

Abstract
Although the CD20-targeted monoclonal antibody rituximab (RTX) has revolutionized the therapeutic landscape for B-cell malignancy, relapsed and refractory disease due to RTX resistance continue to constitute major challenges, illustrating the need for better therapies. Here, we apply drug-free macromolecular therapeutics (DFMT) that amplifies CD20 cross-linking to enhance apoptosis in RTX-resistant cells. Bispecific engager (anti-CD20 Fab′ conjugated with oligonucleotide1) pretargets CD20 and the deletion of Fc-region minimizes its premature endocytosis in resistant cells that rapidly internalize and consume CD20/RTX complexes. Second-step delivery of multivalent polymeric effector (linear copolymer conjugated with multiple copies of complementary oligonucleotide 2) simultaneously hybridizes multiple CD20-bound engagers and strengthens CD20 ligation. Moreover, the restoration of CD20 expression by the pretreatment of cells with a polymer-gemcitabine conjugate, a CD20 expression enhancer, unleashes the full potential of DFMT in the CD20-deficient resistant cells. Hence, amplification of CD20 cross-linking is achieved by (1) the enhancement of surface CD20 accessibility, (2) the increase in CD20 expression, and (3) multimeric CD20 binding, which ultimately translates into the amplified activation of a wide range of innate apoptotic responses. We demonstrated that the altered molecular signaling pathway that originally results in RTX resistance could be circumvented and compensated by other DFMT-augmented pathways. Of note, our preliminary data provide proof-of-concept that CD20 cross-linking amplification emerges as an important strategy for overcoming RTX resistance.
Keywords: drug-free macromolecular therapeutics, receptor cross-linking, rituximab resistance, non-Hodgkin lymphoma, HPMA copolymer
Graphical Abstract
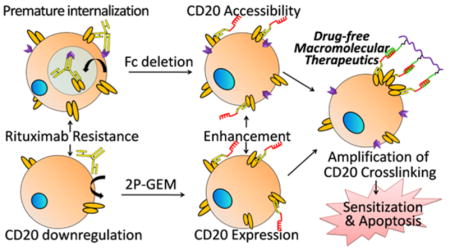
Non-Hodgkin lymphoma (NHL) is the most common hematological malignancy worldwide.1,2 Up to 90% of NHLs originate from B cells where CD20 receptors are exclusively overexpressed.3 The immunotherapy using anti-CD20 monoclonal antibody rituximab (RTX) is the current standard of care alongside conventional chemotherapy.4 However, while <10% of NHL patients receiving RTX-based therapy show complete remission, the relapsed and refractory disease continue to constitute major challenges.5
The cause of RTX resistance is multifactorial but largely ascribed to the abrogation of RTX-mediated immune response. Antibody-dependent cellular cytotoxicity (ADCC) is regarded as the most predominant mechanism underlying the effectiveness of RTX.4,6 It is initiated by CD20-bound RTX and effected by engaging Fcγ receptor (FcγR) on immune effector cells, such as macrophages and natural killer cells. Due to the FcγR polymorphisms, exhaustion of effector cells, or low FcγR phenotype, approximately 50% NHL patients fail initial RTX treatment.7 Intriguingly, while the lack of Fc-FcγR interaction is responsible for intrinsic RTX resistance, Fc-FcγR over involvement partially contributes to acquired RTX resistance. 8–11 Repeated RTX immunotherapy is susceptible to antigenic modulation, an effect described as substantial loss of CD20 from cell surface after RTX stimulation. And it can be divided into (1) “shaving” of RTX/CD20 complexes8–11 and (2) considerable down-regulation of CD20 expression.12,13 RTX/CD20 complexes “shaving” is triggered by the interaction of Fc region in RTX with Fcγ receptor on either B-lymphoma cells (which leads to rapid internalization and degradation of RTX in lysosome)9 or monocytes/macrophages (which leads to the removal of RTX/CD20 complexes from B-cell surface).8,10 Both the “endocytosis” and “trogocytosis” pathways accelerate RTX consumption before the engagement of immune effectors and helps lymphoma B cells escape onslaught from immunotherapy. Meanwhile, decreased CD20 expression results in low surface density of CD20-bound RTX, which greatly attenuates Fc-mediated ADCC.12,13 Consequently, over 50% of patients who initially respond to RTX experience relapse within 5 years, and nearly 60% of them develop resistance to RTX.14,15 As Fc-FcγR interaction plays essential roles in both of the opposing activities: ADCC and RTX resistance, simply enhancing or inhibiting Fc-mediated pathway would potentially compromise the efficacy of RTX. Hence, this dilemma urges the need for therapies that cope with RTX resistance independent of Fc-FcγR-assisted immune function.
Enhancing direct apoptosis in RTX-resistant cells represents an alternative option but still remains far from being fully exploited by RTX-based reagents. One important limitation is the fact that direct binding of RTX to CD20 is unable to induce apoptosis unless CD20 is hyper-cross-linked,16,17 which is a complicated process involving recruitment and presentation by FcγR-bearing effector cells and often appears inefficient.4 Toward this end, we have recently demonstrated that potent cross-linking of CD20 could be stimulated by drug-free macromolecular therapeutics (DFMT).18–20 The approach consists of two components: anti-CD20 Fab′ conjugated with an morpholino oligonucleotide 1 MORF1 (bispecific engager) and linear N-(2-hydroxypropyl)methacrylamide (HPMA) copolymer grafted with multiple copies of complementary morpholino oligonucleotide 2 MORF2 (effector polymer). Pretargeting of Fab′-MORF1 decorates the cells with MORF1 as a result of biorecognition between CD20 and Fab′. Further incubation with effector polymer, multivalent P-(MORF2)x, results in multiple CD20 clustering at the B-lymphoma cell surface, as a result of MORF1-MORF2 hybridization. This triggers apoptosis via calcium influx and mitochondrial pathway in vitro21 and complete eradication of RTX-sensitive lymphoma cells in vivo.22
Herein, we speculate that CD20 cross-linking amplification strategy based on our DFMT system might be especially favorable to overcome RTX resistance for at least two important attributes: (1) Fc deletion in bispecific Fab′-MORF1 engagers and (2) multivalence of polymeric P-(MORF2)x effector. The absence of Fc fragment in Fab′-MORF1 prevents FcγR-promoted “endocytosis” or “trogocytosis” of CD20, thereby maximizing the surface CD20 accessibility for subsequent cross-linking. It also avoids complement activation, an event that is activated by Fc-FcγR interaction and contributes to clinical side effects such as cytokine-release syndrome seen after the first RTX infusion.23 The multivalent linear P-(MORF2)x represents another advantage, because the random coil conformation of the conjugate facilitates to better present targeting moieties grafted to the side chains and the multivalence enables the capacity to simultaneously cross-link multiple CD20-bound engagers. Previously we have demonstrated the higher the valence, the more efficient and pronounced are CD20 cross-linking and apoptosis induction.24 We also anticipate once several closely related apoptosis signals are high enough in magnitude, then the intracellular RTX resistance due to abnormal modulation (e.g., downregulation of Bax and Bak)25 might be compensated. Thus, potentially, the two-step CD20 cross-linking amplifiers are engaged in a two-pronged effort to combat RTX resistance: circumventing Fc-related resistance pathways and confronting abnormal modulations with enhanced apoptosis, in a way that is independent of immune effector functions. At this point, sufficient surface CD20 expression to ensure the efficient pretargeting of the engagers is a prerequisite, which remains to be a bottleneck in CD20 deficient B-lymphoma cells. Alternatively, we propose a combination therapy of CD20 expression enhancers with DFMT. Gemcitabine that upregulates CD20 expression26 will be conjugated to biodegradable, long-circulating HPMA copolymer27 to ensure a sufficient contact with B-lymphoma cells. The increase in CD20 expression, paralleled with the increase in surface CD20 accessibility, is expected to restore and unleash the capability of DFMT to amplify CD20 cross-linking and eventually enhances apoptosis in RTX-resistant cells.
RESULTS AND DISCUSSION
Conjugates and Cell Lines
Bispecific Fab′-MORF1 engager is synthesized following the digestion of RTX to F(ab′)2 (removing the Fc region), reduction of F(ab′)2 to Fab′-SH, and conjugation of Fab′-SH with maleimide-functionalized MORF1 (Figure 1A). The multivalent P-(MORF2)x conjugates were synthesized following reversible addition–fragmentation chain transfer (RAFT) polymerization, side-chain modification with maleimide, and thiol–ene reaction with multiple copies of 3′-thio-modified MORF2 (Figure 1B). Gemcitabine (GEM) was attached to backbone degradable diblock HPMA copolymer via lysosome enzymatically cleavable tetrapeptide GFLG to fabricate 2P-GEM (Figure 1C). The synthesis and characterizations of these conjugates have been previously described18,21,27 and are detailed in Supporting Information, Figures S1–4.
Figure 1.

Conjugates and cell lines. Illustrative structure of (A) Cy5 unlabeled and labeled Fab′-MORF1, (B) Cy3 unlabeled and labeled P-(MORF2)x, and (C) diblock backbone degradable HPMA copolymer-gemcitabine conjugate 2P-GEM. Tetrapeptide GFLG spacer is lysosome enzymatically cleavable. (D) The highly CD20-specific binding of Fab′-MORF1-Cy5 in a co-culture of Raji-GFP cells (expressing green fluorescence) and DG-75 cells. Raji-GFP cells are characterized as CD20 positive cells, and DG-75 cells are characterized as CD20 negative cells. The mixture of Raji-GFP and DG-75 cells were exposed to 1 μM Fab′-MORF1-Cy5 for 1 h. Then Fab′-MORF1-Cy5 binding to different cells (distinguished by GFP expression) were analyzed by flow cytometry and confocal imaging (blue: nuclei; green: GFP; red: Cy5). (E) Surface CD20 expression and RTX binding (at 4 °C) in RTX-sensitive (Raji, RL, U-2932) and RTX-resistant (Raji 4RH, RL 4RH, U-2932 4RH) cell lines. Reductions in CD20 expression and RTX binding were observed in RTX-resistant Raji 4RH and RL 4RH cells, but not U-2932 4RH cells, as compared with their parental RTX-sensitive Raji, RL, and U-2932 cells, respectively.
To verify the selectivity of Fab′-MORF1 toward CD20, we investigated the targeting within a co-culture of CD20 positive Raji cells expressing green fluorescent proteins (Raji-GFP) and CD20 negative DG-75 cells. Raji-GFP and DG-75 cells were mixed and then treated with Cy5-labeled Fab′-MORF1 for 1 h. As the flow cytometry in Figure 1D showed, the majority of Raji-GFP cells were Cy5 positive, while DG-75 cells barely had Cy5 staining. This was further confirmed by confocal microscopy images where Fab′-MORF1-Cy5 mediated apparent binding toward Raji-GFP cells but not DG-75 cells. These results demonstrated that highly specific CD20 targeting function still remained after the Fab′ of RTX was conjugated with MORF1.
To verify the hybridization between the bispecific engager and effector polymer, Raji cells were treated stepwise with Fab′-MORF1-Cy5 and then P-(MORF2)11-Cy3. Confocal imaging in Supporting Information Figure S5 revealed the substantial co-localization of the two components could be significantly inhibited by the addition of excessive free MORF2 before the P-MORF2-Cy3 treatment. This observation validated the selective interaction between the bispecific engager and polymeric effector via MORF1-MORF2 hybridization.
Antigenic modulation is described as the loss of detectable antigen from the surface of a cell after incubation with antibodies.11 The resistant cell lines (Raji 4RH, RL 4RH, and U-2932 4RH) had been generated by repeated exposure of an escalating dose of RTX to their parental cells (Raji, RL, and U-2932).12 Herein, to evaluate antigenic modulation in these cells, differences in surface CD20 expressions and RTX binding between RTX-sensitive and -resistant cells were investigated (Figure 1E). As compared with Raji and RL cells, significant decreases in surface CD20 expression were observed in Raji 4RH and RL 4RH cells, respectively. Due to the lack of surface CD20 expression, Raji 4RH and RL cells experienced a profoundly restricted RTX binding. Meanwhile, U-2932 4RH only expressed a slightly lower amount of surface CD20 than U-2932 cells, and both cells had comparable RTX binding, indicating another mechanism rather than the downregulation of CD20 expression was involved underlying the RTX resistance in U-2932 4RH cells.
DFMT Amplifies CD20 Cross-Linking
Normally CD20 is a non/slow-internalizing receptor on cell surfaces, whereas the clustering of CD20 antigens within the lipid rafts can trigger their rapid intracellular internalization from the cell surface.28,29 To assess whether DFMT only self-assembles at cell surfaces or subsequently triggers CD20 cross-linking, we distinguished the intracellular DFMT following CD20 internalization from the extracellular DFMT just binding on cell surface. By using a condition-optimized protocol of co-incubating with proteinases K, we are able to shave most of the surface CD20 receptors (Figure 2A); meanwhile, it also shaves the surface bound DFMT and only DFMT that enters cells remains.
Figure 2.

DFMT amplifies CD20 cross-linking. (A) Surface CD20 antigen shaving by proteinase K: time and concentration dependence. The arrow indicates the optimized condition selected for the following studies, where co-incubation of Raji cells with 0.4 mg/mL proteinase K for 20 min is able to shave over 80% of surface CD20 receptors. (B) Comparisons of Fab′-MORF1-Cy5 internalizations in the presence and absence of P-(MORF2)11-Cy3 hybridization after surface CD20 shaving. Raji cells were consecutively treated with Fab′-MORF1-Cy5 (1 μM) for 1 h and cell culture medium/P-(MORF2)11-Cy3 (1 μM MORF2) for 5 h, prior to flow cytometry analysis. When surface CD20 is shaved by protease K degradation, the surface CD20-bound Fab′-MORF1-Cy5 is also removed, and only Fab′-MORF1-Cy5 that enters into cells remains. The upper number in flow quadrant indicates the percentage of Cy5 positive cells, while the lower number in flow quadrant indicates the percentage of Cy5 negative cells. (C) The effect of multivalence (x = 0, 3, 10) of P-(MORF2)x on the amplification of CD20 cross-linking. Raji cells were consecutively treated with Fab′-MORF1-Cy5 (1 μM) for 1 h and P-(MORF2)x (1 μM MORF2) for additional 5 h. The overall uptake (without surface CD20 shaving) and intracellular uptake (after surface CD20 shaving) of Cy5 intensity were measured by flow cytometry. β-cyclodextrin (β-CD) works as CD20 cross-linking inhibitor and greatly inhibited Fab′-MORF1-Cy5/P-(MORF2)10 internalization; Cy5-labeled Fab′-MORF1 internalization indicates the level of CD20 cross-linking. CD20 cross-linking activity of Cy5-labeled RTX cross-linked by goat antihuman (GAH) secondary antibodies was also determined.
Fab′-MORF1-Cy5 was first added to Raji cells for 1 h. Without the second-step treatment of P-(MORF2)11-Cy3, a significant reduction in Cy5 positive cell population was observed after surface CD20 shaving (Figure 2B). The corresponding removal of Fab′-MORF1 after removal of surface CD20 antigens indicated the majority of the Fab′-MORF1 engagers initially targeted surface CD20 and only remained on the plasma membrane. By contrast, no obvious reduction was observed after surface CD20 shaving when Raji cells were consecutively exposed to Fab′-MORF1-Cy5 and P-(MORF2)11-Cy3 (Figure 2B), indicating their intracellular internalization. Therefore, P-(MORF2)x effector added to the cells, bound to CD20 pretargeted Fab′-MORF1 engagers on the cell membrane, and initiated effective CD20 cross-linking.
We further investigated whether the two-step delivery amplified the CD20 cross-linking by taking advantage of the multivalence of P-(MORF2)x. Raji cells were incubated with equal concentrations of Fab′-MORF1-Cy5 for 1 h and subsequently treated with P-(MORF2)x with graded MORF2 valences (x = 0, 3, 10) for additional 5 h. Then Fab′-MORF1-Cy5 internalizations were measured by flow cytometry and compared. Results in Figure 2C show, as the MORF2 valence increased, the efficiency of Fab′-MORF1-Cy5 internalization increased, suggesting higher multivalent P-(MORF2)x could simultaneously bind to a higher number of CD20-bound Fab′-MORF1 engagers and amplify the CD20 cross-linking. In addition, the pretreatment with β-cyclodextrin (β-CD), the inhibitor of lipid rafts essential for CD20 cross-linking,30–32 significantly reduced the intracellular uptake of Fab′-MORF1-Cy5 even after the exposure to P-(MORF2)10, thus indicating good correlation between Fab′-MORF1-Cy5 internalization and CD20 cross-linking. Furthermore, Fab′-MORF1/P-(MORF2)10 mediated enhanced degrees of CD20 cross-linking as compared with RTX which was cross-linked by goat antihuman (GAH) secondary antibodies. Hence, we conclude that the two-step DFMT is an efficient CD20 cross-linking amplifier.
DFMT Avoids Premature Fc-Mediated Endocytosis
Although U-2932 and U-2932 4RH cells had comparable CD20 expressions and RTX binding, RTX/GAH could induce moderate apoptosis in U-2932 cells but not U-2932 4RH cells. In particular, DFMT not only outperformed RTX/GAH to induce apoptosis in U-2932 cells but also overcame RTX resistance in U-2932 4RH cells (Figure 3A). Intriguingly, RTX alone internalized into U-2932 4RH significantly more rapidly than U-2932 cells. Moreover, the cross-linking by GAH IgG(Fc) improved RTX internalization in U-2932, but exerted inhibiting effect in U-2932 4RH cells. Without GAH IgG(Fc) addition, only 30% of RTX (after CD20 shaving) entered U-2932 cells, whereas the percentage of internalized RTX increased to 72% in U-2932 4RH cells; with GAH IgG(Fc) cross-linking, 43% RTX entered U-2932, while 53% of RTX entered U-2932 4RH (Figure 3B). The observation in U-2932 4RH cells was in contrast to the widely accepted fact that CD20 is not internalized unless it is cross-linked. As Fcγ receptors on some resistant B lymphoma cells were reported to promote RTX internalization9 and RTX internalization in U-2932 4RH was suppressed to some extent after its Fc binding with GAH IgG(Fc), it is hypothesized that the Fc region of RTX can actively interact with the Fcγ-receptor on U-2932 4RH cells, which results in rapid CD20-RTX complex internalization. By comparison, Fab′-MORF1, without the Fc region, did not internalize into U-2932 4RH efficiently until hyper-cross-linked by P-(MORF2)10 (Figure 3C), which might explain the RTX resistance reversal after treatment of U-2932 4RH cells with DFMT.
Figure 3.

DFMT overcame RTX resistance by avoiding Fc-mediated endocytosis. (A) Apoptosis induction in U-2932 and U-2932 4RH cells after consecutive treatments with RTX (1 μM, 1 h) /GAH (1 μM, 24 h) and Fab′-MORF1 (1 μM, 24 h)/P-(MORF2)10 (1 μM MORF2 equiv, 24 h). The apoptosis level was measured by Annexin V-FITC/PI assay. (B) The overall uptake (before surface CD20 shaving) and intracellular uptake (after surface CD20 shaving) of Cy5-labeled RTX with/without cross-linking by secondary antibody goat antihuman (GAH) IgG(Fc). (C) Intracellular internalization of Cy5-labeled Fab′-MORF1 with/without P-(MORF2)10 cross-linking after surface CD20 shaving. U-2932 and U-2932 4RH cells were consecutively treated with RTX (1 μM) or Fab′-MORF1-Cy5 (1 μM) for 1 h and subsequently cell culture medium (no cross-linking), GAH IgG(Fc) (1 μM), or P-(MORF2)11-Cy3 (1 μM MORF2) for 5 h, prior to CD20 shaving and flow cytometry quantification of Cy5 intensity. (D) Confocal images of internalization and accessible surface amount of RTX, F(ab′)2, and Fab′-MORF1 in U-2932 4RH cells. U-2932 4RH cells were treated with RTX-Cy5, F(ab′)2-Cy5, and Fab′-MORF1-Cy5 for 1 h and further incubated in cell culture medium for 3 h. The surface accessible RTX and F(ab′)2 were detected by AF488-labeled GAH IgG(H+L) staining at 4 °C for 20 min. P-(MORF2)11-Cy3 was also added at 4 °C to biorecognize the surface accessible Fab′-MORF1. Red: Cy5; Green: AF488/ Cy3; Yellow: overlay of red and green. (E) CD20 cycling in U-2932 4RH cells treated with RTX/GAH and Fab′-MORF1/P-(MORF2)10. U-2932 4RH cells were first treated with RTX or Fab′-MORF1 for 1 h, then GAH IgG(Fc) or P-(MORF2)x was added. At 0, 1, 3, 6 h time intervals, the amount of free available CD20 on U-2932 4RH surface were measured.
To further verify the role of Fc fragment in promoting RTX endocytosis, U-2932 4RH cells were treated with RTX-Cy5, F(ab′)2-Cy5 (Fc-deleted RTX-Cy5) and Fab′-MORF1-Cy5 for 1 h and then further incubated in cell culture medium for 3 h. After the treatments, the GAH IgG(H+L)-AF488 secondary antibody was then used at 4 °C to detect the surface RTX, F(ab′)2, and P-(MORF2)11-Cy3 was also used at 4 °C to biorecognize Fab′-MORF1. Confocal images in Figure 3D revealed that RTX-Cy5 substantially entered into the U-2932 4RH cells, while significantly less accessible RTX-Cy5, that could be cross-linked, remained on the cell surface. In contrast, the absence of Fc fragment prevented F(ab′)2-Cy5 from rapid internalization and only resulted in the decoration of cell surface with Cy5 fluorescence. Importantly, the Fab′-MORF1-Cy5 engagers remained on the cell surface with almost no internalization. The addition of P-(MORF2)11-Cy3 effector could access to and hybridize with the CD20 pretargeted Fab′-MORF1-Cy5, as evidenced by their strong surface colocalization, indicating the ability to trigger the subsequent CD20 cross-linking. Consistently, flow cytometry (Supporting Information Figure S6) shows there was no significant difference in the overall cell uptake (before CD20 shaving) of all three groups. However, a majority of RTX-Cy5 endocytosed into the cell, while F(ab′)2-Cy5 and Fab′-MORF1-Cy5 (without Fc regions) had minimal intracellular internalization seen after CD20 shaving. Furthermore, the accessible surface localized RTX was largely limited as compared with F(ab′)2, suggesting RTX internalization was Fc-dependent.
In Supporting Information Figure S7, we show that both CD20-bound RTX-Cy5 and Fab′-MORF1-Cy5 were directed to lysosomes after cross-linked by GAH IgG(Fc) and P-(MORF2)10 for 3 h, respectively. However, different mechanisms may be involved, as indicated by monitoring the surface CD20 recovering processes after the two treatments (Figure 3E). At the beginning, the surface CD20 was partially capped by both RTX and Fab′-MORF1. After the removal of unbound RTX and subsequent addition of GAH IgG(Fc), the amount of free CD20 on cell surface increased rapidly over time and fully recovered within 6 h. This could be partially explained by the previous finding that RTX/CD20 complexes endocytosed with Fcγ receptor to compartment for uncoupling of receptor and ligand (CURL), where RTX dissociated from CD20. Then CD20 recycled back to the surface and left RTX consumed in the lysosome.4,9 Meanwhile, the recovery of surface CD20 was slow and incomplete after P-(MORF2)10 was added to Fab′-MORF1 pretargeted cells. As it is generally accepted that efficient receptor cross-linking induces receptor endocytosis into lysosomes28 and multivalent P-(MORF2)x effector could bind multiple Fab′-MORF1 engagers on the cell surface as guaranteed by the lack of Fc-Fcγ receptor interaction, this strengthened hyper-cross-linking was expected to trigger CD20 endocytosis and redirected CD20 from the recycling pathway primarily to the internalization pathway.
Collectively, U-2932 4RH cells belong to the subtype of RTX-resistant cells that rapidly takes up and consumes the RTX/CD20 complex before RTX is sufficiently cross-linked to initiate signaling pathways. We found DFMT prevents this Fc-dependent process and increases surface accessible CD20 to amplify CD20 cross-linking and apoptosis induction.
DFMT Bypasses Intracellular Abnormal Modulation and Induces Apoptosis via Calcium Influx
Having demonstrated the two-step nanotherapy of bispecific Fab′-MORF1 engager and multivalent P-(MORF2)x effector could effectively cross-link CD20 on U-2932 4RH cells, we then explored the subsequent inductions of the two major apoptotic pathways initiated by CD20 cross-linking: calcium influx and mitochondrial signaling pathways.21 DFMT, Fab′-MORF1/P-(MORF2)10, triggered calcium influx in both U-2932 and U-2932 4RH cells, while RTX could only induce slight calcium influx in U-2932 cells (Figure 4A). DFMT also exerted a higher capability of inhibiting Bcl-2 than RTX, but no significant difference was observed between two cell lines for all of the tested samples (Figure 4B). Similar results were also obtained in mitochondrial membrane potential measurements; DFMT triggered higher degrees of mitochondrial depolarization than RTX in both U-2932 and U-2932 4RH cells (Figure 4C). Intriguingly, U-2932 4RH cells showed an extremely low level of cytochrome c release regardless of the treatments (Figure 4D). The lack of cytochrome c release even after obvious mitochondrial depolarization might block the initiation of caspase-activated apoptosis and account for another reason for RTX resistance. However, although to a lesser extent than in U-2932 cells, DFMT was still able to activate the caspase 3 in U-2932 4RH cells, while RTX failed (Figure 4E). Recent findings have indicated several pathways that lead directly from a calcium signal to caspase activation and apoptosis.33 To validate the participation of calcium influx in caspase activity, U-2932 4RH cells were pretreated with lipid raft inhibitor β-CD (inhibiting CD20 cross-linking) or Ca2+ chelating agent ethylene glycol-bis(β-aminoethyl ether)-N,N,N′,N′-tetraacetic acid (EGTA)34 which had significantly reduced the calcium influx after DFMT treatment (Supporting Information Figure S8). Indeed, the caspase 3 activities were markedly inhibited in the presence of β-CD inhibitor or EGTA chelator (Figure 4E), demonstrating important involvement of calcium signal pathway to trigger apoptosis.
Figure 4.

Comparisons of intracellular signals: (A) calcium influx, (B) Bcl-2 expression, (C) mitochondrial depolarization, (D) cytochrome c release, and (E) caspase 3 activation in U-2932 and U-2932 4RH cells in response to PBS, RTX (1 μM, 1 h)/GAH (1 μM, 24 h) and Fab′-MORF1 (1 μM, 24 h)/P-(MORF2)10 (1 μM MORF2 equiv, 24 h). (F) Proposed schematic illustration of overcoming RTX resistance by DFMT.
In summary, these data revealed the potential RTX resistance in U-2932 4RH cells could be ascribed to (1) Fc-mediated rapid RTX/CD20 endocytosis that resulted in insufficient CD20 cross-linking and (2) cytochrome c deficiency that blocked mitochondrial pathway related apoptosis. In addition, DFMT overcame RTX resistance through: (1) CD20 cross-linking restoration due to the absence of Fc region in Fab′-MORF1; (2) CD20 cross-linking amplification due to the multivalence of P-(MORF2)x; and (3) bypassing the cytochrome c deficiency and inducing apoptosis via calcium influx mediated caspase activation (Figure 4F).
CD20 Upregulation by 2P-GEM Restores CD20 Cross-Linking by DFMT
As validated above, low CD20 expression was observed in Raji 4RH and RL 4RH cells. Although DFMT induced considerably superior apoptosis over RTX/GAH in Raji cells because of CD20 cross-linking amplification, both treatments had negligible therapeutic effects on CD20-deficient Raji 4RH (Supporting Information Figure S9) and RL 4RH cell lines (Supporting Information Figure S10). Due to the lack of surface CD20 expression, DFMT failed to trigger calcium influx (Supporting Information Figure S11) as a result of significantly reduced CD20 cross-linking (Supporting Information Figure S12). Herein, gemcitabine (GEM) was employed to enhance the surface CD20 expression.26 After Raji 4RH and RL 4RH cells were treated with a range of GEM concentrations for variable incubation times, the surface CD20 expressions indeed increased as time prolonged, but the increment did not correlate well with the drug concentrations (Figure 5A). This result indicated sufficient exposure time of GEM rather than the dosage might be a more important factor to upregulate CD20. Hence, in order to prolong the retention of GEM in bloodstream and increase its chance to contact with the lymphoma B cells during circulation, GEM was conjugated onto the long-circulating, backbone-degradable HPMA copolymer forming the nanoconjugate 2P-GEM. Previously, we demonstrated 2P-GEM (32.07 h) had a drastically prolonged half-life as compared with GEM (1.2 h) in mice.27
Figure 5.

2P-GEM restores CD20 cross-linking by DFMT by upregulating CD20 expression. (A) Effect of GEM on surface CD20 upregulation in Raji 4RH and RL 4RH cells. Raji 4RH and RL 4RH cells were incubated with a series concentrations of GEM ranging from 50 to 1000 ng/ mL for 12, 24, and 48 h. After treatments, the CD20 expression on cell surface was investigated. (B) Apoptosis induction in Raji 4RH and RL 4RH cells after sequential treatment of 2P-GEM (100 ng/mL GEM equivalent, 48 h) and DFMT, Fab′-MORF1 (1 μM, 1 h)/P-(MORF2)10 (1 μM MORF2, 24 h). (C) Confocal images of Fab′-MORF1-Cy5 (1 μM, 1 h)/P-(MORF2)x-Cy3(1 μM MORF2, 1 h) in the absence and presence of 2P-GEM pretreatment (100 ng/mL GEM equivalent, 48 h) in Raji 4RH and RL 4RH cells. Blue: nucleus; Red: Cy5; Green: Cy3. (D) Calcium influx in Raji 4RH and RL 4RH cells after sequential treatment of 2P-GEM and DFMT as above.
As shown in Figure 5B, when Raji 4RH and RL 4RH cells were sequentially treated with 2P-GEM and DFMT, the apoptosis induction increased markedly as compared with the cells treated with 2P-GEM or DFMT alone. It also triggered higher apoptosis than the combination of 2P-GEM and RTX/ GAH, demonstrating the superiority of DFMT over RTX. To assess whether 2P-GEM restored and amplified the CD20 cross-linking after DFMT treatment, Raji 4RH and RL 4RH cells were pretreated with 2P-GEM followed by the consecutive treatment of Fab′-MORF1-Cy5 and P-(MORF2)11-Cy3. As confocal imaging (Figure 5C) revealed, in the absence of 2P-GEM pretreatment, exposure to Fab′-MORF1-Cy5 only results in surface decoration of weak Cy5 fluorescence, due to the deficient CD20 in these cells. 2P-GEM markedly facilitated Fab′-MORF1-Cy5 binding to cell surface, because of its ability to upregulate CD20 expression (Supporting Information Figure S13). Consequently, substantial co-localization with second-step multivalent P-(MORF2)11-Cy3 was accomplished (Figure 5C). In addition, the Förster resonance energy transfer (FRET) signal (generated by the close proximity of Cy5 to Cy3) within the cells, indicative of self-assembly of Fab′-MORF1-Cy5 and P-(MORF2)11-Cy3 at CD20 receptor, was profoundly augmented in the cells pretreated with 2P-GEM as compared with the cells that were not pretreated (Supporting Information Figure S14). The calcium influx study (Figure 5D) further suggested that amplification of the CD20 cross-linking after sequential combination treatment of 2P-GEM and DFMT. CD20 acts as a store-operated calcium channel and cross-linking CD20 leads to the extracellular calcium influx.30,31 2P-GEM or DFMT itself only had negligible effect to elicit calcium influx, as a result of lack of surface CD20. Impressively, the sequential combination of 2P-GEM and DFMT dramatically enhanced calcium influx and even generated significant improvement as compared with the combination of 2P-GEM and RTX. This effect correlated with the finding that CD20 upregulation by 2P-GEM restored the CD20 pretargeting function of Fab′-MORF1 and eventually amplified CD20 cross-linking by multivalent P-(MORF2)x effector.
Mutual Potentiation between 2P-GEM and DFMT
Given that CD20 gene promoter has the binding sites to NF-κB that can be activated by GEM,26,35 we then investigated whether 2P-GEM could increase the CD20 expression through NF-κB activating signaling. As shown in Figure 6A, 2P-GEM dramatically enhanced the whole cell expression of CD20 and NF-κB in both Raji 4RH and RL 4RH cell lines as compared with the untreated control, whereas co-incubation with NF-κB inhibitor sulfasalazine36 offset this augmentation. Although NF-κB activation participated in CD20 upregulation, NF-κB signaling is frequently associated with multidrug resistance or other side effects like inflammatory disease.36 Herein, it is important to note that RTX was previously demonstrated to inhibit the NF-κB signaling after binding to CD20 followed by activating Src family kinase and p38 MAPK pathway,4 and we found DFMT maintained this function of RTX. Impressively, sequential delivery of 2P-GEM and DFMT displayed “firstincrease-then-decrease” pattern in NF-κB signaling (Figure 6B) that might potentially sensitize cells to both DFMT (via CD20 upregulation) and GEM (via NF-κB downregulation) while overcoming/minimizing drug resistance.
Figure 6.

Mutual potentiation between 2P-GEM and DFMT. (A) Effect of 2P-GEM on NF-κB signaling activation and CD20 expression. Raji 4RH and RL 4RH cells were treated with 2P-GEM (100 ng/mL GEM equivalent, 48 h) in the presence and absence of 0.1 mM sulfasalazine (NF-κB inhibitor). Whole cell expressions of CD20 and NF-κB were measured by immunostaining and flow cytometry. (B) NF-κB signaling activation and deactivation, (C) cell cycle distribution, (D) Bcl-2/Bax ratio detection, after Raji 4RH and RL 4RH cells were treated with sequential combination of 2P-GEM(100 ng/mL GEM equivalent, 48 h) and DFMT, Fab′-MORF1 (1 μM, 1 h) /P-(MORF2)10 (1 μM MORF2, 24 h). (E) Schematic illustration of overcoming RTX resistance following the sequential treatment.
The major mechanism of action of nucleoside analogues like GEM is through incorporation into DNA, and thus this class of agents shows specificity for cell cycle arrest in S-phase.37 Our results (Figure 6C) showed 2P-GEM could indeed elevate cell population in the S phase, while DFMT had minimal impact on cell cycle. In addition, the sequential combination of 2P-GEM and DFMT could further induce the cell arrest in S-phase, to a higher extent than 2P-GEM alone. This might be due to the fact that NF-κB inhibits the nuclear transporter of GEM, and NF-κB downregulation by DFMT facilitated the localization and DNA intercalation of GEM.
The question whether sequential delivery of 2P-GEM and Fab′-MORF1/P-(MORF2)x could enhance programmed cell death in CD20-deficient RTX-resistant cells was further investigated. The significantly reduced expression in Bcl-2 and Bax accounts for another reason for Raji 4RH and RL 4RH cells resistance.25 When 2P-GEM and DFMT were combined, the ratio of pro-apoptotic Bax to antiapoptotic Bcl-2 increased drastically as compared with the monotherapy (Figure 6D), thus resetting and increasing the cell susceptibility to apoptosis. The enhancement also occurred in inducing the mitochondrial depolarization, which subsequently triggered cytochrome c release and activation of caspase 3 (Supporting Information Figure S15). It should be noted that RTX or DFMT treatment alone had no or only a slight effect on these apoptosis related signaling pathways, due to the lack of surface CD20. Meanwhile, 2P-GEM monotherapy also generated limited alterations, reflecting the multidrug resistance in these cells, probably as a result of down-regulation of pro-apoptotic proteins including Bax. However, combinatorial apoptotic effect after sequential dual-strike therapy of 2P-GEM and DFMT was achieved by their mutual potentiation (Figure 6E). Pretreatment with 2P-GEM acts as surface CD20 expression enhancer. This reserves the prerequisite for receptor-cross-linking mediated apoptosis. Second wave treatment with DFMT exceedingly outperformed the combination with RTX, highlighting the superiority of the multivalence to amplify the CD20 cross-linking. The stronger CD20 cross-linking effect stimulates a cascade of apoptotic events that eventually lead to the upregulation of pro-apoptotic proteins Bax and inhibition of NF-κB, which in turn sensitizes cells to gemcitabine.
In Vivo Validation of Therapeutic Efficacy
To further examine the potential therapeutic effects of the combination of CD20 expression enhancer and CD20 cross-linking amplifier in vivo, NOD/SCID IL2R γnull(NSG) mice bearing systemically disseminated Raji 4RH RTX-resistant lymphomas were sequentially treated with GEM or 2P-GEM on Day 1 and RTX or DFMT (Fab′-MORF1/P-(MORF2)10) on Day 3, as indicated in Figure 7A. When untreated, Raji 4RH lymphomas in the bloodstream of mice would grow, disseminate, and infiltrate in various organs, especially the spinal cord that leads to the onset of hind-limb paralysis and animal death. Thus, the hind-limb paralysis-free survival time after treatment and the presence of residual Raji 4RH cells in bone marrow (BM) would accurately reflect anticancer efficacy.
Figure 7.

In vivo validation of therapeutic efficacy. (A) Schedule of the treatments. (B) Paralysis-free survival and (C) body weight of mice after the treatments (n = 4–5). (D) Flow cytometry analysis of residual Raji 4RH cells (human CD10+CD19+) in the bone marrow (BM). BM cells were isolated from the femur of mice at end point (onset of paralysis), and Raji 4RH cells were dual stained with PE-labeled mouse antihuman CD10 and APC-labeled mouse antihuman CD19 antibodies. Three mice from 2P-GEM → DFMT treatment group that did not undergo paralysis were also sacrificed for antilymphoma efficacy comparison at days 27, 43, and 43, respectively.
As shown in Figure 7B, PBS treated mice paralyzed within 22 days, as expected. The combination therapy of GEM and RTX delayed the cancer progression to a limited degree, but no mice survived over 30 days. 2P-GEM alone exhibited a similar trend as GEM → RTX, and appeared ineffective (all the mice died within 30 days). Impressively, while GEM → DFMT treatment further extended the animal survival by several days (all the mice died within 35 days), no mice receiving the 2P-GEM → DFMT treatment developed paralysis during the 40 days treatments: two mice died at Day 43 but without the onset of paralysis, and still one mouse remained alive over 70 days post-treatment. It should be noted that this is a preliminary in vivo result and the mice were only treated with single dose. To further improve the survival time, the drug dosage and treatment frequency need to be optimized. In addition, no significant weight loss was observed after the injection of 2P-GEM → DFMT (Figure 7C). After mice were sacrificed at their end point (onset of paralysis), BM cells were isolated from the femur, and flow cytometery was used to analyze the residual Raji 4RH B-lymphoma cells (human CD10+CD19+). By comparison, three mice from 2P-GEM → DFMT treatment group that did not undergo paralysis were also sacrificed for the antilymphoma analysis. Results in Figure 7D and Supporting Information Figure S16 show that, compared with other groups of paralyzed mice that bore variably significant amounts of Raji 4RH cells in BM, there was a dramatic decrease of Raji 4RH cells disseminated in BM after treatment of 2P-GEM → DFMT. This result was consistent with the observation that no paralysis occurred in 2P-GEM → DFMT treated mice.
The synergism between RTX and GEM has been demonstrated against the RTX-sensitive NHLs in a previous study.38 However, our finding provides other important implications. First, we used a significantly less dose of GEM (a single dose of 5 mg/kg versus three doses of 120 mg/kg), which is safe and clinically achievable. Second, we conjugated GEM to biodegradable, long-circulating polymer (2P-GEM versus GEM) and used a reversed administration sequence (2P-GEM → DFMT versus RTX → GEM). The pretreatment of 2P-GEM takes the advantage of the ability of GEM to upregulate the CD20 expression, and attaching GEM to 2PGEM prolongs the systemic circulation as well as increases the contact with the Raji 4RH cells thereby maximizing the CD20 upregulation. This is also supported by the superior therapeutic efficacy of 2P-GEM → DFMT over GEM → DFMT in our study. Third, we demonstrated the feasibility of the combination of 2P-GEM and DFMT on RTX-resistant lymphomas (versus RTX-sensitive lymphomas). CD20 expression enhancement by 2P-GEM restores the full potential of DFMT to amplify CD20 cross-linking and eventually enhances antitumor outcome.
CONCLUSIONS
The platform applied herein demonstrates RTX resistance could be overcome using CD20 cross-linking amplifying strategy, which exploits distinctive mechanisms: (1) increasing surface CD20 accessibility (Fc deletion in Fab′-MORF1), (2) increasing surface CD20 expression (pretreatment of 2P-GEM) and (3) increasing surface CD20 ligation (multivalent binding by P-(MORF2)x).
FcγR is implicated in RTX resistance for getting involved either too little (immune effector abrogation) or too much (CD20/RTX “endocytosis” and “trogocytosis”). Recently, efforts have been made to combine RTX with FcγR-blocking antibodies.39 However, FcγR is expressed on both targeted B cells where FcγR-blocking antibody prevents RTX internalization, and on key immune effector cells (e.g., NK cells and macrophages) where FcγR-blocking antibody dampens RTX therapy. Our findings here show DFMT system can address this dilemma in two aspects: reducing the endocytosis while enhancing the apoptosis. Fc-region is deleted in Fab′-MORF1, thereby cutting off FcγR-assisted internalization and maximizing its cell surface accessibility. After pretargeting CD20, Fab′-MORF1 remains dormant on the surface until biorecognition with P-(MORF2)x. Then the multivalence of P-(MORF2)x facilitates the hybridization with multiple CD20-bound engagers and strengthens CD20 cross-linking to augment apoptosis, in a way that is independent of immune functions.
CD20 down-regulation confers another mechanism of RTX resistance. It is no surprise that DFMT appear less effective in the resistant cells that have a low density of surface CD20, but this can be conquered by the pretreatment with 2P-GEM. First, 2P-GEM acts as CD20 expression enhancer. Then, our data reveal the increased surface CD20 expression restores the therapeutic efficacy of DFMT both in vitro and in vivo. Most definitively, amplification of CD20 cross-linking by DFMT stimulates a cascade of apoptotic events including the Bax upregulation and NF-κB inhibition, which in turn sensitize cells to 2P-GEM. This provides evidence that the mutual potentiation of CD20 expression enhancer and CD20 cross-linking amplifier can recapitulate the enhanced apoptosis in RTX-resistant lymphomas.
An additional feature of DFMT system is biomimetic and simultaneous cross-linking of multiple CD20. In contrast to RTX which only triggers limited ligation of CD20 and requires the complicated processes of recruitment and stimulation of effector cells, the cell surface hybridization of CD20-bound Fab′-MORF1 and multivalent P-(MORF2)x is more specific, direct, and potent in clustering numerous CD20. Eventually, this extracellular amplification of CD20 cross-linking translates into a great diversity of enhanced innate responses. Even when a particular pathway is blocked by intracellular abnormal modulation (e.g., cytochrome c deficiency) that originally results in RTX resistance, it can be circumvented and compensated by other DFMT-augmented pathways (e.g., calcium influx and caspase activation).
In summary, the amplification of CD20 cross-linking using an immune-function-independent nanotechnology would be of therapeutic interest to combat RTX resistance. Our platform might be applicable to RTX resistance associated with immune effector dysfunction, CD20 loss, and molecular signal alteration. We might also expand the utility in other B-cell-assisted diseases (e.g., rheumatoid arthritis) and apply this nanotechnology to cross-link a broad range of other non/slowly internalizing receptors (e.g., CD45, death receptor) and amplify their receptor-mediated apoptotic signaling.
MATERIALS AND METHODS
Synthesis and Characterizations
Fab′-MORF1, Fab′-MORF1-Cy5, and polymeric effectors with different valences (P-(MORF2)x and P(-(MORF2)x-Cy3, x = 0, 3, 10, and 11) were synthesized and characterized as previously reported.21 2P-GEM was synthesized and characterized based on previous publication.27 Details are included in the Supporting Information.
Surface CD20 Expression Measurement
Raji, Raji 4RH, RL, RL 4RH, U-2932, and U-2932 4RH cells were cultured in RPMI-1640 medium (Gibco) containing 10% fetal bovine serum (FBS) and a mixture of 0.1 mg/mL streptomycin and 100 units/mL penicillin at 37 °C in a humidified 5% CO2 atmosphere. Surface expression of CD20 antigens on these cells were evaluated by flow cytometry, according to previous report.12 Briefly, 2 × 105 cells were incubated with 10 μg/mL anti-CD20 RTX on ice for 15 min. After washing with 400 μL cold PBS twice and blocking with 400 μL 3% bovine serum albumin (BSA) solution, cells were then resuspended in 3% BSA containing Alexa Fluor-488 goat antihuman IgG(H+L) secondary antibody (1:200) for 15 min in the dark on ice. Afterward, cells were washed with 400 μL cold PBS three times, prior to flow cytometry quantification of Alexa Fluor-488 fluorescence intensity.
Fab′-MORF1 Specificity Investigation
Surface CD20 expressions in Raji-GPF and DG-75 cells were measured by incubating cells with 10 μg/mL RTX-Cy5 for 15 min in the dark on ice. After washing with 400 μL cold PBS twice, Cy5 fluorescence was measured by flow cytometry. For the investigation of Fab′-MORF1 specificity, 8 × 104 Raji-GPF cells (CD20 positive) were mixed and co-cultured with a higher number of 12 × 104 DG-75 cells (CD20 negative). Then 1 μM of Fab′-MORF1-Cy5 was added and incubated with cells in 400 μL cell culture medium for 1 h. Afterward, cells were washed with 400 μL cold PBS twice before the flow cytometry analysis using 488 nm excitation with 530 nm bandpass filter (BLUFL1 channel, for GFP detection) and 640 nm excitation with 670/30 nm bandpass filter (REDFL1 channel, for Cy5 detection). Fab′-MORF1-Cy5 binding in Raji-GPF and DG-75 cells were distinguished by GFP expression. For confocal microscopy imaging, the nuclei of the cells were stained with 5 μg/mL Hoechst 33392 (Thermo Scientific) for 5 min. Then the cells were washed with 400 μL cold PBS three times and resuspended in 400 μLPBS in 4 well chambers prior to confocal visualization.
Hybridization of Fab′-MORF1 and P-MORF2
Two × 105 Raji cells were first treated with 1 μM Fab′-MORF1-Cy5 for 1 h at 37 °C. Then Fab′-MORF1 solution was removed, and cells were washed with 400 μL cold PBS twice. Raji cells were further incubated with P-(MORF2)11-Cy3 (1 μM MORF2 equiv) for 1 h at 37 °C. Afterward the nuclei of the cells were stained with 5 μg/mL Hoechst 33342 at room temperature for 5 min. Then the cells were washed with 400 μL cold PBS three times and resuspended in 400 μL PBS in 4 well chambers prior to confocal visualization. For MORF2 preblocking, Raji cells were pretreated with excess (10 μM) of free MORF2 for 20 min at 37 °C before co-incubation with P-(MORF2)11-Cy3 for 1 h followed by the same procedures.
Surface CD20 Shaving Protocol
After stepwise treatment with Fab′-MORF1-Cy5 (1 μM MORF1 equiv, 1 h) and then P-(MORF2)x-Cy3/P-(MORF2)x (1 μM MORF2 equiv, 5 h), cells were washed twice with 400 μL cold PBS. Afterward, surface CD20 were degraded with 0.4 mg/mL proteinase K (Thermo Scientific) for 20 min at 37 °C. Then, proteinase K was neutralized with the same volume of cell culture medium containing 10% FBS, and cells were washed with 400 μL PBS twice, prior to flow cytometry analysis for intracellular fluorescence of Cy5.
Apoptosis Assay
Annexin V-FITC and PI staining were performed following the RAPID protocol provided by the manufacturer (Oncogene Research Products, Boston, MA). 2.0 × 105 cells pretreated with 2P-GEM (100 ng/mL GEM equivalent) for 48 h were treated with cell culture medium (control), RTX (1 μM, 1 h)/GAH (1 μM, 24 h), or consecutive treatment of Fab′-MORF1 (1 μM, 1 h)/P-(MORF2)10 (1 μM MORF2, 24 h) at 37 °C. Then cells were washed with 400 μL PBS twice and resuspended in 400 μL binding buffer. Cells were stained with 5 μL Annexin V-FITC and 5 μL propidium iodide (PI) solution provided in the kit in dark for 15 min, followed by flow cytometry analysis. All experiments were carried out in triplicate.
Calcium Influx Investigation
After the treatments, 4 × 105 cells were loaded with intracellular calcium indicator Fluo-3AM (5 μM, Thermo Scientific) in 100 μL 1640 cell culture medium (containing 2.5 mM Ca2+) for 30 min at 37 °C. Then flow cytometry was applied, and the fluorescent intensity of intracellular calcium was measured by exciting cells at 488 nm and detecting the emission at 530 nm.
Fc-Dependent Endocytosis Investigation
2.0 × 105 U-2932 4RH cells were treated with 400 μL RTX-Cy5, F(ab′)2-Cy5 and Fab′-MORF1-Cy5 (1 μM Fab′ equivalent) solutions at 37 °C for 1 h. Then cells were washed twice with 400 μL cold PBS to remove the unbound samples and further incubated in cell culture medium at 37 °C for 3 h. Afterward, the surface CD20 on the treated U-2932 4RH cells was either shaved by CD20 shaving protocol (for the investigation of intracellular internalization) or not shaved (for the investigation of overall uptake), prior to flow cytometry analysis for Cy5 intensity. For the assessable surface amount of antibody, U-2932 4RH cells were treated with RTX and F(ab′)2 for 1 h and then further incubated in cell culture medium for 3 h. The GAH IgG(H+L)-AF488 secondary antibody (1:200) was added at 4 °C for 20 min to detect the surface RTX and F(ab′)2 by flow cytometry. For confocal imaging, U-2932 4RH cells were treated with RTX-Cy5, F(ab′)2-Cy5 and Fab′-MORF1-Cy5 for 1 h and then further incubated in cell culture medium for 3 h as above. Afterward, the GAH IgG(H+L)-AF488 secondary antibody (1:50) was used to detect RTX-Cy5 and F(ab′)2-Cy5, and P-(MORF2)11-Cy3 (1 μM MORF2) was used to biorecognize Fab′-MORF1-Cy5 at 4 °C for 20 min, before confocal visualization.
Lysosome Localization Investigation
After U-2932 4RH cells were treated with RTX-Cy5 (1 μM, 1 h)/GAH (1 μM, 3 h) or Fab′-MORF1 (1 μM, 1 h) /P-(MORF2)10-Cy3 (1 μM, 3 h), the lysosomes of the cells were stained with the lysotracker green DND-26 (Thermo Scientific) at 37 °C for 15 min. Then the cells were washed with PBS and resuspended in PBS in 4 well chambers prior to confocal visualization.
Surface CD20 Restoration Investigation
Two ×105 U-2932 4RH cells were incubated with RTX or Fab′-MORF1 (1 μM Fab′ equivalent) at 37 °C for 1 h. Then the unbounded RTX or Fab′-MORF1 was removed, and the cells were incubated with GAH (1 μM) or P-(MORF2)10 (1 μM) for 0, 1, 3, and 6 h. After that, the cell surface CD20 amount was measured as described above.
Mitochondrial Membrane Potential Study
JC-1 mitochondrial membrane potential sensor (Thermo Scientific) was used. After indicated treatments, 2 × 105cells were washed by 400 μL PBS twice and resuspended in 100 μL PBS. Two μL of 200 μM JC-1 was added, and the cells incubated at 37 °C for 30 min. For the positive control group, 1 μL of 50 mM carbonyl cyanide m-chlorophenylhydrazone (CCCP) obtained in the kit was added and incubated simultaneously with JC-1 for 30 min. After washed by PBS twice, cells were resuspended in PBS and analyzed by flow cytometry using 488 nm excitation with 530/30 nm and 585/42 nm band-pass filters. All experiments were carried out in triplicate.
Bcl-2 and Bax Detection
After indicated treatments, the cells were sequentially fixed by 4% paraformaldehyde for 15 min at room temperature, permeabilized by 90% methanol for 30 min on ice, and immunostained by Alexa Fluor 488 conjugated Bcl-2 mAb (1:50, Santa Cruz Biotechnology) and AF647 conjugated Bax mAb (1:50, Santa Cruz Biotechnology) in 3% BSA buffer for 1 h at room temperature. After washing by cold PBS twice, the fluorescence was quantified by flow analysis. All experiments were carried out in triplicate.
Cytochrome c Release Measurement
After the treatments, the cells were collected by centrifugation. Then cells were washed twice by cold PBS, and the cell pellet was lysed by cell extraction buffer (with 1 mM PMSF (phenylmethane sulfonyl fluoride) and 1:100 protease inhibitor cocktail) at 100 μL of 2.0 × 105 cells, for 30 min on ice. Then the extract was transferred to microcentrifuge tube and centrifuged at 13,000 rmp for 10 min at 4 °C. The supernatant was collected for the measurement. The activity was measured using an ELISA kit (R&D Systems) according to manufacturer’s instructions. All experiments were carried out in triplicate.
Caspase 3 Activity Evaluation
A Phi-PhiLux kit (OncoImmunin, Gaithersburg, MD) was used. After treatments cells were washed by PBS and analyzed for caspase-3 activity following the manufacturer’s protocol. All experiments were carried out in triplicate.
Correlation of NF-κB Activation and CD20 Expression
Two ×105 Raji 4RH or RL 4RH cells were treated at 37 °C with 2P-GEM (100 ng/mL GEM equivalent) for 48 h in the presence and absence of NF-κB inhibitor sulfasalazine (0.1 mM or 0.01 mM). Cells treated with PBS only were referred as control. For surface CD20 detection, cells were stained with 10 μg/mL RTX-Cy5 for 20 min at 4 °C. For the detection of whole cell NF-κB and CD20 expressions, cells were fixed by 4% formaldehyde (room temperature, 15 min), permeabilized by 90% methanol (on ice, 30 min), and double immnostained by NF-κB p65 Alexa Fluor 405 Antibody (1:100) and RTX-Cy5 (room temperature, 1 h). Then cells were washed with cold PBS twice, and the fluorescence was quantified by flow analysis.
Cell Cycle Investigation
After the treatments, cells were collected and fixed with 70% alcohol overnight. Cells were stained for 30 min with propidium iodide (PI, 50 μg/mL) in the presence of 1% Rnase A at 37 °C and analyzed by flow cytometry.
In Vivo Therapeutic Efficacy
8-week NOD/SCID IL2R γnull (NSG) mice were intravenously injected with 4 × 106 Raji 4RH cells in 200 μL PBS via the tail vein (day 0). At day 1, a signal dose of 100 μL PBS, GEM (5 mg/kg), 2P-GEM (5 mg/kg GEM equivalent) was given intravenously to the randomly divided mice groups (n = 4–5). At day 3, RTX (1 nmol) or DFMT (1 nmol MORF1/5 nmol P-(MORF2)x with 5 h interval) was intravenously administered into the mice. The body weights of the mice were measured thereafter. The onset of hind-limb paralysis was the experimental end point; in addition, mice were sacrificed when body weight loss was >20%. All animal experiments were performed according to the protocol approved by the Institutional Animal Care and Use Committee (IACUC) of the University of Utah.
Flow Cytometry Analysis of Residual Raji 4RH Cells in Bone Marrow
After mice were sacrificed, fresh femurs from both hind limbs were purged with 5 mL PBS to obtain bone marrow cells. The suspension was filtered through a 70 μm FalconTM cell strainer, followed by centrifugation. Cell pellets were resuspended in 500 μL red blood cell lysis buffer. The suspension was incubated at room temperature for 5 min and washed/resuspended in cold PBS. Allophycocyanin (APC)-labeled mouse antihuman CD19 antibody (10 μL) and PE-labeled mouse antihuman CD10 antibody (10 μL) (BD Biosciences, San Jose, CA) were added to 100 μL single-cell suspension. Cells were incubated for 20 min at 4 °C in the dark and washed with PBS prior to analysis. For flow cytometry, data of 5 × 105 cells were recorded. Raji 4RH cells served as positive control.
Supplementary Material
Acknowledgments
The work was supported in part by NIH grant RO1 GM95606 (to J.K.) from the National Institute of General Medical Sciences, University of Utah Research Foundation, and Bastion Biologics. We acknowledge support of funds in conjunction with grant P30 CS042014 awarded to Huntsman Cancer Institute and to the ET Program at Huntsman Cancer Institute. We thank Drs. F. J. Hernandez-Ilizaliturri and M. S. Czuczman from the Roswell Park Cancer Institute for the kind gift of rituximab-resistant cell lines.
Footnotes
Notes
The authors declare the following competing financial interest(s): J.Y. and J.K. are co-inventors on a pending US patent application (PCT/US2014/023784; assigned to the University of Utah and licensed to Bastion Biologics) related to this work. J.K. is Chief Scientific Advisor and J.Y. is Scientific Advisor for Bastion Biologics. Otherwise, the authors declare no competing financial interests.
ASSOCIATED CONTENT
The Supporting Information is available free of charge on the ACS Publications website at DOI: 10.1021/acsnano.8b00797.
Synthesis and characterization of nanoconjugates; supplementary figures; imaging and mechanistic studies (PDF)
References
- 1.Siegel RL, Miller KD, Jemal A. Cancer Statistics, 2016. Ca-Cancer J Clin. 2016;66:7–30. doi: 10.3322/caac.21332. [DOI] [PubMed] [Google Scholar]
- 2.Torre LA, Bray F, Siegel RL, Ferlay J, Lortet-Tieulent J, Jemal A. Global Cancer Statistics, 2012. Ca-Cancer J Clin. 2015;65:87–108. doi: 10.3322/caac.21262. [DOI] [PubMed] [Google Scholar]
- 3.LeBien TW, Tedder TF. B Lymphocytes: How They Develop and Function. Blood. 2008;112:1570–1580. doi: 10.1182/blood-2008-02-078071. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Seyfizadeh N, Seyfizadeh N, Hasenkamp J, Huerta-Yepez S. A Molecular Perspective on Rituximab: A Monoclonal Antibody for B Cell Non-Hodgkin Lymphoma and Other Affections. Crit Rev Oncol Hemat. 2016;97:275–290. doi: 10.1016/j.critrevonc.2015.09.001. [DOI] [PubMed] [Google Scholar]
- 5.Molina A. A Decade of Rituximab: Improving Survival Outcomes in Non-Hodgkin’s Lymphoma. Annu Rev Med. 2008;59:237–250. doi: 10.1146/annurev.med.59.060906.220345. [DOI] [PubMed] [Google Scholar]
- 6.Clynes RA, Towers TL, Presta LG, Ravetch JV. Inhibitory Fc Receptors Modulate In Vivo Cytoxicity against Tumor Targets. Nat Med. 2000;6:443–446. doi: 10.1038/74704. [DOI] [PubMed] [Google Scholar]
- 7.Taylor RP, Lindorfer MA, Zent CS. Anti-CD20 Antibody Therapy for B-cell Lymphomas. N Engl J Med. 2012;367:876–878. doi: 10.1056/NEJMc1207378. [DOI] [PubMed] [Google Scholar]
- 8.Beum PV, Kennedy AD, Williams ME, Lindorfer MA, Taylor RP. The Shaving Reaction: Rituximab/CD20 Complexes are Removed from Mantle Cell Lymphoma and Chronic Lymphocytic Leukemia Cells by THP-1 Monocytes. J Immunol. 2006;176:2600–2609. doi: 10.4049/jimmunol.176.4.2600. [DOI] [PubMed] [Google Scholar]
- 9.Lim SH, Vaughan AT, Ashton-Key M, Williams EL, Dixon SV, Chan HC, Beers SA, French RR, Cox KL, Davies AJ, Potter KN, et al. Fc Gamma Receptor IIb on Target B Cells Promotes Rituximab Internalization and Reduces Clinical Efficacy. Blood. 2011;118:2530–2540. doi: 10.1182/blood-2011-01-330357. [DOI] [PubMed] [Google Scholar]
- 10.Beers SA, French RR, Chan HTC, Lim SH, Jarrett TC, Vidal RM, Wijayaweera SS, Dixon SV, Kim H, Cox KL, et al. Antigenic Modulation Limits the Efficacy of Anti-CD20 Antibodies: Implications for Antibody Selection. Blood. 2010;115:5191–5201. doi: 10.1182/blood-2010-01-263533. [DOI] [PubMed] [Google Scholar]
- 11.Taylor RP, Lindorfer MA. Antigenic Modulation and Rituximab Resistance. Semin Hematol. 2010;47:124–132. doi: 10.1053/j.seminhematol.2010.01.006. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Czuczman MS, Olejniczak S, Gowda A, Kotowski A, Binder A, Kaur H, Knight J, Starostik P, Deans J, Hernandez-Ilizaliturri FJ. Acquirement of Rituximab Resistance in Lymphoma Cell Lines is Associated with Both Global CD20 Gene and Protein Down-Regulation Regulated at the Pretranscriptional and Posttranscriptional Levels. Clin Cancer Res. 2008;14:1561–1570. doi: 10.1158/1078-0432.CCR-07-1254. [DOI] [PubMed] [Google Scholar]
- 13.Tsai PC, Hernandez-Ilizaliturri FJ, Bangia N, Olejniczak SH, Czuczman MS. Regulation of CD20 in Rituximab-Resistant Cell Lines and B-cell Non-Hodgkin Lymphoma. Clin Cancer Res. 2012;18:1039–1050. doi: 10.1158/1078-0432.CCR-11-1429. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Ujjani C, Cheson BD. The Current Status and Future Impact of Targeted Therapies in Non-Hodgkin Lymphoma. Expert Rev Hematol. 2013;6:191–202. doi: 10.1586/ehm.13.6. [DOI] [PubMed] [Google Scholar]
- 15.Rezvani AR, Maloney DG. Rituximab Resistance. Best Pract Res Clin Haematol. 2011;24:203–216. doi: 10.1016/j.beha.2011.02.009. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Ghetie MA, Bright H, Vitetta ES. Homodimers but not Monomers of Rituxan (Chimeric Anti-CD20) Induce Apoptosis in Human B-lymphoma Cells and Synergize with a Chemotherapeutic Agent and an Immunotoxin. Blood. 2001;97:1392–1398. doi: 10.1182/blood.v97.5.1392. [DOI] [PubMed] [Google Scholar]
- 17.Zhang N, Khawli LA, Hu P, Epstein AL. Generation of Rituximab Polymer may Cause Hyper-Cross-Linking–Induced Apoptosis in Non-Hodgkin’s Lymphomas. Clin Cancer Res. 2005;11:5971–5980. doi: 10.1158/1078-0432.CCR-05-0554. [DOI] [PubMed] [Google Scholar]
- 18.Chu TW, Yang J, Zhang R, Sima M, Kopeček J. Cell Surface Self-Assembly of Hybrid Nanoconjugates via Oligonucleotide Hybridization Induces Apoptosis. ACS Nano. 2014;8:719–730. doi: 10.1021/nn4053827. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Wu K, Liu J, Johnson RN, Yang J, Kopeček J. Drug-Free Macromolecular Therapeutics: Induction of Apoptosis by Coiled-Coil Mediated Crosslinking of Antigens on Cell Surface. Angew Chem, Int Ed. 2010;49:1451–1455. doi: 10.1002/anie.200906232. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Chu TW, Kopeček J. Drug-Free Macromolecular Therapeutics – A New Paradigm in Polymeric Nanomedicines. Biomater Sci. 2015;3:908–922. doi: 10.1039/C4BM00442F. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Li L, Yang J, Wang J, Kopeček J. Drug-Free Macromolecular Therapeutics Induce Apoptosis via Calcium Influx and Mitochondrial Signaling Pathway. Macromol Biosci. 2018;18:1700196. doi: 10.1002/mabi.201700196. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Chu TW, Zhang R, Yang J, Chao MP, Shami PJ, Kopeček J. A Two-Step Pretargeted Nanotherapy for CD20 Crosslinking may Achieve Superior Anti-lymphoma Efficacy to Rituximab. Theranostics. 2015;5:834–846. doi: 10.7150/thno.12040. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Van der Kolk LE, Grillo-López AJ, Baars JW, Hack CE, Van Oers MHJ. Complement Activation Plays a Key Role in the Side-Effects of Rituximab Treatment. Br J Haematol. 2001;115:807–811. doi: 10.1046/j.1365-2141.2001.03166.x. [DOI] [PubMed] [Google Scholar]
- 24.Zhang L, Fang Y, Yang J, Kopeček J. Drug-Free Macromolecular Therapeutics: Impact of Structure on Induction of Apoptosis in Raji B Cells. J Controlled Release. 2017;263:139–150. doi: 10.1016/j.jconrel.2016.12.025. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Olejniczak SH, Hernandez-Ilizaliturri FJ, Clements JL, Czuczman MS. Acquired Resistance to Rituximab is Associated with Chemotherapy Resistance Resulting from Decreased Bax and Bak Expression. Clin Cancer Res. 2008;14:1550–1560. doi: 10.1158/1078-0432.CCR-07-1255. [DOI] [PubMed] [Google Scholar]
- 26.Hayashi K, Nagasaki E, Kan S, Ito M, Kamata Y, Homma S, Aiba K. Gemcitabine Enhances Rituximab-Mediated Complement-Dependent Cytotoxicity to B Cell Lymphoma by CD20 Upregulation. Cancer Sci. 2016;107:682–689. doi: 10.1111/cas.12918. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Zhang R, Yang J, Sima M, Zhou Y, Kopeček J. Sequential Combination Therapy of Ovarian Cancer with Degradable N-(2-hydroxypropyl)methacrylamide Copolymer Paclitaxel and Gemcitabine Conjugates. Proc Natl Acad Sci U S A. 2014;111:12181–12186. doi: 10.1073/pnas.1406233111. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Moody PR, Sayers EJ, Magnusson JP, Alexander C, Borri P, Watson P, Jones AT. Receptor Crosslinking: A General Method to Trigger Internalization and Lysosomal Targeting of Therapeutic Receptor: Ligand Complexes. Mol Ther. 2015;23:1888–1898. doi: 10.1038/mt.2015.178. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Johnson RN, Kopečková P, Kopeček J. Biological Activity of Anti-CD20 Multivalent HPMA Copolymer-Fab′ Conjugates. Biomacromolecules. 2012;13:727–735. doi: 10.1021/bm201656k. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Janas E, Priest R, Wilde JI, White JH, Malhotra R. Rituxan (Anti-CD20 Antibody)-induced Translocation of CD20 into Lipid Rafts is Crucial for Calcium Influx and Apoptosis. Clin Exp Immunol. 2005;139:439–446. doi: 10.1111/j.1365-2249.2005.02720.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Unruh TL, Li H, Mutch CM, Shariat N, Grigoriou L, Sanyal R, Brown CB, Deans JP. Cholesterol Depletion Inhibits Src Family Kinase-Dependent Calcium Mobilization and Apoptosis Induced by Rituximab Crosslinking. Immunology. 2005;116:223–232. doi: 10.1111/j.1365-2567.2005.02213.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Hartley JM, Chu TW, Peterson EM, Zhang R, Yang J, Harris J, Kopeček J. Super-resolution Imaging and Quantitative Analysis of Membrane Protein/Lipid Raft Clustering Mediated by Cell Surface Self-assembly of Hybrid Nanoconjugates. ChemBioChem. 2015;16:1725–1729. doi: 10.1002/cbic.201500278. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Orrenius S, Zhivotovsky B, Nicotera P. Regulation of Cell Death: the Calcium–Apoptosis Link. Nat Rev Mol Cell Biol. 2003;4:552–565. doi: 10.1038/nrm1150. [DOI] [PubMed] [Google Scholar]
- 34.Yao H, Zhang Y, Liu L, Xu Y, Liu X, Lin J, Zhou W, Wei P, Jin P, Wen LP. Inhibition of Lanthanide Nanocrystal-induced Inflammasome Activation in Macrophages by a Surface Coating Peptide through Abrogation of ROS Production and TRPM2-mediated Ca 2+ Influx. Biomaterials. 2016;108:143–156. doi: 10.1016/j.biomaterials.2016.08.036. [DOI] [PubMed] [Google Scholar]
- 35.Shimizu R, Kikuchi J, Wada T, Ozawa K, Kano Y, Furukawa Y. HDAC Inhibitors Augment Cytotoxic Activity of Rituximab by Upregulating CD20 Expression on Lymphoma Cells. Leukemia. 2010;24:1760–1768. doi: 10.1038/leu.2010.157. [DOI] [PubMed] [Google Scholar]
- 36.Arlt A, Gehrz A, Müerköster S, Vorndamm J, Kruse ML, Fölsch UR, Schäfer H. Role of NF-κB and Akt/PI3K in the Resistance of Pancreatic Carcinoma Cell Lines against Gemcitabine-Induced Cell Death. Oncogene. 2003;22:3243–3251. doi: 10.1038/sj.onc.1206390. [DOI] [PubMed] [Google Scholar]
- 37.Shi Z, Azuma A, Sampath D, Li YX, Huang P, Plunkett W. S-Phase Arrest by Nucleoside Analogues and Abrogation of Survival without Cell Cycle Progression by 7-Hydroxystaurosporine. Cancer Res. 2001;61:1065–1072. [PubMed] [Google Scholar]
- 38.Smith MR, Joshi I, Jin F, Obasaju C. Enhanced Efficacy of Gemcitabine in Combination with Anti-CD20 Monoclonal Antibody Against CD20+ Non-Hodgkin’s Lymphoma Cell Lines In Vitro and in SCID Mice. BMC Cancer. 2005;5:103. doi: 10.1186/1471-2407-5-103. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Roghanian A, Teige I, Mårtensson L, Cox KL, Kovacek M, Ljungars A, Mattson J, Sundberg A, Vaughan AT, Shah V, Smyth NR, et al. Antagonistic Human FcγRIIB (CD32B) Antibodies Have Anti-tumor Activity and Overcome Resistance to Antibody Therapy In Vivo. Cancer Cell. 2015;27:473–488. doi: 10.1016/j.ccell.2015.03.005. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.