

Supplemental Digital Content is available in the text.
Keywords: maraviroc, CCR5, HIV, pediatric, pharmacokinetics
Abstract
Background:
Maraviroc is a CC-chemokine receptor 5 antagonist approved to treat adults infected with CC-chemokine receptor 5–tropic (R5) HIV-1. Study A4001031 was conducted to evaluate the pharmacokinetics, safety and efficacy of maraviroc in combination with optimized background therapy in treatment-experienced pediatric patients infected with R5 HIV-1 and support registration of maraviroc for pediatric use.
Methods:
This is an open-label, 2-stage, age-stratified, noncomparative multicenter study. One-hundred and three participants were enrolled into 4 age/formulation cohorts and dosed twice daily. Initial doses were determined by body surface area and optimized background therapy, based on drug interactions with maraviroc in adults. Dose adjustment and pharmacokinetic reevaluation occurred if the average concentrations (Cavg) at Week 2 were <100 ng/mL (Stage 1—dose finding).
Results:
Data from the Week 48 analysis demonstrated that 49/50 Stage 1 participants rolling over into Stage 2 (safety and efficacy) achieved Cavg ≥100 ng/mL. Doses were identified that achieved similar concentration ranges to those seen in adults. The majority (90/103) received optimized background therapy containing potent cytochrome P450 3A inhibitors. Maraviroc was well tolerated and the safety and efficacy were comparable to those of adults. All cohorts had a mean decrease from baseline in HIV-1 RNA of >1 log10. Increases from baseline in the median CD4+ cell count and percentage were seen for all age groups.
Conclusions:
The maraviroc dosing strategy resulted in participants achieving the target Cavg, with exposure ranges similar to those observed in adults on approved doses. The safety and efficacy of maraviroc in this pediatric population were comparable to those seen in adults.
Combination antiretroviral (ARV) therapy has significantly reduced HIV-associated morbidity and mortality in children.1 However, suitable pediatric formulations for many ARVs are lacking, with toxicities and resistance further complicating treatment. Perinatally infected children have often been treated with several drug regimens over several years and may have accumulated multiple resistance mutations.2 The challenging nature of conducting studies in this population results in regulatory approval usually lagging behind adult approvals by several years. HIV-infected children, therefore, have fewer therapeutic options compared with adults and it is important to evaluate all new ARVs in the pediatric population.
The safety and efficacy of maraviroc, a CC-chemokine receptor 5 (CCR5) antagonist for the treatment of HIV-1 infection, have been demonstrated in both treatment-experienced (TE; MOTIVATE 1 and 2 studies) and treatment-naive (MERIT study) adults infected with CCR5-tropic HIV-1.3,4 Five-year follow-up data from these studies have demonstrated favorable long-term safety and durable virologic responses.5,6
Maraviroc, a cytochrome P450 3A (CYP3A) substrate, requires dose adjustment when used in combination with potent CYP3A inhibitors or inducers. The approved dose of maraviroc for adults is 300 mg twice daily (BID) in the absence of potent CYP3A inducers or inhibitors (neutral agents), whereas it is adjusted to 150 mg BID in the presence of potent CYP3A inhibitors and to 600 mg BID in the presence of potent CYP3A inducers (in the absence of potent CYP3A inhibitors).7
Week 48 data from the MOTIVATE studies demonstrated that 43% and 46% of TE adult patients receiving optimized background therapy (OBT) with maraviroc once daily and BID, respectively, achieved an HIV-1 RNA of <50 copies/mL compared with 17% of those receiving OBT only. Maraviroc treatment also provided a significant cluster of differentiation 4 (CD4) benefit.4 Maraviroc was administered at a dose of 300 mg or equivalent (dose reduced to 150 mg when given with potent CYP3A inhibitors) in these studies.4 Assessment of exposure–response at Week 48 demonstrated that the doses used in the MOTIVATE studies delivered plasma concentrations that were high on the exposure–response curve, with near-maximal efficacy achieved at an average concentration (Cavg) of ≥100 ng/mL.8
Study A4001031 evaluated the pharmacokinetics (PK), safety and efficacy of maraviroc in TE pediatric patients to assess pediatric dose formulations and develop dose recommendations for registration of maraviroc in this population. Here, we describe the PK, safety and efficacy data through Week 48.
MATERIALS AND METHODS
Study A4001031 is an ongoing, open-label, multiple-dose trial to assess the PK, safety and efficacy of maraviroc in combination with OBT for the treatment of ARV-experienced CCR5-tropic HIV-1–infected pediatric patients (ClinicalTrials.gov identifier: NCT00791700). It is being conducted in compliance with the Declaration of Helsinki and International Conference on Harmonisation Good Clinical Practice Guidelines, and all local regulatory requirements are being followed. The protocol was approved by institutional review boards and/or independent ethics committees at all sites. Written informed consent was provided by all participants or their parents/caregivers/legal guardians, as appropriate. Where appropriate, assent was also obtained from participants. An independent data monitoring committee reviews the data on a regular basis to ensure the safety of participating patients.
Study Population and Design
Participants were HIV-1–infected children and adolescents (≥2 to <18 years of age) with only CCR5-tropic virus, as identified using the enhanced sensitivity Trofile assay (Monogram Biosciences, San Francisco, CA). Other inclusion criteria included plasma HIV-1 RNA ≥1000 copies/mL and intolerance to, or experience with, at least 2 ARV drug classes for ≥6 months. Key exclusion criteria were the need for more than 5 ARVs (excluding low-dose ritonavir) in an OBT regimen, grade ≥3 neutropenia, thrombocytopenia, anemia, aspartate transaminase, alanine transaminase, creatinine or lipase elevations (defined using 2004 Division of AIDS [DAIDS] toxicity criteria)9 and active opportunistic infections.
Participants were stratified on Day 1 by age and formulation into 1 of 4 cohorts (Fig. 1). Maraviroc was administered BID as tablets or as a 20 mg/mL oral solution, in combination with an OBT regimen selected by the investigator based on susceptibility testing and treatment history and consisting of 3–5 ARVs. The study was divided into 2 stages. Stage 1 was an intensive dose-finding stage, whereas the 48-week safety and efficacy was assessed in Stage 2 once an individual or OBT category dose was identified (Fig. 1). The primary endpoint was evaluated at Week 48, but participants are being followed for up to 5 years to establish the long-term safety of maraviroc.
FIGURE 1.
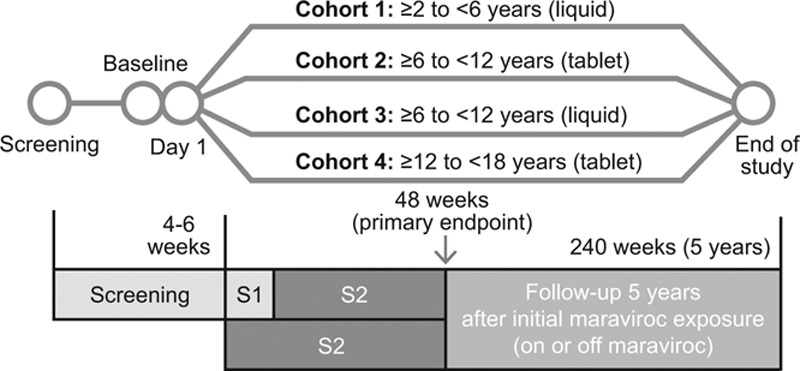
Design of study A4001031. S1, Stage 1: Intensive PK/dose finding (4–12 weeks): Minimum of 12 (Cohort 1) and 10 children (in each of Cohorts 2–4) to complete Stage 1 before entering Stage 2. S2, Stage 2: Safety/Efficacy: Following the minimum numbers being reached for Stage 1, all new patients then directly entered Stage 2.
Dose Selection and Confirmation
Maraviroc doses in Study A4001031 were scaled from adult doses using body surface area (BSA) and assuming a standard adult BSA of 1.73 m2. Initial doses were therefore approximately:
173 mg/m2 in the absence of potent cytochrome P4503A (CYP3A) inhibitors or potent CYP3A inducers (ie, neutral OBT),
87 mg/m2 in the presence of potent CYP3A inhibitors,
347 mg/m2 in the presence of potent CYP3A inducers (in the absence of potent CYP3A inhibitors).
Participants were placed into BSA bands and doses were adjusted for OBT and/or concomitant medication category (Table 1).
TABLE 1.
Maraviroc Pediatric Doses by BSA on Study Entry and OBT Regimen
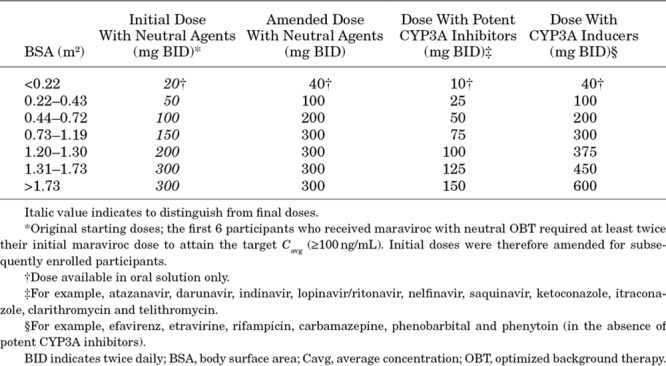
A target Cavg of 100 ng/mL was set based on adult exposure/response data.8 Intensive PK data were obtained for dose optimization at Week 2 for all Stage 1 patients. If dose adjustment was required, intensive PK samples were again obtained after approximately 2 weeks on the new dose. Once it was determined that they were receiving an appropriate maraviroc dose, Stage 1 participants were rolled over into Stage 2. Once a sufficient number of Stage 1 participants were enrolled to allow dose selection by cohort (for OBT containing CYP3A4 inhibitor category only), new participants were enrolled directly into Stage 2.
Participants enrolled in Stage 1 took their maraviroc dose with food on intensive PK sampling days. On all other days maraviroc was taken with or without food.
Assessment of PK
In Stage 1, PK samples were obtained at the following times: pre-dose (0), 1, 2, 4, 6, 8 and 12 hours after dosing at the Week 2 visit; 2 weeks after a dose adjustment at the Week 2 visit (if a dose adjustment occurred), and at the Week 48 week visit in all Stage 1 participants who rolled over into Stage 2 and remained in the study on maraviroc at Week 48. Standard PK parameters were determined by noncompartmental analysis. Sparse PK sampling was also done for all patients who participated in Stage 2 (including participants rolled over from Stage 1) at each visit up to and including Week 48. Samples for sparse PK analysis were obtained within a window of 5–12 hours after a maraviroc dose.
A graphical analysis was performed to compare maraviroc concentration–time data (on initial and optimized doses, and including intensive and sparse sampling), achieved with the doses utilized in Study A4001031 in pediatric patients, with sparse PK data from adult patients studied at recommended adult doses in the approved product labeling on similar background therapy. Data are summarized by the following CYP3A drug interaction dosing categories: potent CYP3A inhibitors, neutral agents and potent CYP3A inducers.
Assessment of Safety
Safety was assessed at all visits with monitoring of adverse events (AEs), serious adverse events (SAEs), vital signs and laboratory parameters. AEs were graded according to 2004 DAIDS toxicity scales.9 All participants who received at least 1 dose of study drug were included in the safety analyses, and data were summarized descriptively. Exposure-adjusted incidence rates were calculated to allow for comparison with data from the MOTIVATE studies in TE adults.
Assessment of Efficacy
HIV-1 RNA (COBAS AmpliPrep/COBAS TaqMan HIV-1 test; Roche, Basel, Switzerland; lower limit of quantification, <48 copies/mL) and CD4+ count (absolute and percentage) were evaluated at all study visits. Efficacy was a secondary endpoint in the study and the efficacy analysis population comprised all participants who received at least 1 dose of the study drug. The proportions (%) of participants with HIV-1 RNA <400 and <48 copies/mL at Week 48 were determined using the US Food and Drug Administration snapshot algorithm, where missing, switch and discontinuation equals failure (MSDF).
Virus susceptibility (PhenoSense GT; Monogram Biosciences, San Francisco, CA) and tropism were assessed at screening and at the time of virologic failure. Patients with virologic failure were defined as those who were discontinued for lack of efficacy and met protocol-defined virologic failure criteria (details in Supplementary Digital Content 1, http://links.lww.com/INF/C856).
Adherence
Adherence to maraviroc and OBT was assessed through reconciliation of the amount of the study drug (pill count and liquid volume) returned at each visit. Caregivers/patients were asked to account for any missed doses.
RESULTS
Study Population and Disposition
Of the 285 patients screened, 103 received at least 1 dose of the study drug. The most common reasons for screen failure were non-R5 virus or failed tropism test (~40%) and HIV-1 RNA <1000 copies/mL (~23%). Participants were enrolled in 8 countries across 24 sites, with the majority enrolled from South Africa (n = 62). The rest were from the United States (n = 12), Thailand (n = 11), Brazil (n = 6), Spain (n = 6), Portugal (n = 4), Italy (n = 1) and Puerto Rico (n = 1).
Demographic and baseline characteristics are summarized in Table 2. There were 49 male and 54 female participants, the majority of whom were Black (68.9%) with a mean age of 10.3 (standard deviation: 4.1) years. Significant previous treatment experience is reflected by the high proportion of participants with resistance-associated mutations, especially to the nucleoside reverse transcriptase inhibitor and non-nucleoside reverse transcriptase inhibitor drug classes.
TABLE 2.
Demographic and Baseline Characteristics
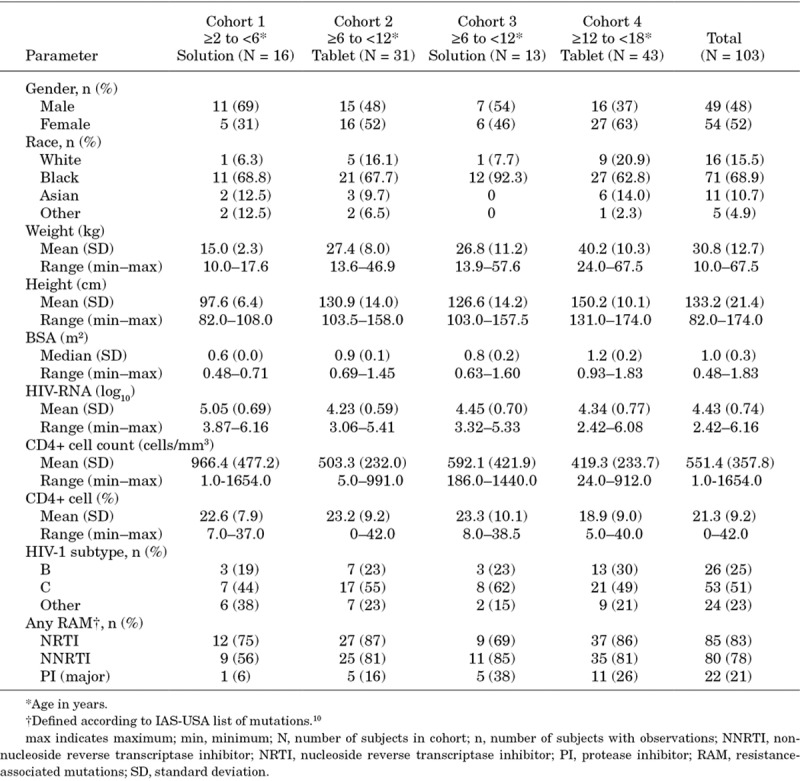
Of the 103 participants treated, 56 were enrolled in Stage 1, with an additional 47 enrolled directly into Stage 2. Seventy-four participants (71.8%) completed Week 48 of treatment; 12/16 (75%) in Cohort 1, 26/31 (83.9%) in Cohort 2, 9/13 (69.2%) in Cohort 3 and 27/43 (62.8%) in Cohort 4. For the 31 participants (30.1%) who discontinued treatment before/at Week 48, the most common reasons were “insufficient clinical response” (determined by the investigator) (23/31 participants; 74.2%), “noncompliance with study treatment” (2/31 participants; 6.5%) and “no longer willing to participate in study” (3/31 participants; 9.7%).
PK Data
The majority of participants (90/103; 87.4%) received maraviroc in combination with an OBT containing potent CYP3A inhibitors. Ten participants (9.7%) received a neutral OBT, and 3 participants (2.9%) received OBT containing potent CYP3A inducers (without potent CYP3A inhibitors).
Six of the 56 patients enrolled into Stage 1 were discontinued before Stage 2 (2 were noncompliant, 2 had AEs, 1 was no longer willing to participate, and blood sampling was unsuccessful in 1). Of the 50 participants who entered Stage 2 after individual dose optimization in Stage 1, 49 achieved the target Cavg of ≥100 ng/mL. Dose adjustments were required for 8 participants (1 on a CYP3A inhibitor, 1 on a CYP3A inducer and 6 receiving a neutral OBT regimen) in Stage 1. One participant on a neutral OBT regimen, who did not reach the required exposure target after 2 upward dose adjustments but clinically responded (<48 copies/mL HIV-1 RNA) on maraviroc 300 mg BID (adult recommended dose), was enrolled into Stage 2. Because the first 6 patients in the neutral OBT group required upward dose adjustments, the initial doses of maraviroc for this OBT group were adjusted upward as shown in Table 1, column 3.
Mean plasma maraviroc concentration time profiles for the 50 Stage 1 participants who enrolled in Stage 2 were similar across cohorts at Week 2 (Fig. 2A; Table, Supplemental Digital Content 2, http://links.lww.com/INF/C857, for detailed PK data) and Week 48. Geometric mean Cavg at Week 48 remained ≥100 ng/mL for all cohorts. There was no evidence of a relationship between maraviroc plasma levels (Cavg at Week 2) and the virologic response rate (MSDF HIV-1 RNA <400 and <48 copies/mL) at Week 48, indicating that higher doses of maraviroc are unlikely to improve response.
FIGURE 2.
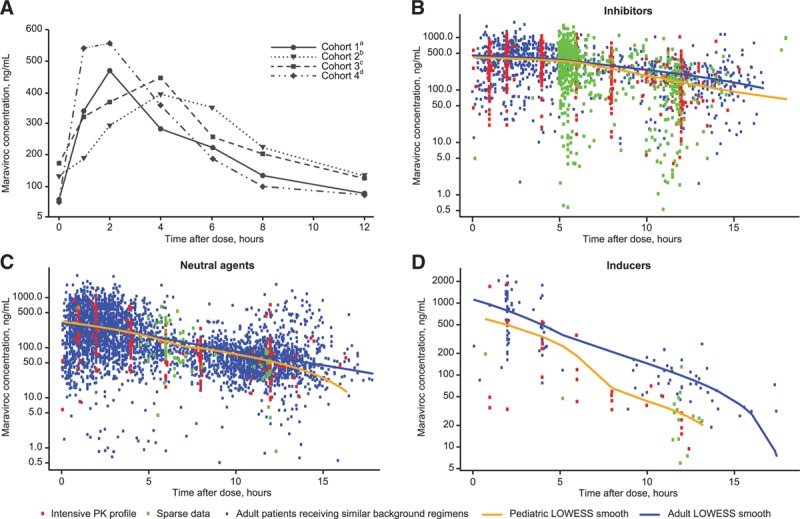
Maraviroc plasma concentration vs. time (A) at Week 2 by cohort; (B–D) by interaction category compared with adult data. A: Median Week 2 concentrations for Stage 1 participants enrolled in Stage 2 (N = 50; participants on final dose). B: Patients receiving maraviroc with potent CYP3A inhibitors. Data from pediatric patients (N = 85) were compared with data from adult patients (N = 125) receiving maraviroc at the approved dose of 150 mg BID in combination with lopinavir/ritonavir, darunavir/ritonavir or atazanavir/ritonavir in Studies A4001027, A4001028 (MOTIVATE 1 and 2),4 A400102911 and A4001098.12 C: Patients receiving maraviroc with neutral agents. Data from pediatric patients (N = 10) were compared with data from adult patients (N = 402) receiving maraviroc at the approved dose of 300 mg BID in combination with neutral agents in studies A4001026 (MERIT),3 A4001027, A4001028 (MOTIVATE 1 and 2),4 A400102911 and A4001098.12 D: Patients receiving maraviroc with potent CYP3A inducers. Data from pediatric patients (N = 2) were compared with data from adult patients (N = 27) receiving maraviroc at the approved dose of 600 mg BID in combination with efavirenz or etravirine in study A4001098.12aCohort 1: ≥2 to <6 years of age, maraviroc oral solution.bCohort 2: ≥6 to <12 years of age, maraviroc tablet formulation.cCohort 3: ≥6 to <12 years of age, maraviroc oral solution.dCohort 4: ≥12 to <18 years of age, maraviroc tablet formulation. BID indicates twice daily; LOWESS, locally weighted scatter plot smoothing.
Concentration–time data for the pediatric patients in Study A4001031 and data from adult HIV-1–infected patients receiving similar therapy demonstrated that across the 3 CYP3A drug interaction categories, the majority of concentrations in pediatric participants fell within the adult observation ranges (Fig. 2B–D).
Safety
Maraviroc was well tolerated. A total of 285 treatment-emergent AEs (all causality) were reported by 74 participants (71.8%). Twelve participants (11.7%) experienced 15 SAEs up to Week 48 in Study A4001031; the majority were infections, and none was considered related to maraviroc treatment by the investigators. There were 2 discontinuations (1 each in Cohorts 3 and 4) and 3 temporary discontinuations (2 in Cohort 3 and 1 in Cohort 4) due to AEs, with no deaths or dose reductions. Category C AIDS-defining events were reported in 2 participants (1 case of pulmonary tuberculosis and 1 case of recurrent episodes of pneumonia and tuberculosis).
The majority of treatment-emergent AEs were grade 1 or 2 in severity, and only 6 participants reported grade 3 and 4 AEs; none of which were considered related to maraviroc by the investigator. Treatment-emergent AEs reported by >10% of participants were diarrhea (17 participants [16.5%]), vomiting (17 participants [16.5%]) and upper respiratory tract infection (14 participants [13.6%]). The overall exposure-adjusted incidence of treatment-emergent AEs was broadly consistent between cohorts. However, the exposure-adjusted incidence of common childhood illnesses, such as diarrhea, vomiting, gastroenteritis, otitis media, otitis media acute and upper respiratory tract infection, was higher in Cohort 1 (children ≥2 years and <6 years of age) compared with the cohorts containing older children. This is consistent with what would be expected in this population.13
Compared with data from the MOTIVATE studies in adults, the exposure-adjusted incidence of most commonly reported AEs was generally similar or lower (Table 3), while AEs with a higher exposure-adjusted incidence in children represent common childhood illnesses, such as infections, or signs/symptoms thereof.
TABLE 3.
Summary of Exposure-adjusted Incidence of All Causality AEs Reported Through Week 48 at >5 Events/100 Patient-Years (in Any Study) for Maraviroc in Adult Studies A4001027, A4001028, and Pediatric Study A4001031
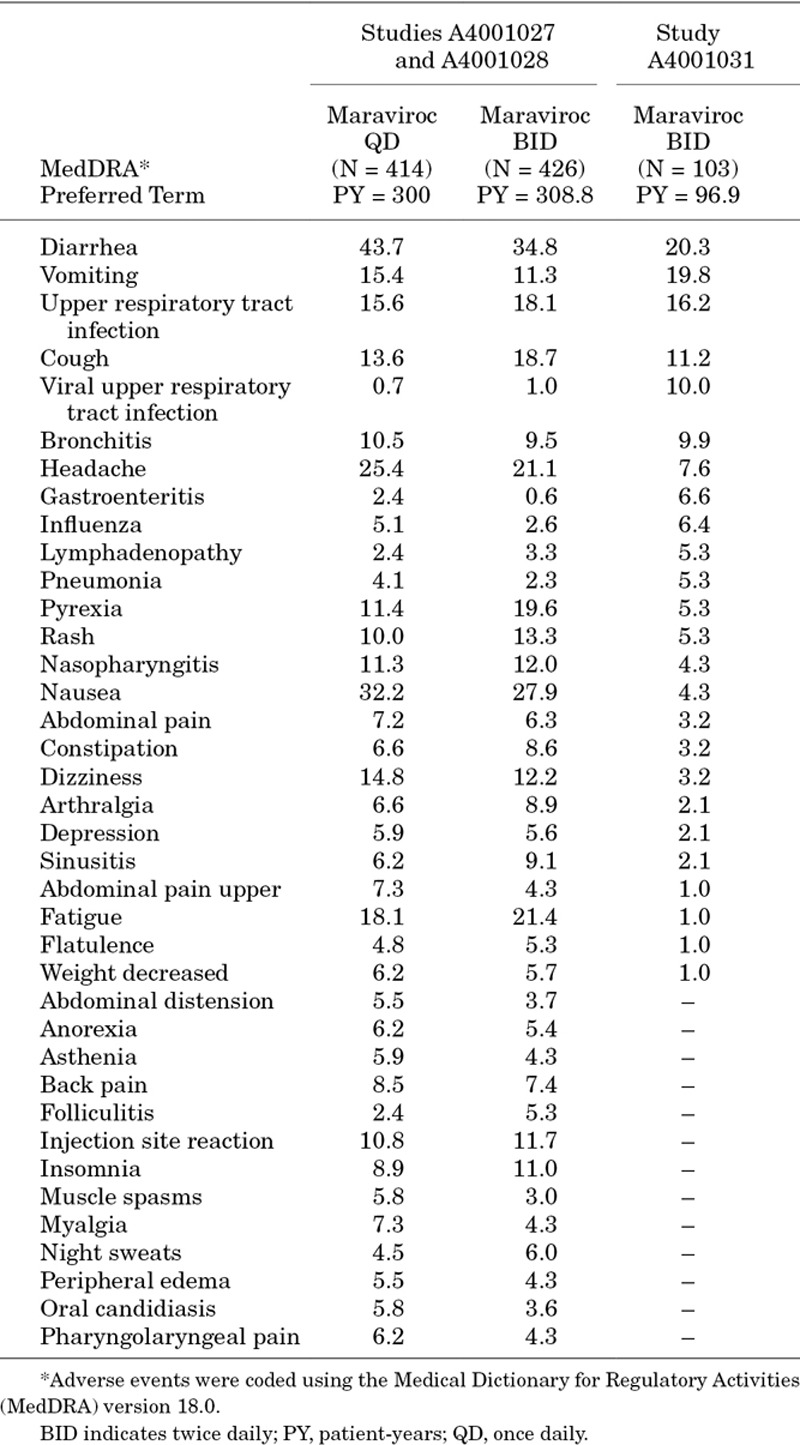
Treatment-emergent grade 3 or 4 laboratory abnormalities (identified using 2004 DAIDS grading criteria)9 were identified in 14 participants. Two experienced grade 4 abnormalities. One had a grade 4 lipase elevation; this event resolved while continuing on maraviroc and was assessed by the investigator as not related to the study drug. No AEs indicative of pancreatitis were reported. Grade 4 aspartate transaminase and alanine transaminase elevations, as well as a grade 3 elevation in total bilirubin, were experienced by 1 participant at the time of discontinuation (for insufficient clinical response), and was attributed to rifampicin/isoniazid treatment by the investigator. Another participant experienced an asymptomatic grade 3 alanine transaminase elevation, as well as grade 2 aspartate transaminase and bilirubin elevations on day 333 of treatment. Maraviroc and OBT (lopinavir/ritonavir and zidovudine/lamivudine) were discontinued and abnormalities resolved after 13 days. All treatments were restarted 6 days after the values returned to normal, and no immediate elevations were observed. The event is unlikely to be related to maraviroc or OBT, given the time to onset and the negative rechallenge.
Seven participants experienced grade 3 neutropenia while on maraviroc, 4 of whom had a low neutrophil count before receiving maraviroc. Six were receiving concurrent zidovudine and/or lopinavir/ritonavir, while the remaining patient received zidovudine for more than 10 years before starting maraviroc and lamivudine/tenofovir. There was no apparent temporal association between the neutropenia and maraviroc treatment, and no discontinuations due to neutropenia. Importantly, none of the participants would have met the criteria for grade 3 neutropenia if severity had been assessed using the 2014 DAIDS criteria14 cutoff for grade 3 absolute neutrophil count decrease (<0.6 × 109/L), rather than the 2004 DAIDS criteria (<0.75 × 109/L).9
Efficacy and Virology
All cohorts had a mean decrease from baseline in HIV-1 RNA of ≥1.5 log10 copies/mL. The proportion of participants achieving HIV-1 RNA <400 copies/mL at Week 48 using the MSDF algorithm was 65.0%, whereas 47.6% achieved <48 copies/mL. The lowest response rate (39.5% achieving HIV-1 RNA <48 copies/mL) was seen in the adolescents in Cohort 4, with all other cohorts achieving a response rate of ≥50%. The absolute CD4+ cell count (cells/mm3) and percentage increased in all Cohorts, with an overall median increase of 192 (interquartile range: 92–352) and 4 (interquartile range: 1–8), respectively. More details are provided in Supplementary Digital Content 3, http://links.lww.com/INF/C858.
The virologic failure population included 23 participants; 5 had dual-mixed virus at failure; 2 of whom had dual-mixed virus identified before maraviroc treatment, but after screening. Resistance to maraviroc (maximum inhibition, 92.1%) was identified in 1 participant. Resistance mutations to background ARV drugs emerged in 8 participants; in 5 only minor protease inhibitor mutations were identified, whereas the remaining 3 had reverse transcriptase mutations. Poor adherence may have contributed to failure, as participants with virologic failure recorded lower adherence to both maraviroc and OBT compared with responders (data not shown).
DISCUSSION
Study A4001031 was designed to evaluate the PK in pediatric patients (≥2 to <18 years of age), allow for dosing recommendations and assess the safety of maraviroc in this population. Efficacy was a secondary endpoint, as the course of HIV infection and the effects of ARV drugs are considered sufficiently similar in pediatric and adult patients to allow for extrapolation of efficacy data.
The BSA-based dosing strategy utilized in study A4001031 achieved similar concentrations to those observed in adults receiving recommended BID doses of maraviroc, using both tablet and oral solution formulations. Forty-nine of 50 participants entering Stage 2 from Stage 1 achieved the target Cavg of ≥100 ng/mL, which was associated with near-maximal efficacy in TE adults in the MOTIVATE studies.8 The range of exposures achieved in the pediatric patients in this study fell within the ranges seen in adults receiving similar background therapies, supporting the bridging between adult and pediatric populations.
Evaluation of safety data through Week 48 demonstrated that maraviroc treatment in combination with other ARVs was generally well tolerated in these pediatric patients with a safety profile comparable to that in adults.4 Overall, the results raise no new safety concerns specific to the pediatric population and, together with the well-established safety profile in adults, support the safety of maraviroc treatment (using oral solution or tablets) as part of combination ARV treatment in HIV-1–infected children.
As this was a noncomparative study where maraviroc was administered in combination with an OBT regimen containing at least 3 other ARVs, it was not designed to evaluate efficacy. However, the virologic and immunologic efficacy, as well as mechanisms of viral escape, were similar to that seen in adult patients in the MOTIVATE studies.4,15 Lack of response appeared to be primarily driven by poor or erratic adherence, consistent with observations in other studies.2,16 The lower response rate in the adolescent patients is similar to what was observed in the DELPHI study with darunavir.16 This is not unexpected given that older children have generally been infected with HIV for a longer time and are therefore likely to have been treated for longer with a higher likelihood of archived resistance mutations.16 Furthermore, adherence to ARVs has been identified as a key concern in the adolescent population.17
In conclusion, the data from study A4001031, supported by the safety and efficacy data in adult studies, demonstrate that age-appropriate formulations/doses of maraviroc are well tolerated, and have safety and efficacy profiles similar to those seen in adults, in TE pediatric patients with CCR5-tropic HIV-1. Consistent with World Health Organization recommendations and pediatric dosing guidelines for several other ARVs,18,19 simplified weight-based dosing regimens for maraviroc in pediatric patients have recently been approved by the US Food and Drug Administration.
ACKNOWLEDGMENTS
This ongoing study is being conducted by Pfizer, Inc, and is funded by ViiV Healthcare. All authors meet the criteria for authorship set forth by the International Committee of Medical Journal Editors. Editorial support was provided by Complete Medical Communications and was funded by ViiV Healthcare. The authors would like to thank all study participants, caregivers, Data Monitoring Committee members and investigators. The invaluable contributions of the study team members (past and present) at Pfizer, Inc, ViiV Healthcare, GlaxoSmithKline and other partner organizations are acknowledged with thanks.
Supplementary Material
Footnotes
The study was sponsored by ViiV Healthcare. C.G. has received grants and personal fees from ViiV Healthcare. K.C. has received grants from Pfizer, Inc. A.F., L.M., J.H., S.R.V. and M.V. are employees of Pfizer, Inc, and may hold stock, stock options or shares in the company. E.R. has served as a consultant for Pfizer, Inc. R.Y.Z.-R. is an employee of GlaxoSmithKline and holds shares in the company. C.C. has served as a consultant for Pfizer, Inc, and ViiV Healthcare, and holds shares in GlaxoSmithKline. A.C. is an employee of ViiV Healthcare and may hold stock or stock options in the company. The other authors have no conflicts of interest to disclose.
Supplemental digital content is available for this article. Direct URL citations appear in the printed text and are provided in the HTML and PDF versions of this article on the journal’s website (www.pidj.com).
REFERENCES
- 1.Gortmaker SL, Hughes M, Cervia J, et al. ; Pediatric AIDS Clinical Trials Group Protocol 219 Team. Effect of combination therapy including protease inhibitors on mortality among children and adolescents infected with HIV-1. N Engl J Med. 2001;345:1522–1528.. [DOI] [PubMed] [Google Scholar]
- 2.Nachman S, Zheng N, Acosta EP, et al. ; International Maternal Pediatric Adolescent AIDS Clinical Trials (IMPAACT) P1066 Study Team. Pharmacokinetics, safety, and 48-week efficacy of oral raltegravir in HIV-1-infected children aged 2 through 18 years. Clin Infect Dis. 2014;58:413–422.. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Cooper DA, Heera J, Goodrich J, et al. Maraviroc versus efavirenz, both in combination with zidovudine-lamivudine, for the treatment of antiretroviral-naive subjects with CCR5-tropic HIV-1 infection. J Infect Dis. 2010;201:803–813.. [DOI] [PubMed] [Google Scholar]
- 4.Gulick RM, Lalezari J, Goodrich J, et al. ; MOTIVATE Study Teams. Maraviroc for previously treated patients with R5 HIV-1 infection. N Engl J Med. 2008;359:1429–1441.. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Cooper DA, Heera J, Ive P, et al. Efficacy and safety of maraviroc vs. efavirenz in treatment-naive patients with HIV-1: 5-year findings. AIDS. 2014;28:717–725.. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Gulick RM, Fatkenheuer G, Burnside R, et al. Five-year safety evaluation of maraviroc in HIV-1-infected treatment-experienced patients. J Acquir Immune Defic Syndr. 2014;65:78–81.. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.ViiV Healthcare. Selzentry [USA Prescribing Information]. 2016. Research Triangle Park, NC: ViiV Healthcare; Available at: https://www.gsksource.com/pharma/content/dam/GlaxoSmithKline/US/en/Prescribing_Information/Selzentry/pdf/SELZENTRY-PI-MG-IFU.PDF. Accessed November 6, 2017. [Google Scholar]
- 8.Jacqmin P, Wade JR, Weatherley B, et al. Assessment of maraviroc exposure-response relationship at 48 weeks in treatment-experienced HIV-1-infected patients in the MOTIVATE studies. CPT Pharmacometrics Syst Pharmacol. 2013;2:e64. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.US Department of Health and Human Services, National Institutes of Health, National Institute of Allergy and Infectious Diseases, Division of AIDS. Division of AIDS table for grading the severity of adult and pediatric adverse events, version 1.0 [NIH web site]. December 2004. Updated August 2009. https://rsc.tech-res.com/docs/default-source/safety/table_for_grading_severity_of_adult_pediatric_adverse_events.pdf?sfvrsn=6. Accessed February 3, 2017.
- 10.Wensing AM, Calvez V, Günthard HF, et al. 2014 update of the drug resistance mutations in HIV-1. Top Antivir Med. 2014;22:642–650.. [PMC free article] [PubMed] [Google Scholar]
- 11.Saag M, Goodrich J, Fätkenheuer G, et al. ; A4001029 Study Group. A double-blind, placebo-controlled trial of maraviroc in treatment-experienced patients infected with non-R5 HIV-1. J Infect Dis. 2009;199:1638–1647.. [DOI] [PubMed] [Google Scholar]
- 12.Rockstroh JK, Soriano V, Plonski F, et al. Hepatic safety in subjects with HIV-1 and hepatitis C and/or B virus: a randomized, double-blind study of maraviroc versus placebo in combination with antiretroviral agents. HIV Clin Trials. 2015;16:72–80.. [DOI] [PubMed] [Google Scholar]
- 13.Gona P, Van Dyke RB, Williams PL, et al. Incidence of opportunistic and other infections in HIV-infected children in the HAART era. JAMA. 2006;296:292–300.. [DOI] [PubMed] [Google Scholar]
- 14.US Department of Health and Human Services, National Institutes of Health, National Institute of Allergy and Infectious Diseases, Division of AIDS. Division of AIDS (DAIDS) table for grading the severity of adult and pediatric adverse events, version 2.0 [NIH web site]. November 2014. Available at: https://rsc.tech-res.com/docs/default-source/safety/daids_ae_grading_table_v2_nov2014.pdf?sfvrsn=8. Accessed February 3, 2017.
- 15.Westby M, van der Ryst E. CCR5 antagonists: host-targeted antiviral agents for the treatment of HIV infection, 4 years on. Antivir Chem Chemother. 2010;20:179–192.. [DOI] [PubMed] [Google Scholar]
- 16.Blanche S, Bologna R, Cahn P, et al. Pharmacokinetics, safety and efficacy of darunavir/ritonavir in treatment-experienced children and adolescents. AIDS. 2009;23:2005–2013.. [DOI] [PubMed] [Google Scholar]
- 17.Murphy DA, Sarr M, Durako SJ, et al. ; Adolescent Medicine HIV/AIDS Research Network. Barriers to HAART adherence among human immunodeficiency virus-infected adolescents. Arch Pediatr Adolesc Med. 2003;157:249–255.. [DOI] [PubMed] [Google Scholar]
- 18.World Health Organization. Consolidated guidelines on the use of antiretroviral drugs for treating and preventing HIV infection. Recommendations for a public health approach [WHO web site]. June 2013. Available at: http://apps.who.int/iris/bitstream/10665/85321/1/9789241505727_eng.pdf. Accessed February 3, 2017. [PubMed]
- 19.Centers for Disease Control. Clinical growth charts [CDC web site]. Available at: http://www.cdc.gov/growthcharts/clinical_charts.htm. Accessed February 3, 2017.