

Abstract
The inverse relationship between gestational age at birth and postviral respiratory morbidity suggests that infants born preterm (PT) may miss a critical developmental window of T cell maturation. Despite a continued increase in younger PT survivors with respiratory complications, we have limited understanding of normal human fetal T cell maturation, how ex utero development in premature infants may interrupt normal T cell development, and whether T cell development has an effect on infant outcomes. In our longitudinal cohort of 157 infants born between 23 and 42 weeks of gestation, we identified differences in T cells present at birth that were dependent on gestational age and differences in postnatal T cell development that predicted respiratory outcome at 1 year of age. We show that naive CD4+ T cells shift from a CD31–TNF-α+ bias in mid gestation to a CD31+IL-8+ predominance by term gestation. Former PT infants discharged with CD31+IL8+CD4+ T cells below a range similar to that of full-term born infants were at an over 3.5-fold higher risk for respiratory complications after NICU discharge. This study is the first to our knowledge to identify a pattern of normal functional T cell development in later gestation and to associate abnormal T cell development with health outcomes in infants.
Keywords: Development, Immunology
Keywords: Adaptive immunity
This study defines normal functional T cell development in later human gestation and associates abnormal T cell development with health outcomes in infants.
Introduction
The immediate postnatal newborn immune system is required to shift abruptly from relative quiescent cell division in utero to active protection against opportunistic pathogens at birth. Successful performance of this singular transition requires that neonates, in the absence of an adapted memory response, sufficiently activate naive T cells to clear infection, while simultaneously maintaining tolerance to commensals. Due to the lack of prospective, longitudinal studies, there has yet to be consensus on what constitutes normal immune development during gestation and, more importantly, which characteristics are essential to sustaining newborn health. The majority of investigations into neonatal adaptive immunity rely on the Th1/Th2 paradigm and consistently show a newborn’s defects in IFN-γ pathways (1, 2). The Th1/Th2 model has proven useful in deciphering protective immune mechanisms in adults, but it has fallen short in its ability to completely summarize a newborn’s response to infection and nonallergic inflammatory states (3, 4). There is a growing body of evidence supporting the notion that a neonate’s adaptive immune system, albeit naive, may have access to alternative mechanisms beyond the canonical Th1/Th2 predominance found in adults (5, 6). These fundamental differences may provide insight into mechanisms governing how a neonate succeeds and fails in achieving the exquisite balance between infection control and immune regulation.
One natural model for understanding how developmentally determined T cell behavior affects neonatal outcomes in humans can be found in extremely low gestational age (GA) newborns (ELGANs; born <29 weeks of gestation). By comparing outcomes of ELGANs, whose postnatal adaptive immune system parallels normal fetal T cell development, to those with postnatal T cell “developmental arrest,” we can begin to understand the impact of normal fetal immune development on an infant’s health. We have previously shown that ELGANs have fewer CD8+ T cells expressing the inhibitory molecule CD31 and are hyperresponsive when compared with T cells from full-term (FT) infants (7). In their study, including 26 infants, Gibbons et al. recently identified the neutrophil chemoattractant IL-8 (CXCL8) as a major effector cytokine produced by CD4+ T cells from neonates born ≤30 weeks after conception and that the majority of IL-8 producers coexpressed CD31 (6). In contrast to the age-dependent T cell CD31 expression found at birth in our own study, authors did not detect an association between IL-8 producers and GA. In their study, increased IL-8+ T cells were detected in 4 infants with active infection. While there is evidence that elevated plasma IL-8 associates with illness severity in FT infants infected with respiratory viruses, this relationship is not well supported in premature infants (8). However, the link between an infant’s clinical outcome and their unique immunophenotype has yet to be established. It may be that CD31+ T cell–associated effector cytokines in neonates contribute to inflammatory conditions in ELGANs, as is seen in adult diseases (9, 10). It is equally possible that the innate-like response from naive CD31+ T cells is protective in neonates who have insufficient passive immunity from maternal antibody, as is the case with ELGANs (11). To date, there are no published studies in humans that summarize major features of fetal T cell development while addressing whether or not such features have any measurable effect on infant health outcomes.
The main objective of this study was to understand the relationship between fetal T cell development and postnatal lung disease. We followed a birth cohort of 157 infants, ranging from 23 weeks to 42 weeks postmenstrual age at birth, recruited to the Prematurity and Respiratory Outcomes Program (PROP) at the University of Rochester Medical Center and Children’s Hospital of Buffalo (12). A smaller number of umbilical cord blood samples from a second, independent cohort of 167 infants (23–42 weeks) recruited at the University of Rochester through the ongoing, longitudinal Prematurity, Respiratory, Immune System and Microbiomes (PRISM) study were used for CD31-dependent cytokine function experiments. Phenotyped T cells at birth, at term-equivalent corrected GA (teCGA), and at 12-months corrected age were correlated with a PROP-validated composite outcome for respiratory morbidity at 12 months (13). teCGA represents the age at which the preterm (PT) infants attained sufficient physiologic stability for discharge to home. teCGA expressed as postmenstrual age, or GA in weeks at birth plus postnatal weeks, occurs in a range from 36 to 42 weeks and can be used to approximate the maturity at birth and discharge of the FT born infants. teCGA, therefore, is a useful time point to assess postnatal immune maturation and compare to that observed in a healthy infant born at term gestation. Results from this large birth cohort will show that mid to late human gestation marks a critical period of continued lymphocyte development, during which accretion of CD31+IL-8+ recent thymic emigrants (RTEs) occurs. ELGANs who failed to achieve an age-appropriate T cell phenotype by teCGA were at greater risk for respiratory morbidity in their first year after NICU discharge. Data presented in the study challenge the field to reconsider the core belief that the neonatal adaptive immune system is in a state of deficiency; rather, neonatal T cells may be uniquely suited to manage the transformation from fetus to newborn.
Results
Patient demographics.
A total of 277 infants were enrolled in the PROP study at the University of Rochester and the State University of New York at Buffalo. 233 completed follow-up and were assessed for evidence of chronic respiratory morbidity. 122 PT subjects were between the ages of 23 0/7 and 28 6/7 weeks at birth, 83 were between 29 0/7 and 35 6/7 weeks, and 28 subjects were healthy FT infants (greater than 36 6/7 weeks at birth). The outcome of chronic respiratory morbidity was based on the previously published postprematurity respiratory disease (PRD) composite PROP outcome (14). PRD was defined by either (a) death due to cardiac or respiratory failure or (b) positive responses on at least 2 of 4 quarterly surveys after discharge in at least one of the following domains: (a) respiratory hospitalization, (b) respiratory medications (bronchodilator, methylxanthine, diuretic, leukotriene receptor antagonist, pulmonary vasodilator), (c) respiratory symptoms (wheezing > once per week per parents, wheezing diagnosed by a doctor, or cough without cold), or (d) home respiratory support (oxygen, continuous positive airway pressure [CPAP], ventilator/tracheostomy).
Subjects were divided into two age cohorts, <29 weeks (ELGANs) and ≥29 weeks, in order define a “fetal” immunophenotype and to enrich the younger cohort with patients for whom prediction of outcome is the most difficult based on clinical factors alone. Demographics for ELGANs and subjects born ≥29 weeks are shown in Supplemental Table 1 (supplemental material available online with this article; https://doi.org/10.1172/jci.insight.96724DS1). Sex, race, and ethnicity were similar between the younger and older cohorts. ELGANs had lower birth weights, less frequent preeclampsia, and higher rates of infection (funisitis, culture-positive sepsis, or necrotizing enterocolitis). Of the 233 subjects with adequate survey follow-up through 1-year corrected age, blood sampling at teCGA and respiratory outcome were sufficient for T cell phenotyping in 68% of subjects (85 and 72 subjects from the ELGANs and ≥29-week cohorts, respectively) (Figure 1). 69 subjects had sufficient samples from all 3 time points for longitudinal T cell analysis. Blood samples from a second independent cohort of 21 ELGANs, 56 PT infants born ≥29 weeks, and 90 FT infants recruited to date through the PRISM study at the University of Rochester were used for validation of the main study findings related to immune development and for follow-up in vitro stimulation experiments.
Figure 1. Consort chart.

Flow chart showing attrition of subjects recruited and samples available at term-corrected age. *PRD, postprematurity respiratory disease.
CD31+CD4+ T cells increase with GA and are higher in females.
Our previously published study investigating CD8+ T cell function in PT infants revealed that CD31 expression on CD8+ T cells was highly dependent on GA at birth (7), suggesting an age-dependent maturation of the naive T cell compartment during the last trimester of fetal immune development. Detailed flow cytometry staining panels are available in Supplemental Table 2. CD31+CD4+ T cell frequencies were identified by manual gating (FSC-A/SSC-A/FSC-H/CD14–/CD3+/CD8–/CD4+/CD31+/CD31–, Figure 2A). There was no significant difference in absolute CD4+ T cell events based on age group, but ELGANs had a greater absolute number of CD31–CD4+ T cell events at birth compared with the older cohort (P < 0.01, Figure 2B). A strong direct correlation existed between the proportion of CD31+CD4+ T cells and GA at birth (r = 0.49, P < 0.0001, Figure 2C). A similar relationship was found at teCGA (r = 0.25, P < 0.001). By 12-months corrected GA (CGA), CD31+CD4+ T cell frequencies were similar across birth age cohorts. Dichotomizing CGA at birth as <29 weeks or ≥29 weeks showed significant differences in CD31+CD4+ T cell events at birth and teCGA. Differences lessened by teCGA time point and were not significant by 12 month (Figure 2D). These results suggest that neonates born earlier in fetal development have an expanded number and proportion of CD31–CD4+ T cells but the balance of CD31+ and CD31– cells normalizes later in infancy.
Figure 2. CD31+CD4+ T cell expression varies by GA at birth and sex.

(A) Dot plots show identification of CD31+ and CD31–CD4+ T cells by sequential gating based on FSC-A/SSC-A/FSC-H, live/CD14–, CD3+, CD4+/CD8–, CD31+/CD31– expression. (B) Total CD4+ cells/ml blood collected, and CD31+/CD31– subsets are shown. (C) Regression lines depict expected relative frequencies and 95% CI of CD31+CD4+ T cells as a function of GA at birth for each of the collected time points and Pearson correlations. (D) Box-and-whisker plots show median ± IQR and minimum/maximum CD31+CD4+ T cells for infants born <29 or ≥29 weeks and (E) males or females for each time point tested (**P < 0.01, ****P < 0.0001, Wilcoxon rank-sum or Wilcoxon matched-pairs signed-rank test). tCGA, term-corrected gestational age.
Clinical factors that associate with both CD31 and GA in the ELGAN cohort were next determined (Supplemental Table 3). Lower CD31+CD4+ T cell frequency (less than median of 60%) at teCGA was highly associated with male sex (P < 0.0001) in both age cohorts and modestly with preeclampsia (P < 0.05) in ELGANs. Males had significantly lower levels of CD31+CD4+ T cells at all time points, including at 12 months, for all age groups when compared with females (Figure 2E). Controlling for clinical exposures, CD31+ proportion from birth through teCGA remained significantly correlated with GA at birth, indicating that duration of gestation and sex are the key determinants of naive CD31+CD4+ T cell frequency in the first year of life.
CD4+CD31+ T cell frequencies and prediction of ELGANS respiratory outcome at 1 year.
In human adults and mouse models, loss of CD31 expression on CD4+ T cells causes immune dysregulation and inflammatory diseases (10, 15). It is conceivable, therefore, that low CD31 expression similarly associates with later inflammation-mediated respiratory morbidity in ELGANs. Predicting respiratory morbidity after NICU discharge in ELGANs based on clinical factors alone has been challenging, and a biomarker would be very useful in improving the surveillance and management of high-risk ELGANs. Utilizing the PROP 1-year respiratory outcomes data, we therefore compared the relative strength of CD31+ T cell balance at birth and at term-equivalent age with clinical risk factors in predicting after PRD outcome in ELGANs.
We first tested the association between commonly associated risk factors with the outcome of PRD across GA cohorts. Consistent with published disease demographics in the PROP study (13), PRD was documented in 71% (CI = 61.6–78.4) of ELGANs and 39% (CI = 29.6–48.5) of subjects ≥29 weeks (Supplemental Table 4). When including all birth age cohorts, the strongest predictor for PRD was younger GA at birth (P < 0.0001), with an AUC of 0.72. Dichotomizing at >29 weeks and <29 weeks of GA alone predicted PRD with a sensitivity of 66.7% (CI = 57.8–74.7) and specificity of 65.4% (CI = 55.4–74.5). However, when restricting analysis to ELGANs, GA was no longer useful for risk stratification (AUC = 0.51, P = 0.71). This suggests that unmeasured variables beyond low GA at birth effect respiratory outcome within the high-risk population of ELGANs.
We next examined clinical exposures that are known to increase respiratory morbidity in premature infants (Supplemental Table 3). In the ELGANs, PRD was associated with greater oxygen exposure at both 7 and 14 days of life (P = 0.03) and need for postnatal steroids (P = 0.008). Interestingly, the intermediate outcome of bronchopulmonary dysplasia (BPD), pulmonary morbidity diagnosed at 36 weeks CGA (often used as a predictor of chronic respiratory morbidity) (13), did not strongly predict the 12-month outcome of PRD (P = 0.07) in our local PROP cohort. Although large epidemiologic studies have shown that males are at higher risk for BPD and respiratory symptoms in the first year (16), our results did not show a significant association between male sex and PRD in our smaller ELGANs age cohort.
Few clinical variables associated with risk for long-term morbidity in ELGANs, prompting further consideration of alternative biomarkers, such as CD31, for PRD prediction. Due to the strength of GA as a risk factor for PRD across all age cohorts, outcome prediction was not improved by including CD31+CD4+ T cells, when including all infants >23 weeks of gestation at birth. However, when we focused the analysis on ELGANs, in whom PRD was not predicted by GA alone, dichotomization of CD31+CD4+ T cells at teCGA to less than or greater than 60% of CD4+ cells identified risk more strongly than did GA (OR = 3.54 and 1.10 for CD31 and GA, respectively) (Figure 3A). CD31+CD4+ T cells <60% at NICU discharge (teCGA) substantially improved the PRD AUC when compared with GA in ELGANs (0.69 and 0.51, respectively [AUC of 1 is considered perfect, greater than 0.80 is considered diagnostic]). Overall, CD31+CD4+ T cells <60% at teCGA predicted PRD with 67% sensitivity and 64% specificity. Although CD31+CD4+ T cell frequency was strongly associated with GA at birth, sampling results at the time of birth did not predict outcome.
Figure 3. Low levels of CD31+CD4+ T cells at term-corrected age in infants born <29 weeks are a stronger predictor of 1-year respiratory outcome than either gestational age at birth or clinical exposures.

Results shown for infants born <29 weeks. (A) Receiver operating characteristic (ROC) curve for gestational age (solid line) and CD31+CD4+ T cell frequency <60% at teCGA (dashed line) and at birth (dotted line). (B) Odds ratio for developing PRD, given exposure to the listed clinical variable, adjusted for CD31+CD4+ T cell frequency of <60% at teCGA (dotted line), and odds ratio for developing PRD, given low CD31 frequency adjusted for listed clinical variable (solid line). NEC, necrotizing enterocolitis.
To test the independent relationship between CD31 and PRD, we controlled for multiple clinical exposures associated with risk for PRD. ELGANs with CD31+CD4+ T cell frequencies <60% at teCGA remained at 3.5-fold higher risk for PRD compared with those with >60% CD31+CD4+ T cells (Figure 3B, P = 0.01 [CI = 1.33–9.44]). Clinical exposures that commonly associate with PRD in ELGANs were distinct from those associated with low CD31 (GA, preeclampsia) (Supplemental Table 3). Moreover, clinical predictors (oxygen exposure and postnatal steroids) were no longer significantly correlated with the outcome of PRD when adjusting for low levels of CD31+CD4+ T cells. Exogenous influences on PRD outcome in ELGANs are therefore independent from those that relate to low levels of CD31+CD4+ T cells at teCGA. Together, this shows that ELGANs and males are born with lower levels of CD31+CD4+ T cells, but ELGANs, regardless of sex, with a persistent “fetal-like” low CD31+CD4+ T cell immunophenotype at teCGA, are at a significantly higher risk for chronic respiratory morbidity after hospital discharge.
Naive CD31+ T cells are enriched for RTEs.
Previous studies have shown enrichment for RTEs in CD31+CD4+ T cell populations in older pediatric subjects (17, 18). Infants born premature have lower circulating RTEs compared with those born at term, but this tends to equalize by the time they reach term equivalent age. Persistent low levels of CD31+CD4+ T cells in ELGANs measured at teCGA therefore may reflect diminished thymic function in a subset of those otherwise well-appearing, discharge-ready patients. To test the hypothesis that CD31 identifies a naive T cell population enriched for RTEs at birth in our cohort, we quantified T cell excision circle (TREC) content in isolated cord blood from a subset of 5 neonates ranging from 32 to 40 weeks of gestation. Naive T cells were then isolated further separated into CD31+ and CD31– fractions using magnetic bead separation. TREC content, which marks RTEs (19), was then compared between the two subpopulations. TREC content was significantly higher in the CD31+ naive T cell fraction when compared with naive CD31– T cells (P < 0.001), supporting our hypothesis that CD31 is a marker of recent thymic egress in umbilical cord naive CD4+ T cells (Figure 4).
Figure 4. Recent thymic emigrants, as measured by T cell excision circle content, in isolated naive CD31+ and CD31– T cell populations at birth.
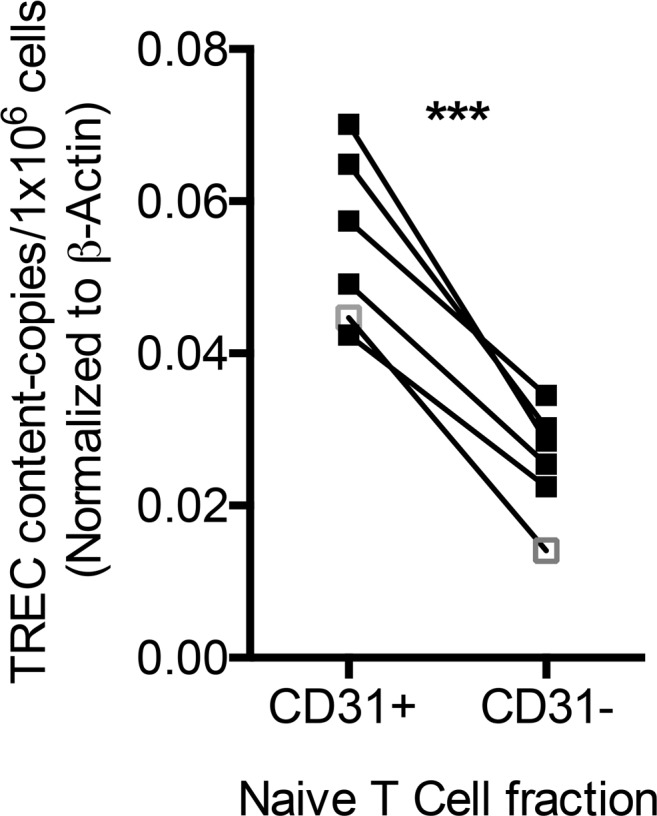
T cell excision circle (TREC) content was quantified in column-sorted CD31+ and CD31– naive T cells and is expressed in above graph normalized to β-actin. Each black square pair connected by lines represents a single-matched UCB sample from a neonatal subject, and the white square outlined in gray shows the result from a representative healthy adult donor (***P < 0.001, paired t test).
Neonatal CD31+ and CD31–CD4+ T cells are primarily naive (CD45RO–), proliferative, and nonsenescent but have different niche-sensing capacities.
Increased CD31–CD4+ T cells predict respiratory disease in ELGANs, with a relationship similar to that found between adult inflammatory diseases and CD31–CD4+ T cells (10, 15). However, it is not known what properties favor their enrichment at younger GAs or if there are specific functional differences in CD31+ and CD31– T cells that confer protection or harm, respectively, to a naive neonatal host. We therefore focused our examination of neonatal CD31+/ CD31– T cell characteristics on those markers related to differentiation, peripheral homeostasis, and effector function. Representative flow plots with CD31 marker costaining can be found in Supplemental Figure 1.
CD45RO coexpression on CD31–CD4+ T cells would suggest that CD31– T cell enrichment in ELGANs could be explained by antigen-induced memory or effector differentiation in vivo related to perinatal exposures. Against this hypothesis, we found no significant differences in CD45RO coexpression patterns on neonatal CD31+ and CD31–CD4+ T cells between the birth time point and teCGA (Figure 5, A and B). While there was greater CD45RO expression on CD31–CD4+ T cells compared with CD31+CD4+ T cells, the majority of CD31–CD4+ T cells from all infants remained CD45RO–. In contrast, the majority of CD31–CD4+ T cells from adult samples were also CD45RO+. Newborns had higher Ki67-expressing (proliferative) CD4+ T cells at birth and teCGA (median [IQR] 0.91 [0.49–1.47] and 0.79 [0.44–1.31], respectively) compared with adults (0.29 [0.16–1.07], P < 0.001). We were unable to detect age-dependent differences within individual CD31+ and CD31– subsets. Neither CD31+ nor CD31– subpopulations contained substantial fractions of senescent cells, as reflected in their low abundance of CD57+ events. These data show that increased proliferation in CD4+ T cells from newborns is not accompanied by greater memory differentiation or cell exhaustion.
Figure 5. CD4+CD31+ and CD31– T cells in neonates display a more naive phenotype, with higher cytokine receptor expression, compared with adults at both birth and teCGA time points.

Boolean combinations of events based on (A and B) CD45RO/Ki67/CD57 expression and (C and D) CD127 and CD215 expression, shown as a percentage of CD31+ and CD31–CD4+ T cell subpopulations. Bars show median (IQR) at birth (black), term-equivalent corrected gestational age (teCGA, gray), and adult (white); dots represent individual subjects (*P < 0.05, **P < 0.01,***P < 0.001, ****P < 0.0001, 2-way ANOVA).
Reduced CD31-expressing CD4+ T cells in newborns could alternatively occur as a result of more robust homeostatic cytokine-stimulated proliferation in the rapidly growing fetus. Greater expression of CD127 (IL-7rα) and CD215 (IL-15rα), which play a central role in the peripheral maintenance of naive and memory CD4+ T cells, respectively (20, 21), at younger GAs could simultaneously support proliferation and maintenance of T cells in a naive state during homeostatic expansion (22). To test age- and T cell subpopulation–related niche-sensing capacity in neonatal T cells, we compared CD127 and CD215 expression on CD31+ and CD31– events across time points and newborn age cohorts (Figure 5, C and D). Neonatal CD4+ T cells expressed higher CD127 at birth and at teCGA compared with adults (P < 0.0001). CD215+ events were similarly low in both CD31+ and CD31– subpopulations across time points and age.
To determine age- and cell-based differences in receptor expression among neonates, we next performed subject-matched phenotype comparisons between CD31+ and CD31–CD4+ T cells within and across age cohorts. Ki67+ events from both CD31+ and CD31– fractions were greater in the ELGAN cohort (P < 0.0001), indicating more basal proliferation in their naive CD31–CD4+ T cells. Basal Ki67+ events were higher in CD31–CD4+ T cells from all infants when compared within subject CD31+ events (P < 0.001). We speculated that, due to their higher rate of proliferation, CD31–CD4+ T cells in ELGANs would also have higher CD45RO expression compared with the older newborn cohort. Contrary to this prediction, CD45RO+ T cells, both CD31+ and CD31–, were lower in ELGANs compared with in those ≥29 weeks (P < 0.01 and P < 0.05, respectively) (Figure 6). Both CD127+ and CD215+ events were more frequent in the CD31– fraction (P < 0.0001), but there were no age-based differences across newborn cohorts in either CD215+ or CD127+CD4+ T cells. Together, these data suggest that both CD31+ and CD31–CD4+ T cells in newborns, and ELGANs in particular, exhibit higher basal proliferation compared with adults, but there is no evidence for associated memory differentiation or cell exhaustion in either cell fraction. While newborn T cells express significantly higher homeostatic cytokine receptors compared with adults, the observed enrichment of CD31–CD4+ T cells in ELGANs is not due to differences in homeostatic cytokine receptor expression.
Figure 6. CD31–CD4+ T cell fractions show a similar pattern of increased proliferation and homeostatic cytokine receptor expression compared with CD31+ T cells in both age cohorts.

Infants <29 weeks have greater proliferation and decreased memory differentiation of both CD31+ and CD31– fractions compared with infants born older than 29 weeks. Line graphs compare subject-matched CD4+CD31+ to CD31– T cell expression of (A) Ki67, (B) CD45RO, (C) CD127, and (D) CD215. Light gray lines indicate subjects born <29 weeks and dark indicates subjects born ≥29 weeks (*P < 0.05, **P < 0.01,***P < 0.001, ****P < 0.0001, Wilcoxon matched-pairs signed-rank test).
CD31 expression identifies CD4+ T cell subsets with unique functional capacities in neonates.
Homeostatic expansion of naive CD4+ T cells is accompanied by changes in effector function, including the gain of TNF-α production (23). Differences in effector potential based on fetal homeostatic proliferation may negatively effect ELGAN health by enhancing or dysregulating their inflammatory responses. We therefore determined if developmental changes in CD31 expression were accompanied by predictable effector functions at varying GAs at birth. In the PRISM study birth cohort of 76 PT (21 ELGANs) and 100 FT infants, we measured T cell intracellular cytokines following in vitro stimulation of cord blood mononuclear cells with staphylococcal enterotoxin B (SEB), a superantigen (staining panel 2, Supplemental Table 2). The predominant cytokines measured by flow cytometry produced in infant T cells were TNF-α, IL-8, and IL-2; IL-10, IL-17, IL-4, and IFN-γ detection was negligible in both age cohorts until the 12-month time point (Supplemental Figure 2). There was a strong positive correlation at birth between GA at birth and IL-8+CD4+ T cells, and a negative correlation existed between TNF-α+ as well as TNF-α/IL-2 double-positive events and GA (r = 0.58, –0.39, –0.43, respectively) (Figure 7A). A similar pattern was found at the teCGA time point but was not present by 12 months of age (Figure 7B).
Figure 7. Cytokine profile of T cells varies by gestational age at birth, and trends persist at term-equivalent corrected gestational age.

Cytokine-producing CD4+ T cells were detected by flow cytometry following in vitro stimulation with staphylococcal enterotoxin B (SEB). (A) Scatter plots show frequency of cytokine-positive umbilical cord blood CD4+ T cells as a function of gestational age at birth (n = 167, linear regression ± 95% CI). (B) Cytokine/marker-positive CD4+ T cell frequency or cell population was analyzed as a linear function of gestational age (± 95% CI) at birth (n = 167), teCGA (n = 176), and 12-month (n = 124) sample collection (asterisk color corresponds to same color subpopulation on the graph, **P < 0.01, ****P < 0.0001, Spearman correlation). TeCGA, term-equivalent corrected gestational age.
The age-dependent expression of IL-8+ and TNF-α+ mirrored the correlation between CD31+ naive CD4+ subsets and gestation, raising the possibility that CD31 marks cell populations with distinct cytokine functions. We therefore performed in vitro stimulation with SEB to detect and CD31 and cytokine costaining on a remaining subset of cord blood samples (10 PT <32 0/7 weeks and 15 FT ≥37 weeks) (Supplemental Figure 3 and staining panel 3, Supplemental Table 2). As expected, the dominant cytokines produced by either the CD31+ or CD31– subpopulation were TNF-α, IL-8, and IL-2, but CD31+ and CD31–CD4+ T cells demonstrated distinct cytokine profiles when stimulated in vitro (Figure 8). Matching CD31+ and CD31– subsets on subjects revealed that significantly more CD31+ T cells expressed IL-8, and more CD31–CD4+ T cells expressed TNF-α and IL-2 (P < 0.001). Boolean analysis, used to compare the permutations of IL-2, IL-8, and TNF-α cytokine production, demonstrated that CD31+CD4+ T cells were higher in single and dual cytokine IL-8+ events, whereas CD31–CD4+ T cells were more frequently single IL-2 or dual TNF-α/IL-2 positive events. Only rare events were positive for both IL-8 and TNF-α, suggesting either counterregulation of these two cytokines at the single-cell level or, alternatively, that naive CD31+IL-8+ and CD31–TNF-α+CD4+ T cells represent separate functional lineages.
Figure 8. Cytokine profile of T cells varies by CD31 expression.

Cytokine-producing umbilical cord blood CD4+ T cells were detected by flow cytometry following in vitro stimulation with SEB. (A) Representative flow plots showing TNF-α and IL-8 expression on CD31+ and CD31– events (sequentially gated as FSC/SSC/live/CD14–/CD3+/CD8–/CD4+/CD69+). (B) Bulk cytokine or (C) Boolean-derived combination events are expressed as a percentage of total CD69+CD4+ of either CD31+ (gray circles) or CD31– (black squares) T cells matched for each subject (*P < 0.05, **P < 0.01, ****P < 0.0001, Wilcoxon matched-pair signed-rank test).
To validate findings based on manual gating, an unsupervised, nonlinear dimensionality reduction of flow cytometry data (staining panel 3, Supplemental Table 2) was performed using the t-distributed stochastic neighbor embedding (t-SNE) dimensionality reduction algorithm (FlowJo v.10.2) (24). t-SNE measures the similarity between pairwise points (single cells) in high dimensional space and embeds them into a reduced, visually rich two-dimensional scatter plot in such a way that the local, high dimensional structure of the single-cell data is maintained. Sixteen samples (7 PT <32 0/7 weeks and 9 FT ≥37 weeks) were first manually pregated to remove debris, doublets, and dead cells and to select for a CD3+ (CD56–CD14–) T cell population; for each sample, the CD3+ population was downsampled (randomly reduced number of input events) to 6,250 events; each downsampled population was then concatenated into a single 100,000 event FCS file. t-SNE was then applied using the following 8 markers: CD4, CD31, CD45RA, CD69, IFN-γ, IL-2, IL-8, and TNF-α. An animated “walk-through” of the resultant local structure, further enriched by PT versus FT sample assignment (focusing on CD69+CD4+ events), revealed a continuum of events that progressed from TNF-α single-positive/CD31– to IL-8 single-positive/CD31+ and also reflected an age progression from PT to FT (Figure 9 and Supplemental Video 1). In summary, these age-distinguished results, validated in two independent cohorts, as well as by an unsupervised t-SNE analysis, support a fetal immune trajectory of CD4+ T cells biased toward TNF-α in early gestation with transition to an IL-8 bias in the third trimester, correlating with a developmental gain in CD31 expression.
Figure 9. t-Stochastic neighbor embedding automated analysis on CD69+CD4+ T cell events.

A subset of 16 study samples was stained to explore the relationship between CD31 and cytokine function in umbilical cord blood. t-SNE was performed within the Flowjo package. (A) Event distribution based on nearest-neighbor analysis and walk through of the IL-8+/TNF-α neighborhood are shown within the circle. (B) Progression of CD31, IL-2, IL-8, and TNF-α (C) and 2-dimensional plots demonstrating a phenotypic continuum associated with age cohort while moving through local structure. t-SNE, t-stochastic neighbor embedding. This figure is available as a Supplemental Video.
Discussion
Results of our study establishes a model for fetal T cell development, in which functional and phenotypic changes that occur later in gestation are dependent on the balance of CD31+ and CD31–CD4+ T cells (model proposed in Figure 10). Furthermore, we present compelling evidence supporting a negative clinical effect of arrested fetal immune maturation signified by failure to increase the percentage of CD4+ T cells expressing CD31. Specifically, ELGANs with persistently low levels of CD31+CD4+ T cells measured at term-corrected age are at over 3.5-fold higher risk for PRD. To our knowledge, this is the first study to associate disruption of age-dependent fetal T cell maturation with long-term morbidity. Though correlative, the association between a developmentally determined immunophenotype and long-term outcome merits discussion of two critical questions: what factors determine CD31+CD4+ T cell numbers during gestation and after early delivery and are there mechanisms by which perturbations in the developmentally determined CD31+ and CD31– T cell balance modifies pulmonary risk in ELGANs?
Figure 10. Fetal T cell development model.

Naive recent thymic emigrants are released as CD31+IL8+CD4+ T cells. Peripheral T cells during fetal development proliferate in response to their expanding niche during rapid fetal growth. Peripherally expanded CD4+ T cells lose CD31 expression and IL-8 effector function and gain TNF-α function. The “fetal” immunophenotype places infants at risk for respiratory morbidity in the first year of life.
There are several potential causes of altered proportions in CD31-expressing CD4+ T cells during development: increased production or expansion of CD31–CD4+ T cells, decreased production or loss of CD31+ cells, and loss of CD31 molecule at the cell surface. Based on our results, developmental differences in CD31+ and CD31– T cell proportions are more likely to be secondary to an overall increase in the CD31– population numbers. Higher Ki67+ and cytokine receptor expression in the CD31– naive CD4+ T cell fraction as well as an increase in their absolute number in the ELGAN cohort support this. In adults and older children, naive low TREC CD31–CD4+ T cells are thought to have undergone proliferation and subsequent peripheral maturation after leaving the thymus (25). The rate of peripheral naive T cell expansion is largely determined by IL-7Rα expression and stromal cell produced IL-7 availability. As T cells increase in number, IL-7 becomes less available and proliferation slows. ELGANS are born immediately following their peak fetal growth velocity, which occurs between 18 and 22 weeks during gestation (26). This rapidly expanding niche may drive homeostatic T cell expansion, and loss of CD31 expression, in the same way lymphopenia exerts proliferation pressure in a host who has completed their growth.
Although our results support a model of fetal T cell phenotype driven by homeostatic expansion, we cannot exclude the “immune layering” hypothesis, in which the source of CD31– T cells in ELGANs is a unique liver-derived progenitor population with distinct phenotypic and functional potential. Mold et al. found that fetal liver HSC-derived T cells were more prone to activation and Treg differentiation compared with bone marrow–derived T cells (27, 28). Consistent with several previous studies (27, 29–31), we observed that the putative Treg population was higher at younger GAs at teCGA. However, we did not identify an association (negative or positive) between Tregs and CD31+CD4+ T cells or between Tregs and clinical outcome (Supplemental Figure 4). In contrast, we found a functional profile strongly biased toward inflammatory TNF-α+ effectors at younger ages. Both Treg and inflammatory T cell differentiation may be effects of the fetal adaptive immune development stage, but they are likely to be independent of one another. Though beyond the scope of our current study, it is also possible that functional defects in Tregs exist at younger GAs. Further studies are necessary to decipher the precise ontogeny and fate of naive CD31–CD4+ T cells in early PT infants and the interrelationship of suppressors and effectors.
The relationship between CD31+CD4+ T cells and sex is consistent with that in previous studies showing that thymic emigrants were lower in males compared with females at any given age (32). Males suffer from more severe respiratory infections and higher rates of prematurity-related lung disease (14, 33–35). When controlling for CD31+CD4+ T cell frequencies, however, sex did not predict PRD, suggesting that sex-related outcomes may actually be secondary to or better predicted by measurements of immune maturation. The interdependent effects of physiologic stress, sex, and fetal growth velocity on CD31 T cell expression, thymic function, and infant clinical outcomes merit further study.
A theoretical consequence to rapid homeostatic expansion of T cells is a break in immune tolerance, which occurs in lymphopenic hosts with inadequate thymic output (36). The mechanism linking homeostatic expansion to autoreactivity appears to be dependent on self-antigen exposure (37). Though our study did not directly test each subject’s RTE concentration, T cell CD31 expression did correlate with TREC content and was lower in ELGANs. Consistent with our hypothesis that lower numbers of CD31+ naive CD4+ T cells represent a relative reduction in RTEs in ELGANs, thymic egress is known to be diminished in neonates born prematurely, experiencing physiologic stress, or with high-dose steroid exposure (38–41). It is plausible, therefore, that sensitivity to homeostatic expansion in neonatal T cells lowers the threshold for self-reactivity in premature neonates experiencing an abundance of self-antigen exposure from insults such as ventilator-induced lung injury. In fact, in their nonhuman primate model of BPD, Rosen et al. showed an association among lung injury, diminished thymic egress, increased lung-reactive CD4+ T cells, and accelerated T cell maturation (42). T cell activation from an abnormal developmental environment during a time of rapid fetal growth and relatively reduced thymic egress may contribute to ELGAN susceptibility to compromised lung function postnatally.
Changes in T cell functional capacity associated with CD31 expression, including the downregulation of IL-8 and licensing of TNF-α in PT infants, could shape the balance between a protective and a pathologic adaptive immune response to primary infection in neonates (25, 43, 44). In a small study of infants hospitalized with respiratory syncytial virus, former PT infants had low nasal wash IL-8 when compared with FT infants, whereas FT infants with higher IL-8 had higher admission clinical severity scores, though less complicated clinical course (27% and 54% of PT versus 0% and 0% of FT required mechanical ventilation and intensive care unit admission) (8). Though our primary study did not conduct viral surveillance, our results would suggest that achieving higher numbers of CD31+CD4+ T cells producing IL-8 by NICU discharge might protect the ELGAN population against severe illness from respiratory infection. In contrast, CD31–TNF-α production, which appears to be counterregulated with CD31+IL-8 in neonatal T cells is likely present during normal fetal development in ELGANs (45). Autocrine TNF-α signaling may be appropriate during early gestation by maintaining peripherally expanded CD31– naive subsets through upregulation of IL-7rα, as seen on naive CD31–CD4+ T cells (46). In an ex utero setting, however, this survival advantage may favor retention of an inflammatory TNF-α licensed T cell population in ELGANs. While TNF-α can play a positive role during embryogenesis in the sterile fetus, it is more likely to cause harm in an inflammatory postnatal environment (47, 48), as is supported by many studies showing elevated TNF-α levels in the lungs of infants with BPD (49). Both IL-8 and TNF-α are considered inflammatory cytokines, however, and their differential effects on PT and FT outcomes is worthy of further investigation.
In addition to varied functional capacity associated with changes in CD31 expression on naive CD4+ T cells, absence of CD31 in itself on CD4+ T cells could contribute to chronic inflammation in ELGANs. CD31 contains two immunoreceptor tyrosine-based inhibitory motifs (ITIMs) on the cytoplasmic tail, which suppresses T cell activation, which is important in shaping a well-regulated, protective immune response. Cleavage of CD31 from the cell surface by metalloprotease enhances TCR signaling but also results in higher activation-induced cell death and decreased memory induction (43, 50). Reduced T cell inhibitory mechanisms in PT neonates with a low frequency of CD31+CD4+ T cells may place them at risk for excessive inflammation during acute infection while biasing against memory differentiation, placing PT infants with low numbers of CD31+ subpopulations at risk for more severe, recurrent respiratory viral infection. If CD31+ cleavage from T cells is found to contribute to PRD in ELGANs, restoration of CD31 signaling at the cell surface represents a therapeutic target to selectively modulate harmful T cell activity and induce protective T cell memory. In fact, investigators have shown that peptide ligation of a truncated CD31 domain remaining on the surface of CD31-cleaved T cells improved T cell regulation and reduced disease burden in an atherosclerosis-prone mouse model (43, 51).
Our results suggest that CD31–CD4+ T cells may identify those ELGANs who appear well enough for discharge, but who may be more vulnerable to respiratory complications after discharge (52, 53). The strength of CD31 at teCGA in ELGANs as a molecular marker exceeds other commonly associated clinical variables for predicting poor respiratory outcome, including oxygen exposure and GA. Infants with low CD31+CD4+ T cell counts may benefit from increased surveillance and perhaps more aggressive strategies to prevent respiratory infection until they demonstrate immune maturity. Although further studies are needed to elucidate mechanisms relating low CD31 frequency to outcome, the CD31 signaling pathway is an opportunity for population-specific drug development, targeting a subset of neonatal patients with high inflammatory and infectious disease burden but few therapeutic options. More broadly, our model challenges the accepted paradigm that newborns are merely in a state of immune deficiency. Rather, the last trimester of human gestation may be a critical window during which the adaptive immune system finalizes its preparation for its singular transition in function from in utero development to ex utero protection.
Methods
Study design and subject recruitment.
PT subjects between the ages of 23 0/7 and 35 6/7 weeks and healthy FT infants at ages greater than 36 6/7 weeks GA at birth were enrolled into National Heart, Lung, and Blood Institute–sponsored (NHLBI-sponsored) PROP at the University of Rochester Medical Center and Women and Children’s Hospital of Buffalo between the years of 2011 and 2015. Subjects were excluded for known congenital anomalies or genetic disorders present or suspected, subject nonviability, and maternal HIV or inherited disorders known to affect immune development. NICU admission was an exclusion criterion for FT subjects in order to identify healthy FT comparison controls. Umbilical cord blood samples from a second independent birth cohort, recruited at the University of Rochester for the PRISM study, were used for in vitro experiments and to validate PROP findings. Study clinical data were collected and managed using Research Electronic Data Capture (REDCap) tools (54).
Outcomes and definitions of clinical variables.
Following hospital discharge, phone surveys were completed at 3 and 9 months of age to determine respiratory symptomatology and changes in medical management or condition. The surveys and physical exams were performed by a pediatric pulmonologist at 6 and 12 months. Patients were diagnosed with PRD based on a composite outcome determined by the PROP working group (14). PRD was diagnosed if subjects died due to cardiac or respiratory failure or if subjects provided a positive response in regard to the following conditions in two or more surveys: (a) respiratory hospitalizations, (b) respiratory medications (bronchodilator, methylxanthine, diuretic, leukotriene receptor antagonist, pulmonary vasodilator), (c) symptoms (wheezing > once per week per parents, wheezing diagnosed by a doctor, and/or cough without cold), and (d) home respiratory support (oxygen, CPAP, ventilator/tracheostomy). Oxygen exposure as AUC at 7 and 14 days was calculated based on methods established by Stevens et al. (55). We chose to apply the modified Shennan definition for BPD, due to its ability to identify the more severe morbidity at 36 weeks after menstrual age as well as its alignment with the NIH workshop definition of moderate and severe BPD (56, 57).
Sample processing, stimulation, and preparation for flow cytometry.
A detailed protocol for sample collection and experimental procedures can be found in our previously published methods (58). Briefly, diluted blood was layered over Ficoll-Paque PLUS for isolation of either umbilical cord mononuclear cells or peripheral blood mononuclear cells (PBMCs). Cell counts were determined manually using trypan blue exclusion. PBMCs were cryopreserved in freezing media. Samples were thawed and prepared for flow cytometry in batches. To reduce misattribution of batch variation to biological differences, each batch included all time points from a given subject (birth, teCGA, and 12 month) as well as a mix of subjects across age cohorts, to reduce variation due to experimental conditions. PBMC recovery and viability were determined using manual count by trypan blue exclusion assay after RBC lysis. Thawed PBMCs were rested overnight in complete medium prior to in vitro stimulation.
Immunocytochemistry was performed with fluorescently tagged antibodies using a micromethod as previously described (58). Staining panel details can be found in Supplemental Table 2. Cells were first stained in PBS with live-dead marker. PBMCs were washed, Fc blocked with 5% normal mouse serum, and then stained in surface antibody cocktail. Cells were permeabilized and Fc blocked again. Cells allocated for intracellular cytokine detection were washed with complete media and plated at 1 × 106 to 2 × 106 PBMCs/200 μl suspension. SEB (final concentration 2 μg/ml) was added to stimulated wells. Samples were incubated in humidified air for 2 hours, after which Golgi blockade was performed for an additional 8 hours. Intracellular cytokine staining cocktail was then added. Stained cells were then fixed in 2% paraformaldehyde.
Events were collected on an 18-color LSR II using BD FACS Diva software. Fluorescence-minus-one healthy adult donor PBMCs were used for standardization across experiments. Gating was performed manually using Flowjo data analysis software (Tree Star Inc.) (59). Additional visualization was accomplished using GraphPad Prism. Flow cytometry data were managed and integrated with REDCap clinical data using the Bio-lab Informatics Server (BLIS, University of Rochester Medical Center) system based on the open-source LabKey Server platform (60).
TREC quantification.
Live naive T cells from a subset of umbilical cord blood (UCB) samples were isolated by negative selection. Thawed PBMCs were incubated with annexin V microbeads (Miltenyi, catalog 130-090-201) at a 1:1 dilution with microbeads from a pan T cell isolation kit (Miltenyi, catalog 130-097-095), and unlabeled cells were then collected following magnetic bead column elution per manufacturer’s recommendations. Methods adhered to the manufacturer’s protocol, with the exception of using 10-μl reactions instead of the recommended 20 μl. Following separation, isolated naive T cells were centrifuged, counted, and resuspended in PBS containing 0.1% BSA and 2 mM EDTA. Live naive CD31+ and CD31– T cell fractions were then similarly separated using CD31 microbeads (Miltenyi, catalog 130-091-935). Quantification of human TRECs was performed using the MyTREC TREC/Beta Actin Real-time qPCR Assay kit (GenenPlus) following the manufacturer’s recommendations. Absolute quantification was determined using standard TREC and β-actin standard curves to generate normalized TREC copies per 1 million cells.
Statistics.
Continuous variables were summarized via means (SD) or medians (IQR), graphically displayed via box plots and scatter plots, and tested using the Wilcoxon test and Spearman’s rank correlation. Dichotomous variables were summarized via counts and proportions and tested using Fisher’s exact test. Two-tailed t tests were applied, and P < 0.05 was considered significant. Logistic regression was used to model PRD as a function of the relative frequency of dichotomized CD31+CD4+ T cells at their median of 60%, with and without adjusting for clinical variables, including GA. Logistic regression was also used to model PRD as a function of GA alone, with and without restricting to <29 weeks. Receiver operating characteristic curves were used to nonparametrically compare biomarker performance to GA. Biomarker and clinical data analyses were performed using SAS version 9 for Windows. In vitro laboratory results were analyzed using Graphpad Prism v 6.0 for Mac.
Study approval.
All study procedures can be found in the previously published protocol (12) and were approved by the University of Rochester Medical Center and State University of New York at Buffalo Institutional Review Boards. As all enrolled subjects were children, written informed consent was obtained from the appropriate surrogate (parent or guardian) prior to inclusion in the study. Families were approached for consent if there were no known barriers to communication or 1 year follow-up, and enrollment occurred before 7 days of age.
Author contributions
KMS, DJT, and GSP performed conception, execution, interpretation, and manuscript preparation. AMR, RMR, GSP, TJM, and MC were Site PIs at University at Buffalo or University of Rochester. KMS, JE, NL, KA, JMA, and KST acquired and analyzed experimental data. EW, HH, TS, and DM performed clinical and sample data collection, study coordination, and recruitment. CLR was the PI for Pulmonary Clinic follow-ups. AMB and DRP performed biostatistical analysis. JHW, SB, and AGS performed clinical and laboratory data integration and management.
Supplementary Material
Acknowledgments
The authors would like to acknowledge the University of Rochester Pediatrics Translational Biospecimen Laboratory, the University of Rochester Flow Cytometry Core, and the University of Rochester Human Immunology Core. This study was dependent on the nurses and staff at the Golisano Children’s Hospital NICU, the Women and Children’s Hospital of Buffalo NICU, and University of Rochester Medical Center Strong Beginnings Maternity Services. We are indebted most of all to the generosity of the NICU families who consented to this and other research studies. This work was supported by NIH National Institute of Allergy and Infectious Diseases (NIAID) 1K08AI108870-01A1 (CD8 T Cell Dysregulation in Premature Infants), NIH NHLBI U01 HL101813-01 (PROP), NIH NIAID HHSN272201200005C (Respiratory Pathogens Research Center), NIH Eunice Kennedy Shriver National Institute of Child Health and Human Development 1K12HD068373-01 (Translational Molecular Pediatrics), and NIH National Center for Advancing Translational Sciences UL1 TR000042 (Clinical and Translational Science Institute).
Version 1. 02/22/2018
Electronic publication
Footnotes
Conflict of interest: The authors have declared that no conflict of interest exists.
Reference information: JCI Insight. 2018;3(4):e96724. https://doi.org/10.1172/jci.insight.96724.
Contributor Information
Jason Emo, Email: Jason_Emo@urmc.rochester.edu.
Nathan Laniewski, Email: Nathan_Laniewski@urmc.rochester.edu.
Jeanne Holden-Wiltse, Email: Jeanne_Wiltse@URMC.Rochester.edu.
Sanjukta Bandyopadhyay, Email: Sanjukta_Bandyopadhyay@URMC.Rochester.edu.
Heidie Huyck, Email: Heidie_Huyck@URMC.Rochester.edu.
Kelly Schooping Tripi, Email: Kelly_Schooping@URMC.Rochester.edu.
Karan Arul, Email: karul@u.rochester.edu.
Elizabeth Werner, Email: Elizabeth_Werner@URMC.Rochester.edu.
Tanya Scalise, Email: Tanya_Scalise@URMC.Rochester.edu.
Deanna Maffett, Email: dandmaff@rochester.rr.com.
Mary Caserta, Email: Mary_Caserta@urmc.rochester.edu.
Rita M. Ryan, Email: ryanr@musc.edu.
Anne Marie Reynolds, Email: areynolds@upa.chob.edu.
Thomas J. Mariani, Email: Tom_Mariani@URMC.Rochester.edu.
Gloria S. Pryhuber, Email: Gloria_Pryhuber@URMC.Rochester.edu.
References
- 1.Wilson CB, Westall J, Johnston L, Lewis DB, Dower SK, Alpert AR. Decreased production of interferon-gamma by human neonatal cells. Intrinsic and regulatory deficiencies. J Clin Invest. 1986;77(3):860–867. doi: 10.1172/JCI112383. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.von Freeden U, Zessack N, van Valen F, Burdach S. Defective interferon gamma production in neonatal T cells is independent of interleukin-2 receptor binding. Pediatr Res. 1991;30(3):270–275. doi: 10.1203/00006450-199109000-00015. [DOI] [PubMed] [Google Scholar]
- 3.Zhang X, Zhivaki D, Lo-Man R. Unique aspects of the perinatal immune system. Nat Rev Immunol. 2017;17(8):495–507. doi: 10.1038/nri.2017.54. [DOI] [PubMed] [Google Scholar]
- 4.Kagina BM, et al. Specific T cell frequency and cytokine expression profile do not correlate with protection against tuberculosis after bacillus Calmette-Guérin vaccination of newborns. Am J Respir Crit Care Med. 2010;182(8):1073–1079. doi: 10.1164/rccm.201003-0334OC. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Galindo-Albarrán AO, et al. CD8+ T cells from human neonates are biased toward an innate immune response. Cell Rep. 2016;17(8):2151–2160. doi: 10.1016/j.celrep.2016.10.056. [DOI] [PubMed] [Google Scholar]
- 6.Gibbons D, et al. Interleukin-8 (CXCL8) production is a signatory T cell effector function of human newborn infants. Nat Med. 2014;20(10):1206–1210. doi: 10.1038/nm.3670. [DOI] [PubMed] [Google Scholar]
- 7.Scheible KM, et al. Developmentally determined reduction in CD31 during gestation is associated with CD8+ T cell effector differentiation in preterm infants. Clin Immunol. 2015;161(2):65–74. doi: 10.1016/j.clim.2015.07.003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Assefa D, Amin N, Dozor AJ, Parton LA. Attenuated interleukin-8/leukocyte immunoresponse in preterm infants compared with term infants hospitalized with respiratory syncytial virus bronchiolitis: a pilot study. Hum Immunol. 2011;72(9):708–711. doi: 10.1016/j.humimm.2011.05.016. [DOI] [PubMed] [Google Scholar]
- 9.Huang L, et al. Decreased frequencies and impaired functions of the CD31+ subpopulation in Treg cells associated with decreased FoxP3 expression and enhanced Treg cell defects in patients with coronary heart disease. Clin Exp Immunol. 2017;187(3):441–454. doi: 10.1111/cei.12897. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Tada Y, et al. Acceleration of the onset of collagen-induced arthritis by a deficiency of platelet endothelial cell adhesion molecule 1. Arthritis Rheum. 2003;48(11):3280–3290. doi: 10.1002/art.11268. [DOI] [PubMed] [Google Scholar]
- 11.Malek A, Sager R, Kuhn P, Nicolaides KH, Schneider H. Evolution of maternofetal transport of immunoglobulins during human pregnancy. Am J Reprod Immunol. 1996;36(5):248–255. doi: 10.1111/j.1600-0897.1996.tb00172.x. [DOI] [PubMed] [Google Scholar]
- 12.Pryhuber GS, et al. Prematurity and respiratory outcomes program (PROP): study protocol of a prospective multicenter study of respiratory outcomes of preterm infants in the United States. BMC Pediatr. 2015;15:37. doi: 10.1186/s12887-015-0346-3. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Keller RL, et al. Bronchopulmonary dysplasia and perinatal characteristics predict 1-year respiratory outcomes in newborns born at extremely low gestational age: A prospective cohort study. J Pediatr. 2017;187:89–97.e3. doi: 10.1016/j.jpeds.2017.04.026. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Keller RL, et al. Bronchopulmonary dysplasia and perinatal characteristics predict 1-year respiratory outcomes in newborns born at extremely low gestational age: A prospective cohort study. J Pediatr. 2017;187:89–97.e3. doi: 10.1016/j.jpeds.2017.04.026. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Wong MX, Hayball JD, Hogarth PM, Jackson DE. The inhibitory co-receptor, PECAM-1 provides a protective effect in suppression of collagen-induced arthritis. J Clin Immunol. 2005;25(1):19–28. doi: 10.1007/s10875-005-0354-7. [DOI] [PubMed] [Google Scholar]
- 16.Peacock JL, Marston L, Marlow N, Calvert SA, Greenough A. Neonatal and infant outcome in boys and girls born very prematurely. Pediatr Res. 2012;71(3):305–310. doi: 10.1038/pr.2011.50. [DOI] [PubMed] [Google Scholar]
- 17.Douaisi M, et al. CD31, a valuable marker to identify early and late stages of T cell differentiation in the human thymus. J Immunol. 2017;198(6):2310–2319. doi: 10.4049/jimmunol.1500350. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Junge S, et al. Correlation between recent thymic emigrants and CD31+ (PECAM-1) CD4+ T cells in normal individuals during aging and in lymphopenic children. Eur J Immunol. 2007;37(11):3270–3280. doi: 10.1002/eji.200636976. [DOI] [PubMed] [Google Scholar]
- 19.Hazenberg MD, Verschuren MC, Hamann D, Miedema F, van Dongen JJ. T cell receptor excision circles as markers for recent thymic emigrants: basic aspects, technical approach, and guidelines for interpretation. J Mol Med. 2001;79(11):631–640. doi: 10.1007/s001090100271. [DOI] [PubMed] [Google Scholar]
- 20.Azevedo RI, et al. IL-7 sustains CD31 expression in human naive CD4+ T cells and preferentially expands the CD31+ subset in a PI3K-dependent manner. Blood. 2009;113(13):2999–3007. doi: 10.1182/blood-2008-07-166223. [DOI] [PubMed] [Google Scholar]
- 21.Lodolce J, et al. Interleukin-15 and the regulation of lymphoid homeostasis. Mol Immunol. 2002;39(9):537–544. doi: 10.1016/S0161-5890(02)00211-0. [DOI] [PubMed] [Google Scholar]
- 22.Marleau AM, Sarvetnick N. T cell homeostasis in tolerance and immunity. J Leukoc Biol. 2005;78(3):575–584. doi: 10.1189/jlb.0105050. [DOI] [PubMed] [Google Scholar]
- 23.Priyadharshini B, Welsh RM, Greiner DL, Gerstein RM, Brehm MA. Maturation-dependent licensing of naive T cells for rapid TNF production. PLoS One. 2010;5(11):e15038. doi: 10.1371/journal.pone.0015038. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Jamieson AR, Giger ML, Drukker K, Li H, Yuan Y, Bhooshan N. Exploring nonlinear feature space dimension reduction and data representation in breast Cadx with Laplacian eigenmaps and t-SNE. Med Phys. 2010;37(1):339–351. doi: 10.1118/1.3267037. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Kohler S, Thiel A. Life after the thymus: CD31+ and CD31- human naive CD4+ T-cell subsets. Blood. 2009;113(4):769–774. doi: 10.1182/blood-2008-02-139154. [DOI] [PubMed] [Google Scholar]
- 26.Bertino E, et al. Fetal growth velocity: kinetic, clinical, and biological aspects. Arch Dis Child Fetal Neonatal Ed. 1996;74(1):F10–F15. doi: 10.1136/fn.74.1.F10. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Mold JE, et al. Fetal and adult hematopoietic stem cells give rise to distinct T cell lineages in humans. Science. 2010;330(6011):1695–1699. doi: 10.1126/science.1196509. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Mold JE, McCune JM. At the crossroads between tolerance and aggression: Revisiting the “layered immune system” hypothesis. Chimerism. 2011;2(2):35–41. doi: 10.4161/chim.2.2.16329. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Cupedo T, Nagasawa M, Weijer K, Blom B, Spits H. Development and activation of regulatory T cells in the human fetus. Eur J Immunol. 2005;35(2):383–390. doi: 10.1002/eji.200425763. [DOI] [PubMed] [Google Scholar]
- 30.Dirix V, Vermeulen F, Mascart F. Maturation of CD4+ regulatory T lymphocytes and of cytokine secretions in infants born prematurely. J Clin Immunol. 2013;33(6):1126–1133. doi: 10.1007/s10875-013-9911-4. [DOI] [PubMed] [Google Scholar]
- 31.Weitkamp JH, et al. Ontogeny of FOXP3 (+) regulatory T cells in the postnatal human small intestinal and large intestinal lamina propria. Pediatr Dev Pathol. 2009;12(6):443–449. doi: 10.2350/08-09-0533.1. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Pido-Lopez J, Imami N, Aspinall R. Both age and gender affect thymic output: more recent thymic migrants in females than males as they age. Clin Exp Immunol. 2001;125(3):409–413. doi: 10.1046/j.1365-2249.2001.01640.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Caserta MT, Yang H, Gill SR, Holden-Wiltse J, Pryhuber G. Viral Respiratory Infections in Preterm Infants during and after Hospitalization. J Pediatr. 2017;182:53–58.e3. doi: 10.1016/j.jpeds.2016.11.077. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Falagas ME, Mourtzoukou EG, Vardakas KZ. Sex differences in the incidence and severity of respiratory tract infections. Respir Med. 2007;101(9):1845–1863. doi: 10.1016/j.rmed.2007.04.011. [DOI] [PubMed] [Google Scholar]
- 35.Klein MI, et al. Differential gender response to respiratory infections and to the protective effect of breast milk in preterm infants. Pediatrics. 2008;121(6):e1510–e1516. doi: 10.1542/peds.2007-1757. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Datta S, Sarvetnick N. Lymphocyte proliferation in immune-mediated diseases. Trends Immunol. 2009;30(9):430–438. doi: 10.1016/j.it.2009.06.002. [DOI] [PubMed] [Google Scholar]
- 37.Le Saout C, Mennechet S, Taylor N, Hernandez J. Memory-like CD8+ and CD4+ T cells cooperate to break peripheral tolerance under lymphopenic conditions. Proc Natl Acad Sci USA. 2008;105(49):19414–19419. doi: 10.1073/pnas.0807743105. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Gruver AL, Sempowski GD. Cytokines, leptin, and stress-induced thymic atrophy. J Leukoc Biol. 2008;84(4):915–923. doi: 10.1189/jlb.0108025. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Chen CM, Yu KY, Lin HC, Yeh GC, Hsu HH. Thymus size and its relationship to perinatal events. Acta Paediatr. 2000;89(8):975–978. doi: 10.1111/j.1651-2227.2000.tb00420.x. [DOI] [PubMed] [Google Scholar]
- 40.Kong FK, Chen CL, Cooper MD. Reversible disruption of thymic function by steroid treatment. J Immunol. 2002;168(12):6500–6505. doi: 10.4049/jimmunol.168.12.6500. [DOI] [PubMed] [Google Scholar]
- 41.Kwan A, et al. Newborn screening for severe combined immunodeficiency in 11 screening programs in the United States. JAMA. 2014;312(7):729–738. doi: 10.1001/jama.2014.9132. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42.Rosen D, Lee JH, Cuttitta F, Rafiqi F, Degan S, Sunday ME. Accelerated thymic maturation and autoreactive T cells in bronchopulmonary dysplasia. Am J Respir Crit Care Med. 2006;174(1):75–83. doi: 10.1164/rccm.200511-1784OC. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43.Fornasa G, et al. TCR stimulation drives cleavage and shedding of the ITIM receptor CD31. J Immunol. 2010;184(10):5485–5492. doi: 10.4049/jimmunol.0902219. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44.Newman DK, Hamilton C, Newman PJ. Inhibition of antigen-receptor signaling by Platelet Endothelial Cell Adhesion Molecule-1 (CD31) requires functional ITIMs, SHP-2, and p56 (lck) Blood. 2001;97(8):2351–2357. doi: 10.1182/blood.V97.8.2351. [DOI] [PubMed] [Google Scholar]
- 45.Zhang X, et al. CD4 T cells with effector memory phenotype and function develop in the sterile environment of the fetus. Sci Transl Med. 2014;6(238):238ra72. doi: 10.1126/scitranslmed.3008748. [DOI] [PubMed] [Google Scholar]
- 46.Abe A, et al. An enhancer of the IL-7 receptor α-chain locus controls IL-7 receptor expression and maintenance of peripheral T cells. J Immunol. 2015;195(7):3129–3138. doi: 10.4049/jimmunol.1302447. [DOI] [PubMed] [Google Scholar]
- 47.Toder V, Fein A, Carp H, Torchinsky A. TNF-alpha in pregnancy loss and embryo maldevelopment: a mediator of detrimental stimuli or a protector of the fetoplacental unit? J Assist Reprod Genet. 2003;20(2):73–81. doi: 10.1023/A:1021740108284. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48.Borsini A, Zunszain PA, Thuret S, Pariante CM. The role of inflammatory cytokines as key modulators of neurogenesis. Trends Neurosci. 2015;38(3):145–157. doi: 10.1016/j.tins.2014.12.006. [DOI] [PubMed] [Google Scholar]
- 49.Xu L, Yoon H, Zhao MQ, Liu J, Ramana CV, Enelow RI. Cutting edge: pulmonary immunopathology mediated by antigen-specific expression of TNF-alpha by antiviral CD8+ T cells. J Immunol. 2004;173(2):721–725. doi: 10.4049/jimmunol.173.2.721. [DOI] [PubMed] [Google Scholar]
- 50.Ross EA, et al. CD31 is required on CD4+ T cells to promote T cell survival during Salmonella infection. J Immunol. 2011;187(4):1553–1565. doi: 10.4049/jimmunol.1000502. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 51.Fornasa G, et al. A CD31-derived peptide prevents angiotensin II-induced atherosclerosis progression and aneurysm formation. Cardiovasc Res. 2012;94(1):30–37. doi: 10.1093/cvr/cvs076. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 52.Filippone M, Bonetto G, Cherubin E, Carraro S, Baraldi E. Childhood course of lung function in survivors of bronchopulmonary dysplasia. JAMA. 2009;302(13):1418–1420. doi: 10.1001/jama.2009.1419. [DOI] [PubMed] [Google Scholar]
- 53.Baraldi E, Carraro S, Filippone M. Bronchopulmonary dysplasia: definitions and long-term respiratory outcome. Early Hum Dev. 2009;85(10 Suppl):S1–S3. doi: 10.1016/j.earlhumdev.2009.08.002. [DOI] [PubMed] [Google Scholar]
- 54.Harris PA, Taylor R, Thielke R, Payne J, Gonzalez N, Conde JG. Research electronic data capture (REDCap)--a metadata-driven methodology and workflow process for providing translational research informatics support. J Biomed Inform. 2009;42(2):377–381. doi: 10.1016/j.jbi.2008.08.010. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 55.Stevens TP, Dylag A, Panthagani I, Pryhuber G, Halterman J. Effect of cumulative oxygen exposure on respiratory symptoms during infancy among VLBW infants without bronchopulmonary dysplasia. Pediatr Pulmonol. 2010;45(4):371–379. doi: 10.1002/ppul.21199. [DOI] [PubMed] [Google Scholar]
- 56.Ehrenkranz RA, et al. Validation of the National Institutes of Health consensus definition of bronchopulmonary dysplasia. Pediatrics. 2005;116(6):1353–1360. doi: 10.1542/peds.2005-0249. [DOI] [PubMed] [Google Scholar]
- 57.Poindexter BB, et al. Comparisons and limitations of current definitions of bronchopulmonary dysplasia for the prematurity and respiratory outcomes program. Ann Am Thorac Soc. 2015;12(12):1822–1830. doi: 10.1513/AnnalsATS.201504-218OC. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 58.Scheible K, et al. Stability of T cell phenotype and functional assays following heparinized umbilical cord blood collection. Cytometry A. 2012;81(11):937–949. doi: 10.1002/cyto.a.22203. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 59.Parks DR, Roederer M, Moore WA. A new “Logicle” display method avoids deceptive effects of logarithmic scaling for low signals and compensated data. Cytometry A. 2006;69(6):541–551. doi: 10.1002/cyto.a.20258. [DOI] [PubMed] [Google Scholar]
- 60.Nelson EK, et al. LabKey Server: an open source platform for scientific data integration, analysis and collaboration. BMC Bioinformatics. 2011;12:71. doi: 10.1186/1471-2105-12-71. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.