholar]
- 24. Sarafeddinov A, Schmidt S, Adolf F, Mainusch M, Bender A, Fuchsbauer HL (2009) A novel transglutaminase substrate from Streptomyces mobaraensis triggers autolysis of neutral metalloproteases. Biosci Biotechnol Biochem 73:993–999. [DOI] [PubMed] [Google Scholar]
- 25. Batra J, Szabó A, Caulfield TR, Soares AS, Sahin‐Tóth M, Radisky ES (2013) Long‐range electrostatic complementary governs substrate recognition by human chymotrypsin C, a key regulator of digestive enzyme activation. J Biol Chem 288:9848–9859. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26. Gerber U, Jucknischke U, Putzien S, Fuchsbauer HL (1994) A rapid and simple method for the purification of transglutaminase from Streptoverticillium mobaraense . Biochem J 299:825–829. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27. Kabsch W (2010) XDS. Acta Cryst 66:125–132. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28. Bunkóczi G, Echols N, McCoy AJ, Oeffner RD, Adams PD, Read RJ (2013) Phaser.MRage: automated molecular replacement. Acta Cryst 69:2276–2286. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29. Afonine PV, Grosse‐Kunstleve RW, Echols N, Headd JJ, Moriarty NW, Mustyakimov M, Terwilliger TC, Urzhumtsev A, Zwart PH, Adams PD (2012) Towards automated crystallographic structure refinement with phenix.refine. Acta Cryst 68:352–367. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30. Baker NA, Sept D, Joseph S, Holst MJ, McCammon JA (2001) Electrostatics of nanosystems: application to microtubules and the ribosome. Proc Natl Acad Sci U S A 98:10037–10041. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Supporting Information