

Abstract
CHD7, an ATP-dependent chromatin remodeler, is disrupted in CHARGE syndrome, an autosomal dominant disorder characterized by variably penetrant abnormalities in craniofacial, cardiac, and nervous system tissues. The inner ear is uniquely sensitive to CHD7 levels and is the most commonly affected organ in individuals with CHARGE. Interestingly, upregulation or downregulation of retinoic acid (RA) signaling during embryogenesis also leads to developmental defects similar to those in CHARGE syndrome, suggesting that CHD7 and RA may have common target genes or signaling pathways. Here, we tested three separate potential mechanisms for CHD7 and RA interaction: (a) direct binding of CHD7 with RA receptors, (b) regulation of CHD7 levels by RA, and (c) CHD7 binding and regulation of RA-related genes. We show that CHD7 directly regulates expression of Aldh1a3, the gene encoding the RA synthetic enzyme ALDH1A3 and that loss of Aldh1a3 partially rescues Chd7 mutant mouse inner ear defects. Together, these studies indicate that ALDH1A3 acts with CHD7 in a common genetic pathway to regulate inner ear development, providing insights into how CHD7 and RA regulate gene expression and morphogenesis in the developing embryo.
Keywords: Development, Neuroscience
Keywords: Embryonic development, Genetic diseases
CHD7, disrupted in human CHARGE syndrome, regulates retinoic acid synthetic enzyme Aldh1a3, and loss of Aldh1a3 rescues Chd7-mutant mouse inner ear defects.
Introduction
Basic morphogenetic processes, including cellular proliferation and differentiation, must be tightly regulated for proper organismal development. Proliferation and differentiation of cells correlate with changes in gene expression that are heritable and specific to distinct cellular lineages. Proteins that regulate chromatin remodeling and nucleosome positioning govern these heritable spatiotemporal gene expression profiles. The underlying mechanisms linking their functions in the cell nucleus to cellular phenotypes are not well understood (1). Importantly, human genetic conditions that exhibit overlapping phenotypic features often share underlying pathogenic mechanisms and can provide important insights into the regulation of both cellular and chromatin level processes.
All-trans retinoic acid (ATRA), a metabolite of vitamin A1 (retinol), was discovered in 1987 as the first candidate vertebrate morphogen (2). ATRA is fat soluble and diffuses into the cell nucleus where it interacts with cellular fatty acid–binding proteins (Fabps) or cellular retinol-binding proteins (Crabps) (3). Retinol undergoes oxidation to become retinaldehyde (retinal) by alcohol dehydrogenases and retinol dehydrogenase enzymes. Retinol is subsequently oxidized to form retinoic acid (RA) by cytoplasmic retinaldehyde dehydrogenase enzymes (ALDH1As; formerly called RALDH enzymes) (3). RA acts in the cell nucleus where it binds to RA receptors (RARs) that form heterodimers with retinoid receptors (RXRs). RAR/RXR heterodimers bind to gene promoter regions, where they generally repress downstream target genes in the absence of RA and activate expression in the presence of RA (4). Embryonic exposure to either excessive or diminished levels of RA leads to abnormalities in neurogenesis, ocular morphogenesis, inner ear development, cardiogenesis, and limb development (5). The complexity of phenotypes related to vitamin A metabolism and RA signaling is similar to the highly variable birth defects associated with human mutations in genes encoding RA-related enzymes, receptors, and binding proteins (6–8).
CHARGE syndrome is a multiple congenital anomaly condition caused by heterozygous loss-of-function pathogenic variants in the chromodomain helicase DNA-binding protein 7 (CHD7) gene (9). Individuals with CHARGE syndrome present with ocular coloboma, heart defects, atresia of the choanae, retarded growth and development, genital hypoplasia, and ear defects, including deafness and semicircular canal dysgenesis (9, 10). Interestingly, clinical features of in utero vitamin A deficiency (hypovitaminosis A) and excess (hypervitaminosis A) overlap with those observed in CHARGE syndrome, suggesting common underlying pathogenic mechanisms (5, 11, 12). This phenotypic overlap led to the early hypothesis that CHARGE may be mechanistically related to vitamin A embryopathy (13).
CHD7 is the causative gene in approximately 90% of cases of CHARGE, and in most cases, the underlying genetic mechanism is thought to be haploinsufficiency (14). CHD7 encodes an ATP-dependent chromatin-remodeling protein that regulates downstream target gene expression via changes in nucleosome accessibility (15). However, CHD7 and RA may also act together to regulate gene expression and embryonic development. In previous studies using Chd7-deficient mice, we showed that inner ear semicircular canal malformations could be rescued by citral, an inhibitor of RA synthesis, whereas forebrain neurogenesis defects were rescued by ATRA supplementation (16). These diametrically opposed effects of RA in the inner ear and brain, combined with significant phenotypic overlap among CHARGE syndrome, RA embryopathy (17), and vitamin A deficiency, suggest tissue- or cell-type-specific interactions between CHD7 and RA signaling (18, 19).
Here, we tested three possible mechanisms for interactions of RA and CHD7 during development: (a) direct interactions between RAR and CHD7, (b) RA regulation of CHD7 expression, and (c) CHD7 regulation of RA-related signaling genes and proteins. Our results provide evidence against direct interactions between CHD7 and RAR/RXR and do not support RA-mediated regulation of CHD7 gene expression or protein levels. Instead, we found that CHD7 acts upstream of the RA synthetic enzyme ALDH1A3 in the mouse inner ear and neural progenitor cells and that loss of Aldh1a3 compensates for Chd7 deficiency in semicircular canal development. Together, these data suggest that Chd7 and Aldh1a3 act in a common genetic pathway in inner ear and neuronal lineages, providing insights for exploration of the pathogenic mechanisms underlying CHARGE syndrome.
Results
Loss of Chd7 disrupts RA-related gene expression.
Previous studies from our laboratory showed that citral, a RA inhibitor, rescues Chd7 heterozygous inner ear phenotypes (16). To further investigate genetic mechanisms of this rescue, we performed genome-wide mRNA sequencing on Chd7+/+, Chd7Gt/+, and Chd7Gt/Gt microdissected inner ears from E10.5 embryos (n = 4 ears per genotype). Chd7Gt/+ inner ears showed significant downregulation of 95 transcripts and upregulation of 43 transcripts. Chd7Gt/Gt inner ears showed significant downregulation of 669 transcripts and upregulation of 416 transcripts. Differentially expressed RA-related genes were visualized using a volcano plot comparing magnitude and significance of changes in gene expression between Chd7+/+ and Chd7Gt/+ ears (Figure 1A) and between Chd7+/+ and Chd7Gt/Gt ears (Figure 1B). We found that multiple RA signaling–related genes were differentially expressed in E10.5 Chd7Gt/Gt inner ears compared with wild-type ears, including genes encoding the RA synthesis enzymes, Aldh1a1, Aldh1a2, and Aldh1a3; the RA degradation enzyme, Cyp26b1; and the RARs, Rarb and Rarg (Figure 1C). Of these, Aldh1a3 was the most dramatically upregulated in both Chd7Gt/+ and Chd7Gt/Gt inner ears.
Figure 1. Misregulation of retinoic acid–related genes with Chd7 deficiency.

(A and B) RNA-seq data from Chd7Gt/+ (A) and Chd7Gt/Gt (B) E10.5 inner ears, visualized by volcano plot. Chd7Gt/+ inner ears show significant downregulation of 95 transcripts and upregulation of 43 transcripts. Chd7Gt/Gt inner ears show significant downregulation of 669 transcripts and upregulation of 416 transcripts. Chd7 and retinoic acid (RA) signaling–related genes are labeled. n = 2 for each genotype. (C) RNA-seq data from E10.5 inner ears showing fold change in mRNA levels of RA-related genes. *P < 0.05. (D and E) Gene ontology term analysis of RNA-seq results describing processes (D) and functions (E) disrupted in E10.5 Chd7Gt/+ inner ears compared with Chd7+/+ inner ears. Significance was determined using DESeq (v. 1.22.1).
Gene ontology term analysis showed that inner ear development, neurogenesis, central nervous system development, and cellular differentiation are all highly affected by heterozygous loss of Chd7 (Figure 1D). Analysis for gene function also confirmed that Chd7 is involved in modulation of regulatory region DNA binding, chromatin binding, and RNA polymerase II regulatory region sequence-specific DNA binding, consistent with known roles for Chd7 in transcription process regulation (Figure 1E).
To validate our RNA-sequencing (RNA-seq) results, we performed quantitative RT-PCR on independent samples of Chd7 mutant mouse ears at E10.5. We found that inner ears from Chd7Gt/+ mice showed an approximately 2-fold upregulation of Aldh1a3 expression compared with inner ears from wild-type mice (Figure 2A). We also performed qRT-PCR on inner ears from E10.5 Aldh1a3 mutant mice. Aldh1a3 mutant mice were previously generated by deletion of exon 11 (20), and our TaqMan probe targeted exons 2–3; thus, some expression of Aldh1a3 in Aldh1a3–/– mice was expected. Our results showed no change in Chd7 levels with reduced Aldh1a3 dosage (Figure 2B). We also asked whether Chd7 deficiency affected Aldh1a3 expression in other tissues. To test this, we performed qRT-PCR on neural progenitor cells differentiated from Chd7+/+ and Chd7Gt/Gt mouse embryonic stem cells (ESCs) and found that Aldh1a3 expression was dramatically upregulated in Chd7Gt/Gt cells (Figure 2C).
Figure 2. Aldh1a3 is misregulated in Chd7 mutant mouse ears.

(A) Representative qRT-PCR data showing fold change in expression of Chd7 and Aldh1a3 in Chd7Gt/+ E10.5 mouse otocysts compared with Chd7+/+ otocysts. This experiment was repeated 6 times. (B) Representative qRT-PCR data showing fold change in expression of Aldh1a3 and Chd7 in Aldh1a3+/– and Aldh1a3–/– E10.5 mouse otocysts compared with Aldh1a3+/+ otocysts. This experiment was repeated 13 times. (C) Representative qRT-PCR data showing fold change in Chd7 and Aldh1a3 expression in neural progenitors differentiated from Chd7Gt/+ and Chd7Gt/Gt mouse embryonic stem cells compared with those differentiated from Chd7+/+ mouse embryonic stem cells. This experiment was repeated twice. Significance was determined by Student’s t tests, and P values were corrected using the Bonferroni method. ***P ≤ 0.001. (D–F) In situ hybridization of sectioned littermate E10.5 inner ears shows increased Aldh1a3 expression in Chd7Gt/Gt inner ears compared with Chd7Gt/+ and Chd7+/+ inner ears (n = 6 ears per genotype). Regions of high expression in the otocyst are demarcated by solid bars. Scale bars: 100 μm.
Our qRT-PCR results were further confirmed by in situ hybridization in E10.5 inner ears, which showed minimal or no Aldh1a3 expression in Chd7+/+ inner ears and dosage-dependent increases in expression of Aldh1a3 in Chd7Gt/+ and Chd7Gt/Gt inner ears (Figure 2, D–F). Immunostaining for ALDH1A3 and CHD7 showed complementary expression at E12.5 in both vestibular and auditory regions of the ear, including the semicircular canals, cochlea, and vestibular/spiral ganglia (Supplemental Figure 1; supplemental material available online with this article; https://doi.org/10.1172/jci.insight.97440DS1). Together, these results indicate that Chd7 and Aldh1a3 are expressed in the developing vestibular and auditory regions of the inner ear and that loss of Chd7 is associated with upregulated Aldh1a3 in both the inner ear and in neural progenitor cells.
Overexpression of CHD7 inhibits ALDH1A3 expression in human cells.
Because loss of Chd7 affected expression of several RA signaling genes, we asked whether overexpression of CHD7 would have the opposite effect on those genes. To test this, we transfected 293T cells with a pcDNA 3.1-Flag vector containing full-length human CHD7 cDNA and confirmed overexpression by Western blotting (Figure 3, A and B). We then assayed for changes in a panel of RA-signaling genes using qRT-PCR (Figure 3C). Following overexpression of CHD7, expression of several RA signaling genes was altered. Most notably, ALDH1A3 transcripts were reduced 35% in cells overexpressing CHD7 compared with empty vector control cells. This result is consistent with our RNA-seq and qRT-PCR data from mouse inner ears and neural progenitor cells. It also suggests that the inhibitory effect of CHD7 on ALDH1A3 expression is conserved across species, cell types, and tissues.
Figure 3. Increased CHD7 affects expression of retinoic acid–related genes.

(A and B) 293T cells transfected with a vector containing human CHD7 and FLAG. CHD7 and FLAG levels were assayed by Western blot (A) and quantified (B) using ImageJ software. This experiment was repeated twice. Images were derived from duplicate samples run contemporaneously on parallel gels. (C) Expression of retinoic acid–related genes examined using qPCR of mRNA extracted from 293T cells transfected with CHD7 overexpression or empty vector. This experiment was repeated 2 times. Significance was determined by Student’s t tests. *P ≤ 0.05.
CHD7 occupies genomic regions near ALDH1A3.
Given the robust changes in ALDH1A3 expression with altered Chd7 dosage, we asked whether ALDH1A3 is a direct target of CHD7 binding. To test this, we performed CHD7 ChIP-qPCR on chromatin isolated from human SH-SY5Y neuroblastoma cells, which are known to express CHD7 (21) and to differentiate into neurons in response to RA (Supplemental Figure 2) (21, 22). Four candidate genomic regions were selected to detect direct binding of CHD7 on the ALDH1A3 locus based on publicly available ChIP-sequencing (ChIP-seq) data gathered from H1-hES cells (GEO accession GSM1003473) and vertebrate conservation (Figure 4A). Regions 1 and 2 are upstream of the transcription start site, while regions 3 and 4 fall within intron 1. ChIP-qPCR showed that the two sites upstream of the transcription start site, particularly region 2, were substantially enriched for CHD7, suggesting that CHD7 directly binds to and regulates expression of the human ALDH1A3 gene.
Figure 4. CHD7 binds the region upstream of the ALDH1A3 gene in human and mouse cells.

(A) ChIP-qPCR analysis in human SH-SY5Y neuroblastoma cells targeting CHD7 upstream of ALDH1A3. Four candidate sites (red squares) were selected based on publicly available CHD7 ChIP-seq data from human embryonic stem cells (GEO accession GSM1003473) and the high degree of sequence conservation among vertebrates (67). (B) Chd7 ChIP-qPCR analysis in mouse E10.5 microdissected otocysts upstream of Aldh1a3. BLAST was used to identify the homologous sequences upstream of mouse Aldh1a3; site 2′ (purple square) was selected for Chd7 ChIP-qPCR analysis. qPCR reactions were repeated two times in triplicates. Data are represented as mean ± SEM, and significance was determined by 2-tailed unpaired t tests.
To determine whether CHD7 also binds the Aldh1a3 locus in the mouse inner ear, we performed ChIP-qPCR on E10.5 mouse microdissected otocysts. A BLAST search was performed to locate regions with sequence similarity between the sites identified to be bound by CHD7 in human cells and mouse Aldh1a3 (23). Candidate site 2′ (Figure 4B) was selected for further analysis. As a negative control, we included the transcription start site of the mouse Aldh1a3 gene. We also included upstream site 5 (Figure 4B), since it has been shown to be occupied by CHD2 in the CH12-LX murine cell line (Stanford/Yale Transcription Factor Binding Site track in UCSC browser). Our ChIP-qPCR results showed significant enrichment of CHD7 at upstream site 5 compared with the TSS and with site 2′. Taken together, these results suggest that CHD7 directly interacts with genomic sites upstream of ALDH1A3 in both human and mouse cells.
CHD7 is insensitive to RA levels.
In Chd7 mutant mice, neuronal and inner ear abnormalities are influenced by altered RA levels (16), raising the possibility that RA may directly regulate Chd7 gene expression. To test this biochemically, we again used SH-SY5Y neuroblastoma cells that express high levels of CHD7 and readily differentiate into neurons upon RA treatment (24, 25). We selected CHD5, a CHD7-related chromatin remodeler, as a positive control, since it is highly upregulated in response to 13-cis-RA but not ATRA (26). Histone deacetylase 2 (HDAC2) was used as a loading control, as it was previously shown to be unaffected by 13-cis-RA (26). Unlike CHD5, CHD7 levels were stable in response to both ATRA and 13-cis RA (Figure 5, A–E). CHD7 levels were also insensitive to the ALDH1A inhibitors citral and DEAB (27) (Figure 5, E and F).
Figure 5. Retinoic acid does not affect CHD7 levels in SH-SY5Y cells.

Western blotting of CHD7 and CHD5 levels in various cell types. HDAC2 is used a loading control. Each experiment was performed at least 2 times. (A) Treatment with 10 μM all-trans retinoic acid (ATRA) increased CHD5 levels and had no effect on CHD7 levels over 12 days. (B) Treatment with 1, 5, or 10 μM ATRA for 7 or 12 days mildly induced CHD5 levels and had no effect on CHD7. (C) Treatment with 10 μM 13-cis retinoic acid (13-cis-RA) for 6 days increased CHD5 levels but had no effect on CHD7 levels relative to treatment with DMSO only. (D) Treatment with 1, 5, or 10 μM 13-cis-RA for 7 or 12 days induced CHD5 levels and had no effect on CHD7 levels. (E) Treatment with ATRA (1, 5, or 10 μM) or citral (1, 5, or 10 μM) for 3 days had no effect on CHD7 levels. (F) Treatment with 10 μM DEAB for 3 days had no major effect on CHD7 levels. This experiment was repeated twice. (G) CHD7 levels were lower in neural progenitor cells derived from forebrains of E12.5 Chd7Gt/+ embryos compared with Chd7+/+ embryos. (H) Treatment with 10 μM ATRA or 10 μM citral had no effect on CHD7 levels relative to DMSO vehicle control in neural progenitors derived from forebrains of E12.5 Chd7+/+ or Chd7Gt/+ mice.
To assay CHD7 protein levels in other neuronal cells, we established neural progenitor cell lines from two Chd7+/+ and two Chd7Gt/+ mouse E12.5 embryonic forebrains. CHD7 protein levels were reduced in Chd7Gt/+ neural progenitor cells compared with Chd7+/+ neural progenitor cells (Figure 5G). Notably, CHD7 levels in forebrain-derived neural progenitors from Chd7+/+ and Chd7Gt/+ mice were also stable after ATRA or citral treatment (Figure 5H). Together, these results indicate that CHD7 protein levels are insensitive to RA signaling in human SH-SY5Y cells and mouse forebrain-derived neural progenitors.
Evidence against interactions between CHD7 and RAR.
RA acts in the cell nucleus by binding to RAR, which induces RAR heterodimerization with RXR on RA response elements (RAREs) to activate downstream gene expression (28). We next asked whether CHD7 might physically interact with RAR/RXR heterodimers and act as a corepressor or coactivator of downstream RA gene expression. To test this, we again used human SH-SY5Y neuroblastoma cells, which express all isoforms of RAR and RXR (22). We used pan-RAR antibody for CHD7 coimmunoprecipitation, since anti-RAR antibody also interacts with heterodimers containing RXR. In untreated SH-SY5Y cells, coimmunoprecipitation with anti-CHD7 and pan anti-RAR antibodies showed the expected enrichment of CHD7 and RAR proteins, respectively (Figure 6A). In contrast, CHD7 was absent in anti-RAR immunoprecipitates, and RAR was absent in CHD7 immunoprecipitates. Similar results were observed in SH-SY5Y cells after 7 days of treatment with 10 μM ATRA (Figure 6B). These results provide evidence against direct interactions between CHD7 and RAR-containing complexes as a means for promoting SH-SY5Y neuronal differentiation and regulation of RA-responsive gene expression.
Figure 6. CHD7 does not directly bind RAR in human SH-SY5Y neuroblastoma cells, and there is minimal overlap in genomic binding sites in mouse embryonic stem cells.
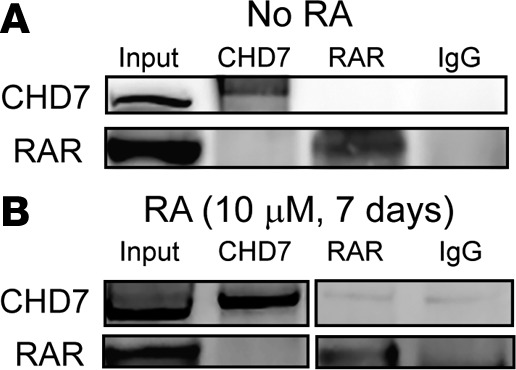
Representative coimmunoprecipitation blots of CHD7 and RAR show no direct binding between CHD7 and RAR in (A) untreated (n = 2) or (B) all-trans retinoic acid–treated (ATRA-treated) (10 μM for 7 days) SH-SY5Y cells (n = 2). Images in B were derived from duplicate samples run contemporaneously on parallel gels.
CHD7 and RAR exhibit restricted colocalization in mouse ESCs.
CHD7 is known to directly associate with several proteins during development, including p300, OCT4, NANOG, and SOX2 (29, 30). To further test the hypothesis that CHD7 may associate with RAR/RXR heterodimers at RAREs located throughout the genome, we undertook a computational analysis of CHD7 and RAR colocalization in mouse ESCs using published ChIP-seq data sets. Specifically, we obtained data from a CHD7 ChIP-seq experiment (30) and a pan-RAR ChIP-seq experiment, both performed in mouse ESCs (31) (see the Methods for GEO/SRA accession information). To best replicate the conditions in which we would expect CHD7 to colocalize with RAR, we used RAR ChIP-seq data acquired from mouse ESCs 8 hours after treatment with RA, which was shown to significantly enhance pan-RAR antibody binding across the genome (32). Following intersection of CHD7- and RAR-associated reads, we determined that co-occupancy by both proteins was restricted to approximately 0.6% of all CHD7-bound sequences and 0.9% of all RAR-bound sequences, suggesting that direct interaction between CHD7 and RAR is not a primary reason for the high phenotypic overlap between CHARGE syndrome and RA embryopathy.
CHD7 does not alter global embryonic RA activity.
Given our evidence against direct interactions between CHD7 and RARs, as well as unchanged CHD7 levels with RA treatment or inhibition, we next tested whether CHD7 regulates RA signaling. To assay for changes in RA activity in vivo and in vitro upon Chd7 loss, we used transgenic mice with RARE-lacZ (33) and cells with RARE-luciferase (34) and varying dosage of Chd7. RARE-lacZ–transgenic mice express lacZ as early as E10.5 in the neural tube, heart, eye, and ear. Thus, we established crosses of RARE-lacZ;Chd7+/+ and RARE-lacZ;Chd7+/– mice and performed whole-mount X-gal staining and staining of sections from E10.5–E12.5 embryos (Figure 7, A–G). At E10.5, X-gal staining was present throughout the neural tubes of RARE-lacZ mice containing Chd7 wild-type, heterozygous, and null alleles (Figure 7, A–C). Interestingly, there were no observable differences in X-gal staining among these embryos, indicating that global embryonic RA activity does not depend on proper Chd7 dosage.
Figure 7. Chd7 dosage does not affect global retinoic acid reporter activity.

(A–G) E10.5, E11.5, or E12.5 embryos containing RARE-lacZ transgene and 2 (Chd7+/+), 1 (Chd7+/–), or no (Chd7–/–) wild-type alleles for Chd7 were stained with X-gal to reflect RA activity. Chd7–/– embryos were hypoplastic and did not survive beyond E10.5; otherwise, no differences in X-gal staining pattern between wild-type and Chd7 mutant embryos were noted. Embryo counts are as follows: Chd7+/+ — E10.5, n = 4; E11.5, n = 4; E12.5, n = 5; Chd7+/– — E10.5, n = 8; E11.5, n = 5; E12.5, n = 7; Chd7–/– — E10.5, n = 1. (H) Overexpression of FLAG-HA-hCHD7 in 293T cells has no effect on RARE-luciferase reporter in the presence or absence of 1 μM all-trans retinoic acid (RA). K998R CHD7 missense mutant construct also has no effect on RARE-luciferase reporter activity. Data are shown as luciferase normalized to Renilla. (I) siRNA knockdown of hCHD7 in 293T cells does not alter RARE-luciferase reporter activity, in the presence or absence of 1 μM RA. Control siRNA against cyclophilin B enhances RA activity as expected (35). Data are shown as luciferase normalized to Renilla. Significance was determined by ordinary 1-way ANOVA tests. **P ≤ 0.001.
To determine whether the central nervous system or inner ear exhibits differential RA activity with varying Chd7 dosage, we sectioned X-gal–stained embryos at E10.5, E11.5, and E12.5. We found enrichment of X-gal stain in the neural tube, hindbrain, and cochleovestibular ganglion in all embryos, with no qualitative differences in staining between genotypes or stages (data not shown). X-gal staining of inner ear sections from E13.5 RARE-lacZ;Chd7+/+ and RARE-lacZ;Chd7+/– embryos showed robust RARE-lacZ activity in specific regions throughout the inner ear, including the semicircular canals, VIII nerve ganglion, and utricle that was similar in both genotypes (Supplemental Figure 3). We also performed coculture of dissociated E8.5 Chd7+/+, Chd7+/–, and Chd7–/– mouse embryos together with F9 cells expressing RARE-lacZ (Supplemental Figure 4) (34). This assay showed no measurable differences in RA activity across the three genotypes.
To test whether CHD7 is sufficient to alter RA activity, we performed transient transfections in 293T cells with RARE-luciferase and a CHD7 overexpression vector (for gain of function) or CHD7 siRNA (for loss of function). For all luciferase analyses, Renilla was used as a control. In CHD7 gain-of-function studies, 293T cells were transfected with empty pcDNA 3.1 vector or pcDNA 3.1-Flag-CHD7 or pcDNA 3.1-Flag-CHD7 K998R (Figure 7H). In a loss-of-function test, siRNA was used to knockdown CHD7 in 293T cells (Figure 7I). Cyclophilin B siRNA was used as a positive control, as it is known to repress RARE activity (35). Overexpression or knockdown of CHD7 had no effect on luciferase, with or without RA treatment, indicating that CHD7 is insufficient to alter RA signaling in cultured cells. Together, these results suggest that loss of Chd7 has no major effects on RA activity based on X-gal staining of whole-mounted and sectioned embryos and coculture assays.
Loss of Aldh1a3 rescues Chd7Gt/+ inner ear malformations.
Because inner ear development is sensitive to RA and because CHD7 regulates Aldh1a3 expression, we tested the functional relationship between Chd7 and Aldh1a3 in vivo by crossing Chd7Gt/+ and Aldh1a3+/– mice and paint-filling E14.5 embryonic inner ears (Figure 8). Heterozygous loss of Chd7 results in highly penetrant semicircular canal abnormalities in a variety of genetic backgrounds (36, 37). Similarly, all Chd7Gt/+ inner ears exhibited variable semicircular canal abnormalities, primarily affecting the lateral and posterior canals, with no specific differences between right and left ears (Figure 8, A and D). Aldh1a3+/– and Aldh1a3–/– ears showed no detectable semicircular canal abnormalities, whereas Chd7Gt/+;Aldh1a3+/– double heterozygous ears exhibited semicircular canal defects strikingly similar to those present in Chd7Gt/+ mice (Figure 8, B, C, and E). Interestingly, loss of both copies of Aldh1a3 in Chd7Gt/+ mice led to normal-appearing semicircular canals in 42% (12 of 28) of ears examined (Table 1), suggesting partial rescue of Chd7Gt/+ related inner ear malformations.
Figure 8. Aldh1a3 loss rescues Chd7-deficient inner ears.

Paint-filled inner ears from E14.5 embryos obtained by crossing Chd7Gt/+ and Aldh1a3+/– mice. (A) Shown are the cochlea (COC), endolymphatic duct (ED), posterior semicircular canal (PC), anterior semicircular canal (AC), lateral semicircular canal (LC), and saccule (SAC). Ears with loss of 1 (Chd7+/+;Aldh1a3+/–; B) or both (Chd7+/+;Aldh1a3–/–; C) copies of Aldh1a3 appear similar to wild-type ears. Ears from Chd7Gt/+;Aldh1a3+/+ (D) and Chd7Gt/+;Aldh1a3+/– (E) embryos exhibit lateral canal truncation (*) and posterior canal defects (#), whereas 42% (12 of 28) of ears from Chd7Gt/+;Aldh1a3–/– (F) mice appear normal. (A–C) Left ears; (D–F) right ears. There were no differences in left-right laterality of phenotypes. Table 1 shows the number of affected ears for each genotype, as well as n values. Scale bars: 500 μm.
Table 1. Phenotypes of paint-filled mouse inner ears.

Discussion
In this study, we explored the underlying mechanisms by which CHD7 and RA regulate inner ear and neural development. Our results show that CHD7 directly binds to and represses Aldh1a3 in both developing mouse inner ear and human neuroblastoma cells. Interestingly, loss of Chd7 has no apparent effect on RA activity at the level of the whole embryo or in sectioned inner ears, and does not alter RA activity in vitro. Neither does RA stimulate CHD7 expression in human neuroblastoma cells (unlike CHD5, which is positively regulated by RA, ref. 26), nor does RA promote direct interaction between CHD7 and RAR, either before or after rapid induction of neuronal differentiation. Nevertheless, genetic loss of Aldh1a3 partially rescues the lateral and posterior semicircular canal malformations observed in Chd7Gt/+ embryos, consistent with CHD7 acting upstream of RA signaling in the ear or with RA-independent effects of CHD7. Together, these data indicate complex interactions between RA and CHD7, and support direct repression of Aldh1a3 by CHD7, as shown in our model (Figure 9).
Figure 9. Model depicting the influences of CHD7 and RA signaling on gene expression.

In the cytoplasm, ALDH1A3 catalyzes the oxidation of retinaldehyde to retinoic acid (RA). RA then enters the nucleus and binds RA receptor (RAR). RAR forms a heterodimer with retinoid receptor (RXR), and the complex binds RA responsive elements (RAREs) to regulate transcription of downstream genes. Shown are three potential interactions between CHD7 and RA that we investigated in this study. In the first potential mechanism, CHD7 forms a complex with RAR/RXR heterodimers that acts to activate transcription of downstream target genes. In the second potential mechanism, RAR/RXR heterodimers bind upstream of CHD7 to regulate its transcription. In the third potential mechanism, CHD7 binds upstream of ALDH1A3, repressing its transcription. Our results provide evidence against the first two mechanisms, in favor of the third.
CHD7 effects on RA pathway genes.
Our RNA-seq analysis of Chd7Gt/+ embryonic ears showed mildly reduced expression of Aldh1a1, Aldh1a2, and Cyp26b1, suggesting that these genes may be additional targets of CHD7. Dynamic expression of multiple RA-related genes has been described in inner ear development. All three Aldh1a genes (Aldh1a1, Aldh1a2, and Aldh1a3) and Cyp26b1 genes are expressed in the developing ear and undergo complex changes in expression patterns throughout embryogenesis (6). Notably, Aldh1a and Cyp26b genes are expressed in complementary regions of the ear and surrounding areas, suggesting cross-regulatory mechanisms that control RA levels. These findings are consistent with the distribution of RA across the anterior-posterior axis of the developing ear. For example, Aldh1a2 is highly expressed in the mesenchyme posterior to the otic region, whereas Cyp26b1 is highly expressed in the ectoderm anterior to the otic region (6). The complex misregulation of RA metabolism genes upon Chd7 loss suggests there may be compensatory mechanisms that explain the lack of major effects on RARE activity observed in our Chd7 mutant embryos. CHD7 could also have additional RA-related targets that have yet to be characterized. Our results represent what we believe to be the first report of a genetic rescue for Chd7 mutant inner ears in mice and the first identification of a RA-related gene as a direct transcriptional target of CHD7 in the ear.
Mechanisms of CHD7 and RA signaling.
At transcriptionally silent genes, RAR/RXR heterodimers bind RARE sequences and form repressive complexes that include polycomb-related proteins (38). RA treatment stimulates release of polycomb and recruitment of transcription factors that activate or repress downstream gene expression (16). Given the high phenotypic overlap among CHARGE syndrome, RA embryopathy, and vitamin A deficiency, we predicted that CHD7 may directly interact with RAR-RXR heterodimers to regulate gene expression. However, coimmunoprecipitation showed no direct interaction of CHD7 and RAR. We also hypothesized that Chd7 may directly regulate RA levels. Whole-mount embryos and inner ear sections of E10.5–E13.5 RARE-lacZ mice, however, exhibited no change in RA reporter levels with loss of Chd7. Consistent with these results, mRNA sequencing of Chd7 heterozygous inner ears at E10.5 showed no change in expression of other genes known to be downstream of RA, such as Pitx2, Gad1, Hoxa1, Hoxb1, Hoxa3, Hoxd4, Hnf1b, Pax6, Olig2, Hoxa5, Tgfb1, Fgf8, Cdx1, and Pdx1. Furthermore, altered Chd7 dosage by overexpression and siRNA knockdown had no effect on RA-induced RARE-luciferase activity in vitro. These results suggest that CHD7 regulates the expression of the RA synthase gene Aldh1a3 but does not affect global RA signaling, suggesting that the Chd7/Aldh1a3 pathway may have a different function in development.
CHD7-containing complexes have been characterized in a variety of cell types, including human ESCs (PBAF), HeLa cells (WDR4, FAM124B), mouse neural progenitors (SOX2 and other proteins), and cardiomyocytes (SMAD1/5/8 and Brg1) (29, 32, 39–42). The lack of interaction between CHD7 and RA suggests that instead of functioning in the same large complex, CHD7 may regulate nucleosome accessibility independent of RAR/RXR heterodimers. This could occur prior to or concomitant with heterodimer binding via movement of nucleosomes as a means for increasing or decreasing access to RARE sites in promoter or enhancer regions. Importantly, CHD7 may exhibit RAR/RXR-dependent functions in other cell types, embryonic stages, or specific organs not assayed in our studies.
CHD7 binds to thousands of promoter and regulatory regions in the mammalian genome (30) and has been shown to activate expression of several downstream target genes, including Twist1, Reelin, Sox4/Sox11, Otx2, Gbx2, Osterix, and Creb3l2 (39, 43–46). To our knowledge, this is the first report of a confirmed target gene that is repressed by CHD7.
We also intersected CHD7-bound sequences with RAR-bound sequences obtained from two previously published ChIP-seq data sets (30, 31). We found that fewer than 1% of assayed sequences exhibited co-occupancy by both CHD7 and RAR. By contrast, nearly 60% of SOX2-bound sites in the human NSC genome exhibit CHD7 co-occupancy (29). These data indicate that CHD7 does not colocalize as extensively with RAR (and, by proxy, with RAR/RXR heterodimers) as with other transcription factors and cofactors. Thus, co-occupancy of genomic sites by CHD7 and RAR/RXR heterodimers is not a primary mechanism by which RA signaling is regulated in the developing mouse or in human tissues. Further experimentation is needed to determine how CHD7 regulation of downstream genes leads to the phenotypic consequences observed in CHARGE syndrome.
Roles for Chd7 and Aldh1a3 in inner ear development.
Homozygous loss of Chd7 in mice is embryonic lethal after E10.5, whereas heterozygous loss of CHD7 in humans causes CHARGE syndrome (47, 48). Chd7 heterozygous and conditional-deletion mice exhibit highly penetrant abnormalities in semicircular canals, ranging from hypoplasia to truncation or complete absence (36). In Chd7 heterozygous mice, lateral canals are commonly affected, with variable defects in posterior canals, whereas conditional knockouts exhibit complete loss of vestibular structures, with only rudimentary endolymphatic ducts (36, 49). Treatment of Chd7Gt/+ mice with citral, an inhibitor of ALDH1A enzymes, rescues inner ear semicircular canal abnormalities in roughly one-third of embryos (16). Here, we found that loss of Aldh1a3 also rescues the lateral and posterior semicircular canal malformations in a significant proportion of Chd7Gt/+ mice, consistent with genetic epistasis. Interestingly, this degree of rescue by Aldh1a3 loss is similar to that previously observed with citral treatment (16). Our results also predict that increased Aldh1a3 dosage may mimic Chd7 heterozygous mutant inner ear malformations, but this has not been tested. Previous studies have shown that Aldh1a3–/– mice exhibit subtle structural inner ear malformations that are detectable only by 3D reconstruction from serial sections (50). Such minor differences in anatomy in our mutant mice may have been missed by the paint-fill approach. Nevertheless, the mouse inner ear is highly sensitive to dosage of both Chd7 and Aldh1a3 (16).
In humans, recessive pathogenic variants in ALDH1A3 are associated with a variety of ocular malformations (microphthalmia, anophthalmia, displaced pupils, and coloboma), structural cardiac defects, developmental delay, and autistic features (51, 52). Although Aldh1a3–/– mice show subtle inner ear abnormalities, there are no reports of hearing or balance disturbance in humans with heterozygous or homozygous pathogenic ALDH1A3 variants, suggesting that there may be compensatory mechanisms, differences in phenotypes between humans and mice, reduced penetrance of inner ear malformations, or incomplete clinical evaluations for reported individuals. Future studies of humans with pathogenic or presumed pathogenic variants in ALDH1A3 should help determine whether these individuals exhibit auditory or vestibular abnormalities.
Our RNA-seq and qRT-PCR data from microdissected mouse inner ears, combined with ChIP experiments in inner ears and SH-SY5Y cells, indicate that CHD7 acts upstream of Aldh1a3 via direct binding and repression of Aldh1a3. The rescue of Chd7 mutant inner ears by loss of Aldh1a3 is also consistent with genetic epistasis. Our data provide insights into the roles for RA and CHD7 during development and suggest avenues for exploring the effects of Chd7 haploinsufficiency in other tissues and cell types.
Methods
All reagents and culture media were purchased from Life Technologies/Invitrogen (Carlsbad) unless otherwise indicated.
Mice.
Chd7Gt/+ (47) (JAX stock 030659) mice were maintained by backcrossing B6129SF1/J (JAX stock 101043) mice to N7. Aldh1a3+/– mice (20) (JAX stock 018846) were maintained by backcrossing with B6129SF1/J (JAX stock 101043) and intercrossing with Chd7Gt/+. Chd7+/– mice were generated by crossing the Chd7flox/flox mice (53) (JAX stock 030660) with EIIa-cre mice (JAX stock 003724). A homozygous breeding pair of RARE-lacZ mice was obtained from JAX (stock 008477) for maintenance of the RARE-lacZ line. Chd7+/– mice were crossed with RARE-lacZ mice to generate Chd7+/–;(RARE-lacZ) mice, which were then backcrossed to the homozygous RARE-lacZ strain in order to generate Chd7+/–;(RARE-lacZ × 2) mice. Chd7+/– mice were maintained by backcrossing with B6129SF1/J mice (JAX stock 101043) and intercrossing with Chd7+/– mice. Chd7flox/flox mice (53) (JAX stock 030660) and Chd7Gt/+ mice (47) (JAX stock 030659) have been recently archived at Jackson Laboratory.
Cell culture.
Human SH-SY5Y neuroblastoma cells and 293T cells were purchased from ATCC and cultured in DMEM with 20% FBS. E12.5 forebrain neural stem cell cultures were established from Chd7Gt/+ or Chd7+/+ littermate embryos, as previously described (54). Left and right forebrains were dissected under a microscope, digested with Accutase (Invitrogen) to single-cell suspension for 15 minutes at 37°C, and cultured into individual wells of a 12-well plate in 5% CO2 at 37°C. Yolk sacs were collected for genotyping, as previously described (47), and neural stem cells were cultured in NSCM (DMEM/F, B27 N2, 20 ng/ml EGF, 20 ng/ml bFGF).
Neural progenitor cell differentiation from mouse ESCs.
Neural progenitor cell differentiation from ESCs followed a previously published protocol (55). Mouse Chd7+/+ and Chd7Gt/Gt ESCs were harvested and dissociated into single cells using 0.05% Trypsin, seeded at a density of 3 × 105 cells/ml in embryoid body medium (EBM) (ESCM without LIF) onto petri dishes to form embryoid bodies, and cultured for 4 days in EBM. Embryoid bodies were then cultured with 5 μM RA for 4 days, dissociated, and plated into cell culture dishes with DMEM/F12 supplemented with B27, EGF, and bFGF to form neurospheres and cell lines.
Inner ear dissections.
Chd7Gt/+ male mice were crossed with Chd7Gt/+ female mice to generate timed pregnancies. Crosses were also established between Aldh1a3+/– and Aldh1a3+/– male and female mice in order to generate timed pregnancies. The day of vaginal plug identification was designated as E0.5. Pregnant dames were euthanized by cervical dislocation at E10.5, and embryos were removed for further dissection. The yolk sac and caudal third of each embryo was saved for DNA isolation and genotyping as previously described (47). Inner ears were dissected from E10.5 embryos into cold PBS. After brief centrifugation at 200 g, PBS was removed and samples stored at –80 °C.
RNA-seq.
RNA-seq was performed on E10.5 Chd7+/+, Chd7Gt/+, and Chd7Gt/Gt ears (n = 2 pairs of ears per genotype). Polyadenylated RNA was extracted from each microdissected ear sample using the RNAqueous-Micro RNA Isolation Kit (Ambion). RNA was reverse transcribed, and cDNA libraries were prepared with the Illumina TruSeq RNA-seq v2 kit. Libraries were sequenced using the Illumina HiSeq 2500 platform with paired-end 100-bp reads. Raw reads were aligned to the mouse reference genome (NCBI mm9) using Bowtie2 (v.2.0.6) (56). Unique reads were retained and normalized to the total of mapped reads for each sample. Expression levels of mapped reads were calculated in units of fragments per kilobase of transcript per million mapped reads (FPKM). Genes with low FPKM ( < 0.3) were removed from analysis. Differential expression between genotypes was calculated using DESeq (v.1.22.1), and genes were considered statistically significant with a Benjamini-Hochberg P < 0.05 (57).
Gene ontology analysis.
Gene ontology analysis was performed using GOTermFinder on differentially expressed genes identified in the E10.5 RNA-seq analyses (58). Gene ontology terms associated with process, function, and component were obtained using a significance threshold of P < 0.05 and FDR < 0.05.
Bioinformatic analysis.
RARE motifs were identified within the mouse Chd7 gene sequence (mm10), including 10,000 bp upstream and downstream inclusive of relevant regulatory regions, using the FIMO script contained within the MEME Suite (59). Using the Galaxy tool suite (60), RAR-bound regions contained within the mouse Chd7 gene sequence were identified by reanalysis of pan-RAR ChIP-seq data (31). ChIP-seq sites were uploaded into the UCSC Genome Browser, and RAR-bound sites at the Chd7 locus were cross-referenced with RAREs.
BED files from a previously published CHD7 ChIP data set (30) were obtained from the NCBI GEO database (accession GSE22341). Coordinates of the BED files were uploaded to custom tracks within the UCSC Genome Browser to allow mapping of the putative CHD7 protein localization site both within the coding sequence of RA-related genes and within a 10-kb window surrounding each gene.
Quantitative real-time PCR.
For quantitative RT-PCR in cells, total RNA was extracted from cell pellets with chloroform (ACROS Organics) and isopropanol (ACROS Organics, NJ) followed by DNAse I treatment (Roche) to avoid genomic DNA contamination. For qPCR of inner ears, total RNA was isolated from frozen otic tissue samples using the RNAqueous-Micro Total RNA Isolation Kit (AM1931, ThermoFisher Scientific). Total RNA was quantified by Nanodrop 2000 (ThermoFisher Scientific), and 2 μg RNA per sample was reverse transcribed using SuperScript First-Strand Synthesis System. Quantitative real-time PCR analysis was performed using SYBR master mix (ABI) on an ABI 7500 qPCR machine (ABI) using a standard protocol. Primers are listed in Table 2.
Table 2. Human qPCR primers.

To measure gene expression in E10.5 otocysts, embryos were harvested into 1X PBS on ice. Immediately following collection, embryos were bisected along the sagittal midline. Otic tissue was microdissected bilaterally (with care taken to remove periotic mesenchyme) into 100 μl ×1 PBS, spun at 200 g for 10 seconds, micropipette aspirated, and flash frozen on dry ice. The remaining tissue was collected for genotyping. Total RNA was isolated using the RNAqueous-Micro RNA Isolation Kit (Ambion) and treated with DNaseI prior to cDNA synthesis. cDNA was generated using the SuperScript First-Strand cDNA Synthesis System with oligo(dT) primers (Invitrogen). Gene expression levels were assayed using the TaqMan system (Applied Biosystems). Expression was assayed using TaqMan probes for Chd7 (product Mm01219527_m1) and Aldh1a3 (product Mm00474056_m1). Atp5b (product Mm01160389_g1) was used as a reference control. Each sample was run in triplicate using a StepOne-Plus Real-Time qPCR System (Applied Biosystems). Fold change of the assayed gene relative to Gapdh was defined as 2–ΔΔCT. An unpaired t test (for comparisons between two genotypes) or an ordinary 1-way ANOVA (for comparisons among three genotypes) was performed on Atp5b-normalized fold change values to determine statistical significance (P < 0.05).
In situ hybridization.
In situ hybridization was performed on sections from paraffin-embedded embryos using digoxigenin-labeled riboprobes as previously described (61), at annealing temperature 65°C. Aldh1a3 in situ probe was generously provided by Kenneth Campbell (Cincinnati Children’s Hospital) (62). Multiple sections from at least 4 ears of each genotype were tested.
Immunofluorescence.
Immunofluorescence was performed on sections from paraffin-embedded embryos as previously described (61). Anti-CHD7 (D3F5 6505) (Cell Signaling) and anti-ALDH1A3 (ab129815) (Abcam) were used at final concentrations of 1:7,500. Secondary biotin-conjugated donkey anti-Rabbit antibody (Jackson Immunochemicals) was processed using tyramide signal amplification 555 (T30955) (ThermoFisher) and then visualized on a Leica DMB upright fluorescence microscope.
CHD7 overexpression.
Flag-HA-pcDNA3.1 (Addgene) was used as a control vector. pcDNA3.1-Flag-6xHis v.1 hCHD7 WT and pcDNA3.1-Flag-6xHis v.1 hCHD7 K998R vectors, donated by Joanna Wysocka (Stanford University, Stanford, California, USA), were as previously published (39). For mRNA detection, 5 × 106 HEK293T cells were plated in 10-cm2 dishes the day before transfection. Vectors were transfected with Lipofectamine 2000 Transfection Reagent (ThermoFisher Scientific). Empty Flag-HA-pcDNA3.1 was used as a control. Forty-eight hours after transfection, cells were collected and lysed with Trizol. mRNA was extracted with chloroform (ACROS Organics) and isopropanol (ACROS Organics) followed by DNAse I treatment (Roche) to avoid genomic DNA contamination.
ChIP-qPCR.
Putative human CHD7-binding sites were identified by analysis of CHD7 ChIP-seq data in hESCs using the UCSC genome browser. Primers were designed to target 4 sites around ALDH1A3: two upstream of the transcription start site and two in the first intron (63) (ENCODE accession ENCSR000AVA) (primer sequences are listed in Table 3). For chromatin preparation, SH-SY5Y cells were plated in 10-cm2 dishes and cultured in DMEM and 10% FBS. Confluent cells were washed twice with PBS while still adherent. Cells were cross-linked in 1% paraformaldehyde for 30 minutes at room temperature and quenched by 250 mM glycine. Cells were then collected in 15 ml conical tubes and harvested by centrifugation. All cell pellets were washed with PBS and frozen on dry ice. Cell pellets were treated with 100 mM Tris (pH 7.5), 10 mM EDTA, 1% Triton X-100, 1% sodium deoxycholate, and 10% glycerol and then sheared by sonication. Chromatin was then immunoprecipitated by incubating with 8 μl CHD7 antibody (Cell Signaling) or 4 mg rabbit normal IgG antibody (Jackson ImmunoResearch) prebound to DYNAbeads M-280 (Life Technologies) overnight at 4°C. CHD7-bound beads were washed 3 times with 150 mM NaCl in IP wash buffer, 3 times with 500 mM LiCl (MilliporeSigma) in IP wash buffer, and once with TE. Harvested chromatin was then eluted from the beads, crosslinks were reversed, and DNA was purified with the Minelute PCR purification kit (Life Technologies/Invitrogen). qPCR was carried out with SYBR Master mix on a 7500 ABI qPCR machine.
Table 3. ChIP-qPCR primers.

For mouse E10.5 otocysts, 4 CD-1 timed pregnant females were obtained from Charles River Breeding Labs. A total of 50 embryos were dissected after cervical dislocation and hysterectomy. Both (left and right) otocysts (total of 100) were microdissected from each embryo, collected in 200 μl PBS, and gently spun and the supernatant was discarded. The otocysts were frozen on dry ice and stored at –80°C. Crosslinking and nucleus preparation was performed as described previously (30) (truChIP Chromatin Shearing Reagent Kit, Covaris). Chromatin was sheared by sonication (Covaris S220 Focused-ultrasonicator) to generate 400- to 800-bp chromatin fragments. Chromatin samples were incubated with anti-CHD7 antibody (catalog 6505, Cell Signaling) and immunoprecipitated with Protein A Magnetic Beads (Fisher Scientific). Reversal of cross-linking was performed overnight followed by elution of DNA fragments. BLAST was used to search for regions with sequence similarity between the identified human CHD7-bound sites and mouse upstream regions of Aldh1a3 (23). For qPCR, SYBR Green Master Mix (Applied Biosciences) was used on a StepOne-Plus Real-Time PCR machine (Applied Biosciences), and results were analyzed using the percent input method. Technical triplicates with standard errors were reported, and statistical differences were determined by Student’s t test.
Cell growth and differentiation assays.
SH-SY5Y cells (5 × 105 or 1 × 106; n = 3 wells per condition) were plated in 6-well plates and allowed to grow for 2, 4, or 6 days; digested in 0.05% Trypsin-EDTA; resuspended in culture media; and collected and counted in trypan blue with a hemacytometer. Cell numbers were analyzed by Student’s t test, and results were graphed in Excel. For cell differentiation assays, anti-Tuj1 (neuron-specific class III β-tubulin) was used as a marker of differentiated neurons. SH-SY5Y cells were plated onto coverslips in 12-well plates at a density of 5 × 105 cells per well. After 7 days of treatment with DMSO (vehicle), RA (10 μM), or citral (10 μM), cells were washed with PBS and then fixed in 4% paraformaldehyde (MilliporeSigma) for 30 minutes. Cells were then washed with PBS and blocked in 2% BSA blocking solution (MilliporeSigma) for 2 hours at room temperature. Cells were then incubated with anti-Tuj1 antibody (1:100, catalog MRB-435P, Berkeley Antibody Company) overnight at 4°C, washed in PBS, and incubated with goat anti-rabbit Alexa Fluor 555 (1:1,000, catalog A21137, Invitrogen ) for 1 hour at room temperature. Cells were then washed with PBS, incubated with DAPI (1:1,000) for 5 minutes, washed with PBS, and mounted on slides with 50% glycerol. Cells were imaged on a Leica DM5000 B microscope (Leica). For counting, 10 visual fields (×20) per well were selected at random, and the number of Tuj1-positive cells was counted and expressed as a percentage of the total number of DAPI-positive cells.
Treatment of cells with RA or RA inhibitors.
For RA-induced SH-SY5Y cell differentiation, 3 × 106 cells were plated into 10-cm2 dishes and cultured in DMEM and 10% FBS. Cells were treated with 1 μM, 5 μM, or 10 μM ATRA (MilliporeSigma) or 13-cis-RA (MilliporeSigma) for 1–12 days. Cells were also treated with the RA inhibitors citral (10 μM) or N, N-diethylaminobenzaldehyde (DEAB) (10 μM, MilliporeSigma). Culture media were changed every 2 days. ATRA, 13-cis-RA, citral, and DEAB were diluted in DMSO. E12.5 neural progenitor cells were treated with 10 μM ATRA or citral for 3 days, and cell pellets were collected for CHD7 level analysis by Western blotting.
For Western blotting, total nuclear protein was quantified with the BCA Protein Assay Kit (Life Technologies) (10–20 μg per sample), electrophoresed in 4%–12% Bis-Tris gels, and transferred to nitrocellulose membrane at 4°C overnight. Primary antibodies used for Western blotting were anti-CHD7 antibody (catalog 6505, Cell Signaling Technology), anti-CHD5 (catalog H-185, Santa Cruz), anti-FLAG (catalog F7425, MilliporeSigma), and anti-HDAC2 antibody (Santa Cruz). Secondary antibodies were goat anti-rabbit IgG Horseradish Peroxidase (catalog 31461, Thermo Fisher Scientific) and rabbit anti-goat IgG Horseradish Peroxidase (catalog 31403, Thermo Fisher Scientific).
Coimmunoprecipitation.
SH-SY5Y cells (5 × 106) were plated in 10-cm2 dishes and grown in DMEM, 10% FBS. For nuclear protein extraction, culture media were removed and washed with cold PBS for 3 times on ice. Cells were then incubated on ice in 3 ml cold Buffer I (15 mM HEPES, 10 mM KCl [MilliporeSigma]), 0.1 mM EDTA (MilliporeSigma), 1 mM MgCl2 (MilliporeSigma), and 350 mM sucrose (MilliporeSigma) and collected with a cell scraper. Cell lysates were then centrifuged at 8,000 g for 5 minutes. Supernatants were discarded, and cell pellets were lysed with Buffer II (15 mM HEPES, 110 mM KCl [MilliporeSigma], 250 mM NaCl [MilliporeSigma], 0.1 mM EGTA [MilliporeSigma]), 5% glycerol (MilliporeSigma), and PMSF (MilliporeSigma). After centrifugation for 10 minutes, supernatants were collected, flash frozen in liquid nitrogen, and stored at –80°C.
For coimmunoprecipitation, nuclear extracts (350 μg per sample) were incubated with specific antibodies overnight and then incubated with protein A/G plus-Agarose Beads (Santa Cruz) for 1 hour at 4°C (see complete unedited blots in the supplemental material). Anti-rabbit normal IgG (Jackson ImmunoResearch) was used as a control antibody for coimmunoprecipitation. Beads were then washed with buffer II (0.01% Tween 20 [MilliporeSigma]) and added to ×4 LDS sample buffer. Samples and bound proteins were detected by Western blotting. Antibodies used for coimmunoprecipitation were anti-CHD7 (catalog 6505, Cell Signaling) and anti-RAR (catalog M-454, Santa Cruz).
Analysis of CHD7 and RAR localization in mouse ESCs.
DNA sequences associated with CHD7 and RAR were compared with determine commonly bound regions within the mouse ESC genome. CHD7 ChIP-seq reads were obtained from Schnetz et al. (30) (GEO, GSE22341; SRA, SRP002695) while pan-RAR ChIP-seq reads were obtained from Mahony et al. (31) (GEO, GSE19409; SRA, SRP002214). All data were aligned to the NCBI36 (mm8) build of the mouse genome. BED files containing significant reads were uploaded to Galaxy (v.17.05). For CHD7 ChIP-seq, the high-threshold peak set was used for downstream analysis. For pan-RAR ChIP-seq, the data set corresponding to the +RA treatment group was used. These two data sets were intersected using BEDTools (64) with the following commands: “-wa” and “-u” to enable identification of all CHD7 reads that overlapped at least once with pan-RAR reads; the required minimum overlap (“-f”) between data set reads was set to 50%. Results were plotted as a quantitative Venn diagram using the Venn Diagram Generator available from the Whitehead Institute for Biomedical Research (http://jura.wi.mit.edu/bioc/tools/venn.php) and edited in Illustrator (Adobe).
X-gal staining.
Timed pregnancies were established between Chd7+/–;(RARE-lacZ)×2 and Chd7+/–;(RARE-lacZ)×2 or (RARE-lacZ ×2) to ensure similar lacZ copy number between embryos. Litters were collected at E10.5, E11.5, E12.5, and E13.5 following cervical dislocation and hysterectomy. Embryos were fixed and either (a) processed for cryosections, sectioned and stained as previously described (65), or fixed and processed for whole-mount staining, as described previously (66).
RARE-lacZ cell and embryo assays.
F9 RARE-lacZ cells were provided by James H. Millonig (Robert Wood Johnson Medical School, New Brunswick, New Jersey, USA) (34), and cultured in 0.4 mg/ml G418 culture medium (10% FBS, Atlas Biologicals]) with 1% penicillin/streptomycin DMEM/F12). For coculture assays, 1 day prior to coculture, F9 cells (105 cells/well) were plated on 0.2% gelatin-coated (MilliporeSigma) 96-well plates in culture media without G418. Separate wells were plated in triplicate for standard ATRA concentration response curves (0, 100 fM, 1 pM, 10 pM, 100 pM, 1 nM, 10 nM, 100 nM), as previously described (34). ATRA was dissolved in DMSO. Timed pregnancies were established between Chd7+/– male and female mice. E8.5 mouse embryos were dissected, washed in ice-cold PBS, and incubated in 1.5-ml Eppendorf tubes containing 10 μl 0.05% Trypsin-EDTA on ice. Yolk sacs were saved and processed for Chd7+/+, Chd7+/–, and Chd7–/– genotyping, as previously described (47). Embryos were digested for 5 minutes at 37°C. Single-cell suspensions were then transferred to the F9 cell plated 96-well plate (1 embryo per well). Plates were cultured overnight at 37°C and 5% CO2 and then processed for β-gal histochemistry and detection using a SpectraMax M2 multimode microplate reader (Molecular Devices). ATRA-treated F9 RARE cells were used to generate a standard curve for comparison with experimental samples.
Luciferase assays.
HEK-293T cells (3 × 104) were plated in each well of a 96-well plate the day before transfection. For each combination of plasmids, 8 replicate wells were plated. Flag-HA-pcDNA3.1, pcDNA3.1-Flag-CHD7, and pcDNA3.1-Flag-CHD7 K998R were separately cotransfected with pGL3-RARE-luciferase vector (Addgene) and pRL-SV40P (Addgene) using Lipofectamine 2000 transfection reagent and opti-MEM (Life Technologies). After 24 hours, media were changed to include vehicle (DMSO) with or without 1 μM ATRA and treated overnight before luciferase detection.
For siRNA transfection, DharmaFECT Duo transfection reagent (GE Healthcare Dharmacon Inc.) was used. siGENOME human CHD7 siRNA-SMARTpool (GE healthcare) and siGENOME Cyclophilin B control siRNA were separately cotransfected with pGL3-RARE-luciferase and pRL-SV40P vectors. Culture media were changed 24 hours after transfection followed by addition of DMSO with and without 1 μM ATRA. Luciferase assays were carried out 48 hours after transfection of CHD7 expression or siRNA plasmids. Luciferase and Renilla signals were detected with the Dual Luciferase reporter assay system (Promega) and GloMax multi detection system (Promega). Data were analyzed by Student’s t test.
Inner ear paint-filling.
Timed pregnancies were established between Chd7Gt/+;Aldh1a3+/– and Aldh1a3+/– mice. Embryos were collected at E14.5 after cervical dislocation and hysterectomy. Embryos were washed in PBS and fixed in Bodian’s fixative (5% glacial acetic acid, 1.85% formaldehyde, and 75% ethanol) and then cleared using methylsalicylate. Heads were bisected and the brain was removed prior to injection of 3% Whiteout (BIC) solution as previously described (47).
Statistics.
Two-tailed Student’s t tests were used to determine significant differences between 2 data sets. The resulting P values were corrected for multiple comparisons using the Bonferroni method. Ordinary 1-way ANOVA tests were used to determine statistical significance among 3 or more data sets. A P value of less than 0.05 was considered significant. All graphs show the mean ± SD, unless otherwise indicated.
Study approval.
All mouse husbandry practices and procedures were approved by the University of Michigan Institutional Animal Care and Use Committee.
Author contributions
HY, SFH, JMS, and EDS performed experiments, prepared figures, and drafted and edited the manuscript. DLS performed inner ear paint-fills and edited the manuscript. GJS performed experiments and prepared graphs. YR conceived experiments and edited the manuscript. PCS performed experiments and edited the manuscript. DMM conceived and performed experiments, analyzed data, and drafted and edited figures and the manuscript. CFB assisted with mouse inner ear ChIP assays. SI contributed reagents and helped with experimental design and troubleshooting of Western blots, coimmunoprecipitation, and ChIP-qPCR.
Supplementary Material
Acknowledgments
We thank Ruchi Bajpai (University of Southern California) and Joanna Wysocka for the CHD7 overexpression plasmid, James Millonig for the F9 RARE-lacZ cell line, Kenneth Campbell (Cincinnati Children’s Hospital) for Aldh1a3 in situ probe, and Doris Wu and Gregg Duester for the Aldh1a3 mutant mice. We thank Anthony Antonellis for assistance with the luciferase experiments. YR is supported by the NIH (R01 DC009410, DC014456, and P30-DC05188) and by the R. Jamison and Betty Williams Professorship. PCS is supported by the NIH (R01 CA193677, CA204279, and DC009410). SI is supported by the March of Dimes Foundation, Cooley’s Anemia Foundation, and the NIH (R01 NS089896). DMM is supported by the NIH (R01 DC009410, DC014456, and T32-GM007863) and by Donita B. Sullivan, MD Professor in Pediatrics at the University of Michigan.
Version 1. 02/22/2018
Electronic publication
Footnotes
Conflict of interest: The authors have declared that no conflict of interest exists.
Reference information: JCI Insight. 2018;3(4):e97440. https://doi.org/10.1172/jci.insight.97440.
Contributor Information
Hui Yao, Email: huyao@umich.edu.
Yehoash Raphael, Email: yoash@umich.edu.
Peter C. Scacheri, Email: pxs183@case.edu.
Shigeki Iwase, Email: siwase@umich.edu.
References
- 1.Allis CD, Jenuwein T. The molecular hallmarks of epigenetic control. Nat Rev Genet. 2016;17(8):487–500. doi: 10.1038/nrg.2016.59. [DOI] [PubMed] [Google Scholar]
- 2.Thaller C, Eichele G. Identification and spatial distribution of retinoids in the developing chick limb bud. Nature. 1987;327(6123):625–628. doi: 10.1038/327625a0. [DOI] [PubMed] [Google Scholar]
- 3.Rhinn M, Dollé P. Retinoic acid signalling during development. Development. 2012;139(5):843–858. doi: 10.1242/dev.065938. [DOI] [PubMed] [Google Scholar]
- 4.Evans RM, Mangelsdorf DJ. Nuclear receptors, RXR, and the big bang. Cell. 2014;157(1):255–266. doi: 10.1016/j.cell.2014.03.012. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Rothman KJ, Moore LL, Singer MR, Nguyen US, Mannino S, Milunsky A. Teratogenicity of high vitamin A intake. N Engl J Med. 1995;333(21):1369–1373. doi: 10.1056/NEJM199511233332101. [DOI] [PubMed] [Google Scholar]
- 6.Romand R, Dollé P, Hashino E. Retinoid signaling in inner ear development. J Neurobiol. 2006;66(7):687–704. doi: 10.1002/neu.20244. [DOI] [PubMed] [Google Scholar]
- 7.Romand R, Kondo T, Fraulob V, Petkovich M, Dollé P, Hashino E. Dynamic expression of retinoic acid-synthesizing and -metabolizing enzymes in the developing mouse inner ear. J Comp Neurol. 2006;496(5):643–654. doi: 10.1002/cne.20936. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Pennimpede T, Cameron DA, MacLean GA, Li H, Abu-Abed S, Petkovich M. The role of CYP26 enzymes in defining appropriate retinoic acid exposure during embryogenesis. Birth Defects Res Part A Clin Mol Teratol. 2010;88(10):883–894. doi: 10.1002/bdra.20709. [DOI] [PubMed] [Google Scholar]
- 9.Vissers LE, et al. Mutations in a new member of the chromodomain gene family cause CHARGE syndrome. Nat Genet. 2004;36(9):955–957. doi: 10.1038/ng1407. [DOI] [PubMed] [Google Scholar]
- 10.Hale CL, Niederriter AN, Green GE, Martin DM. Atypical phenotypes associated with pathogenic CHD7 variants and a proposal for broadening CHARGE syndrome clinical diagnostic criteria. Am J Med Genet A. 2016;170A(2):344–354. doi: 10.1002/ajmg.a.37435. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Sanlaville D, et al. Phenotypic spectrum of CHARGE syndrome in fetuses with CHD7 truncating mutations correlates with expression during human development. J Med Genet. 2006;43(3):211–217. doi: 10.1136/jmg.2005.036160. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Moerike S, Pantzar JT, De Sa D. Temporal bone pathology in fetuses exposed to isotretinoin. Pediatr Dev Pathol. 2002;5(4):405–409. doi: 10.1007/s10024-001-0258-0. [DOI] [PubMed] [Google Scholar]
- 13.Warburg M. Update of sporadic microphthalmos and coloboma. Non-inherited anomalies. Ophthalmic Paediatr Genet. 1992;13(2):111–122. doi: 10.3109/13816819209087611. [DOI] [PubMed] [Google Scholar]
- 14.Janssen N, et al. Mutation update on the CHD7 gene involved in CHARGE syndrome. Hum Mutat. 2012;33(8):1149–1160. doi: 10.1002/humu.22086. [DOI] [PubMed] [Google Scholar]
- 15.Bouazoune K, Kingston RE. Chromatin remodeling by the CHD7 protein is impaired by mutations that cause human developmental disorders. Proc Natl Acad Sci USA. 2012;109(47):19238–19243. doi: 10.1073/pnas.1213825109. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Micucci JA, et al. CHD7 and retinoic acid signaling cooperate to regulate neural stem cell and inner ear development in mouse models of CHARGE syndrome. Hum Mol Genet. 2014;23(2):434–448. doi: 10.1093/hmg/ddt435. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Lammer EJ, et al. Lott IT, et al. Retinoic acid embryopathy. N Engl J Med. 1985;313(14):837–841. doi: 10.1056/NEJM198510033131401. [DOI] [PubMed] [Google Scholar]
- 18.Frenz DA, et al. Retinoid signaling in inner ear development: A “Goldilocks” phenomenon. Am J Med Genet A. 2010;152A(12):2947–2961. doi: 10.1002/ajmg.a.33670. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Frenz DA, Liu W, Galinovic-Schwartz V, Van De Water TR. Retinoic acid-induced embryopathy of the mouse inner ear. Teratology. 1996;53(5):292–303. doi: 10.1002/(SICI)1096-9926(199605)53:5<292::AID-TERA3>3.0.CO;2-3. [DOI] [PubMed] [Google Scholar]
- 20.Molotkov A, Molotkova N, Duester G. Retinoic acid guides eye morphogenetic movements via paracrine signaling but is unnecessary for retinal dorsoventral patterning. Development. 2006;133(10):1901–1910. doi: 10.1242/dev.02328. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Schnetz MP, et al. Genomic distribution of CHD7 on chromatin tracks H3K4 methylation patterns. Genome Res. 2009;19(4):590–601. doi: 10.1101/gr.086983.108. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Joshi S, Guleria R, Pan J, DiPette D, Singh US. Retinoic acid receptors and tissue-transglutaminase mediate short-term effect of retinoic acid on migration and invasion of neuroblastoma SH-SY5Y cells. Oncogene. 2006;25(2):240–247. doi: 10.1038/sj.onc.1209027. [DOI] [PubMed] [Google Scholar]
- 23.Altschul SF, Gish W, Miller W, Myers EW, Lipman DJ. Basic local alignment search tool. J Mol Biol. 1990;215(3):403–410. doi: 10.1016/S0022-2836(05)80360-2. [DOI] [PubMed] [Google Scholar]
- 24.Påhlman S, et al. Differentiation and survival influences of growth factors in human neuroblastoma. Eur J Cancer. 1995;31A(4):453–458. doi: 10.1016/0959-8049(95)00033-f. [DOI] [PubMed] [Google Scholar]
- 25.Sidell N, Chang B, Bhatti L. Upregulation by retinoic acid of interleukin-2-receptor mRNA in human T lymphocytes. Cell Immunol. 1993;146(1):28–37. doi: 10.1006/cimm.1993.1003. [DOI] [PubMed] [Google Scholar]
- 26.Higashi M, et al. Retinoic acid-induced CHD5 upregulation and neuronal differentiation of neuroblastoma. Mol Cancer. 2015;14:150. doi: 10.1186/s12943-015-0425-y. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Chute JP, et al. Inhibition of aldehyde dehydrogenase and retinoid signaling induces the expansion of human hematopoietic stem cells. Proc Natl Acad Sci USA. 2006;103(31):11707–11712. doi: 10.1073/pnas.0603806103. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Niederreither K, Dollé P. Retinoic acid in development: towards an integrated view. Nat Rev Genet. 2008;9(7):541–553. doi: 10.1038/nrg2340. [DOI] [PubMed] [Google Scholar]
- 29.Engelen E, et al. Sox2 cooperates with Chd7 to regulate genes that are mutated in human syndromes. Nat Genet. 2011;43(6):607–611. doi: 10.1038/ng.825. [DOI] [PubMed] [Google Scholar]
- 30.Schnetz MP, et al. CHD7 targets active gene enhancer elements to modulate ES cell-specific gene expression. PLoS Genet. 2010;6(7):e1001023. doi: 10.1371/journal.pgen.1001023. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Mahony S, Mazzoni EO, McCuine S, Young RA, Wichterle H, Gifford DK. Ligand-dependent dynamics of retinoic acid receptor binding during early neurogenesis. Genome Biol. 2011;12(1):R2. doi: 10.1186/gb-2011-12-1-r2. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Li W, et al. Brg1 governs distinct pathways to direct multiple aspects of mammalian neural crest cell development. Proc Natl Acad Sci USA. 2013;110(5):1738–1743. doi: 10.1073/pnas.1218072110. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Rossant J, Zirngibl R, Cado D, Shago M, Giguère V. Expression of a retinoic acid response element-hsplacZ transgene defines specific domains of transcriptional activity during mouse embryogenesis. Genes Dev. 1991;5(8):1333–1344. doi: 10.1101/gad.5.8.1333. [DOI] [PubMed] [Google Scholar]
- 34.Ababon MR, Li BI, Matteson PG, Millonig JH. Quantitative measurement of relative retinoic acid levels in e8.5 embryos and neurosphere cultures using the F9 RARE-Lacz cell-based reporter assay. J Vis Exp. 2016;(115):54443. doi: 10.3791/54443. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Richards B, et al. Genetic selection for modulators of a retinoic-acid-responsive reporter in human cells. Genetics. 2003;163(3):1047–1060. doi: 10.1093/genetics/163.3.1047. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Adams ME, Hurd EA, Beyer LA, Swiderski DL, Raphael Y, Martin DM. Defects in vestibular sensory epithelia and innervation in mice with loss of Chd7 function: implications for human CHARGE syndrome. J Comp Neurol. 2007;504(5):519–532. doi: 10.1002/cne.21460. [DOI] [PubMed] [Google Scholar]
- 37.van Ravenswaaij-Arts C, Martin DM. New insights and advances in CHARGE syndrome: Diagnosis, etiologies, treatments, and research discoveries. Am J Med Genet C Semin Med Genet. 2017;175(4):397–406. doi: 10.1002/ajmg.c.31592. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Cunningham TJ, Duester G. Mechanisms of retinoic acid signalling and its roles in organ and limb development. Nat Rev Mol Cell Biol. 2015;16(2):110–123. doi: 10.1038/nrm3932. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Bajpai R, et al. CHD7 cooperates with PBAF to control multipotent neural crest formation. Nature. 2010;463(7283):958–962. doi: 10.1038/nature08733. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Liu Y, Harmelink C, Peng Y, Chen Y, Wang Q, Jiao K. CHD7 interacts with BMP R-SMADs to epigenetically regulate cardiogenesis in mice. Hum Mol Genet. 2014;23(8):2145–2156. doi: 10.1093/hmg/ddt610. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.Batsukh T, et al. CHD8 interacts with CHD7, a protein which is mutated in CHARGE syndrome. Hum Mol Genet. 2010;19(14):2858–2866. doi: 10.1093/hmg/ddq189. [DOI] [PubMed] [Google Scholar]
- 42.Batsukh T, et al. Identification and characterization of FAM124B as a novel component of a CHD7 and CHD8 containing complex. PLoS ONE. 2012;7(12):e52640. doi: 10.1371/journal.pone.0052640. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43.Whittaker DE, et al. The chromatin remodeling factor CHD7 controls cerebellar development by regulating reelin expression. J Clin Invest. 2017;127(3):874–887. doi: 10.1172/JCI83408. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44.He D, et al. Chd7 cooperates with Sox10 and regulates the onset of CNS myelination and remyelination. Nat Neurosci. 2016;19(5):678–689. doi: 10.1038/nn.4258. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45.Yu T, et al. Deregulated FGF and homeotic gene expression underlies cerebellar vermis hypoplasia in CHARGE syndrome. Elife. 2013;2:e01305. doi: 10.7554/eLife.01305. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46.Feng W, et al. The chromatin remodeler CHD7 regulates adult neurogenesis via activation of SoxC transcription factors. Cell Stem Cell. 2013;13(1):62–72. doi: 10.1016/j.stem.2013.05.002. [DOI] [PubMed] [Google Scholar]
- 47.Hurd EA, et al. Loss of Chd7 function in gene-trapped reporter mice is embryonic lethal and associated with severe defects in multiple developing tissues. Mamm Genome. 2007;18(2):94–104. doi: 10.1007/s00335-006-0107-6. [DOI] [PubMed] [Google Scholar]
- 48.Bosman EA, Penn AC, Ambrose JC, Kettleborough R, Stemple DL, Steel KP. Multiple mutations in mouse Chd7 provide models for CHARGE syndrome. Hum Mol Genet. 2005;14(22):3463–3476. doi: 10.1093/hmg/ddi375. [DOI] [PubMed] [Google Scholar]
- 49.Hurd EA, et al. Mature middle and inner ears express Chd7 and exhibit distinctive pathologies in a mouse model of CHARGE syndrome. Hear Res. 2011;282(1-2):184–195. doi: 10.1016/j.heares.2011.08.005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 50.Romand R, et al. Retinoic acid deficiency impairs the vestibular function. J Neurosci. 2013;33(13):5856–5866. doi: 10.1523/JNEUROSCI.4618-12.2013. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 51.Semerci CN, et al. Novel splice-site and missense mutations in the ALDH1A3 gene underlying autosomal recessive anophthalmia/microphthalmia. Br J Ophthalmol. 2014;98(6):832–840. doi: 10.1136/bjophthalmol-2013-304058. [DOI] [PubMed] [Google Scholar]
- 52.Fares-Taie L, et al. ALDH1A3 mutations cause recessive anophthalmia and microphthalmia. Am J Hum Genet. 2013;92(2):265–270. doi: 10.1016/j.ajhg.2012.12.003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 53.Hurd EA, Poucher HK, Cheng K, Raphael Y, Martin DM. The ATP-dependent chromatin remodeling enzyme CHD7 regulates pro-neural gene expression and neurogenesis in the inner ear. Development. 2010;137(18):3139–3150. doi: 10.1242/dev.047894. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 54.Currle DS, Hu JS, Kolski-Andreaco A, Monuki ES. Culture of mouse neural stem cell precursors. J Vis Exp. 2007;(2):152. doi: 10.3791/152. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 55.Klincumhom N, et al. Generation of neuronal progenitor cells and neurons from mouse sleeping beauty transposon-generated induced pluripotent stem cells. Cell Reprogram. 2012;14(5):390–397. doi: 10.1089/cell.2012.0010. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 56.Langmead B, Salzberg SL. Fast gapped-read alignment with Bowtie 2. Nat Methods. 2012;9(4):357–359. doi: 10.1038/nmeth.1923. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 57.Benjamini Y, Hochberg Y. Controlling the false discovery rate: a practical and powerful approach to multiple testing. J R Stat Soc Series B Stat Methodol. 1995;57(1):289–300. [Google Scholar]
- 58.Boyle EI, Weng S, Gollub J, Jin H, Botstein D, Cherry JM, Sherlock G. GO::TermFinder--open source software for accessing Gene Ontology information and finding significantly enriched Gene Ontology terms associated with a list of genes. Bioinformatics. 2004;20(18):3710–3715. doi: 10.1093/bioinformatics/bth456. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 59.Grant CE, Bailey TL, Noble WS. FIMO: scanning for occurrences of a given motif. Bioinformatics. 2011;27(7):1017–1018. doi: 10.1093/bioinformatics/btr064. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 60.Afgan E, et al. The Galaxy platform for accessible, reproducible and collaborative biomedical analyses: 2016 update. Nucleic Acids Res. 2016;44(W1):W3–W10. doi: 10.1093/nar/gkw343. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 61.Martin DM, Skidmore JM, Fox SE, Gage PJ, Camper SA. Pitx2 distinguishes subtypes of terminally differentiated neurons in the developing mouse neuroepithelium. Dev Biol. 2002;252(1):84–99. doi: 10.1006/dbio.2002.0835. [DOI] [PubMed] [Google Scholar]
- 62.Waclaw RR, Wang B, Campbell K. The homeobox gene Gsh2 is required for retinoid production in the embryonic mouse telencephalon. Development. 2004;131(16):4013–4020. doi: 10.1242/dev.01272. [DOI] [PubMed] [Google Scholar]
- 63.ENCODE Project Consortium. An integrated encyclopedia of DNA elements in the human genome. Nature. 2012;489(7414):57–74. doi: 10.1038/nature11247. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 64.Quinlan AR, Hall IM. BEDTools: a flexible suite of utilities for comparing genomic features. Bioinformatics. 2010;26(6):841–842. doi: 10.1093/bioinformatics/btq033. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 65.Sclafani AM, Skidmore JM, Ramaprakash H, Trumpp A, Gage PJ, Martin DM. Nestin-Cre mediated deletion of Pitx2 in the mouse. Genesis. 2006;44(7):336–344. doi: 10.1002/dvg.20220. [DOI] [PubMed] [Google Scholar]
- 66.Skidmore JM, Waite MR, Alvarez-Bolado G, Puelles L, Martin DM. A novel TaulacZ allele reveals a requirement for Pitx2 in formation of the mammillothalamic tract. Genesis. 2012;50(1):67–73. doi: 10.1002/dvg.20793. [DOI] [PubMed] [Google Scholar]
- 67.ENCODE Project Consortium. An integrated encyclopedia of DNA elements in the human genome. Nature. 2012;489(7414):57–74. doi: 10.1038/nature11247. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.