

 A new, mild, functional-group tolerant approach to the construction aryl- and heteroaryl sulfones via Ni/photoredox dual catalysis is described.
A new, mild, functional-group tolerant approach to the construction aryl- and heteroaryl sulfones via Ni/photoredox dual catalysis is described.
Abstract
This report details the development and implementation of a strategy to construct aryl- and heteroaryl sulfones via Ni/photoredox dual catalysis. Using aryl sulfinate salts, the C–S bond can be forged at room temperature under base-free conditions. An array of aryl- and heteroaryl halides are compatible with this approach. The broad tolerance and mild nature of the described reaction could potentially be employed to prepare sulfones with biological relevance (e.g., in bioconjugation, drug substance synthesis, etc.) as demonstrated in the synthesis of drug-like compounds or their precursors. When paired with existing Ni/photoredox chemistry for Csp3–Csp2 cross-coupling, an array of diverse sulfone scaffolds can be readily assembled from bifunctional electrophiles. A mechanistic manifold consistent with experimental and computational data is presented.
Introduction
Sulfones are prominent motifs that are of particular relevance to both the medicinal chemistry and agrochemical communities (Scheme 1A).1 Aryl- and heteroaryl sulfones are excellent electronic modifiers, a property that has been exploited in drug design, various synthetic methods, as well as in the material sciences (e.g., polymer synthesis).2 Various tactics can be used to install this valuable functional group (Scheme 1B), although the most commonly employed strategy is the exhaustive oxidation of the corresponding sulfide.3 The most apparent limitations of this approach are the notable sensitivity of a variety of functional groups to the strongly oxidizing conditions necessary for sulfone formation and the possibility of incomplete oxidation (affording the sulfoxide). More generally, this approach requires the C–S bond to be forged4 prior to sulfone formation and thus has the added drawback of using odious, more toxic thiols as part of the synthetic sequence. Alternatively, nucleophilic aromatic substitution processes using sulfinate salts (easily prepared, bench-top stable, non-odorous solids) does allow direct access to diaryl- or heteroaryl sulfones.5 However, this route is limited by the inherent restrictions of SNAr chemistry (electron-deficient arenes with specific substitution patterns and elevated temperatures).
Scheme 1. Importance of sulfones and synthetic approaches.

In light of the limitations of both of these classical methods, transition metal-catalyzed cross-coupling processes have emerged as an alternate means of assembling these types of sulfones (Scheme 1B).6–8 Indeed, in these reactions, sulfinate salts can be cross-coupled with an array of electrophilic partners and even with organoboron species using either copper- or palladium-based catalysts. Unfortunately, these approaches are dependent on strongly alkaline conditions and high temperature to facilitate ArO2S–Csp2 bond formation. Willis and co-workers have employed DABSO {1,4-diazabicyclo[2.2.2]octane bis(sulfur dioxide) adduct} as a SO2 surrogate to construct sulfones de novo from two distinct aryl halides, thus proffering an elegant means to construct unsymmetrical sulfones (Scheme 1B).8 However, to do so, main group organometallics and/or elevated temperatures/strong bases must be employed. Thus, this approach does not overcome the existing limitation on functional group compatibility and sulfinate scope.
Given that, to the best of our knowledge, analogous Ni-based processes were not documented in the literature nor were there general conditions established for executing this type of coupling under mild conditions, we considered whether Ni/photoredox dual catalysis9,10 might be uniquely poised for facile ArO2S–Csp2 bond formation. In this process, radicals are generated by photoredox-mediated SET events and subsequently undergo facile single electron transmetalation with transition metal catalysts. Ultimately, this allows myriad Y–Csp2 bonds (Y = Csp3, N, O, S, and P) to be forged under remarkably mild conditions using an array of functional group-rich electrophiles (aryl/alkenyl halides, aryl/alkenyl sulfonate esters, and acyl chlorides).10 During the course of this work and while this manuscript was under review, two papers detailing similar transformations using Ni/photoredox dual catalysis were reported by Rueping and Manolikakes, respectively.11
In our consideration of this approach, we envisioned that aryl sulfinate salts that have low redox potentials (e.g., E1/2 = –0.37 V vs. SCE for 2a)12 could undergo SET oxidation by the appropriate photocatalyst to furnish sulfonyl radicals (Scheme 2A). The ultimate fate of these S-centered radicals does vary (Scheme 2B). Whereas certain alkyl sulfinates can undergo C–S bond dissociation to give alkyl radicals and SO2 evolution at this point,13 aryl sulfonyl radicals are much less prone to C–S scission.14 Thus, these types of radicals could be intercepted by Ni0 species A to generate a NiI–SO2R species B.15 Subsequent oxidative addition to an aryl halide would yield a NiIII complex C. Reductive elimination at this point would forge the ArO2S–Csp2 bond and yield a NiI species D, which could undergo SET reduction by the reduced state of the appropriate photocatalyst to regenerate A.16
Scheme 2. Mechanistic proposal for cross-coupling.

Model studies focused on the cross-coupling of aryl bromide 1a and sodium sulfinate 2a. Using a slight modification of conditions previously optimized for alkylsilicates and 2 equiv. of 2a, the desired cross-coupling was initially realized (Table 1, entry 1). Control studies confirmed that this was indeed a dual catalytic process (entries 2–5) and that all the components of the reaction were necessary to ensure cross-coupling. Early screens assessing the role of solvent and photocatalyst revealed that Ru(bpy)3(PF6)2 (Eox1/2 [RuII/RuI] = 0.77 V vs. SCE)10d in conjunction with highly polar solvents such as DMSO, DMF, or DMA were ideal for the generation of sulfone 3a. Surprisingly, a major by-product of this reaction was the corresponding aryl sulfide 4a, which may arise from disproportionation of sulfonyl radicals to give thiols/thiolates or thiyl radicals (Scheme 2B).17,18 Such species are known to engage in cross-coupling under Ni/photoredox conditions.19 Higher temperatures appeared to increase sulfide formation and diminish overall conversion (entry 14). To improve conversion and minimize formation of this byproduct, High-Throughput Experimentation (HTE) screening of an array of Ni sources and ligands was employed (see ESI†). Ultimately, a combination of 1,10 phenanthroline (phen) with Ni(py)4Cl2 was identified as an appropriate cross-coupling system. For convenience, we assessed a bench-top stable, pre-formed nickel complex [Ni(phen)·(H2O)4]Cl2 in place of Ni(py)4Cl2/phen and obtained virtually identical results. Ultimately, we were also able to reduce the nickel loading by half without compromising yield or drastically increasing reaction time. Control experiments under these optimized conditions confirmed that this was indeed a dual catalytic process; no reaction occurred in the absence of irradiation, photocatalyst, or nickel (see the ESI† for details). Analysis of these conditions over time indicated that the relative ratio of sulfide to sulfone did not appreciably change over extended reaction time (see the ESI† for details).
Table 1. Initial attempts of sulfinate cross-coupling a b c .
| |||
| Entry | Deviation from initial conditions | 3a/IS ratio | 4a/IS ratio |
| 1 | None | 3.37 | 0.26 |
| 2 | No light (dark) | n.d. | n.d. |
| 3 | No Ru photocatalyst | n.d. | n.d. |
| 4 | No Ni catalyst | n.d. | n.d. |
| 5 | No Ni and Ru photocatalyst | n.d. | n.d. |
| 6 | MeOH, acetone, dioxane, or MeCN | n.d. | n.d. |
| 7 | DMA | 2.73 | 0.23 |
| 8 | DMF | 3.10 | 0.30 |
| 9 | DMF/H2O (9 : 1) | 2.57 | n.d. |
| 10 | [Ir(dFCF3ppy)2(bpy)](PF6) | 1.67 | 0.24 |
| 11 | 4CzlPN | 0.44 | 0.32 |
| 12 | Ru(bpz)3(PF6)2 | n.d. | n.d. |
| 13 | Lower loading of 2a (1.2 equiv.) | 2.64 | 0.28 |
| 14 | Reaction performed at 50 °C | 2.40 | 0.21 |
aOptimization reactions performed using 0.1 mmol of 1a in the presence of 4,4′-di-tert-butylbiphenyl as internal standard (IS) (0.01 mmol) for 24 h at 27 °C.
bRatios of 3a or 4a to IS determined by HPLC analysis of crude reaction mixture; n.d. = not detected.
c4CzlPN: 2,4,5,6-tetra-9H-carbazol-9-yl-1,3-benzenedicarbonitrile.
With suitable conditions in hand, the scope of the transformation was next explored (Table 2). An evaluation of the reaction in the context of various aryl halide coupling partners was first conducted. Gratifyingly, the scope of the transformation was quite broad, and the yields were moderate to good. In practically all cases, small amounts of the undesired thioether byproduct were formed but were easily removed via column chromatography. Generally, both aryl iodides and aryl bromides performed well, with aryl iodides proving to be superior when electron-rich substituents were present or when significant amounts of the thioether byproduct was observed. Indeed, Ni(phen)-type species have been shown to be more competent catalysts when using aryl iodides under Ni/photoredox conditions.20 The reaction tolerates an array of functional groups, including nitrogen-based heterocycles, amides, lactones, phenols, and species that may be prone to H-atom abstraction events. Many of these systems would not be amenable to the aforementioned classical approaches for sulfone construction or even those that employ transition metal catalysts. Indeed, systems such as 3o would give an alternate product under SNAr-type conditions, and systems such as 3c would likely undergo off-target oxidation under the conditions required for oxidative sulfone synthesis from thioethers. The reaction was electrophile-specific, enabling a selective functionalization of bifunctional electrophiles (e.g., 3o, 3v, 3w, 3aa). In addition, the process described here tolerates functional group-rich medicinal chemistry intermediates such as 3ad. The absence of any thioether by-product and the known propensity for radical addition–elimination reactions when preparing indene 3p prompted us to investigate whether Ni was indeed necessary for this substrate. As suspected, this reaction proceeds without Ni in excellent yield.
Table 2. Scope of diaryl sulfone synthesis via Ni/photoredox dual catalysis a b .
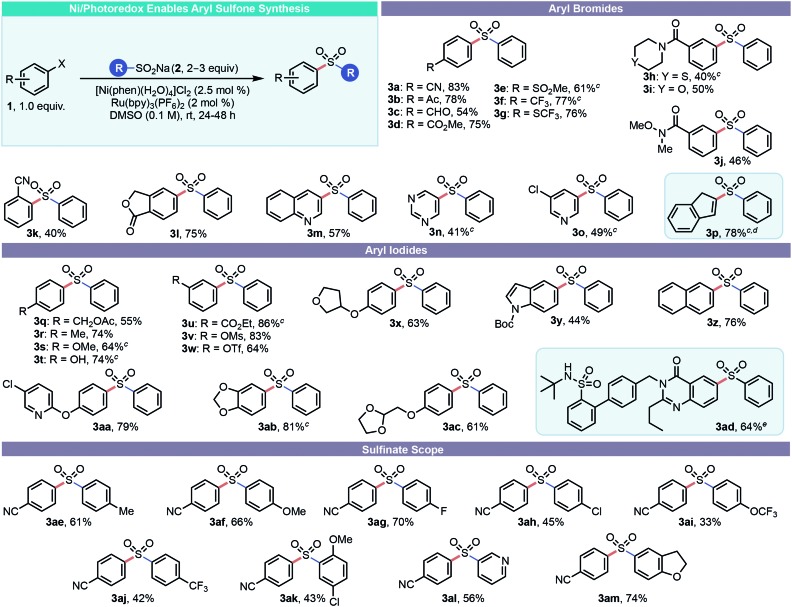
|
aUnless otherwise noted reactions were performed using aryl halide (1.0 equiv., 0.5 mmol), sulfinate salt (2 equiv.), [Ni(phen)·(H2O)4]Cl2 (2.5 mol%) and Ru(bpy)3(PF6)2 (2 mol%) in DMSO (0.1 M) at rt with irradiation with blue LEDs.
bAll yields are isolated yields after purification.
c3 equiv. of sulfinate salt were used.
dNo Ni was used for this example.
eReaction performed on 0.15 mmol scale of aryl halide.
After surveying a broad range of electrophilic partners, a variety of aryl sulfinate salts were next assessed. Although some are commercially available, these salts are readily accessed from commodity chemicals (sulfonyl chlorides and sodium sulfite) in a single chemical step and are bench-top stable, crystalline solids.21 An array of sulfinate salts readily succumbed to cross-coupling. Both electron-rich and electron-poor sulfinates were compatible, as were heterocyclic and polycyclic sulfinates. Of note is pyridyl substrate 3al, where an umpolung disconnection is drawn as compared to the more typical SNAr-type approach. In addition, chemoselective cross-coupling ensured 3ah and 3ak retained their functional handles for further diversification. Sulfonate esters, especially electron-deficient aryl triflates, readily undergo Ni/photoredox Csp3–Csp2 cross coupling with alkylsilicates.22 Thus, 3w represents an ideal substrate for rapid diversification, although sulfonate ester-bearing sulfones are not explicitly known to engage in Ni/photoredox cross-coupling.
To highlight the synthetic value of the approach described here for the rapid assembly of complex molecules, a series of tandem Csp3–Csp2 cross-coupling reactions were attempted (Scheme 3). In addition, the requisite amount of the sulfone needed for these studies presented the opportunity to assess the scalability of the process. Gratifyingly, not only could 3w be prepared on scale (5 mmol, a 10 fold scale up), but this material subsequently underwent Csp3–Csp2 cross coupling, yielding an array of complex sulfones in good yield from a common intermediate.
Scheme 3. Sequential Ni/photoredox cross-coupling.

As a further demonstration of the utility of this approach for assembling sulfones, a short synthetic sequence was used to prepare the precursor for the 5-HT6 receptor antagonist RVT-101 (Scheme 4).23 During our evaluation of the scope of this sulfonylation process, conditions optimized here were found not to work with aryl chlorides. It is likely that oxidative addition using these recalcitrant electrophiles is an insurmountable task for the Ni complex, which was optimized for sulfonylation. We took advantage of this innate selectivity for the synthesis of RVT-101. NIS-mediated iodination of 8-chloroquinoline proceeded smoothly, giving the desired iodide 1af. Treatment of 1af with 2a under the optimized Ni/photoredox conditions gave sulfone 3aq in 56% yield.
Scheme 4. Synthesis of a therapeutic agent precursor.

To gain insight into the mechanism and origin of product selectivity, we turned to quantum mechanical calculations.24 Previously, we demonstrated that dual Ni/photoredox cross-coupling reactions involving radicals converged to an alkyl–NiIII intermediate that can undergo reversible radical dissociation prior to reductive elimination.15 Similarly, DFT calculations (Scheme 5) in the current system support formation of NiIII intermediate C (via Ni0/NiI/NiIII or Ni0/NiII/NiIII pathway; not calculated) and reversible radical dissociation (12.4 kcal mol–1viaE-TSRA) to form NiIIE and ˙SO2Ph species prior to reductive elimination. Reductive elimination of NiIII intermediate C (viaC-TSRE) will generate sulfone product 3 and NiID (Scheme 5A). Alternatively, prior to reductive elimination, the ˙SO2Ph radical can undergo a complex disproportionation (not calculated) to generate ˙SPh radical, which can engage with the NiII intermediate E (viaE′-TSRA) to form the sulfide adduct 4, after reductive elimination of C′ (viaC′-TSRE). Based on experiments, we favor competition between reductive elimination (viaC-TSRE) and complex disproportionation, presumably via nickel/PC-promoted pathway, as product selectivity determining steps (vide infra). Overall, the highest barrier for sulfone formation is the reductive elimination (15.4 kcal mol–1viaC-TSRE; Scheme 5A), while thiyl radical addition to the Ni complex (14.6 kcal mol–1) is the highest barrier for sulfide 4 formation pathway (Scheme 5A). If disproportionation of ˙SO2Ph to ˙SPh radical is fast (vide infra), the latter pathway is favored by ca. 1 kcal mol–1, which will lead to sulfide as major product. These results are not consistent with the observed results, in which the sulfone is the major product. Given that formation of sulfone 3 was accompanied by significant sulfide by-product 4 for a series of aryl bromides (Scheme 5B), at this level of theory, we estimate that the barrier for disproportionation is ca. 16 kcal mol–1. If the barriers for reductive elimination (C to D) are ∼16 kcal mol–1, it will therefore be in competition with ˙SO2Ar radical disproportionation. In turn, the ˙SAr will compete for addition to NiII to form NiIII–SAr intermediate C′ which will quickly undergo reductive elimination to form thioether 4. Consistent with experiment, the formation of sulfone is only slightly favored for R = H, OMe, and Me, in which the reductive elimination barriers are estimated to be >15 kcal mol–1 (Scheme 5C; bold, RED). However, in the presence of electron deficient aryl bromides (e.g., R = CN; Scheme 5C) the reductive elimination barrier leading to sulfone is much lower (13.4 kcal mol–1) than this complex disproportionation threshold, leading to a higher sulfone to sulfide ratio. In this regard, we found a linear correlation (R2 = 0.85) between sulfone/sulfide ratio and Hammett σp for a series of para-substituted aryl bromides (See ESI†).
Scheme 5. Mechanistic analysis of Csp2–SO2R cross-coupling via DFT calculations. Free energies (kcal mol–1) are with respect to nickel(iii) C and PhS˙ radical.

Notably, DFT calculations rule against generation of ˙SPh from ˙SO2Ph radical prior to formation of Ni(ii) phenyl bromide intermediate. In this scenario, and in contrast to experiment, DFT calculations predict Ph–S–Ar 4 as the major product for a variety of aryl bromides because the overall barriers for sulfide formation (Scheme 5C, bold, BLUE) are lower than sulfone formation (Scheme 5C, bold, RED). Further, DFT calculations rule against rapid equilibration between ˙SAr and ˙SO2Ar. If so, the equilibrium will strongly tilt toward formation of Ph–S–Ar 4 for all systems.
Conclusions
In summary, Ni/photoredox dual catalysis enables the construction of aryl- and heteroaryl sulfones from aryl halides and sulfinate salts. The base-free, room-temperature reaction conditions described here permit a wide array of functional groups to be tolerated. The broad tolerance and mild nature of the described reaction will likely translate to its use in the preparation of sulfones with biological relevance (e.g., in bioconjugation, drug substance synthesis, etc.) as demonstrated in the synthesis of drug-like compounds or their precursors. A mechanistic manifold consistent with experimental and computational data is presented that will aid in rational reaction design and provides a holistic understanding of the reaction that is currently absent in the literature. Finally, sequential functionalization using the process outlined here and existing Ni/photoredox methods allows an array of diverse scaffolds to be assembled from bifunctional electrophiles.
Conflicts of interest
There are no conflicts of interest to declare.
Supplementary Material
Acknowledgments
The authors are grateful for the financial support provided by NIGMS (R01 GM 113878) and NIH S10 OD011980. C. B. K. is grateful for an NIH NRSA postdoctoral fellowship (F32 GM 117634). OG is grateful to the University of Maryland College Park for start-up funds and computational resources from UMD DT2 and XSEDE (CHE160082 and CHE160053). We sincerely thank Ms Rebecca Wiles, Dr James Phelan, and Dr Álvaro Gutierrez-Bonet of the University of Pennsylvania (UPenn) for useful discussions. We thank Dr Charles W. Ross, III (UPenn) for assistance in obtaining HRMS data.
Footnotes
†Electronic supplementary information (ESI) available: Experimental procedures, characterization data for all new products, copies of NMR spectra, Cartesian coordinates for computational data. See DOI: 10.1039/c7sc05402e
References
- (a) Feng M., Tang B., Liang S. H., Jiang X. Curr. Top. Med. Chem. 2016;16:1200. doi: 10.2174/1568026615666150915111741. [DOI] [PMC free article] [PubMed] [Google Scholar]; (b) Dörwald F. Z., Sulfones in Lead Optimization for Medicinal Chemists: Pharmacokinetic Properties of Functional Groups and Organic Compounds, Wiley-VCH Verlag GmbH & Co. KGaA, Weinheim, Germany, 2012. [Google Scholar]
- El-Hibri M. J. and Weinberg S. A., Polysulfones in Encyclopedia of Polymer Science and Technology, Wiley: New York, 2002. [Google Scholar]
- (a) Su W. Tetrahedron Lett. 1994;35:4955. [Google Scholar]; (b) Rayner C. M. Contemp. Org. Synth. 1995;2:409. [Google Scholar]
- (a) Hartwig J. F. Acc. Chem. Res. 2008;41:1534. doi: 10.1021/ar800098p. [DOI] [PMC free article] [PubMed] [Google Scholar]; (b) Beletskaya I. P., Ananikov V. P. Chem. Rev. 2011;111:1596. doi: 10.1021/cr100347k. [DOI] [PubMed] [Google Scholar]
- (a) Liang S., Zhang R.-Y., Xi L. Y., Chen S.-Y., Yu X.-Q. J. Org. Chem. 2013;78:11874. doi: 10.1021/jo401828b. [DOI] [PubMed] [Google Scholar]; (b) Maloney K. M., Kuethe J. T., Linn K. Org. Lett. 2011;13:102. doi: 10.1021/ol102629c. [DOI] [PubMed] [Google Scholar]
- Representative Pd-based approaches: ; (a) Cacchi S., Fabrizi G., Goggiamani A., Parisi L. M., Bernini R. J. Org. Chem. 2004;69:5609. doi: 10.1021/jo0493469. [DOI] [PubMed] [Google Scholar]; (b) Reeves D. C., Rodriguez S., Lee H., Haddad N., Krishnamurthy D., Senanayake C. H. Tetrahedron Lett. 2009;50:2870. [Google Scholar]
- Representative Cu-based approaches: ; (a) Beaullieu C., Guay D., Wang Z., Evans D. A. Tetrahedron Lett. 2004;45:3233. [Google Scholar]; (b) Baskin J. M., Wang Z. Org. Lett. 2002;4:4423. doi: 10.1021/ol0269190. [DOI] [PubMed] [Google Scholar]; (c) Kar A., Sayyed I. A., Lo W. F., Kaiser H. M., Beller M., Tse M. K. Org. Lett. 2007;9:3405. doi: 10.1021/ol071396n. [DOI] [PubMed] [Google Scholar]
- (a) Chen Y., Willis M. C. Chem. Sci. 2017;8:3249. doi: 10.1039/c6sc05483h. [DOI] [PMC free article] [PubMed] [Google Scholar]; (b) Deeming A. S., Russell C. J., Henessy A. J., Willis M. C. Org. Lett. 2014;16:150. doi: 10.1021/ol403122a. [DOI] [PMC free article] [PubMed] [Google Scholar]; (c) Emmett E. J., Hayter B. R., Willis M. C. Angew. Chem., Int. Ed. 2013;52:12679. doi: 10.1002/anie.201305369. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Seminal reports: ; (a) Tellis J. C., Primer D. N., Molander G. A. Science. 2014;345:433. doi: 10.1126/science.1253647. [DOI] [PMC free article] [PubMed] [Google Scholar]; (b) Zuo Z., Ahneman D. T., Chu L., Terrett J. A., Doyle A. G., MacMillan D. W. C. Science. 2014;345:437. doi: 10.1126/science.1255525. [DOI] [PMC free article] [PubMed] [Google Scholar]
- For reviews see: ; (a) Tellis J. C., Kelly C. B., Primer D. N., Jouffroy M., Patel N. R., Molander G. A. Acc. Chem. Res. 2016;49:1429. doi: 10.1021/acs.accounts.6b00214. [DOI] [PMC free article] [PubMed] [Google Scholar]; (b) Gui Y.-Y., Sun L., Lu Z.-P., Yu D.-G. Org. Chem. Front. 2016;3:522. [Google Scholar]; (c) Skubi K. L., Blum T. R., Yoon T. P. Chem. Rev. 2016;116:10035. doi: 10.1021/acs.chemrev.6b00018. [DOI] [PMC free article] [PubMed] [Google Scholar]; (d) Kelly C. B., Patel N. R., Primer D. N., Jouffroy M., Tellis J. C., Molander G. A. Nat. Protoc. 2017;12:472. doi: 10.1038/nprot.2016.176. [DOI] [PMC free article] [PubMed] [Google Scholar]; (e) Matsui J. K., Lang S. B., Heitz D. R., Molander G. A. ACS Catal. 2017;7:2563. doi: 10.1021/acscatal.7b00094. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (a) Yue H., Zhu C., Rueping M. Angew. Chem., Int. Ed. 2017;57:1371. doi: 10.1002/anie.201711104. [DOI] [PMC free article] [PubMed] [Google Scholar]; (b) Lui N.-W., Hofman K., Herbert A., Manolikakes G. Org. Lett. 2018;20:760. doi: 10.1021/acs.orglett.7b03896. [DOI] [PubMed] [Google Scholar]
- Meyer A. U., Jäger S., Hari D. P., König B. Adv. Synth. Catal. 2015;357:2050. [Google Scholar]
- , see: ; (a) Chatgilialoglu C., Lunazzi L., Ingold K. U. J. Org. Chem. 1983;48:3588. [Google Scholar]; (b) For a recent example of SO2 extrusion from sulfonyl radicals under photoredox conditions see: Gualandi A., Mazzarella D., Ortega-Martínez A., Mengozzi L., Calcinelli F., Matteucci E., Monti F., Armaroli N., Sambri L., Cozzi P. G., ACS Catal., 2017, 7 , 5357 . [Google Scholar]
- Hering T., Meyer A. U., König B. J. Org. Chem. 2016;81:6927. doi: 10.1021/acs.joc.6b01050. [DOI] [PubMed] [Google Scholar]
- Gutierrez O., Tellis J. C., Primer D. N., Molander G. A., Kozlowski M. C. J. Am. Chem. Soc. 2015;137:4896. doi: 10.1021/ja513079r. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Alternatively, a mechanism involving energy transfer of a Ni-sulfinate species would give rise to a similar outcome, see: ; (a) Heitz D. R., Tellis J. C., Molander G. A. J. Am. Chem. Soc. 2016;138:12715. doi: 10.1021/jacs.6b04789. [DOI] [PMC free article] [PubMed] [Google Scholar]; (b) Welin E. R., Le C., Arias-Rotondo D. M., McCusker J. K., MacMillan D. W. C. Science. 2017;355:380. doi: 10.1126/science.aal2490. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Interestingly, the formation of the sulfide by-product is not discussed in the very recent report by Rueping (ref. 11a). However, the trends of reported yields in their work and ours are nearly identical. At the suggestion of a reviewer, we confirmed sulfide formation using the conditions outlined in their report using a set of representative substrates that overlapped between the two reports (3a–d, 3m). Thus, the trend in yields may be partially attributed to the formation of the sulfide byproduct. Indeed, the Manolikakes report corroborates thioether formation under similar conditions to those outlined here
- (a) Kice J. L., Guaraldi G., Venier C. G. J. Org. Chem. 1966;31:3561. [Google Scholar]; (b) Koch P., Ciuffarin E., Fava A. J. Am. Chem. Soc. 1970;92:5971. [Google Scholar]; (c) Hoyle J., The Oxidation and Reduction of Sulphinic Acids and their Derivatives, in Sulphinic Acids, Esters and Derivatives, John Wiley & Sons, Inc., Chichester, UK, 1990. [Google Scholar]
- (a) Jouffroy M., Kelly C. B., Molander G. A. Org. Lett. 2016;18:876. doi: 10.1021/acs.orglett.6b00208. [DOI] [PMC free article] [PubMed] [Google Scholar]; (b) Oderinde M. S., Frenette M., Robbins D. W., Aquila B., Johannes J. W. J. Am. Chem. Soc. 2016;138:1760. doi: 10.1021/jacs.5b11244. [DOI] [PubMed] [Google Scholar]; (c) Santandrea J., Minozzi C., Cruch C., Collins S. K. Angew. Chem., Int. Ed. 2017;56:12255. doi: 10.1002/anie.201705903. [DOI] [PubMed] [Google Scholar]
- Lin K., Wiles R. J., Kelly C. B., Davies G. H. M., Molander G. A. ACS Catal. 2017;7:5129. doi: 10.1021/acscatal.7b01773. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (a) Aziz J., Messaoudi S., Alami M., Hamze A. Org. Biomol. Chem. 2014;12:9743. doi: 10.1039/c4ob01727g. [DOI] [PubMed] [Google Scholar]; (b) Pinnick H. W., Reynolds M. A. J. Org. Chem. 1979;44:160. [Google Scholar]
- Patel N. R., Molander G. A. J. Org. Chem. 2016;81:7271. doi: 10.1021/acs.joc.6b00800. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (a) Rosse G., Schaffhauser H. Curr. Top. Med. Chem. 2010;10:207. doi: 10.2174/156802610790411036. [DOI] [PubMed] [Google Scholar]; (b) Ivachtchenko A. V., Lavrovsky Y., Okun I. J. Alzheimer's Dis. 2016;53:583. doi: 10.3233/JAD-151146. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Frisch M. J., et al., Gaussian 09, revision D.01, Gaussian, Inc., Wallingford, CT, 2004, All calculations were computed at the UM06/6-311+G(d,p)-SDD (for Br, S, and Ni) in THF (SMD)//UB3LYP/6-31G(d)-LANL2DZ (for Br, S, and Ni) level of theory using Guassian09. See ESI† for computational details.
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.