

Abstract
We investigated the effects of granulocyte-macrophage colony stimulating factor (GM-CSF) on behavioral and pathological outcomes in Alzheimer’s disease (AD) and non-transgenic mice. GM-CSF treatment in AD mice reduced brain amyloidosis, increased plasma Aβ, and rescued cognitive impairment with increased expression of calbindin and synaptophysin in the hippocampus and increased levels of doublecortin-positive cells in the dentate gyrus. These data extend pleiotropic neuroprotection mechanisms for GM-CSF in AD and include regulatory T cell-mediated immunomodulation of microglial function, Aβ clearance, stabilizing synaptic integrity, and induction of neurogenesis. Together these data support further development of GM-CSF as a neuroprotective agent for AD.
Keywords: APP/PS1 mice, cognitive function, Tregs, calbindin, synaptophysin, doublecortin, Aβ plaques, hippocampus, amyloidosis
Graphical Abstract
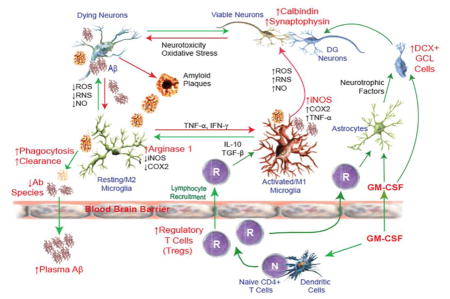
1. Introduction
Innate and adaptive immunity are operative during progression of neurodegenerative disorders that include Alzheimer’s disease (AD) and Parkinson’s disease (PD), multiple sclerosis (MS), amyotrophic lateral sclerosis (ALS) and stroke (Achim et al., 2009, Anderson et al., 2014, Olson and Gendelman, 2016). These disorders show an inflammatory component linked to neural impairment or repair of the central nervous system (CNS). Immune competent microglia, macrophages, and dendritic cells produce factors that affect the brain’s microenvironment with adaptive immunity, in part, also serving as a disease driver (Gendelman and Mosley, 2015, Huang et al., 2009). As such, misfolded proteins represented by α-synuclein neural inclusions in PD and amyloid-β (Aβ) accumulation in AD can alter immunologic tolerance and affect microglial activation leading to robust production of pro-inflammatory neurotoxins, oxidative stress, and excitotoxicity. All are linked to a spectrum of disorders beyond neurodegenerative disease that include nutritional deficiencies, drugs of abuse, neuroinfections, and depression (Calcia et al., 2016, DiSabato et al., 2016, Hurley and Tizabi, 2013, Maes et al., 2012, Mahgoub and Alexopoulos, 2016, Minihane et al., 2015). On the one hand, disease-induced factors can be also harnessed for diagnostic and therapeutic gain in improving neural function. Hence researches from our laboratory and others have aimed immune modulating strategies targeting immune-based activities as common outcomes of neurodegenerative disorders (Kosloski et al., 2010, Olson and Gendelman, 2016, Puligujja et al., 2015). While effector T cells (Teffs) are neurodestructive and exacerbate immune activation and perpetuate neurodegenerative disease progression, regulatory T cells (Tregs) orchestrate self-tolerance, modulate microglial activation and support neuronal protection (Banerjee et al., 2008, Reynolds et al., 2007a, Reynolds et al., 2007b, Sakaguchi, 2000, Saresella et al., 2011, Takahashi et al., 2000). Indeed, the balance between Treg- and Teff-mediated immunomodulation can affect the tempo and progression of diseases. Thus induction of Tregs by controlling the balance between CD4+ T cell subsets is recognized as a potential therapeutic approach for neurodegenerative diseases. For instance, Tregs mediate neuroprotection by reduction in human immunodeficiency virus type one (HIV-1) release, killing virus-infected macrophages, and transforming virus-infected macrophages from an M1 to an M2 phenotype (Huang et al., 2010).
Granulocyte macrophage-colony stimulating factor (GM-CSF) has been used as an adjunctive therapy of chemotherapy-induced granulocytopenia (Buchsel et al., 2002). The 23-kDa glycoprotein functions as a growth factor capable of generating granulocytes and macrophages from myeloid progenitor cells, but does not appear to be indispensable for those functions and also serves to regulate immune responses in a pleiotropic fashion (Becher et al., 2016). It stimulates mobilization of hematopoietic progenitor cells, and thus induces differentiation of granulocyte, macrophage, and eosinophil precursor cells as well as Tregs in the peripheral circulation (Rowin et al., 2012, Ruef and Coleman, 1990, Sheng et al., 2011). In the CNS, GM-CSF is produced by resident astroglia and T cells, and can also cross the blood–brain barrier (BBB) where it engages receptors on both immune and neural cells (McLay et al., 1997). GM-CSF and its receptor, GM-CSFRα, are expressed by neurons. GM-CSF signaling plays a major role in structural plasticity relevant to learning and memory (Krieger et al., 2012, Schabitz et al., 2008), controls cell death following spinal cord injury (Ha et al., 2005), and protects neurons in traumatic brain injury (TBI) mice (Shultz et al., 2014). Recently we showed that therapeutic immune transformation by GM-CSF induces neuroprotection in models of PD and TBI (Kelso et al., 2015, Kosloski et al., 2013). These findings led to our recent proof-of-concept phase I clinical trial, which demonstrated benefits in improving motor skill sets in PD patients (Gendelman et al., 2017). Moreover, GM-CSF immune transformation is associated with neuroprotection and improved motor function in animals and humans (Gendelman HE et al., 2017, PMID: 28649610; Kosloski LM et al., 2013, PMID: 24210793, Bergamini A et al., 2000, PMID: 11012779). These works have now been extended to investigate how GM-CSF affects AD pathogenesis. Previous studies investigated how mixed pro- and anti-inflammatory cytokine profiles induced by macrophage-, granulocyte- and granulocyte macrophage- colony stimulating factors (M-CSF, G-CSF and GM-CSF) affect amyloidosis and behavioral impairments in AD mice. In these studies associations between neurogenesis, microglial activation, cytokine production and neuroprotective responses were identified. These were seen as the mechanisms underlying the positive therapeutic benefits of CSFs for both neuroinflammatory and neurodegenerative disorders (Sanchez-Ramos J et al., 2009, PMID: 19500657; Boyd TD et al., 2010, PMID: 20555144; Doi Y et al., 2014, PMID: 25062013). Based upon these prior observations we now posit an additional mechanisms for GM-CSF immune transformation that include both innate microglial and adaptive immune responses that affect Aβ clearance in AD. The work serves to extend GM-CSF beneficial effects in rheumatoid arthritis (RA) by improving disease outcomes by control of inflammation (Avci et al., 2016). Moreover, our current observations support inverse associations between RA and AD where non-steroidal anti-inflammatory drug (NSAID) use in RA attenuates brain injury. While NSAIDs fail to ameliorate AD-associated memory impairments, GM-CSF-enhanced memory supports its therapeutic benefits in aged, cognitively-impaired AD models (Moss and Hamilton, 2000).
One way that GM-CSF facilitates improvements in brain function is by recruiting monocyte-macrophages from the peripheral blood into the brain and affects clearance of Aβ plaques (Butovsky et al., 2007, Darlington et al., 2015, El Khoury et al., 2007, Fu et al., 2016, Hohsfield and Humpel, 2015, Malm et al., 2005, Savchenko et al., 2016, Simard et al., 2006, Zuroff et al., 2017). Additionally, increases in neural cell connections in brains of GM-CSF-treated subjects may provide insights into the association between memory improvements and control of innate and adaptive immune responses. We posit that GM-CSF transforms immune responses and as such leads to neuroprotection and control of amyloid biology. Altered induction of microglia and Tregs is associated with clinical benefit, and support additional studies as well as on-going clinical trials (NCT01409915).
2. Materials and methods
2.1. Transgenic mice and GM-CSF administration
Transgenic (Tg) mice that overexpress the Swedish mutation of human amyloid precursor protein (APP) (designated as the Tg2576 strain) were obtained from Drs. G. Carlson and K. Hsiao-Ashe through Mayo Medical Venture (Hsiao et al., 1996). Presenilin 1 (PS1) mice overexpressing the mutant protein of human PS1 (M146L line 6.2) were provided by Dr. K. Duff through the University of South Florida (Duff et al., 1996). Both Tg strains were maintained in a B6/129 hybrid background (Kiyota et al., 2009). Non-Tg B6/129 and APP/PS1 double-Tg mice were developed in parallel as described previously (Kiyota et al., 2013, Kiyota et al., 2011, Kiyota et al., 2010); the latter are hereafter referred to as AD mice. Recombinant murine GM-CSF (Peprotech, Rocky Hill, NJ) was reconstituted in sterile Dulbecco’s PBS (DPBS). For these studies, female non-Tg and AD mice were administered GM-CSF at 50 μg/kg body weight, i.p. (Kelso, Elliott, 2015, Kosloski, Kosmacek, 2013) once daily for 10 consecutive days at 4 months of age, followed by 2 rounds of 5 daily injections given at 3-week intervals. These doses were previously considered safe and efficacious (Sanchez-Ramos J et al., 2009, PMID: 19500657 (for G-CSF); Boyd TD et al., 2010, PMID: 20555144; Kosloski LM et al., 2013, PMID: 24210793). In the current studies, non-Tg and AD mice treated with DPBS served as vehicle controls. All animal studies adhered to the guidelines established by the Institutional Animal Care and Use Committee at University of Nebraska Medical Center.
2.2. Flow cytometric analyses
Flow cytometric analyses was performed on peripheral blood samples taken by submandibular vein puncture with a 5 mm animal lancet (MEDIpoint INC, Mineola, NY, USA) and 100 μl of blood collected into K3EDTA collection tubes (Greiner BioOne North America, Monroe, NC, USA, cat. 450475). Each 50 μl sample of whole blood was diluted with 50 μl FACS staining buffer (FSB) and monoclonal antibodies (Abs) added (all from eBioscience/Thermo Fisher Scientific, Waltham, MA, USA) that included 400 μg of PerCP-Cyanine5.5-conjugated anti-CD3ε (clone 145-2C11, cat. 45-0031-80), 500 μg of PE-Cyanine7-anti-CD4 (clone RM4-5, cat. 25-0042-81), and 500 μg of PE-anti-CD25 (clone PC61.5, cat. 12-0251-82). Samples were incubated for 30 min at room temperature, centrifuged at 400xg for 5 min at 4°C, and resuspended in 2 ml FSB containing 0.1% BSA. The samples were fixed, permeabilized, and stained with APC-anti-mouse/rat Foxp3 staining set (clone FJK-16s, cat. 77-5775) according to the manufacturer’s protocol. After labeling, cell suspensions were washed by centrifugation, fixed, and analyzed using BD LSR II Flow Cytometer (BD Biosciences, San Jose, CA, USA) at the University of Nebraska Medical Center Flow Cytometer Research Facility by personnel not associated with the experimental protocol. Data analysis was performed with FACSDiva software (BD Biosciences, San Jose, CA, USA) by an investigator that was blinded to the experimental groups. Lymphocytes were electronically bit mapped and CD3+CD4+ T cells were gated to determine frequencies of CD25hiFoxP3+ Tregs within the CD4+ T cell population.
2.3. Radial arm water maze test
The radial arm water maze (RAWM) task was performed in a blinded fashion at 7 months of age as previously described with minor modifications (Kiyota, Gendelman, 2013). Animals from masked cages were introduced into the perimeter of a circular water-filled tank 110 cm in diameter and 91 cm in height (San Diego Instruments, San Diego, CA) with triangular inserts placed in the tank to produce six swim paths radiating from a central area. Spatial cues for mouse orientation were present on the tank walls. At the end of one arm, a 10 cm circular Plexiglas platform was submerged 1 cm deep and as such hidden from the mice. The platform was located in the same arm for four consecutive acquisition trials (T1 through T4), and one 30 min delayed retention trial (T5), but in a different arm on different days. For T1–T4, the mouse started the task from a different randomly chosen arm, excluding the arm with the platform. After four trials, the mouse was returned to its cage for 30 min, and then administered the retention trial (T5) starting from the same arm as in T4. Each trial lasted 1 min, and an error was scored each time the mouse entered the wrong arm; entered the arm with the platform, but did not climb on it; or did not make a choice for 20 sec. The trial ended when the mouse climbed onto and remained on the hidden platform for 10 sec. The mouse was given 20 sec to rest on the platform between trials. The time taken by the mouse to reach the platform was recorded as trial latency. If the mouse did not reach the platform, 60 sec was recorded as latency, and the mouse was gently guided to the submerged platform. The errors over 6-day test were divided into two blocks, and the errors in each block consisting of 3-day test were averaged for statistical analysis.
2.4. Brain tissue preparation for immunohistochemistry
Mice were deeply euthanized with isoflurane and peripheral blood collected by cardiac puncture and transferred into K3EDTA collection tubes (Greiner BioOne North America, Monroe, NC, USA, cat. 450475). After collection, mice were transcardially perfused with 25 ml of ice-cold PBS and brains rapidly removed. The left hemisphere was dissected and immediately frozen in dry ice for biochemical testing. The right hemisphere was immersed in freshly depolymerized 4% paraformaldehyde for 48hr at 4°C, and protected by successive 24hr immersions in 15% and 30% sucrose in 1×PBS. Fixed, cryopreserved brains were sectioned coronally using a Cryostat (Leica, Bannockburn, IL, USA) with sections serially collected and stored at −80°C for immunohistochemical tests. Immunohistochemistry was performed using specific Abs to identify pan-Aβ (1:100, rabbit polyclonal, Thermo Fisher Scientific, Waltham, MA, USA, cat. PA5-20737), Iba1 (1:1000, rabbit polyclonal, Wako, Richmond, VA, USA, cat. 019-19741), and doublecortin (Dcx) (1:500, goat polyclonal, Santa Cruz Biotechnology, Santa Cruz, CA, USA, cat. sc-8067). Immunodetection was visualized using biotin-conjugated anti-rabbit or anti-goat IgG was used as a secondary Ab, followed by a tertiary incubation with Vectastain ABC Elite kit (Vector Laboratories, Burlingame, CA, USA, cat. PK-6100). One percent thioflavin S (Sigma, St. Louis, MO, USA) in 50% EtOH was used for counterstaining of compact plaque For quantification analysis, the slides were masked and coded, and areas of Aβ load and morphology of Iba1-positive microglia were analyzed by investigators blinded to strain and treatment using ImageJ software (NIH, Bethesda, MD, USA) at 300 μm intervals in ten 30 μm coronal sections from each mouse. Seven mouse brains per group were analyzed.
Immunofluorescence was performed in brain sections with anti-calbindin D-28k (1:20,000, rabbit polyclonal, Swant, Bellinzona, Switzerland, cat. CB38) and anti-synaptophysin (1:1000, clone SY38, EMD Millipore, Billerica, MA, USA, cat. MAB5258-I) Abs, followed by incubation with Alexa Fluor 568-conjugated anti-rabbit IgG (H+L) (1:1,000) and Alexa Fluor 488-conjugated anti-mouse IgG (H+L) (1:1,000) secondary Abs (Kiyota et al., 2015). Sections were mounted with Vectashield-DAPI (Vector Laboratories, Burlingame, CA, USA). Images in the dentate gyrus (DG) of the hippocampus were captured using a Nuance EX multispectral imaging system (Cambridge Research & Instruments, Woburn, MA, USA). Calbindin/DAPI or synaptophysin/DAPI images were pre-processed with standard outputs to a spectral library using Nuance software. Data were quantified with ImageJ software (NIH, Bethesda, MD, USA) by separating color channels and converting to grayscale to obtain red (calbindin)/green (synaptophysin) and blue (DAPI) staining intensities in the DG. Calbindin/synaptophysin positive areas were normalized to DAPI intensity against the non-Tg-PBS group to ensure standardization of differences amongst experimental conditions. Eight brains per group were analyzed.
2.5. Stereological quantification
Stereological quantification for Dcx in the hippocampus was performed as described (Kiyota, Morrison, 2015). In brief, immune-positive cells were counted in a blinded fashion in every 8th section through the entire anterio–posterior extent of the DG (total 12 sections per hippocampus) and estimated using stereological analysis with Stereo Investigator system with an optical fractionator module (MBF Bioscience, Williston, VT). The system consisted of a high sensitivity digital camera (OrcaFlash2.8, Hamamatsu C11440-10C, Hamamatsu, Japan) interfaced with a Nikon Eclipse 90i microscope (Nikon, Melville, NY, USA). Within the Stereo Investigator program, the contour of DG of each section was delineated using a tracing function. While sections showed shrinkage along the anterio–posterior axis, the extent of shrinkage between different animals was similar. The dimensions for the counting frame (450 × 450 um) and the grid size (500 × 500 um) were set. The z-plane focus was adjusted at each section for clarity, and images were automatically acquired according to each setting. The data file containing all slice pictures of the DG was quantified using the fractionator application and Stereo Investigator computed the estimated cell population. Total cell counts and the Gunderson (m=1) values were recorded for each animal and compared between groups using a statistical software (Prism 4.0, GraphPad Software, San Diego, CA).
2.6. Immunoprecipitation and Western blot
Protein extraction and immunoblot analysis for Aβ oligomers were performed as described previously (Kiyota, Gendelman, 2013). Briefly, 100 μg of protein was incubated with unconjugated pan-Aβ monoclonal Ab (6E10, 2 μg/ml; Biolegend, San Diego, CA, USA, cat. 803001) in radioimmunoprecipitation assay buffer (RIPA) buffer (25mM Tris-HCl (pH 7.6), 150mM NaCl, 1% NP-40, 1% sodium deoxycholate, 0.1% SDS, 1×protease inhibitor cocktail; ThermoFisher Scientific, Rockford, IL, USA, cat. 89901) at 4°C for 1hr, followed by incubation with 40 μl Protein A/G Plus agarose (Santa Cruz Biotechnology, Dallas, TX, USA, cat. sc2003) at 4°C overnight. Precipitates were collected by centrifugation at 3000 rpm, 4°C for 5 min, reconstituted with sample buffer, incubated at 95°C for 5 min, and then analyzed by electrophoresis on 16% sodium dodecyl sulfate (SDS)-polyacrylamide Tris-Tricine gel. Proteins were transferred on 0.2 μm pore size polyvinylidine fluoride membrane (Immobilon-P, Millipore, Billerica, MA, USA, cat IPVH00010). Membranes were blocked in 3% BSA/Tris-buffered saline Tween 20 (TBST), and incubated with biotinylated 6E10 monoclonal Ab (1:1000; Biolegend, San Diego, CA, USA, cat. 803001), followed by incubation with HRP-conjugated streptavidin (ThermoFisher Scientific, Rockford, IL, USA, cat. N100). For other Western blot analysis tissue proteins were incubated with β-mercaptoethanol at 100°C for 5 min, followed by electrophoresis on SDS-polyacrylamide Tris-glycine gel and transferred to polyvinylidine fluoride membrane. Membranes were blocked in 5% skim milk/TBST and incubated with primary Abs to low density lipoprotein receptor-related protein 1 (LRP1) (1:500, mouse monoclonal, Millipore Sigma, MA, USA, cat. 438192), receptor for advanced glycation end products (RAGE) (1:1000, rabbit polyclonal, Abcam, Cambridge, MA, USA, cat. ab37647), arginase 1 (1:300, rabbit monoclonal, Cell Signaling Technology, Denver, MA, USA, cat. 93668S), nitric oxide synthase-2 (NOS-2) (1:300, rabbit monoclonal, Cell Signaling Technology, Denver, MA, USA, cat. 13120S), and β-actin (1:2000, Sigma, St. Louis, MO, USA, cat. A3854), at 4°C overnight, followed by 60 min incubation with HRP conjugated anti-rabbit or mouse secondary Abs (1:2000, Santa Cruz Biotechnology, Santa Cruz, CA, USA). Immunoreactive bands were detected using SuperSignal West Pico or Femto Chemiluminescent substrate and images were captured using a myECL Imager (Thermo Fisher Scientific, Waltham, MA, USA). Immunoblots were quantified using ImageJ software (NIH, Bethesda, MD, USA) relative to β-actin expression.
2.7. Enzyme-Linked Immunosorbent Assay (ELISA)
Concentrations of Aβ40 and Aβ42 in plasma were quantified using human Aβ40 and Aβ42 ELISA kits (cat. KHB3482 and KHB3442, respectively) according to the manufacturer’s protocols (Thermo Fisher Scientific, Waltham, MA, USA). The ELISA for Aβ40 does not cross react with Aβ42, and the Aβ42 ELISA does not cross react with Aβ40. Moreover, no cross-reactivity is detected with the following peptides or proteins: Aβ [1–12], Aβ [1–20], Aβ [12–28], Aβ [22–35], Aβ [1–43], Aβ [42–1], α-synuclein, APP and tau. Cross reactivity with rodent Aβ [1–40] is limited to 0.5% by the manufacturer’s specifications or a maximum of 0.024 ng/ml plasma in these studies.
2.8. Statistics
All data are presented as means ± standard errors of the mean (SEM). Comparisons of means for two groups were analyzed by Student’s t-test and multiple mean comparisons were analyzed by one-way ANOVA with appropriate post-hoc tests or two-way repeated measures ANOVA followed by appropriate multiple comparison tests (Prism 4.0, Graphpad Software, San Diego, CA). A value of p ≤ 0.05 was regarded as a significant difference.
3. Results
3.1. Treg induction
We chose to investigate a potential therapeutic effect of GM-CSF for AD based on the cytokines known neuroprotective effects in TBI and PD (Kelso, Elliott, 2015, Kosloski, Kosmacek, 2013). In the first step to address a role for GM-CSF in AD, we evaluated changes in Treg numbers in AD mice. Both non-Tg and AD mice received daily i.p. injections of either PBS or GM-CSF in PBS (50 μg/kg/day) for 10 days at 4 months of age. Flow cytometry analyses of peripheral blood samples to detect CD3+CD4+CD25hiFoxP3+ Tregs (Fig. 1A) were performed before (day 0), and 5 and 11 days after the first GM-CSF administration. In untreated AD mice, frequencies of Tregs were significantly diminished compared to non-Tg controls (Fig. 1B). GM-CSF treatment for 10 days increased Treg frequencies, but not at 5 days post-treatment compared to PBS-treated controls in non-Tg (Fig. 1C) and AD (Fig. 1D) mice, thus demonstrating that GM-CSF boosts Treg populations in AD as well as non-Tg mice.
Fig. 1.

GM-CSF increases Treg frequencies in non-Tg and AD mice. Peripheral blood samples were obtained at 4 months of age and stained for expression of CD3ε, CD4, and CD25, permeabilized, and then stained for intracellular FoxP3. Frequencies of Tregs were determined by flow cytometric analysis. (A) Lymphocytes were gated by electronic bit maps defined by forward-angle light scatter (FALS) and side-scatter (SSC) and by expression of CD3 and CD4. Percentages of CD25hiFoxP3+ T cells were determined within the CD3+CD4+ population. Samples were obtained from non-Tg and AD mice that were untreated (B) or treated with PBS or GM-CSF for 10 days (Day 0–10) (C, D). Mean percentages of Tregs (± SEM) were calculated for (B) 19 or 23 naïve non-Tg or AD mice, respectively; (C) n = 9–10 treated non-Tg mice per group; and (D) n = 11–12 treated AD mice per group. Significant differences were assessed by Student’s t-test (B) or Kruskal-Wallis non-parametric ANOVA and post-hoc multiple comparisons of mean ranks (C, D).
3.2. Assay of learning and memory
AD mice show impaired hippocampal function, memory acquisition and retention at 6–7 months of age (Diamond et al., 1999, Jensen et al., 2005, Kiyota, Ingraham, 2011). As Tregs were recognized as neuroprotective in mouse models of human neurodegenerative and neuroinflammatory diseases (Gendelman and Mosley, 2015, Mosley et al., 2012) and GM-CSF induces Treg we assessed whether learning and memory function would be affected by cytokine administration. In these experiments, GM-CSF- or PBS-treated AD mice were subjected to the RAWM task. Non-Tg mice served as controls of the task. Two 3-day blocks for trial 1 (T1; randomized initial trial), T2 and T3 (memory acquisition trials), T4 (final acquisition trial), and T5 (delayed retention trial) were used to evaluate the memory function at 7 months of age (Fig. 2). All animal groups showed reduced error numbers by T3 and T4 during day 4–6 trials. By T5, while PBS-treated AD mice showed the highest number of errors with significant differences compared to the other groups, GM-CSF-treated AD mice showed lower number of errors similar to non-Tg controls. The data supports the idea that impaired memory acquisition and retention are operative, but GM-CSF administration significantly improves memory function in AD mice.
Fig. 2.
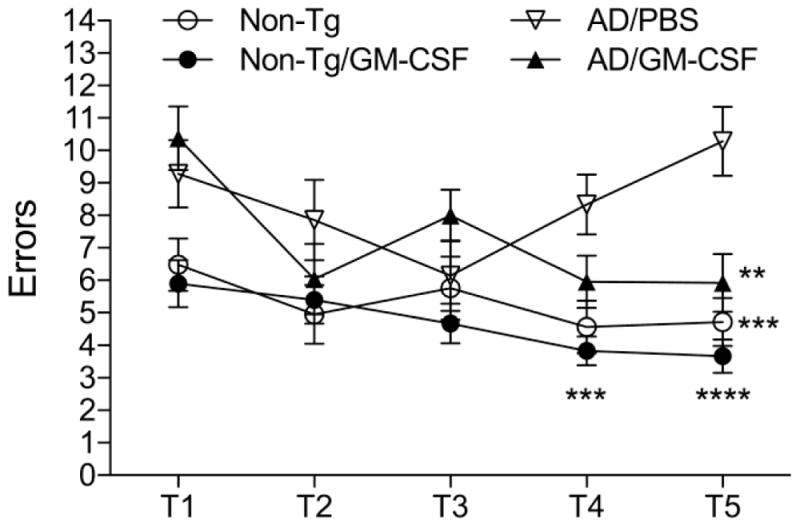
GM-CSF administration improves memory retention in AD mice. Non-Tg mice treated with PBS (Non-Tg/PBS) or GM-CSF (Non-Tg/GM-CSF) and AD mice treated with PBS (AD/PBS) or GM-CSF (AD/GM-CSF) were tested by the RAWM task at 7 months of age. Non-Tg mice served as normal behavior controls for the spatial learning task. The compiled mean errors for day 4–6 are shown. Data are presented as mean number of errors ± SEM (n = 8 per group), and **p ≤ 0.01, ***p ≤ 0.001, ****p ≤ 0.0001 compared to AD/PBS treated group as determined by two-way ANOVA and Bonferroni post-hoc test.
3.3. Effects of Aβ deposition and plasma Aβ40
We examined whether GM-CSF-mediated improvement of memory function is associated with β-amyloidosis (Fig. 3A). Unexpectedly, both cortex and hippocampal Aβ load were unchanged between PBS and GM-CSF-treated groups (1.62% and 1.44% of occupancy for Aβ load in cortex, 0.62% and 0.58% in hippocampus, respectively, Fig. 3B–C). Next, we evaluated plasma Aβ levels in PBS and GM-CSF-treated AD mice using Aβ40 or Aβ42 ELISA. Aβ40 level was significantly increased in GM-CSF-treated AD mice as compared to PBS-injected AD mice (Fig. 3D, p = 0.0498), while plasma Aβ42 level was unchanged (Fig. 3E). That neither ELISA assay cross reacts substantially with mouse Aβ species, we ruled out the possibility that increased plasma Aβ40 was of murine origin, however the origin of Aβ40 from other tissues cannot be ruled out. We also evaluated Aβ species of different molecular weight from extracellular-enriched brain homogenates of non-Tg and AD mice by immunoprecipitation and Western blot. Treatment with GM-CSF diminished Aβ monomers (21.7%, p = 0.0215), dimers (17.0%, p=0.0454), pentamers (22.5%, p = 0.0460), hexamers (17.6%, p = 0.0472), and 75 kDa Aβ species (Aβ75) (30.6%, p = 0.0015) (Fig. 4B). Interestingly, GM-CSF treatment diminished sAPPα in homogenates of AD mice by 14% (p = 0.0251). These results point to the ability of GM-CSF to ameliorate different forms of soluble Aβ oligomeric species. Together, these results suggest that GM-CSF administration reduces soluble Aβ oligomers as well as sAPPα and promotes clearance of Aβ40 from the brain to the vascular circulation, which may contribute to the alteration of overall soluble forms of Aβ in the brain.
Fig. 3.

GM-CSF facilitates Aβ40 peripheral influx in AD mice. (A) Representative images of Aβ staining in cortex and hippocampus of PBS- or GM-CSF-treated AD mice at 7 months of age. Scale bar = 200 μm. (B, C) Quantification of total Aβ load in the cortex (B) and the hippocampus (C) (n=7 per group, 12 sections per brain). (D, E) Levels of Aβ40 (D) and Aβ42 (E) in plasma were measured by human Aβ40- or Aβ42-specific ELISA (n=7 per group). Data represent mean plasma concentrations ± SEM, and *p ≤ 0.05 compared to AD/PBS as determined by Student’s t-test.
Fig. 4.

GM-CSF attenuates different Aβ oligomer levels in AD mice. (A) Immunoblot image showing different Aβ oligomer species in non-Tg and AD mice brain homogenates. (B) Densitometric analysis revealed expression of monomer, dimer, pentamer, hexamer, and Aβ75 oligomer species of Aβ as well as sAPPα (n=6 per group). Data are presented as mean intensity of expression ± SEM, and *p ≤ 0.05 and **p ≤ 0.01 compared to AD/PBS as determined by Student’s t-test.
3.4. Aβ transporter modification
The BBB located Aβ transporters control cerebral Aβ levels that ultimately govern AD progression (Do et al., 2016, Herring et al., 2016). To support the ability of GM-CSF to facilitate Aβ clearance across the brain to the vascular circulation, we determined expression in the brain of the Aβ efflux-conducting receptor, LRP1, and the Aβ influx receptor, RAGE (Fig. 5A). LRP1 expression was significantly reduced in vehicle-treated AD mice as compared to non-Tg mice (Fig. 5B, p < 0.05). However, GM-CSF treatment increased LRP1 levels in non-Tg (14.5%) and AD mice (24.4%, p < 0.05) compared to vehicle-treated AD mice (Fig. 5B), supporting the finding of increased Aβ efflux from the brain. RAGE level was significantly increased in vehicle-treated AD mice compared to non-Tg mice (Fig. 5C, p < 0.01) and was significantly ameliorated by GM-CSF treatment (Fig. 5C, p < 0.05). Overall these data support the notion that GM-CSF administration facilitates Aβ clearance across the brain to the vascular circulation as evidenced by ELISA assay.
Fig. 5.

GM-CSF modifies Aβ transporter levels in AD mice brain. (A) The expression of of LRP1 and RAGE in non-Tg and AD animal groups was determined by Western blot analysis. (B, C) Densitometric analysis of LRP1 (B) and RAGE (C) expression in GM-CSF-treated or untreated mice (n = 4 per group). Data are presented as mean intensity of expression ± SEM, and aap ≤ 0.01, bp≤0.05, bbp ≤ 0.01 and cp ≤ 0.05 compared to aanon-Tg/PBS, b,bbnon-Tg/GM-CSF, or cAD/PBS as determined by one-way ANOVA and Newman-Keuls post-hoc test.
3.5. Alteration of microgliosis surrounding Aβ plaques
Aβ peptides and deposition induce reactive microgliosis and subsequent neuroinflammation linked to neuronal injury (Heneka et al., 2015). In contrast, microglia also affect Aβ clearance (Butovsky, Kunis, 2007, Doens and Fernandez, 2014, El Khoury, Toft, 2007, Malm, Koistinaho, 2005, Simard, Soulet, 2006, Zuroff, Daley, 2017); yet experimental diminution of microglia has been shown to yield little effect on Aβ plaques (Grathwohl et al., 2009, Jankowsky et al., 2005, Spangenberg et al., 2016). To determine whether GM-CSF affects microglial accumulation in the AD brain, Iba1-immunoreactive cells surrounding Aβ plaques were assessed. Immunohistochemistry revealed reactive microglia surrounding thioflavin-S-positive Aβ plaques (Fig. 6A). Quantitation of those microglia revealed numbers of Iba1-immunoreactive microglia proximate to Aβ plaques were increased in GM-CSF-treated AD mice compared to PBS control (Fig. 6B, p = 0.0412), suggesting that GM-CSF administration facilitates microglial cell accumulation surrounding Aβ plaques. Further, we quantified total areas containing Iba1-positive microglia in non-Tg and AD mice brains. In the cortex (Fig. 7A), GM-CSF-treated AD mice showed significant increase (p < 0.05) in the area of microglia-positive brain compared to untreated or treated non-Tg and PBS-treated AD mice (Fig. 7C). In the hippocampus (Fig. 7B), similar results were obtained showing greater areas of Iba1-positive microglia in GM-CSF-treated AD mice than control animal groups (Fig. 7D, p < 0.01). These results support the ability of GM-CSF to alter microglial morphology and densities in AD mice.
Fig. 6.
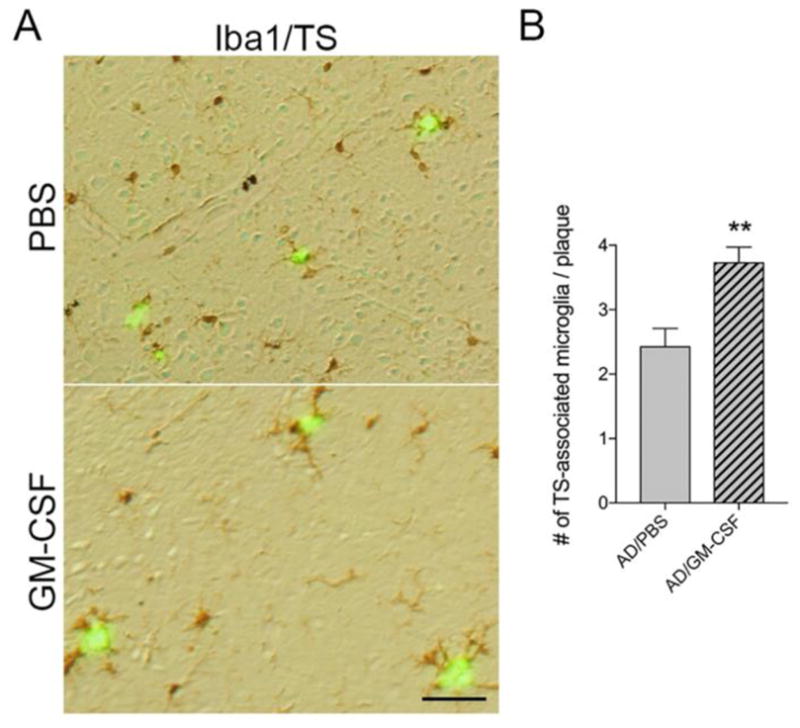
GM-CSF facilitates accumulation of microglia surrounding Aβ plaques in AD mice. (A) Representative images of Iba1 staining in the cortex of PBS- or GM-CSF-treated AD mice. Scale bar = 100μm. (B) Quantification of the mean number of Iba1-positive cells adjacent to thioflavin S (TS)-stained Aβ plaques (n=7 per group, 12 sections per brain, and 3 plaques per section). Data represent mean microglia number ± SEM, and **p < 0.01 compared to AD/PBS as determined by Student’s t-test.
Fig. 7.

GM-CSF treatment increases area occupied by Iba1-positive microglia in AD mice brains. (A, B) Representative images of Iba1 staining in the cortex (A) and the hippocampus (B) of the non-Tg and AD mice brains received PBS or GM-CSF. Scale bar = 200 μm. (C, D) Quantification of area occupied by Iba1-positive cells including cell processes per area (μm2) in cortex (C) and hippocampus (D) (n = 5 per group, 12 sections per brain, 3 areas per section). Data represent mean percentage of Iba1 occupied area/μm2 ± SEM, and ap ≤ 0.05, aap ≤ 0.01, bp ≤ 0.05, bbp ≤ 0.01, cp ≤ 0.05 and ccp ≤ 0.01 compared to a,aanon-Tg/PBS, b,bbnon-Tg/GM-CSF, or c,ccAD/PBS as determined by one-way ANOVA and Newman-Keuls post-hoc test.
3.6. Amelioration of microglial phenotype
As discussed earlier, microglia can play role in neuroinflammation as well as Aβ clearance. These neurodestructive and neurotropic effects are attributed to the ability of microglia to polarize to M1 (classical activation) and M2 (alternative activation) phenotypes, respectively, depending upon the surrounding microenvironment signals (Cherry et al., 2014, 2015). To efficiently combat against disease progression, balanced pro-inflammatory M1 and anti-inflammatory M2 microglia are required (Song and Suk, 2017, Tang and Le, 2016). Western blot analysis was performed to determine the expression of NOS-2 and arginase 1 in the non-Tg and AD mice brains to evaluate microglial M1 and M2 phenotypes, respectively (Fig. 8). NOS-2 expression was significantly increased in the vehicle-treated AD mice as compared to the non-Tg mice (Fig. 8B, p < 0.01). However, GM-CSF administration significantly ameliorated NOS-2 expression to control levels in AD mice (Fig. 8B, p < 0.01). Surprisingly, arginase 1 expression was not significantly changed among all treatment groups (Fig. 8C). Overall, GM-CSF treatment significantly diminished the NOS-2/arginase 1 ratio in AD mice (Fig. 8D, p < 0.01). Together these data suggest that GM-CSF administration attenuates pro-inflammatory microglial phenotype in AD-like conditions.
Fig. 8.

GM-CSF treatment attenuates pro-inflammatory microglial phenotype in AD mice. (A) Immunoblot images of NOS-2 and arginase 1 expression assessed in PBS- or GM-CSF-treated non-Tg and AD mice brain homogenates. (B, C, D) Densitometric analysis of NOS-2 (B) and arginase 1 (C) expression and NOS-2/arginase 1 ratio (D) in non-Tg and AD mice brain (n = 4 per group). Data are presented as mean intensity of expression ± SEM, and aap ≤ 0.01, bbp ≤ 0.01 and ccp ≤ 0.01 compared to aanon-Tg/PBS, bbnon-Tg/GM-CSF, or ccAD/PBS as determined by one-way ANOVA and Newman-Keuls post-hoc test.
3.7. Modulation of synaptic molecules
We next explored calbindin- and synaptophysin-associated mechanisms of GM-CSF-improved memory function. The calcium-binding protein calbindin-D-28k regulates intracellular calcium levels that are essential for hippocampal learning and memory (Molinari et al., 1996). Synaptophysin is an integral membrane glycoprotein of presynaptic vesicles, and correlated to cognitive decline in AD (Masliah et al., 1989, Sze et al., 1997). The expression of synaptophysin is associated with memory acquisition and retention in animal models of human disease (Frick and Fernandez, 2003, Rutten et al., 2005). Therefore, we assessed by immunofluorescence the expression of calbindin and synaptophysin (Fig. 9A–B) in the DG. Measures of fluorescent intensities revealed that levels of calbindin were unchanged in PBS- and GM-CSF-treated non-Tg mice, while levels in PBS-injected AD mice were significantly reduced (Fig. 9C, p < 0.05). Administration of GM-CSF in AD mice returned calbindin expression to that of non-Tg mice. Similarly, synaptophysin expression in non-Tg animals was unchanged regardless of treatment, whereas levels were reduced by 30% in PBS-treated AD mice (Fig. 9D, p < 0.05). Treatment of AD mice with GM-CSF rescued levels of synaptophysin to those of non-Tg mice. Thus, these data support the notion that improved memory function in GM-CSF-treated AD mice is facilitated with improved expression of calbindin and synaptophysin.
Fig. 9.

GM-CSF increases expression of calbindin and synaptophysin in AD mice. (A, B) Expression of calbindin (A) and synaptophysin (B) was determined in the dentate gyrus (DG) of the hippocampus for non-Tg and AD mice treated with PBS or GM-CSF. Scale bar = 200 μm for primary figures and 50 μm for insets (C, D). Quantification of calbindin (C) and synaptophysin (D) expression levels (n = 8 per group, 12 sections per brain). Data represent mean fluorescence intensity ± SEM and p ≤ 0.05 compared to anon-Tg/PBS, bnon-Tg/GM-CSF, or dAD/GM-CSF as determined by one-way ANOVA and Newman-Keuls post-hoc test.
3.8. Restoration of hippocampal neurogenesis
A number of studies correlate memory function and hippocampal neurogenesis in the DG (Bruel-Jungerman et al., 2007, Deng et al., 2010, Kiyota, Morrison, 2015). To uncover potential links between the effects of GM-CSF, memory function, and neurogenesis, we examined the expression of Dcx within the hippocampal DG (Fig. 10A). Dcx is a marker for newly generated premature neurons in the subgranular zone of the DG and serves as a reliable screen for neurogenesis (Rao and Shetty, 2004). The numbers of Dcx-positive (Dcx+) cells in the DG of PBS-treated mice were significantly reduced compared to non-Tg mice treated with PBS or GM-CSF (Fig 10B, p < 0.01). Treatment of AD mice with GM-CSF produced an 87% increase in the number of Dcx+ cells compared to PBS-treated AD mice, but did not reach statistical significance. These data indicate that pre-mature neuronal differentiation is in part protected by GM-CSF.
Fig. 10.

GM-CSF partially rescues hippocampal neurogenesis of AD mice. (A) Immunohistochemical detection of Dcx-labeled cells in the DG of the hippocampi from 7-month-old non-TG and AD mice treated with PBS or GM-CSF. Scale bar = 200 μm. (B) Quantification of numbers of Dcx-labeled cells in the granular cell layer (GCL) of the DG (n = 7 per group, 12 sections per brain). Data represent mean number of Dcx+ cells ± SEM, and p ≤ 0.05 compared to anon-Tg/PBS, bnon-Tg/GM-CSF, or cAD/PBS as determined by one-way ANOVA with Newman-Keuls post-hoc test.
4. Discussion
M-CSF, G-CSF and GM-CSF were previously used in the treatment of schizophrenia, addiction and neurodegenerative disorders with clear pharmacological end-points (Maurer et al., 2008, Kamigaki et al., 2016, Shin and Cho, 2016, Tsai et al., 2017). In AD mice, CSFs reverse amyloidosis, microglial toxicities and behavioral impairments through enhanced neurogenesis and altered cytokine production (Sanchez-Ramos J et al., 2009, PMID: 19500657; Boyd TD et al., 2010, PMID: 20555144; Doi Y et al., 2014, PMID: 25062013). Notably, intrahippocampal injections of GM-CSF was shown to reduce amyloidosis and improve cognitive function, hippocampal synaptic integrity and microglial densities (Boyd et al., 2010). GM-CSF was shown to affect better disease outcomes that G-CSF or M-CSF, Moreover, prior research performed with GM-CSF and used in a various clinical care settings support its safety and abilities to traverse the blood brain barrier (Banks, 2016, Mietelska-Porowska and Wojda, 2017, Muszynski et al., 2017, Zenaro et al., 2017). This coupled with an ongoing phase II clinical trial (NCT01409915) support GM-CSF-based strategies for AD.
The findings cited above coupled with a number of recent studies demonstrate a significant role of innate and adaptive immunity in the pathophysiology of AD (Boutajangout and Wisniewski, 2013, Gendelman and Mosley, 2015, VanItallie, 2017). These work, taken together, support the idea that GM-CSF with significant effects on both arms of the immune system can elicit substantive beneficial roles for the nervous system by the transformation of neurodestructive microglia and Teffs to neuroprotective glia and anti-inflammatory Tregs in neurodegenerative, aging, or inflammatory disease models (Daria et al., 2017, Kelso, Elliott, 2015, Kosloski, Kosmacek, 2013, Thomaty et al., 2017) and in human disease (Gendelman, Zhang, 2017). In support of this notion, a recently completed phase I safety study in PD patients highlighted the translational value of Teff to Treg transformation by GM-CSF and improvements in motor skill sets in treated patients (Gendelman, Zhang, 2017). One mechanism surrounding neuroprotective immunity was linked, in large measure, to the increased number and function of Tregs as demonstrated by flow cytometry, metabolomic, and genomic arrays. Earlier studies showing neuroprotective effects of induced or adoptively transferred Tregs in different neurodegenerative disease models further support our findings that Tregs modulate brain microglial function (Baek et al., 2016, Li et al., 2013, Reynolds, Banerjee, 2007b). Thus recruitment of microglia subsequent to Aβ deposition and aggregation was attributable to production of monocyte chemoattractant protein (MCP)-1and chemoattractant gradients after signaling by TLR and NLR inflammatory receptors after binding by cognate pattern recognition receptors (PRRs), such as pathogen-associated molecular patterns (PAMPs) and damage-associated molecular patterns (DAMPs). Reports also suggest that GM-CSF induces expression of MHC class II and co-stimulatory molecules in microglia, which further facilitate recruitment (Codarri et al., 2011, Ponomarev et al., 2007). Additionally, GM-CSF stimulates microglia to induce proliferation and reactivation of CD4+ T cells (Aloisi et al., 2000, Fischer et al., 1993). Therefore, GM-CSF stimulated microglia can stimulate adoptive immune responses in different neurodegenerative conditions.
Diminished Treg number and function correlate well with AD progression (Duffy et al., 2017, Gendelman and Mosley, 2015, Larbi et al., 2009, Le Page et al., 2017, Saresella et al., 2010). GM-CSF treatment induces Treg frequency as assessed in periphery. Also exogenous GM-CSF can easily transverse the BBB relatively unrestricted given its small molecular weight and an increased BBB permeability in AD (Greenberg et al., 2004, Kinnecom et al., 2007, Takeda et al., 2014). Tregs modulate functional differentiation of mononuclear phagocytes through direct interaction within the brain parenchyma. More T cells infiltrate the brain of AD patients and AD transgenic models than found in control subjects and wildtype mice (Laurent et al., 2017, McManus et al., 2015, Mietelska-Porowska and Wojda, 2017, Togo et al., 2002). Tregs impart beneficial effects through microglia, independent of their Aβ clearing effect. Apart from phagocytic activity, microglia can produce multiple effects, including production of neurotrophic factors and/or inflammatory mediators, regulate neuronal activity by microglia-astrocyte-synapse interactions, and contribute to synaptic remodeling and plasticity (Salter and Beggs, 2014). These effects together contribute to modulate cognitive functions, independent of Aβ phagocytosis. For instance, microglia-derived BDNF can reverse synapse loss, partially normalize aberrant gene expression, improve cell signaling, and restore learning and memory; all independent of amyloid deposition levels (Nagahara et al., 2013, Nagahara et al., 2009). These data support observations found in this study that showed the tandem increase in Tregs, diminished proinflammatory microglia, and rescue of synaptic integrity. Thus, Treg-mediated modulation of microglial function may affect AD-related cognitive deficits without altering amyloid load.
Microglia are known to induce multiple effector functions apart from their phagocytic activity which may contribute to modulation of cognitive functions independent of Aβ phagocytosis. Although, inverse correlations between Aβ deposition and cognitive deficit suggest that the deficits may rather be related to some diffusible or even intracellular Aβ species other than the fibrillar Aβ peptide sequestered into plaques (Duyckaerts et al., 2008). Thus, we cannot rule out that Treg-mediated protective effects are partially associated with phagocytic activity of microglia in the current study wherein GM-CSF induced Treg, significantly reduced soluble Aβ species, increased peripheral Aβ levels, and improved cognitive function. Previous studies also reported altered expression of microglia activation markers such as IL-1 receptor-like 3 (IL-1RL2) and ubiquitin-specific protease 18 (USP18), and reduced recruitment after Treg depletion in APP/PS1 mice, thus supporting the ability of Tregs to modulate microglial function (Boraschi and Tagliabue, 2013, Goldmann et al., 2015).
Underpinning these results are older reports showing that a number of well-known growth factors that affect hematopoiesis and angiogenesis show therapeutic benefit in diverse neurodegenerative disorders. These disorders include, but are not limited to, amyotrophic lateral sclerosis (ALS) (Banerjee, Mosley, 2008, Beers et al., 2017, Henkel et al., 2013), stroke (Liesz et al., 2013), PD (Gendelman and Mosley, 2015, Olson and Gendelman, 2016), and TBI (Kelso, Elliott, 2015).
The beneficial action of each of the neuroprotective factors are linked to neuroplasticity and stem cell growth and differentiation (Baek, Ye, 2016, Duffy, Keating, 2017, Ye et al., 2016). Beyond Teff-Treg transformation a central player in immune responses within the CNS and studied for decades in the setting of neurodegenerative disease and in particular AD is the microglia. The prior paradigm that microglial cells produce only pro-inflammatory neurotoxic responses in response to injury is no longer believed to be correct. The microglial cell rapidly evolves in structure and function, and can produce a cadre of neuroprotective and homeostatic factors in disease. Thus microglia are unique immune cells in the CNS having the ability to polarize into two different phenotypes, M1 (classical activation) and M2 (alternative activation). In early stages of AD, anti-inflammatory M2 microglia predominate surrounding Aβ plaques for phagocytosis and degradation, likely to shift more towards the pro-inflammatory M1 state with disease progression. This altered M1 and M2 microglia balance correlates well with progression of different neurodegenerative and neuroinflammatory conditions (Song and Suk, 2017, Tang and Le, 2016). In regards to cytokines, activated microglia are known to induce a high level expression of GM-CSF mRNA and protein, while M-CSF and IL-34 levels are unchanged (Kamigaki, Hide, 2016). This is associated with phosphorylation of STAT5 and its translocation to the nucleus. Moreover, the JAK2/STAT5 pathway is not simply involved in microglial survival, but secondarily affects the transcription of survival-related genes (Kamigaki, Hide, 2016). Such microglial survival commonly occurs in response to injury and or inflammation and affects more widespread neuroprotective functions as neighboring neurons also survive for extended time periods. Thus, a disease-activated microglia produces endogenous GM-CSF that acts not simply to transform immunity to a neuroprotective signature, but changes the phenotype of the microglial itself to nourish and support neuronal function and integrity (Gendelman and Mosley, 2015, Kosloski, Kosmacek, 2013, Nakagawa et al., 2006).
While vaccines targeting Aβ have shown encouraging results in mouse models of human disease, significant adverse events were shown in humans to be linked to T cell responses (Nicoll et al., 2003, Schenk et al., 2012). This was seen in the first clinical trial AN1792 underlined the need for better appreciation of adaptive immunity in AD pathobiology. Tregs are known to control Aβ-specific CD4+ T cell responses (Baek, Ye, 2016). In support of the current study, early depletion of Tregs has been shown to accelerate cognitive deficits in APP/PS1 mice and without altering Aβ deposition (Toly-Ndour et al., 2011). Early onset cognitive impairment was more strongly correlated to recruitment of microglia to deposits of amyloid. In contrast Treg induction through IL-2 treatment, increased numbers of plaque-associated microglia and improved cognitive functions in APP/PS1 mice (Dansokho et al., 2016). Thus, there is ample evidence that Tregs play a beneficial role in AD by affecting microglial response to Aβ and ensuring the sustenance of a neuroprotective brain microenvironment (Baek, Ye, 2016, Dansokho, Ait Ahmed, 2016).
5. Conclusion
In summary, we report that GM-CSF rescues cognitive decline in AD mice, increases numbers of Aβ plaque-adjacent microglia, and reduces Aβ oligomers with enhanced efflux of Aβ40 from brain to the peripheral circulation. Moreover, we provide evidence for GM-CSF-mediated rescue of several neuropathological manifestations within the hippocampi of AD mice, including the loss of calbindin and synaptophysin expression and diminution of Dcx-expressing cells in the DG. Finally as an immune modulating agent, GM-CSF corrected deficits in peripheral Treg frequencies in AD mice to levels found in non-Tg mice. Together, these data serve to suggest that neuroprotection afforded by GM-CSF functions by pleiotropic mechanisms that involve immune modulation, synaptic plasticity, and neurogenesis to achieve beneficial outcomes in AD. Thus, both innate (microglia) and adoptive (Treg) effectors play significant roles in GM-CSF neuroprotective in AD mice.
Highlights.
Treatment of AD mice with GM-CSF:
Rescued learning and memory function.
Increased calbindin and synaptophysin expression in hippocampus.
Rescued doublecortin-positive cells in the dentate gyrus.
Increased Treg numbers and reduced proinflammatory microglial responses.
Numbers of Iba1-reactive microglia adjoining Aβ plaques increased.
Reduced Aβ oligomers, increased plasma Aβ, and altered Aβ transporters suggested clearance.
Acknowledgments
The authors thank the Flow Cytometry Research Facility at the University of Nebraska Medical Center for assistance with flow cytometric analyses.
Funding
This work was supported in part by NIH Grant AG043540, DA028555, NS036126, NS034239, MH064570, NS043985, MH062261, and DOD Grant 421-20-09A to HEG, the Carol Swarts Emerging Neuroscience Fund, start-up funds from the Department of Pharmacology and Experimental Neuroscience, and the Shoemaker Award for Neurodegenerative Research to TK.
Abbreviations
- AD
Alzheimer’s disease
- APP
amyloid precursor protein
- Aβ
amyloid-beta
- ALS
amyotrophic lateral sclerosis
- Abs
antibodies
- BBB
blood-brain barrier
- CNS
central nervous system
- CSF
colony stimulating factor
- DG
dentate gyrus
- Dcx
doublecortin
- Teffs
effector T cells
- ELISA
enzyme-linked immunosorbent assay
- EPO
erythropoietin
- FSB
FACS staining buffer
- G-CSF
granulocyte colony-stimulating factor
- GM-CSF
granulocyte-macrophage colony-stimulating factor
- GM-CSFR
GM-CSF receptor
- M-CSF
macrophage colony-stimulating factor
- NSAID
non-steroidal anti-inflammatory drug
- PD
Parkinson’s disease
- PS1
presenilin 1
- RA
rheumatoid arthritis
- RAWM
radial arm water maze
- SEM
standard error of the mean
- TBI
traumatic brain injury
- Tg
transgenic
- Tregs
regulatory T cells
Footnotes
Authors’ contributions
TK and JM designed the supervised, designed and analyzed the data. TK and JM wrote the manuscript with HEG and RLM. YL and BD performed the experiments and analyzed the data. MN and IY performed the experiments and assisted in the data analysis. RLM wrote and edited the manuscript and assisted in data analyses and edited the manuscript. HEG supervised the research, developed the experimental design and wrote and edited the manuscript. All authors discussed the results and conclusions, reviewed and commented the manuscript. All listed authors read and approved the final manuscript.
Conflicts of interest
The authors declare no conflicts of interest
Publisher's Disclaimer: This is a PDF file of an unedited manuscript that has been accepted for publication. As a service to our customers we are providing this early version of the manuscript. The manuscript will undergo copyediting, typesetting, and review of the resulting proof before it is published in its final form. Please note that during the production process errors may be discovered which could affect the content, and all legal disclaimers that apply to the journal pertain.
References
- Achim CL, Adame A, Dumaop W, Everall IP, Masliah E Neurobehavioral Research C. Increased accumulation of intraneuronal amyloid beta in HIV-infected patients. J Neuroimmune Pharmacol. 2009;4:190–9. doi: 10.1007/s11481-009-9152-8. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Aloisi F, De Simone R, Columba-Cabezas S, Penna G, Adorini L. Functional maturation of adult mouse resting microglia into an APC is promoted by granulocyte-macrophage colony-stimulating factor and interaction with Th1 cells. J Immunol. 2000;164:1705–12. doi: 10.4049/jimmunol.164.4.1705. [DOI] [PubMed] [Google Scholar]
- Anderson KM, Olson KE, Estes KA, Flanagan K, Gendelman HE, Mosley RL. Dual destructive and protective roles of adaptive immunity in neurodegenerative disorders. Transl Neurodegener. 2014;3:25. doi: 10.1186/2047-9158-3-25. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Avci AB, Feist E, Burmester GR. Targeting GM-CSF in rheumatoid arthritis. Clin Exp Rheumatol. 2016;34:39–44. [PubMed] [Google Scholar]
- Baek H, Ye M, Kang GH, Lee C, Lee G, Choi DB, et al. Neuroprotective effects of CD4+CD25+Foxp3+ regulatory T cells in a 3xTg-AD Alzheimer’s disease model. Oncotarget. 2016;7:69347–57. doi: 10.18632/oncotarget.12469. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Banerjee R, Mosley RL, Reynolds AD, Dhar A, Jackson-Lewis V, Gordon PH, et al. Adaptive immune neuroprotection in G93A-SOD1 amyotrophic lateral sclerosis mice. PLoS One. 2008;3:e2740. doi: 10.1371/journal.pone.0002740. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Banks WA. From blood-brain barrier to blood-brain interface: new opportunities for CNS drug delivery. Nat Rev Drug Discov. 2016;15:275–92. doi: 10.1038/nrd.2015.21. [DOI] [PubMed] [Google Scholar]
- Becher B, Tugues S, Greter M. GM-CSF: From growth factor to central mediator of tissue inflammation. Immunity. 2016;45:963–73. doi: 10.1016/j.immuni.2016.10.026. [DOI] [PubMed] [Google Scholar]
- Beers DR, Zhao W, Wang J, Zhang X, Wen S, Neal D, et al. ALS patients’ regulatory T lymphocytes are dysfunctional, and correlate with disease progression rate and severity. JCI Insight. 2017;2:e89530. doi: 10.1172/jci.insight.89530. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Boraschi D, Tagliabue A. The interleukin-1 receptor family. Semin Immunol. 2013;25:394–407. doi: 10.1016/j.smim.2013.10.023. [DOI] [PubMed] [Google Scholar]
- Boutajangout A, Wisniewski T. The innate immune system in Alzheimer’s disease. Int J Cell Biol. 2013;2013:576383. doi: 10.1155/2013/576383. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Boyd TD, Bennett SP, Mori T, Governatori N, Runfeldt M, Norden M, et al. GM-CSF upregulated in rheumatoid arthritis reverses cognitive impairment and amyloidosis in Alzheimer mice. J Alzheimers Dis. 2010;21:507–18. doi: 10.3233/JAD-2010-091471. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bruel-Jungerman E, Rampon C, Laroche S. Adult hippocampal neurogenesis, synaptic plasticity and memory: facts and hypotheses. Rev Neurosci. 2007;18:93–114. doi: 10.1515/revneuro.2007.18.2.93. [DOI] [PubMed] [Google Scholar]
- Buchsel PC, Forgey A, Grape FB, Hamann SS. Granulocyte macrophage colony-stimulating factor: current practice and novel approaches. Clin J Oncol Nurs. 2002;6:198–205. doi: 10.1188/02.CJON.198-205. [DOI] [PubMed] [Google Scholar]
- Butovsky O, Kunis G, Koronyo-Hamaoui M, Schwartz M. Selective ablation of bone marrow-derived dendritic cells increases amyloid plaques in a mouse Alzheimer’s disease model. Eur J Neurosci. 2007;26:413–6. doi: 10.1111/j.1460-9568.2007.05652.x. [DOI] [PubMed] [Google Scholar]
- Calcia MA, Bonsall DR, Bloomfield PS, Selvaraj S, Barichello T, Howes OD. Stress and neuroinflammation: a systematic review of the effects of stress on microglia and the implications for mental illness. Psychopharmacology (Berl) 2016;233:1637–50. doi: 10.1007/s00213-016-4218-9. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Cherry JD, Olschowka JA, O’Banion MK. Neuroinflammation and M2 microglia: the good, the bad, and the inflamed. J Neuroinflammation. 2014;11:98. doi: 10.1186/1742-2094-11-98. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Cherry JD, Olschowka JA, O’Banion MK. Arginase 1+ microglia reduce Abeta plaque deposition during IL-1beta-dependent neuroinflammation. J Neuroinflammation. 2015;12:203. doi: 10.1186/s12974-015-0411-8. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Codarri L, Gyulveszi G, Tosevski V, Hesske L, Fontana A, Magnenat L, et al. RORgammat drives production of the cytokine GM-CSF in helper T cells, which is essential for the effector phase of autoimmune neuroinflammation. Nat Immunol. 2011;12:560–7. doi: 10.1038/ni.2027. [DOI] [PubMed] [Google Scholar]
- Dansokho C, Ait Ahmed D, Aid S, Toly-Ndour C, Chaigneau T, Calle V, et al. Regulatory T cells delay disease progression in Alzheimer-like pathology. Brain. 2016;139:1237–51. doi: 10.1093/brain/awv408. [DOI] [PubMed] [Google Scholar]
- Daria A, Colombo A, Llovera G, Hampel H, Willem M, Liesz A, et al. Young microglia restore amyloid plaque clearance of aged microglia. EMBO J. 2017;36:583–603. doi: 10.15252/embj.201694591. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Darlington D, Li S, Hou H, Habib A, Tian J, Gao Y, et al. Human umbilical cord blood-derived monocytes improve cognitive deficits and reduce amyloid-beta pathology in PSAPP mice. Cell Transplant. 2015;24:2237–50. doi: 10.3727/096368915X688894. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Deng W, Aimone JB, Gage FH. New neurons and new memories: how does adult hippocampal neurogenesis affect learning and memory? Nat Rev Neurosci. 2010;11:339–50. doi: 10.1038/nrn2822. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Diamond DM, Park CR, Heman KL, Rose GM. Exposing rats to a predator impairs spatial working memory in the radial arm water maze. Hippocampus. 1999;9:542–52. doi: 10.1002/(SICI)1098-1063(1999)9:5<542::AID-HIPO8>3.0.CO;2-N. [DOI] [PubMed] [Google Scholar]
- DiSabato DJ, Quan N, Godbout JP. Neuroinflammation: the devil is in the details. J Neurochem. 2016;139(Suppl 2):136–53. doi: 10.1111/jnc.13607. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Do TM, Dodacki A, Alata W, Calon F, Nicolic S, Scherrmann JM, et al. Age-dependent regulation of the blood-brain barrier influx/efflux equilibrium of amyloid-beta peptide in a mouse model of Alzheimer’s disease (3xTg-AD) J Alzheimers Dis. 2016;49:287–300. doi: 10.3233/JAD-150350. [DOI] [PubMed] [Google Scholar]
- Doens D, Fernandez PL. Microglia receptors and their implications in the response to amyloid beta for Alzheimer’s disease pathogenesis. J Neuroinflammation. 2014;11:48. doi: 10.1186/1742-2094-11-48. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Duff K, Eckman C, Zehr C, Yu X, Prada CM, Perez-tur J, et al. Increased amyloid-beta42(43) in brains of mice expressing mutant presenilin 1. Nature. 1996;383:710–3. doi: 10.1038/383710a0. [DOI] [PubMed] [Google Scholar]
- Duffy SS, Keating BA, Perera CJ, Moalem-Taylor G. The role of regulatory T cells in nervous system pathologies. J Neurosci Res. 2017 doi: 10.1002/jnr.24073. [DOI] [PubMed] [Google Scholar]
- Duyckaerts C, Potier MC, Delatour B. Alzheimer disease models and human neuropathology: similarities and differences. Acta Neuropathol. 2008;115:5–38. doi: 10.1007/s00401-007-0312-8. [DOI] [PMC free article] [PubMed] [Google Scholar]
- El Khoury J, Toft M, Hickman SE, Means TK, Terada K, Geula C, et al. Ccr2 deficiency impairs microglial accumulation and accelerates progression of Alzheimer-like disease. Nat Med. 2007;13:432–8. doi: 10.1038/nm1555. [DOI] [PubMed] [Google Scholar]
- Fischer HG, Nitzgen B, Germann T, Degitz K, Daubener W, Hadding U. Differentiation driven by granulocyte-macrophage colony-stimulating factor endows microglia with interferon-gamma-independent antigen presentation function. J Neuroimmunol. 1993;42:87–95. doi: 10.1016/0165-5728(93)90215-k. [DOI] [PubMed] [Google Scholar]
- Frick KM, Fernandez SM. Enrichment enhances spatial memory and increases synaptophysin levels in aged female mice. Neurobiol Aging. 2003;24:615–26. doi: 10.1016/s0197-4580(02)00138-0. [DOI] [PubMed] [Google Scholar]
- Fu Y, Hsiao JH, Paxinos G, Halliday GM, Kim WS. ABCA7 mediates phagocytic clearance of amyloid-beta in the brain. J Alzheimers Dis. 2016;54:569–84. doi: 10.3233/JAD-160456. [DOI] [PubMed] [Google Scholar]
- Gendelman HE, Mosley RL. A perspective on roles played by innate and adaptive immunity in the pathobiology of neurodegenerative disorders. J Neuroimmune Pharmacol. 2015;10:645–50. doi: 10.1007/s11481-015-9639-4. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gendelman HE, Zhang Y, Santamaria P, Olson KE, Schutt CR, Bhatti D, et al. Evaluation of the safety and immunomodulatory effects of sargramostim in a randomized, double-blind phase 1 clinical Parkinson’s disease trial. npj Parkinson’s Disease. 2017;3:10. doi: 10.1038/s41531-017-0013-5. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Goldmann T, Zeller N, Raasch J, Kierdorf K, Frenzel K, Ketscher L, et al. USP18 lack in microglia causes destructive interferonopathy of the mouse brain. EMBO J. 2015;34:1612–29. doi: 10.15252/embj.201490791. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Grathwohl SA, Kalin RE, Bolmont T, Prokop S, Winkelmann G, Kaeser SA, et al. Formation and maintenance of Alzheimer’s disease beta-amyloid plaques in the absence of microglia. Nat Neurosci. 2009;12:1361–3. doi: 10.1038/nn.2432. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Greenberg SM, Gurol ME, Rosand J, Smith EE. Amyloid angiopathy-related vascular cognitive impairment. Stroke. 2004;35:2616–9. doi: 10.1161/01.STR.0000143224.36527.44. [DOI] [PubMed] [Google Scholar]
- Ha Y, Park HS, Park CW, Yoon SH, Park SR, Hyun DK, et al. Synthes award for resident research on spinal cord and spinal column injury: granulocyte macrophage colony stimulating factor (GM-CSF) prevents apoptosis and improves functional outcome in experimental spinal cord contusion injury. Clin Neurosurg. 2005;52:341–7. [PubMed] [Google Scholar]
- Heneka MT, Carson MJ, El Khoury J, Landreth GE, Brosseron F, Feinstein DL, et al. Neuroinflammation in Alzheimer’s disease. Lancet Neurol. 2015;14:388–405. doi: 10.1016/S1474-4422(15)70016-5. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Henkel JS, Beers DR, Wen S, Rivera AL, Toennis KM, Appel JE, et al. Regulatory T-lymphocytes mediate amyotrophic lateral sclerosis progression and survival. EMBO Mol Med. 2013;5:64–79. doi: 10.1002/emmm.201201544. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Herring A, Munster Y, Akkaya T, Moghaddam S, Deinsberger K, Meyer J, et al. Kallikrein-8 inhibition attenuates Alzheimer’s disease pathology in mice. Alzheimers Dement. 2016;12:1273–87. doi: 10.1016/j.jalz.2016.05.006. [DOI] [PubMed] [Google Scholar]
- Hohsfield LA, Humpel C. Intravenous infusion of monocytes isolated from 2-week-old mice enhances clearance of Beta-amyloid plaques in an Alzheimer mouse model. PLoS One. 2015;10:e0121930. doi: 10.1371/journal.pone.0121930. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hsiao K, Chapman P, Nilsen S, Eckman C, Harigaya Y, Younkin S, et al. Correlative memory deficits, Abeta elevation, and amyloid plaques in transgenic mice. Science. 1996;274:99–102. doi: 10.1126/science.274.5284.99. [DOI] [PubMed] [Google Scholar]
- Huang X, Reynolds AD, Mosley RL, Gendelman HE. CD 4+ T cells in the pathobiology of neurodegenerative disorders. J Neuroimmunol. 2009;211:3–15. doi: 10.1016/j.jneuroim.2009.04.006. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Huang X, Stone DK, Yu F, Zeng Y, Gendelman HE. Functional proteomic analysis for regulatory T cell surveillance of the HIV-1-infected macrophage. J Proteome Res. 2010;9:6759–73. doi: 10.1021/pr1009178. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hurley LL, Tizabi Y. Neuroinflammation, neurodegeneration, and depression. Neurotox Res. 2013;23:131–44. doi: 10.1007/s12640-012-9348-1. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Jankowsky JL, Slunt HH, Gonzales V, Savonenko AV, Wen JC, Jenkins NA, et al. Persistent amyloidosis following suppression of Abeta production in a transgenic model of Alzheimer disease. PLoS Med. 2005;2:e355. doi: 10.1371/journal.pmed.0020355. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Jensen MT, Mottin MD, Cracchiolo JR, Leighty RE, Arendash GW. Lifelong immunization with human beta-amyloid (1-42) protects Alzheimer’s transgenic mice against cognitive impairment throughout aging. Neuroscience. 2005;130:667–84. doi: 10.1016/j.neuroscience.2004.09.055. [DOI] [PubMed] [Google Scholar]
- Kamigaki M, Hide I, Yanase Y, Shiraki H, Harada K, Tanaka Y, et al. The Toll-like receptor 4-activated neuroprotective microglia subpopulation survives via granulocyte macrophage colony-stimulating factor and JAK2/STAT5 signaling. Neurochem Int. 2016;93:82–94. doi: 10.1016/j.neuint.2016.01.003. [DOI] [PubMed] [Google Scholar]
- Kelso ML, Elliott BR, Haverland NA, Mosley RL, Gendelman HE. Granulocyte-macrophage colony stimulating factor exerts protective and immunomodulatory effects in cortical trauma. J Neuroimmunol. 2015;278:162–73. doi: 10.1016/j.jneuroim.2014.11.002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kinnecom C, Lev MH, Wendell L, Smith EE, Rosand J, Frosch MP, et al. Course of cerebral amyloid angiopathy-related inflammation. Neurology. 2007;68:1411–6. doi: 10.1212/01.wnl.0000260066.98681.2e. [DOI] [PubMed] [Google Scholar]
- Kiyota T, Gendelman HE, Weir RA, Higgins EE, Zhang G, Jain M. CCL2 affects beta-amyloidosis and progressive neurocognitive dysfunction in a mouse model of Alzheimer’s disease. Neurobiol Aging. 2013;34:1060–8. doi: 10.1016/j.neurobiolaging.2012.08.009. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kiyota T, Ingraham KL, Jacobsen MT, Xiong H, Ikezu T. FGF2 gene transfer restores hippocampal functions in mouse models of Alzheimer’s disease and has therapeutic implications for neurocognitive disorders. Proc Natl Acad Sci U S A. 2011;108:E1339–48. doi: 10.1073/pnas.1102349108. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kiyota T, Morrison CM, Tu G, Dyavarshetty B, Weir RA, Zhang G, et al. Presenilin-1 familial Alzheimer’s disease mutation alters hippocampal neurogenesis and memory function in CCL2 null mice. Brain Behav Immun. 2015;49:311–21. doi: 10.1016/j.bbi.2015.06.014. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kiyota T, Okuyama S, Swan RJ, Jacobsen MT, Gendelman HE, Ikezu T. CNS expression of anti-inflammatory cytokine interleukin-4 attenuates Alzheimer’s disease-like pathogenesis in APP+PS1 bigenic mice. FASEB J. 2010;24:3093–102. doi: 10.1096/fj.10-155317. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kiyota T, Yamamoto M, Schroder B, Jacobsen MT, Swan RJ, Lambert MP, et al. AAV1/2-mediated CNS gene delivery of dominant-negative CCL2 mutant suppresses gliosis, beta-amyloidosis, and learning impairment of APP/PS1 mice. Mol Ther. 2009;17:803–9. doi: 10.1038/mt.2009.44. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kosloski LM, Ha DM, Hutter JA, Stone DK, Pichler MR, Reynolds AD, et al. Adaptive immune regulation of glial homeostasis as an immunization strategy for neurodegenerative diseases. J Neurochem. 2010;114:1261–76. doi: 10.1111/j.1471-4159.2010.06834.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kosloski LM, Kosmacek EA, Olson KE, Mosley RL, Gendelman HE. GM-CSF induces neuroprotective and anti-inflammatory responses in 1-methyl-4-phenyl-1,2,3,6-tetrahydropyridine intoxicated mice. J Neuroimmunol. 2013;265:1–10. doi: 10.1016/j.jneuroim.2013.10.009. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Krieger M, Both M, Kranig SA, Pitzer C, Klugmann M, Vogt G, et al. The hematopoietic cytokine granulocyte-macrophage colony stimulating factor is important for cognitive functions. Sci Rep. 2012;2:697. doi: 10.1038/srep00697. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Larbi A, Pawelec G, Witkowski JM, Schipper HM, Derhovanessian E, Goldeck D, et al. Dramatic shifts in circulating CD4 but not CD8 T cell subsets in mild Alzheimer’s disease. J Alzheimers Dis. 2009;17:91–103. doi: 10.3233/JAD-2009-1015. [DOI] [PubMed] [Google Scholar]
- Laurent C, Dorothee G, Hunot S, Martin E, Monnet Y, Duchamp M, et al. Hippocampal T cell infiltration promotes neuroinflammation and cognitive decline in a mouse model of tauopathy. Brain. 2017;140:184–200. doi: 10.1093/brain/aww270. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Le Page A, Garneau H, Dupuis G, Frost EH, Larbi A, Witkowski JM, et al. Differential phenotypes of myeloid-derived suppressor and T regulatory cells and cytokine levels in amnestic mild cognitive impairment subjects compared to mild Alzheimer diseased patients. Front Immunol. 2017;8:783. doi: 10.3389/fimmu.2017.00783. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Li P, Gan Y, Sun BL, Zhang F, Lu B, Gao Y, et al. Adoptive regulatory T-cell therapy protects against cerebral ischemia. Ann Neurol. 2013;74:458–71. doi: 10.1002/ana.23815. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Liesz A, Zhou W, Na SY, Hammerling GJ, Garbi N, Karcher S, et al. Boosting regulatory T cells limits neuroinflammation in permanent cortical stroke. J Neurosci. 2013;33:17350–62. doi: 10.1523/JNEUROSCI.4901-12.2013. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Maes M, Berk M, Goehler L, Song C, Anderson G, Galecki P, et al. Depression and sickness behavior are Janus-faced responses to shared inflammatory pathways. BMC Med. 2012;10:66. doi: 10.1186/1741-7015-10-66. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Mahgoub N, Alexopoulos GS. Amyloid hypothesis: Is there a role for antiamyloid treatment in late-life depression? Am J Geriatr Psychiatry. 2016;24:239–47. doi: 10.1016/j.jagp.2015.12.003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Malm TM, Koistinaho M, Parepalo M, Vatanen T, Ooka A, Karlsson S, et al. Bone-marrow-derived cells contribute to the recruitment of microglial cells in response to beta-amyloid deposition in APP/PS1 double transgenic Alzheimer mice. Neurobiol Dis. 2005;18:134–42. doi: 10.1016/j.nbd.2004.09.009. [DOI] [PubMed] [Google Scholar]
- Masliah E, Terry RD, DeTeresa RM, Hansen LA. Immunohistochemical quantification of the synapse-related protein synaptophysin in Alzheimer disease. Neurosci Lett. 1989;103:234–9. doi: 10.1016/0304-3940(89)90582-x. [DOI] [PubMed] [Google Scholar]
- Maurer MH, Schabitz WR, Schneider A. Old friends in new constellations--the hematopoetic growth factors G-CSF, GM-CSF, and EPO for the treatment of neurological diseases. Curr Med Chem. 2008;15:1407–11. doi: 10.2174/092986708784567671. [DOI] [PubMed] [Google Scholar]
- McLay RN, Kimura M, Banks WA, Kastin AJ. Granulocyte-macrophage colony-stimulating factor crosses the blood--brain and blood--spinal cord barriers. Brain. 1997;120(Pt 11):2083–91. doi: 10.1093/brain/120.11.2083. [DOI] [PubMed] [Google Scholar]
- McManus RM, Mills KH, Lynch MA. T cells-protective or pathogenic in Alzheimer’s disease? J Neuroimmune Pharmacol. 2015;10:547–60. doi: 10.1007/s11481-015-9612-2. [DOI] [PubMed] [Google Scholar]
- Mietelska-Porowska A, Wojda U. T lymphocytes and inflammatory mediators in the interplay between brain and blood in Alzheimer’s disease: potential pools of new biomarkers. J Immunol Res. 2017;2017:4626540. doi: 10.1155/2017/4626540. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Minihane AM, Vinoy S, Russell WR, Baka A, Roche HM, Tuohy KM, et al. Low-grade inflammation, diet composition and health: current research evidence and its translation. Br J Nutr. 2015;114:999–1012. doi: 10.1017/S0007114515002093. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Molinari S, Battini R, Ferrari S, Pozzi L, Killcross AS, Robbins TW, et al. Deficits in memory and hippocampal long-term potentiation in mice with reduced calbindin D28K expression. Proc Natl Acad Sci U S A. 1996;93:8028–33. doi: 10.1073/pnas.93.15.8028. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Mosley RL, Hutter–Saunders JA, Stone DK, Gendelman HE. Inflammation and adaptive immunity in Parkinson’s disease. Cold Spring Harb Perspect Med. 2012;2:a009381. doi: 10.1101/cshperspect.a009381. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Moss ST, Hamilton JA. Proliferation of a subpopulation of human peripheral blood monocytes in the presence of colony stimulating factors may contribute to the inflammatory process in diseases such as rheumatoid arthritis. Immunobiology. 2000;202:18–25. doi: 10.1016/S0171-2985(00)80048-0. [DOI] [PubMed] [Google Scholar]
- Muszynski P, Kulczynska-Przybik A, Borawska R, Litman-Zawadzka A, Slowik A, Klimkowicz-Mrowiec A, et al. The relationship between markers of inflammation and degeneration in the central nervous system and the blood-brain barrier impairment in Alzheimer’s disease. J Alzheimers Dis. 2017;59:903–12. doi: 10.3233/JAD-170220. [DOI] [PubMed] [Google Scholar]
- Nagahara AH, Mateling M, Kovacs I, Wang L, Eggert S, Rockenstein E, et al. Early BDNF treatment ameliorates cell loss in the entorhinal cortex of APP transgenic mice. J Neurosci. 2013;33:15596–602. doi: 10.1523/JNEUROSCI.5195-12.2013. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Nagahara AH, Merrill DA, Coppola G, Tsukada S, Schroeder BE, Shaked GM, et al. Neuroprotective effects of brain-derived neurotrophic factor in rodent and primate models of Alzheimer’s disease. Nat Med. 2009;15:331–7. doi: 10.1038/nm.1912. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Nakagawa T, Suga S, Kawase T, Toda M. Intracarotid injection of granulocyte-macrophage colony-stimulating factor induces neuroprotection in a rat transient middle cerebral artery occlusion model. Brain Res. 2006;1089:179–85. doi: 10.1016/j.brainres.2006.03.059. [DOI] [PubMed] [Google Scholar]
- Nicoll JA, Wilkinson D, Holmes C, Steart P, Markham H, Weller RO. Neuropathology of human Alzheimer disease after immunization with amyloid-beta peptide: a case report. Nat Med. 2003;9:448–52. doi: 10.1038/nm840. [DOI] [PubMed] [Google Scholar]
- Olson KE, Gendelman HE. Immunomodulation as a neuroprotective and therapeutic strategy for Parkinson’s disease. Curr Opin Pharmacol. 2016;26:87–95. doi: 10.1016/j.coph.2015.10.006. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ponomarev ED, Maresz K, Tan Y, Dittel BN. CNS-derived interleukin-4 is essential for the regulation of autoimmune inflammation and induces a state of alternative activation in microglial cells. J Neurosci. 2007;27:10714–21. doi: 10.1523/JNEUROSCI.1922-07.2007. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Puligujja P, Balkundi SS, Kendrick LM, Baldridge HM, Hilaire JR, Bade AN, et al. Pharmacodynamics of long-acting folic acid-receptor targeted ritonavir-boosted atazanavir nanoformulations. Biomaterials. 2015;41:141–50. doi: 10.1016/j.biomaterials.2014.11.012. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Rao MS, Shetty AK. Efficacy of doublecortin as a marker to analyse the absolute number and dendritic growth of newly generated neurons in the adult dentate gyrus. Eur J Neurosci. 2004;19:234–46. doi: 10.1111/j.0953-816x.2003.03123.x. [DOI] [PubMed] [Google Scholar]
- Reynolds A, Laurie C, Mosley RL, Gendelman HE. Oxidative stress and the pathogenesis of neurodegenerative disorders. Int Rev Neurobiol. 2007a;82:297–325. doi: 10.1016/S0074-7742(07)82016-2. [DOI] [PubMed] [Google Scholar]
- Reynolds AD, Banerjee R, Liu J, Gendelman HE, Mosley RL. Neuroprotective activities of CD4+CD25+ regulatory T cells in an animal model of Parkinson’s disease. J Leukoc Biol. 2007b;82:1083–94. doi: 10.1189/jlb.0507296. [DOI] [PubMed] [Google Scholar]
- Rowin J, Thiruppathi M, Arhebamen E, Sheng J, Prabhakar BS, Meriggioli MN. Granulocyte macrophage colony-stimulating factor treatment of a patient in myasthenic crisis: effects on regulatory T cells. Muscle Nerve. 2012;46:449–53. doi: 10.1002/mus.23488. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ruef C, Coleman DL. Granulocyte-macrophage colony-stimulating factor: pleiotropic cytokine with potential clinical usefulness. Rev Infect Dis. 1990;12:41–62. doi: 10.1093/clinids/12.1.41. [DOI] [PubMed] [Google Scholar]
- Rutten BP, Van der Kolk NM, Schafer S, van Zandvoort MA, Bayer TA, Steinbusch HW, et al. Age-related loss of synaptophysin immunoreactive presynaptic boutons within the hippocampus of APP751SL, PS1M146L, and APP751SL/PS1M146L transgenic mice. Am J Pathol. 2005;167:161–73. doi: 10.1016/S0002-9440(10)62963-X. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sakaguchi S. Regulatory T cells: key controllers of immunologic self-tolerance. Cell. 2000;101:455–8. doi: 10.1016/s0092-8674(00)80856-9. [DOI] [PubMed] [Google Scholar]
- Salter MW, Beggs S. Sublime microglia: expanding roles for the guardians of the CNS. Cell. 2014;158:15–24. doi: 10.1016/j.cell.2014.06.008. [DOI] [PubMed] [Google Scholar]
- Saresella M, Calabrese E, Marventano I, Piancone F, Gatti A, Alberoni M, et al. Increased activity of Th-17 and Th-9 lymphocytes and a skewing of the post-thymic differentiation pathway are seen in Alzheimer’s disease. Brain Behav Immun. 2011;25:539–47. doi: 10.1016/j.bbi.2010.12.004. [DOI] [PubMed] [Google Scholar]
- Saresella M, Calabrese E, Marventano I, Piancone F, Gatti A, Calvo MG, et al. PD1 negative and PD1 positive CD4+ T regulatory cells in mild cognitive impairment and Alzheimer’s disease. J Alzheimers Dis. 2010;21:927–38. doi: 10.3233/JAD-2010-091696. [DOI] [PubMed] [Google Scholar]
- Savchenko E, Malm T, Konttinen H, Hamalainen RH, Guerrero-Toro C, Wojciechowski S, et al. Abeta and inflammatory stimulus activate diverse signaling pathways in monocytic cells: Implications in retaining phagocytosis in Abeta-laden environment. Front Cell Neurosci. 2016;10:279. doi: 10.3389/fncel.2016.00279. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Schabitz WR, Kruger C, Pitzer C, Weber D, Laage R, Gassler N, et al. A neuroprotective function for the hematopoietic protein granulocyte-macrophage colony stimulating factor (GM-CSF) J Cereb Blood Flow Metab. 2008;28:29–43. doi: 10.1038/sj.jcbfm.9600496. [DOI] [PubMed] [Google Scholar]
- Schenk D, Basi GS, Pangalos MN. Treatment strategies targeting amyloid beta-protein. Cold Spring Harb Perspect Med. 2012;2:a006387. doi: 10.1101/cshperspect.a006387. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sheng JR, Muthusamy T, Prabhakar BS, Meriggioli MN. GM-CSF-induced regulatory T cells selectively inhibit anti-acetylcholine receptor-specific immune responses in experimental myasthenia gravis. J Neuroimmunol. 2011;240–241:65–73. doi: 10.1016/j.jneuroim.2011.10.010. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Shin YK, Cho SR. Exploring erythropoietin and G-CSF combination therapy in chronic stroke patients. Int J Mol Sci. 2016;17:463. doi: 10.3390/ijms17040463. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Shultz SR, Tan XL, Wright DK, Liu SJ, Semple BD, Johnston L, et al. Granulocyte-macrophage colony-stimulating factor is neuroprotective in experimental traumatic brain injury. J Neurotrauma. 2014;31:976–83. doi: 10.1089/neu.2013.3106. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Simard AR, Soulet D, Gowing G, Julien JP, Rivest S. Bone marrow-derived microglia play a critical role in restricting senile plaque formation in Alzheimer’s disease. Neuron. 2006;49:489–502. doi: 10.1016/j.neuron.2006.01.022. [DOI] [PubMed] [Google Scholar]
- Song GJ, Suk K. Pharmacological modulation of functional phenotypes of microglia in neurodegenerative diseases. Frontiers in aging neuroscience. 2017;9:139. doi: 10.3389/fnagi.2017.00139. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Spangenberg EE, Lee RJ, Najafi AR, Rice RA, Elmore MR, Blurton-Jones M, et al. Eliminating microglia in Alzheimer’s mice prevents neuronal loss without modulating amyloid-beta pathology. Brain. 2016;139:1265–81. doi: 10.1093/brain/aww016. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sze CI, Troncoso JC, Kawas C, Mouton P, Price DL, Martin LJ. Loss of the presynaptic vesicle protein synaptophysin in hippocampus correlates with cognitive decline in Alzheimer disease. J Neuropathol Exp Neurol. 1997;56:933–44. doi: 10.1097/00005072-199708000-00011. [DOI] [PubMed] [Google Scholar]
- Takahashi T, Tagami T, Yamazaki S, Uede T, Shimizu J, Sakaguchi N, et al. Immunologic self-tolerance maintained by CD25(+)CD4(+) regulatory T cells constitutively expressing cytotoxic T lymphocyte-associated antigen 4. J Exp Med. 2000;192:303–10. doi: 10.1084/jem.192.2.303. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Takeda S, Sato N, Morishita R. Systemic inflammation, blood-brain barrier vulnerability and cognitive/non-cognitive symptoms in Alzheimer disease: relevance to pathogenesis and therapy. Frontiers in aging neuroscience. 2014;6:171. doi: 10.3389/fnagi.2014.00171. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Tang Y, Le W. Differential roles of M1 and M2 microglia in neurodegenerative diseases. Mol Neurobiol. 2016;53:1181–94. doi: 10.1007/s12035-014-9070-5. [DOI] [PubMed] [Google Scholar]
- Thomaty S, Pezard L, Xerri C, Brezun JM. Acute granulocyte macrophage-colony stimulating factor treatment modulates neuroinflammatory processes and promotes tactile recovery after spinal cord injury. Neuroscience. 2017;349:144–64. doi: 10.1016/j.neuroscience.2017.02.035. [DOI] [PubMed] [Google Scholar]
- Togo T, Akiyama H, Iseki E, Kondo H, Ikeda K, Kato M, et al. Occurrence of T cells in the brain of Alzheimer’s disease and other neurological diseases. J Neuroimmunol. 2002;124:83–92. doi: 10.1016/s0165-5728(01)00496-9. [DOI] [PubMed] [Google Scholar]
- Toly-Ndour C, Lui G, Nunes MM, Bruley-Rosset M, Aucouturier P, Dorothee G. MHC-independent genetic factors control the magnitude of CD4+ T cell responses to amyloid-beta peptide in mice through regulatory T cell-mediated inhibition. J Immunol. 2011;187:4492–500. doi: 10.4049/jimmunol.1003953. [DOI] [PubMed] [Google Scholar]
- Tsai ST, Chu SC, Liu SH, Pang CY, Hou TW, Lin SZ, et al. Neuroprotection of granulocyte colony-stimulating factor for early stage Parkinson’s disease. Cell Transplant. 2017;26:409–16. doi: 10.3727/096368916X694247. [DOI] [PMC free article] [PubMed] [Google Scholar]
- VanItallie TB. Alzheimer’s disease: Innate immunity gone awry? Metabolism. 2017;69S:S41–S9. doi: 10.1016/j.metabol.2017.01.014. [DOI] [PubMed] [Google Scholar]
- Ye M, Chung HS, Lee C, Yoon MS, Yu AR, Kim JS, et al. Neuroprotective effects of bee venom phospholipase A2 in the 3xTg AD mouse model of Alzheimer’s disease. J Neuroinflammation. 2016;13:10. doi: 10.1186/s12974-016-0476-z. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zenaro E, Piacentino G, Constantin G. The blood-brain barrier in Alzheimer’s disease. Neurobiol Dis. 2017;107:41–56. doi: 10.1016/j.nbd.2016.07.007. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zuroff L, Daley D, Black KL, Koronyo-Hamaoui M. Clearance of cerebral Abeta in Alzheimer’s disease: reassessing the role of microglia and monocytes. Cell Mol Life Sci. 2017;74:2167–201. doi: 10.1007/s00018-017-2463-7. [DOI] [PMC free article] [PubMed] [Google Scholar]