

Abstract
In the thousands of years of rice domestication in Asia, many useful genes have been lost from the gene pool. Wild rice is a key source of diversity for domesticated rice. Genome sequencing has suggested that the wild rice populations in northern Australia may include novel taxa, within the AA genome group of close (interfertile) wild relatives of domesticated rice that have evolved independently due to geographic separation and been isolated from the loss of diversity associated with gene flow from the large populations of domesticated rice in Asia. Australian wild rice was collected from 27 sites from Townsville to the northern tip of Cape York. Whole chloroplast genome sequences and 4,555 nuclear gene sequences (more than 8 Mbp) were used to explore genetic relationships between these populations and other wild and domesticated rices. Analysis of the chloroplast and nuclear data showed very clear evidence of distinctness from other AA genome Oryza species with significant divergence between Australian populations. Phylogenetic analysis suggested the Australian populations represent the earliest‐branching AA genome lineages and may be critical resources for global rice food security. Nuclear genome analysis demonstrated that the diverse O. meridionalis populations were sister to all other AA genome taxa while the Australian O. rufipogon‐like populations were associated with the clade that included domesticated rice. Populations of apparent hybrids between the taxa were also identified suggesting ongoing dynamic evolution of wild rice in Australia. These introgressions model events similar to those likely to have been involved in the domestication of rice.
Keywords: Australian wild rice, nuclear genes chloroplast sequence, Oryza AA genome, phylogenetic analysis
1. INTRODUCTION
Rice (Oryza sativa L.) is a critically important cereal crop being a key source of carbohydrates (calories) and an important source of many other nutrients for more than half of the world's people (Civáň, Craig, Cox, & Brown, 2015; Huang et al., 2012). The wild relatives of rice represent a valuable resource for rice improvement and adaptation to meet the needs of a growing human population in a changing environment (Henry, 2016; Henry et al., 2010; Mickelbart, Hasegawa, & Bailey‐Serres, 2015).
Wild Oryza species are widespread in northern Australia (Henry et al., 2010). This is an area without a long history of rice cultivation, implying that the wild populations have remained largely isolated from the impacts of gene flow from domesticated crops that has apparently been widespread in Asia (Brozynska et al., 2017). The AA genome species of rice include cultivated species and their close relatives (Choi, Platts, Fuller, Wing, & Purugganan, 2017). Draft genome sequences of the AA genome populations from Australia have recently been reported indicating that these populations may be an important genetic resource for rice because of their high diversity and phylogenetic relationship to domesticated rice (Brozynska, Furtado, & Henry, 2015; Brozynska et al., 2014, 2017; Sotowa et al., 2013; Wambugu, Brozynska, Furtado, Waters, & Henry, 2015).
We now report on an analysis of the genomes of rice collected from sites over a wide area in northeastern Australia allowing analysis of the diversity and relationships within and between these wild populations.
2. MATERIAL AND METHODS
2.1. Field collections
Samples and data were collected during May 2015, 2016, and 2017, from northeastern Queensland, Australia. Collections ranged from south of Townsville to the most northerly parts of Cape York Peninsula (Figure 1). Seeds and vegetative material were collected from 29 sites. GPS coordinates, observations of plant spike form, awn length, an herbarium voucher, and photographs of flowers (where possible) were obtained at each site (Appendix S1, Table S1, Figure S1).
Figure 1.
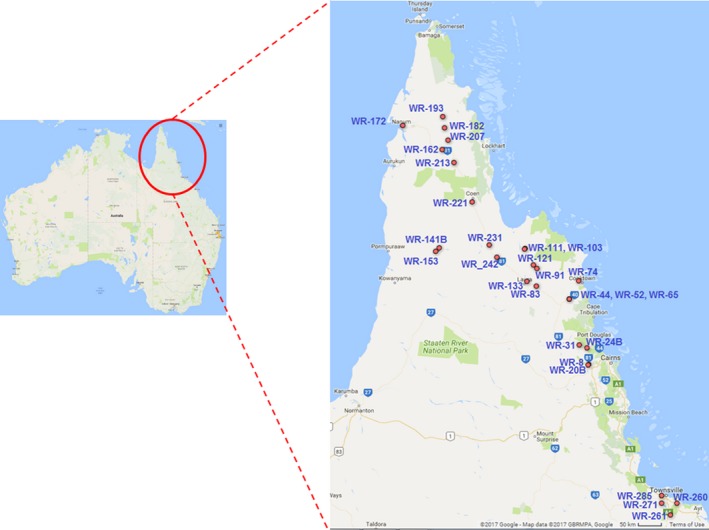
Australian wild rice collection sites. Red dots indicate collection sites
2.2. Morphological measurement
Anther and awn measurements were recorded in the field. For anther length, 4–8 flowers from 3 to 6 immature panicles were selected at random from each population, photographed against a standard background with a scale, and measurements obtained later in the laboratory using Image‐Pro Plus software (Media Cybernetics, MD, USA, http://www.mediacy.com/index.aspx?page=IPP). The awn length was measured for ten different plants from each population selected at random.
2.3. DNA extraction and sequencing
Vegetative tissue from 29 samples (representing each of the collection sites) was prepared and DNA extracted as described by Furtado (2014). Three approaches were used to assess the quality and quantity of the extracted DNA: Nano Drop (Thermo Fisher Scientific), agarose gel electrophoresis, and Qubit (Thermo Fisher Scientific). Multiplex sequencing of the 29 wild rice samples was conducted using a Hiseq 4000 (Illumina) using 2 × 150 paired end technique, aiming to produce approximately 10× whole genome coverage on average. Reference chloroplast genome sequences were obtained as described in (Appendix S1, Table S9).
2.4. Chloroplast genome assembly
The sequence reads were analyzed using CLC Genomic workbench V.9, Geneious V.9.1.5 and Clone Manager Professional 9 (Kim et al., 2015). A quality check (QC) was applied to all raw data. Based on the results of the QC report, reads were trimmed. A dual pipeline approach was used to assemble the chloroplast genome sequences: mapping reads to reference and de novo assembly. The outputs of both pipelines were combined, and all discrepancies were resolved and corrected manually.
2.5. Chloroplast phylogenetic analysis
The assembled chloroplast genome sequences together with those that were obtained from earlier studies (a total of 42) were analyzed using Geneious V 9.1.5 (geneious.com). Chloroplast genomes were aligned using the MAFFT (MAFFT v7.308 Algorithm: auto, scoring matrix: 1PAM/k = 2 gap open penalty:1.53 offset value:0.123) plugin tool (Katoh, Misawa, Kuma, & Miyata, 2002). The alignment file was inspected physically. Bayesian inference (BI), maximum likelihood (ML), and maximum parsimony (MP) approaches, using the software packages MrBayes (Huelsenbeck & Ronquist, 2001), PHYLM (Carbonell‐Caballero et al., 2015; Guindon & Gascuel, 2003), PAUP (Swofford, 2002), respectively, were utilized to infer the evolutionary relationships. (Appendix S1, Table S6). Genetic diversity for the whole chloroplast calculated using DnaSP software (Rozas, Sánchez‐DelBarrio, Messeguer, & Rozas, 2003).
2.6. Chloroplast genome annotation
All chloroplast sequences were annotated using the CpGAVAS website (http://www.herbalgenomics.org/0506/cpgavas/analyzer/home), using the default parameters as recommended. The outcome was imported directly into Geneious software to allow comparison with the reference O. sativa japonica NC_001320 to identify polymorphisms.
2.7. Phylogenetic analysis of nuclear genes
Phylogenetic analysis was based upon a set of 4,643 genes that were found in all include Oryza species (Brozynska et al., 2017). These sequences were obtained from the sequence data pool for each field sample and reference genome using the software packages FastQC, BWA, Samtools, bcftools, and MUMmer. The accession identifiers of the reference samples used were as follows: O. sativa japonica AA GCA_000005425.2, O. sativa indica AA GCA_000004655.2, O. rufipogon AA GCA_000817225.1, O. nivara AA GCA_000576065.1, O. barthii AA GCA_000182155.3, O. glaberrima AA GCA_000147395.2, O. glumaepatula AA GCA_000576495.1, O. meridionalis AA GCA_000338895.2, Taxon A AA LONB00000000, Taxon B AA LONC00000000, and O. punctata BB GCA_000573905.1. A total of 4,555 genes were obtained from all samples and references. These genes were divided into groups based upon the chromosomal location in O. sativa japonica. Multiple sequence alignment was performed at the gene level using MAFFT (Katoh et al., 2002). Following this individual gene alignment, files were concatenated into single alignment for each chromosome; then, all chromosomes were combined into a whole genome alignment of 8,179,015 base pairs (Figure 3b).
Figure 3.
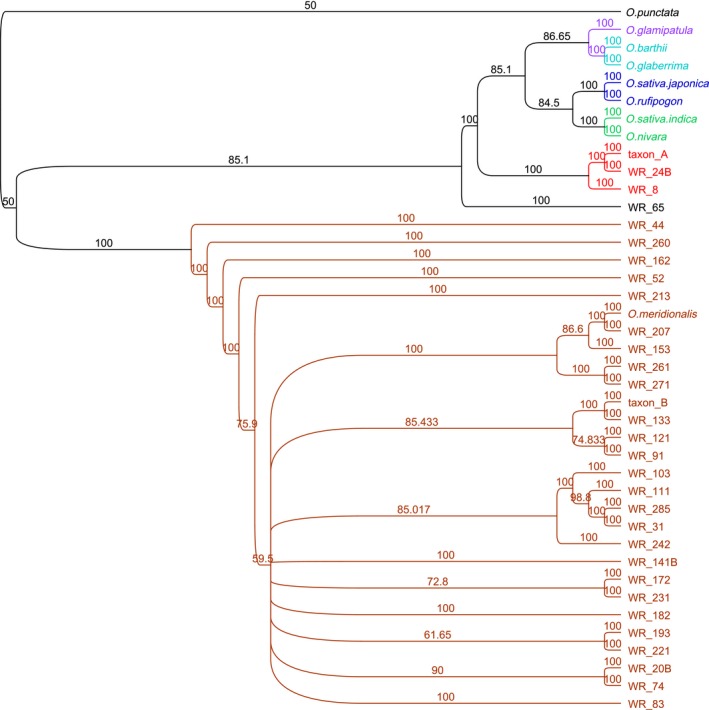
Diversity of nuclear genomes. (a) Phylogenetic tree based on MP analysis of the concatenated alignment of all nuclear genes. Colors relate to the main clades. Red and brown clades are from Australia. Bootstrap values (maximum parsimony, 1,000 replicates) are shown on the branches; (b) individual chromosome analysis showing
Phylogenetic trees were reconstructed using three analytical approaches: ML, MP, and BI. For the ML analysis, PHYML version 20131022 was used with the following settings: tree topology search: NNIs, initial tree = parsimony, model of nucleotide substitution = GTR (Guindon & Gascuel, 2003). For the MP analysis, PAUP 4.0 was used with the following setting: stepwise taxon addition with random seed, heuristic tree search strategy, and 1,000 bootstrap (Swofford, 2002). For the BI analysis, MrBayes was used with same as reported in Brozynska et al. (2017).
3. RESULTS AND DISCUSSION
Wild AA genome rice was collected from 27 sites in north Queensland, Australia (Figure 1 and Appendix S1, Table S1). Plants were found around the margins of lakes and creeks (Appendix S1, Figure S1) where for the most part, water was available to support their growth. Wild rice was not located on Cape York north of the Jardine River (−11.103665, 142.283901) or on the Islands of Torres Strait, consistent with Herbarium records (AVH, accessed 30 June 2017). Although the cause of this distributional gap, and its temporal dynamics, is unclear, it may represent a contemporary barrier to gene flow with populations to the north in New Guinea and South East Asia.
Wild plants in the field showed significant morphological variation (Table S1), particularly in spike morphology, awn length, and anther length. Awn length varied more than threefold between sites with the open panicle types (O. rufipogon‐like, Taxon A) having shorter awns than the closed panicle types (O. meridionalis‐like, Taxon B). The shortest anthers (c. 1.5 mm) were found in plants resembling O. meridionalis or taxon B. In contrast, the longest anthers (4.5 mm) were found in plants resembling O. rufipogon or taxon A. Both awn and anther length showed highly significant (p < .01) differences between sites. The results agree with previous studies of these Australian populations. (Brozynska et al., 2014; Sotowa et al., 2013; Waters, Nock, Ishikawa, Rice, & Henry, 2012).
All regions of the chloroplasts were successfully sequenced. The high sequence coverage ensured a complete genome sequence was obtained for all sites in the assembly pipeline that was used. The average coverage of the total chloroplast for all samples was 683× while the highest and lowest coverages were 2,063× and 10×, respectively (Appendix S1, Table S2). Compared to the reference sequence, an average of 129.6 variants (deletions, insertions, and SNPs) per sample were found (Appendix S1, Table S3), which agrees with the results reported by Brozynska et al. (2014). A total of 18 functional polymorphisms were found in the chloroplasts with six of them common to all samples (Appendix S1, Tables S4, S5).
The aligned sequence comprised 135,532 bp. Of the variable sites, 227 were parsimony‐informative and 661 were uninformative (427 were unique). The phylogenetic trees constructed using different approaches (Appendix S1, Table S6) were highly congruent (Brozynska et al., 2014; Kim et al., 2015; Wambugu et al., 2015). As in earlier work (Wambugu et al., 2015), a clade including O. glumipatula and O. longistaminata was sister to all other AA genome rices which were divided into an Australian clade, and a clade with Asian and African taxa including the two domesticated species. The Australian clade contained two main clades: a small clade (7 populations) containing Taxon A and a much larger clade (20 populations) containing the majority of the samples including Taxon B and O. meridionalis. This result confirms that the chloroplast genome of Taxon A is not closely related to that of Asian O. rufipogon despite the plants having a similar appearance. Eight unique chloroplast molecular makers were found in all members of the clade that includes Taxon A (Appendix S1, Table S7) (Kim et al., 2015). The chloroplasts of the different Australian AA genome taxa showed significant genetic differences (Figure 2). The concatenated alignment of 4,555 nuclear genes comprised 8,179,015 bp of which 44.1% were invariant. The minimum and maximum lengths were 5,916,081 and 7,013,653 bp, respectively, slightly longer than reported previously (Brozynska et al., 2017). The nuclear analysis (as one full length sequence and by chromosomes) grouped the Australian samples into two main clades. One of these included Taxon A and the other much larger group (27 samples) included Taxon B and O. meridionalis types (Figure 3). This analysis confirmed the nuclear genomes of the diverse O. meridionalis group including Taxon B are sister to those of all other AA genome taxa. However, four other Australian samples including Taxon A grouped within the clade that includes all other AA genome species as suggested by the single genome analysis of (Brozynska, et al. 2017). The phylogeny based upon individual chromosomes (Appendix S1, Figure [Link], [Link], [Link], [Link], [Link], [Link], [Link], [Link], [Link], [Link], [Link], [Link]) shows that these populations were a sister to all Asian and African rices (chromosomes 4, 5, 6, 7, 8) or the Asian rices (chromosome 9, 10), O. indica/O. nivara (1, 2, 3, 11) or Australian (12) clades indicating significant introgression between the different populations of wild rice.
Figure 2.
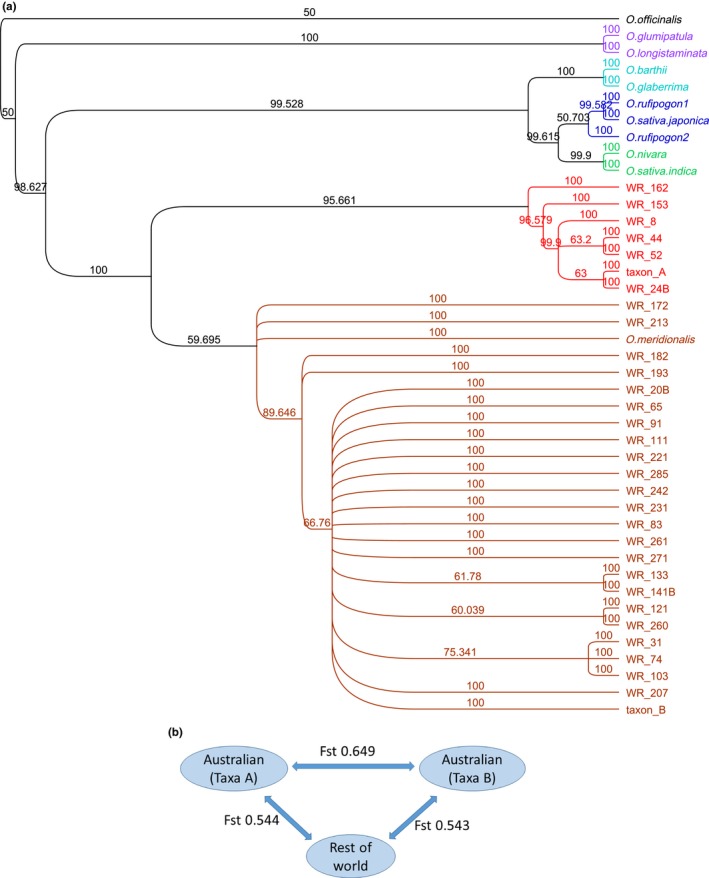
Diversity of chloroplast genomes. (a) Phylogenetic tree based on MP analysis of whole chloroplast genome sequences. Colors relate to the main clades. Red and brown clades are from Australia. Bootstrap values (MP 1,000 replicates) are shown on the branches; (b) genetic distances between populations in Australia and elsewhere
The chloroplast genomes of Taxon B are diverse and include a small number (populations WR‐44, WR‐52. WR‐153, WR‐162) that showed close relationships to the chloroplast genome found in the plants with an A genome. These included the most divergent B types (e.g., WR‐44, WR‐52 and WR‐162). Some of these were from sites where morphological traits were somewhat intermediate between the Taxon A and Taxon B types. For example, the populations found on the Lakeland‐Cooktown road had large anthers and panicles that varied from open to close. The divergent B nuclear genome and A chloroplast genome suggests plants in these populations may be hybrids. Population WR‐65 had a B‐type chloroplast but an A‐type nuclear genome.
Both chloroplast and nuclear gene analysis suggest a high diversity of AA genome wild rice in Australia. This supports the view that Australia might be a center of diversity for the AA genome clade. The populations with a morphology similar to O. meridionalis are diverse and may include both annual and perennial types (Brozynska et al., 2014; Sotowa et al., 2013). These populations could all be considered part of one diverse species, O. meridionalis. The nuclear genome analysis of the O. rufipogon‐like (Taxon A) populations places them in the Asian clade together with domesticated rices. This suggests these Australian populations should be considered as a distinct, undescribed taxon (Brozynska et al., 2017). Analysis of the chloroplast genomes placed Australian plants with O. rufipogon‐like morphology in the Australian clade, distant from the Asian O. rufipogon which were placed in the Asian clade. Some populations with a nuclear genome similar to O. meridionalis had a chloroplast genome that was closer to the O. rufipogon‐like plants (Taxon A) suggesting that their evolutionary history involved some introgression or hybridization and chloroplast capture (Brozynska et al., 2014, 2017; Wambugu et al., 2015). One example of chloroplast capture in the other direction was also detected (WR‐65). This illustrates a dynamic state of evolution of wild Oryza in Australia. This type of ongoing introgression is demonstrated by the analysis of the individual chromosomes in these populations and similar events may explain the domestication of wild indica by introgression of domestication alleles from domesticated japonica (Civan 2015). Extensive evidence shows distinct wild progenitors populations for indica and japonica rice that require separate domestication (Civan 2015) while the presence of common domestication related alleles suggests a single domestication event (Huang 2012). The discovery of natural hybrids between taxa with greater divergence than indica and japonica demonstrates the potential for similar hybridization events to be associated with the transfer of domestication‐related alleles during rice domestication.
Further research should determine the diversity of useful alleles in these populations that might be incorporated into domesticated rice to improved stress tolerance and grain quality. The need for increased efforts to conserve these species in situ and ex situ is suggested by the very limited collection of this material in seed collections and the more limited distribution of the O. rufipogon‐like populations in the wild in locations that may be threatened by the incursion of weeds.
CONFLICT OF INTERESTS
The authors declare no competing financial interests.
AUTHOR CONTRIBUTIONS
R.H. conceived the project. I.C., D.C. G.F., A.M.M, A.F., and R.H. collected field samples. A.M.M., A.F., and R.H. analyzed the data and wrote manuscript. All authors read and edited the manuscript.
Supporting information
ACKNOWLEDGMENTS
We thanks John Thurlow and Alan Lambert (Mareeba Shire Council), Kerry Walsh (Department of Environment and Heritage Protection), Annabelle Olsson and Tim Nevard (Mareeba Tropical Savanna and Wetland Reserve Wildlife Conservancy of Tropical Queensland), Ernest Madua (Traditional Owner and Ranger Napranum), Les Harrigan and Conrad Yeatman (Land Trust Traditional Owners of Rinyirru), Gil Hainey (Northern Peninsula Area Regional Council), Louise Stone (Mapoon Aboriginal Shire Council), Uncle Shorty (Traditional Owner, Far North Cape Area), Peta Standley (Cape York Natural Resource Management), Paul Ryan (Paul Ryan Global), Warren Strevens (Northern Peninsula Area Regional Council/Injinoo Ranger Base), Janie White (Shire of Cook QPWS Office), Tony Cockburn and Matt Wallace (Rinyirru National Park), Ray Byers, Lisa Hamilton, Tiparat Tikapunya, Xavier Tierney, Andrew Geering for assistance with sample and data collection. We also thank the HCED Iraq program for providing a PhD scholarship to Ali Mohammad Moner.
Moner AM, Furtado A, Chivers I, Fox G, Crayn D, Henry RJ. Diversity and evolution of rice progenitors in Australia. Ecol Evol. 2018;8:4360–4366. https://doi.org/10.1002/ece3.3989
REFERENCES
- Brozynska, M. , Copetti, D. , Furtado, A. , Wing, R. A. , Crayn, D. , Fox, G. , … Henry, R. J. (2017). Sequencing of Australian wild rice genomes reveals ancestral relationships with domesticated rice. Plant Biotechnology Journal, 15, 765–774. https://doi.org/10.1111/pbi.12674 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Brozynska, M. , Furtado, A. , & Henry, R. J. (2015). Genomics of crop wild relatives: Expanding the gene pool for crop improvement. Plant Biotechnology Journal, 14, 1070–1085. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Brozynska, M. , Omar, E. S. , Furtado, A. , Crayn, D. , Simon, B. , Ishikawa, R. , & Henry, R. J. (2014). Chloroplast genome of novel rice germplasm identified in northern Australia. Tropical Plant Biology, 7, 111–120. https://doi.org/10.1007/s12042-014-9142-8 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Carbonell‐Caballero, J. , Alonso, R. , Ibañez, V. , Terol, J. , Talon, M. , & Dopazo, J. (2015). A phylogenetic analysis of 34 chloroplast genomes elucidates the relationships between wild and domestic species within the genus Citrus . Molecular Biology and Evolution, 32, 2015–2035. https://doi.org/10.1093/molbev/msv082 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Choi, J. Y. , Platts, A. E. , Fuller, D. Q. , Wing, R. A. , & Purugganan, M. D. (2017). The rice paradox: Multiple origins but single domestication in Asian rice. Molecular Biology and Evolution, 34, 969–979. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Civáň, P. , Craig, H. , Cox, C. J. , & Brown, T. A. (2015). Three geographically separate domestications of Asian rice. Nature Plants, 1, 15164. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Furtado, A. (2014). DNA extraction from vegetative tissue for next‐generation sequencing. Methods in Molecular Biology, 1099, 1–5. [DOI] [PubMed] [Google Scholar]
- Guindon, S. , & Gascuel, O. (2003). A simple, fast, and accurate algorithm to estimate large phylogenies by maximum likelihood. Systematic Biology, 52, 696–704. https://doi.org/10.1080/10635150390235520 [DOI] [PubMed] [Google Scholar]
- Henry, R. J. (2016). Genomics strategies for germplasm characterization and the development of climate resilient crops In Kjmar S. (Ed.), Crop breeding: Bioinformatics and preparing for climate change (pp. 3–10). Waretown, NJ: Apple Academic Press; CRC Press; https://doi.org/10.1201/9781315365084 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Henry, R. J. , Rice, N. , Waters, D. L. E. , Kasem, S. , Ishikawa, R. , Hao, Y. , … Vaughan, D. (2010). Australian Oryza: Utility and conservation. Rice, 3, 235–241. https://doi.org/10.1007/s12284-009-9034-y [Google Scholar]
- Huang, X. , Kurata, N. , Wei, X. , Wang, Z. X. , Wang, A. , Zhao, Q. , … Han, B. (2012). A map of rice genome variation reveals the origin of cultivated rice. Nature, 490, 497–501. https://doi.org/10.1038/nature11532 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Huelsenbeck, J. P. , & Ronquist, F. (2001). MRBAYES: Bayesian inference of phylogenetic trees. Bioinformatics, 17, 754–755. https://doi.org/10.1093/bioinformatics/17.8.754 [DOI] [PubMed] [Google Scholar]
- Katoh, K. , Misawa, K. , Kuma, K. , & Miyata, T. (2002). MAFFT: A novel method for rapid multiple sequence alignment based on fast Fourier transform. Nucleic Acids Research, 30, 3059–3066. https://doi.org/10.1093/nar/gkf436 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kim, K. , Lee, S. C. , Lee, J. , Yu, Y. , Yang, K. , Choi, B. S. , … Yang, T. J. (2015). Complete chloroplast and ribosomal sequences for 30 accessions elucidate evolution of Oryza AA genome species. Scientific Reports, 5, 15655 https://doi.org/10.1038/srep15655 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Mickelbart, M. V. , Hasegawa, P. M. , & Bailey‐Serres, J. (2015). Genetic mechanisms of abiotic stress tolerance that translate to crop yield stability. Nature Reviews Genetics, 16, 237–251. https://doi.org/10.1038/nrg3901 [DOI] [PubMed] [Google Scholar]
- Rozas, J. , Sánchez‐DelBarrio, J. C. , Messeguer, X. , & Rozas, R. (2003). DnaSP, DNA polymorphism analyses by the coalescent and other methods. Bioinformatics, 19, 2496–2497. https://doi.org/10.1093/bioinformatics/btg359 [DOI] [PubMed] [Google Scholar]
- Sotowa, M. , Ootsuka, K. , Kobayashi, Y. , Hao, Y. , Tanaka, K. , Ichitani, K. , … Ishikawa, R. (2013). Molecular relationships between Australian annual wild rice, Oryza meridionalis, and two related perennial forms. Rice, 6, 26 https://doi.org/10.1186/1939-8433-6-26 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Swofford, D. L . (2002). PAUP*. Phylogenetic analysis using parsimony (*and other methods). Version 4. Sunderland, MA: Sinauer Associates. [Google Scholar]
- Wambugu, P. W. , Brozynska, M. , Furtado, A. , Waters, D. L. , & Henry, R. J. (2015). Relationships of wild and domesticated rices (Oryza AA genome species) based upon whole chloroplast genome sequences. Scientific Reports, 5, 13957 https://doi.org/10.1038/srep13957 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Waters, D. L. , Nock, C. J. , Ishikawa, R. , Rice, N. , & Henry, R. J. (2012). Chloroplast genome sequence confirms distinctness of Australian and Asian wild rice. Ecology and Evolution, 2, 211–217. https://doi.org/10.1002/ece3.66 [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials