

Abstract
BACKGROUND
Nitric oxide (NO) has been heavily implicated in migraine. Nitroglycerin is a prototypic NO-donor, and triggers migraine in humans. However, nitroglycerin also induces oxidative/nitrosative stress and is a source of peroxynitrite - factors previously linked with migraine etiology. Soluble guanylyl cyclase (sGC) is the high affinity NO receptor in the body, and the aim of this study was to identify the precise role of sGC in acute and chronic migraine.
METHODS
We developed a novel brain-bioavailable sGC stimulator (VL-102), and tested its hyperalgesic properties in mice. We also determined the effect of VL-102 on c-fos and calcitonin gene related peptide (CGRP) immunoreactivity within the trigeminovascular complex. In addition, we also tested the known sGC inhibitor, ODQ, within the chronic nitroglycerin migraine model.
RESULTS
VL102-evoked acute and chronic mechanical cephalic and hind-paw allodynia in a dose-dependent manner, which was blocked by the migraine medications sumatriptan, propranolol, and topiramate. In addition, VL-102 also increased c-fos and CGRP expressing cells within the trigeminovascular complex. Importantly, ODQ completely inhibited acute and chronic hyperalgesia induced by nitroglycerin. ODQ also blocked hyperalgesia already established by chronic nitroglycerin, implicating this pathway in migraine chronicity.
CONCLUSIONS
These results indicate that nitroglycerin causes migraine-related pain through stimulation of the sGC pathway, and that super-activation of this receptor may be an important component for the maintenance of chronic migraine. This work opens the possibility for negative sGC modulators as novel migraine therapies.
Keywords: trigeminovascular pain, CGRP, cGMP, mouse model, nitric oxide
Introduction
Despite the high incidence of migraine, there is an enormous number of migraine sufferers for whom currently available therapies are ineffective or poorly tolerated (1-3). Chronic migraine patients are particularly affected by the limited therapeutic options. This chronic condition is defined as 15 headache days/month lasting at least 3 months (4); and less than 50% of chronic migraine patients reported satisfaction with their treatment (2). The development of novel therapeutics has been limited by the incomplete understanding of the mechanisms that regulate migraine, and chronic migraine particularly. A better understanding of the molecular determinants that drive this condition from an episodic to a chronic state would open up new avenues for pharmacological intervention and preventative therapies.
Nitric oxide (NO) is an important signaling molecule in the brain and periphery. The NO-donor, nitroglycerin (NTG), is a known human migraine trigger and has been used extensively as an initiator in human experimental models of migraine (5, 6). NTG needs to be bioactivated in the body to yield NO, although NO-independent actions of NTG have also been proposed (7, 8). It is commonly believed that depletion of the bioactivation apparatus and concomitant induction of oxidative stress cause the phenomenon of clinical NTG tolerance, possibly through peroxynitrite formation (9-11). Both oxidative stress and peroxynitrite have been associated with migraine etiology (12, 13). Furthermore, NO may elicit biological responses through cyclic guanosine monophosphate (cGMP)-dependent and cGMP-independent pathways; and the phosphodiesterase 5 inhibitor, sildenafil, can evoke headache in migraine patients (14, 15). Thus, binding of NO to its high affinity receptor, soluble guanylyl cyclase (sGC), eliciting cGMP production, is only one of a number of feasible pathways through which NTG might induce migraine.
The aim of this study was to test the hypothesis that the sGC pathway mediates migraine induction by NTG and thereby to define this pathway as an appropriate drug target for migraine therapy. Pharmacological blockade by the sGC inhibitor, ODQ, was used as one approach, but ODQ is known to interact with other heme-containing proteins other than sGC. A more specific approach to probe the intermediacy of sGC is to use a NO-independent activator of sGC. Fortunately, such chemical probes exist and have been optimized for therapeutic use in pulmonary arterial hypertension (16). To this end we have developed and characterized a novel brain-penetrant sGC stimulator, VL-102. Our work identifies the precise role of sGC activation in acute and chronic migraine pain, and identifies it as a novel therapeutic strategy for the treatment of this disorder.
Materials and Methods
cGMP live cells assay
Intracellular levels of cGMP were measured using the GloSensor cGMP assay (Promega). Briefly, HEK293 cells (RRID CVCL_0045) stably expressing pGloSensor-42F cGMP (Promega) were seeded in a 96-well plate and incubated overnight in Dulbecco’s Modified Eagle Medium high glucose supplemented with 10% foetal bovine serum (FBS), 20 mM L-glutamine, 1% sodium pyruvate, 1% penicillin and streptomycin, and 0.2 mg/mL hygromycin. Before measurements, cells were equilibrated for 2 hours in the dark in CO2 independent medium (Gibco) containing 2mM D-luciferin and 100 µM IBMX (sigma). Then cells were treated for 30 min with 10 µM VL102, YC-1 or ODQ in presence or absence of increasing concentration of DETA-NO (Cayman). Intracellular cGMP level was monitored in live cells by measuring luminescence generated from firefly luciferase GloSensor cGMP construction using a Synergy microplate reader (BioTek).
qPCR
Total RNA was isolated from SH-SY5Y cells (RRID CVCL_0019) 24 h post-treatment. RNA was isolated using the RNeasy Plus Mini kit from Quiagen. RNA samples were then reverse transcribed to single-stranded cDNA. cDNA pre-amplification and qPCR were performed following the manufacturer’s guidelines using Superscript III (life technologies) and the TaqMan® Gene Expression Assay system (Applied Biosystems), respectively. Actin beta (ACTB: Hs01060665_g1), Glyceraldehyde-3-phosphate dehydrogenase (GAPDH, Hs02758991_g1) and hypoxanthine phosphoribosyltransferase 1 (HPRT1, Hs02800695_m1) were used as housekeeping genes. The TaqMan Gene Expression assay reference number Hs04194186_s1 was used to capture cFOS transcripts. The threshold cycle (CT) of each target product was determined and ΔCT values between cFOS transcripts and housekeeping genes were calculated using the ABI StepOnePlus software. The fold change (2−ΔΔCt) for each sample was calculated relative to the median ΔCT in control samples.
Animals
Subjects were male and female C57BL6/J mice (Jackson Laboratories, Bar Harbor, ME, USA), between 8-20 weeks old. Animals were group housed in a 12-12 light-dark cycle, and food was available ad libitum. All experimental procedures were approved by the University of Illinois at Chicago Office of Animal Care and Institutional Biosafety Committee, in accordance with AALAC guidelines and the Animal Care Policies of the University of Illinois at Chicago, as well as with the European Union directive on the subject of animal rights. Animals were weighed daily during treatment, and no adverse effects of treatment were observed.
Hippocampal and aortic sGC activity
Individual aortae, or hippocampi pooled from 4 rats, were homogenized in TEDS buffer (10 mM Tris-HCl, pH 7.6, 0.32 M sucrose, 2 mM EDTA, 1 mM DTT) containing protease inhibitors. Aortic homogenates were centrifuged at 105,000 × g for 60 min and the supernatant fraction obtained. The hippocampal homogenate was centrifuged at 12,000 × g for 15 min to obtain the supernatant fraction and pellet (synaptosomes). Synaptosomes were resuspended in Locke’s buffer. Enzyme activation was assessed for DEA/NO (10 nM-10 μM), in the presence or absence of 100 μM YC-1 or VL-102. Reactions were initiated by the addition of hippocampal protein or aortic supernatant protein and terminated by the addition of 50 mM sodium acetate (pH 4.0) followed by heating at 90° C for 3 min. The cGMP content was quantified by radioimmunoassay (17). Values for enzyme activity in the presence of VL-102 or YC-1 alone were subtracted in order to show the potentiation by VL-102 and YC-1 of NO-dependent activation of sGC. Data points shown are mean of determinations from two animals.
Pharmacokinetic studies
Three-month-old male C57BL/6 mice were administered VL-102 by single i.p. injection (1 mg/kg). At the appropriate time point, mice were sacrificed using CO2 asphyxiation. Blood was immediately collected from the dorsal aorta in 1 mL K3EDTA tubes (Greiner Vacuette) and kept on ice. After centrifugation at (3200×g for 20 min at 4 °C), the plasma supernatant was collected and immediately processed for analyses. Remaining plasma was stored at 80 °C. Following blood collection, each mouse was intracardially perfused with ice-cold PBS buffer (pH 7.4) and was decapitated. Brain was separated by hemisphere and half hemisphere was immediately processed for analyses while the other half was flash frozen with liquid nitrogen to be stored at −80 °C. Briefly, the reconstituted brain and plasma samples were analyzed after addition of the internal standard. Each sample was analyzed in triplicate using LC-MS/MS and each sample set was analyzed with a set of calibration standards. Peak areas for analytes and standards were calculated and the amount of each compound in each sample was determined using the calibration curves.
The data were fitted to a 2-compartment open model using PKSolutions software (Summit Research Services, Montrose, CO) to derive pharmacokinetic.
Sensory sensitivity testing
For all behavioral experiments, animals were counterbalanced into groups following the first basal test for mechanical sensitivity. The experimenter was blinded to the drug condition being tested. No adverse events were observed in any of the experiments. All animal testing occurred in low-light conditions, between 8-15h. To determine mechanical responses, the threshold for response to punctate mechanical stimuli (mechanical hyperalgesia) was tested according to the up-and-down method (18). Animals were habituated to the testing boxes and/or racks for 2 days prior to testing. For hindpaw sensitvity, the plantar surface of the animal hindpaw was stimulated with a series of eight von Frey filaments (bending force ranging from 0.008 to 2 g). A response was defined as a lifting or shaking of the paw upon stimulation. The first filament tested was 0.4 g. In the absence of a response, a heavier filament (up) was tried, and in the presence of a response, a lighter filament (down) was tested. This pattern was followed for a maximum of four filaments following the first response. For cephalic testing, mice were tested in 4 oz paper cups, to which they had been previously habituated. The periorbital region caudal to the eyes and near the midline was tested, similar to the up-down method described above. For all experiments, groups were counterbalanced based on their naïve baselines (basal responses taken on day 1).
Drug administration
All injections were administered as a 10 mL/kg volume, intraperitoneally, unless otherwise indicated. VL-102 was dissolved in a 5% DMSO saline solution, which was also used as the vehicle control. ODQ was diluted in 5% DMSO, 10% Tween-80, PBS and injected at 5 ml/kg volume; which was also used for the vehicle conditions for the control group. All other drugs were dissolved in 0.9% saline solution, which was used as the corresponding vehicle control. The treatment of the mice on testing days was as follows: habituation to the test rack, 15-20 minutes later measurement of baseline mechanical thresholds, administration of VL-102, and 2h later test again for mechanical responses. To test the effect of acute migraine medication, sumatriptan (0.6 mg/kg, Sandoz) was injected 1h15min post-VL-102, and mice were tested 45 min later. For preventative treatment, mice were treated with topiramate (30 mg/kg, Johnson & Johnson) or propranolol (20 mg/kg, Sigma) daily for 11 days, and on days 3,5,7,9, and 11 they were tested with VL-102 as above. For chronic ODQ experiments, 1 mg/kg ODQ was administered immediately prior to NTG (10 mg/kg, American Reagents) injections. In the acute experiment, ODQ was inject 2h prior to testing.
Immunohistochemistry & Quantification
For c-fos experiments, tissue was collected 2 hours after an acute injection of VL-102 (1 mg/kg, i.p.) or vehicle (0.05% DMSO/0.9% saline, i.p.). For CGRP experiments, mice were treated chronically as described above with vehicle or VL-102, and sacrificed 24h after the last injection. Mice were anesthetized with Somnasol (100 µL/mouse; 390 mg/mL pentobarbital sodium; Henry Schein, SKU#024352), and perfused intracardially with 15 mL of ice-cold phosphate-buffered saline (0.1M PBS, pH 7.2) and subsequently 50mL of ice-cold 4% paraformaldehyde (PFA)/0.1M PBS (pH 7.4). Whole brain and trigeminal ganglia were harvested from the mice and post-fixed overnight in 4% PFA/0.1M PBS at 4°C. Tissue was cryoprotected in 30% sucrose/0.1M PBS for 24-36 hours, or until it sank. Brains were flash frozen in 2-methyl butane on dry ice, coronal sections of the trigeminal nucleus caudalis were sliced at 14 µM, and sagittal sections of the trigeminal ganglia were sliced at 16 µM. All sections within Bregma −0.747mm to −8.15mm were collected for c-fos immunohistochemistry, and entire trigeminal ganglia were sliced and collected for CGRP immunohistochemistry. For c-fos staining, free-floating sections were washed in 0.1M phosphate-buffered saline with 0.2% Triton X-100 (PBST), and incubated in 0.3% H2O2 for 30 minutes. Free-floating sections were washed in PBST, and blocked with 10% normal rabbit serum in PBST (10% NRST) for 2 hours. Sections were incubated overnight at 4°C in a primary anti-cFos solution (RRID AB_2629503; 5C-52-G; Santa Cruz; 1:1000 dilution) made in 2% normal rabbit serum in PBST (2% NRST). Sections were washed with 10% NRST before incubating with a secondary antibody solution (biotinylated rabbit anti-goat; RRID AB_2336126; Vector Laboratories; 1:200 dilution) in 2% NRST for 2 hours at room temperature. Sections were washed in PBST, incubated in avidin-biotin complex reagent using a kit (ABC Vectastain, Buringame, CA) for 1 hour at room temperature, and washed 2 times in PBST. Nickel-enhanced diaminobenzadine (DAB) (Sigma) was used to visualize primary antibody staining. Sections were mounted, dried, and dehydrated by immersing slides in 70-80-90-100% EtOH for 2 minutes each. Slides were cover-slipped, and images taken by a single observer in a blinded manner using an EVOS FL Auto Cell Imaging System using a 20× objective. TNC region was determined based upon the morphological appearance of the section under bright field, and comparison with the Mouse Brain Atlas (19). Expression of cFos was blindly performed, and quantified by averaging 6-10 whole brain sections containing right and left hemispheres of the trigeminal nucleus caudalis per mouse (n=6 mice/group). For CGRP histology, sections of the trigeminal ganglia were immediately mounted onto slides after cryoslicing. Slides were blocked with 5% normal donkey serum in 0.1M phosphate-buffered saline with 0.3% Triton X-100 (NDST) for 1 hour at room temperature. Slides were incubated overnight at room temperature with primary sheep anti-CGRP antibody (RRID AB_725809; ab22560; Abcam; 1:1000 dilution) made in 1% NDST. Slides were washed with 1% NDST before incubating with a secondary antibody solution (Alexa Fluor 555 Donkey anti-Sheep; Life Technologies; 1:1000) made in 1% NDST for 2 hours at room temperature. Slides were washed with 0.1M phosphate buffer, and cover slipped with Mowiol-DAPI mounting solution. Images for quantification were taken by 2 observers in a blinded manner using an EVOS FL Auto Cell Imaging System, using a 40× objective. All images collected were used for analysis. Expression of CGRP was blindly performed, and quantified by counting total CGRP-positive cells from all sections containing both right and left ganglia per mouse (n=4/group). Portions of this work were carried out in the Confocal Microscopy Facility of the Research Resources Center at the University of Illinois at Chicago; confocal images were taken by a Zeiss Laser Scanning Microscope (LSM) 710.
Statistical Analysis
Data are expressed as mean ± s.e.m. Outliers were determined as 1.5 outside of the interquartile range (quartile 1 and 3). All statistical analyses were performed by Sigmastat software, and graphs were generated using GraphPad Prism. The dose and time response to VL-102, and the effect of sumatriptan was analyzed using a 1-way ANOVA. For all chronic experiments a two-way repeated measures ANOVA was performed, with drug and time as factors. When a significant interaction occurred, subsequent Holm-Sidak post-hoc analysis was performed. For immunohistochemical experiments data was analyzed using unpaired t-test. A significance level of p<0.05 was used.
Results
VL-102 is a novel sGC stimulator with brain bioavailability
Small molecules that bind to sGC to increase cGMP production from GTP are termed NO-independent or heme-independent and coined as either sGC stimulators or sGC activators, respectively (16). The so-called sGC stimulators are positive allosteric modulators of sGC, which (i) increase NO-independent sGC activity and (ii) potentiate the activation of sGC caused by NO binding to the heme-regulatory site (20). To develop a brain-bioavailable sGC stimulator we synthesized and assayed sGC activity of a library based upon the benzyl indazole chemical scaffold of YC-1, the prototype sGC stimulator (data not shown). VL-102, was identified as a brain bioavailable chemical probe that stimulates sGC and activates cGMP release in living cells both independent of NO and by potentiating NO action (Figure 1A). As anticipated, the actions of VL-102 and YC-1 were ablated by the sGC inhibitor ODQ, and were potentiated by NO generated by the NO-donor DETA/NO. Bioavailability of VL-102 was tested in male C57BL/6 mice after IP administration at two concentrations and two time points. The observed brain/plasma ratio of VL-102 was greater than unity under all conditions, with concentrations at the lower dose, in brain tissues at 15 min and 2 h, measured at 250 nM and 80 nM, respectively (Figure 1B). This low dose was used predominantly in testing acute and sustained hyperalgesia, emphasizing the potency of VL-102 in vivo. A final comparison of VL-102 with YC-1, in the presence and absence of a related NO-donor, DEA/NO, was made in rat aorta and hippocampus (Figure 1C). This comparison is important, because the α1β1-sGC isoform is found centrally and peripherally; whereas the α2β1-sGC isoform is enriched in the brain and synapses (21). Rat aortic homogenates were high in sGC specific activity and although the activity of hippocampal fractions (supernatant and synaptosomes) was lower, the fold increase in basal sGC activity was similar in each tissue fraction. Notably, VL-102 and YC-1 produce marked increases in cGMP levels at very low levels of NO. These results show that we have developed a novel sGC stimulator that is brain bioavailable.
Figure 1.
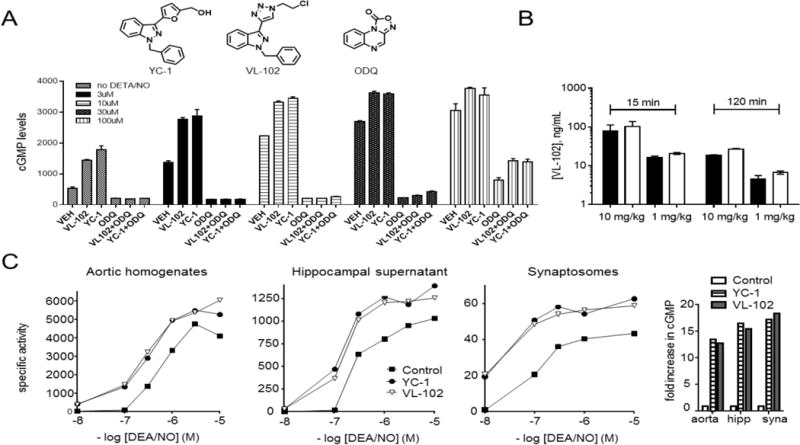
Characterization of a novel brain-bioavailable sGC stimulator. A) Increasing levels of cGMP measured in living cells, stimulated by VL102 and YC-1 (both 10 µM), in the absence, or presence, of varied concentrations of the NO-donor DETA/NO; and inhibited by ODQ (10 µM). DETA/NO (500 µM) is known to yield 600 nM [NO] under similar cell culture conditions. B) Bioavailability of VL-102 in male C57BL/6 mice treated with single dose VL-102 (1 or 10 mg/kg IP) showing concentrations measured in plasma (black bars) or brain cortex (white bar. C) Activation of sGC by NO-donor, DEA/NO, and potentiation of NO by sGC stimulators YC-1 or VL-102 (100 µM) in rat aortic tissue homogenates and rat hippocampal supernatant and synaptosomal fractions. The right hand panel shows the fold increase in cGMP by YC-1 and VL-102 in the absence of DEA/NO. Specific activity is expressed in nmol/min/mg protein. VL-102 is a novel sGC stimulator that crosses the blood brain barrier.
Repeated administration of VL-102 produces acute and sustained hyperalgesia which is dose dependent
We have previously reported that systemic administration of NTG can produce acute hyperalgesia, as well as a progressive and sustained basal hypersensitivity (22-24). We used the same protocol to test VL-102 to determine if selective sGC stimulation would produce a similar effect. Our preliminary characterization of VL-102 showed that like NTG, peak hyperalgesic responses were observed 2h post-injection, and in all other experiments we continued to assess post-treatment responses at this time point. For the chronic experiment, we administered VL-102 every second day for 9 days, resulting in a total of 5 VL-102 injection-test days. Basal testing on each day (i.e. prior to injection of VL-102/veh) revealed that like NTG, chronic VL-102 treatment produced a significant time and dose-dependent basal mechanical hypersensitivity over 9 days (Figure 2A). In addition, VL-102 significantly evoked mechanical hyperalgesia in a dose- and time-dependent manner, as compared to vehicle controls (Figures 2A,B). Although VL-102 showed significant effects on post-treatment response at the lowest dose of 0.01 mg/kg, the largest and most robust decreases in basal and post-treatment responses were observed with 1 mg/kg of VL-102. In another group of mice we again observed a marked basal mechanical hypersensitivity following chronic treatment (Figure 2C, treatment). Following the final treatment day (day 9), mechanical responses in mice were assessed daily to determine the recovery time for this basal hypersensitivity. Sensory responses returned to the level of naïve mechanical thresholds (day 1) 5 days following the final injection of VL-102 (Figure 2C, day 14, RECOVERY).
Figure 2.
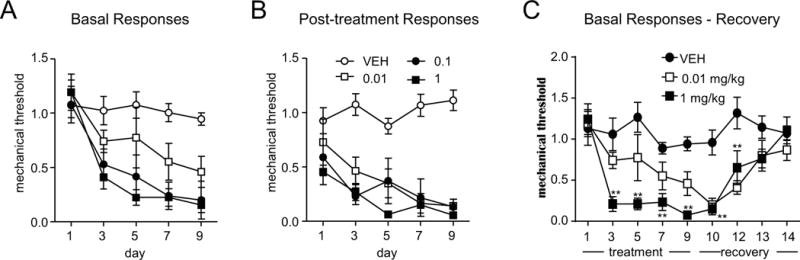
Chronic administration of the sGC stimulator, VL-102, produces both acute and chronic mechanical hyperalgesia in the hindpaw. C57Bl/6J mice were treated every second day with different doses of VL-102 (0-1 mg/kg, ip) for 9 days. (A) Basal responses, as assessed prior to vehicle or VL-102 administration, significantly decreased in groups receiving 0.1 and 1 mg/kg, starting from day 3 onwards (p<0.01), and in 0.01 mg/kg treated animals on days 7 and 9 (p<0.05). p<0.001 effect of dose, time and interaction, 2-way RM ANOVA and Holm-Sidak post-hoc analysis. (B) VL-102 produced significant mechanical hyperalgesia in mice tested 2 hours post-drug or vehicle administration. p<0.01 effect of dose and time, and interaction, 2-way RM ANOVA and Holm-Sidak post-hoc analysis. n=6-12/group. (C) Basal mechanical responses, as assessed prior to vehicle or VL-102 (0.01 and 1 mg/kg, IP) treatment, significantly decreased in the VL-102 group during the treatment period, and took 5 days to recover following the final VL-102 injection. For 1 mg/kg p<0.001 effect of drug, time and interaction, for 0.01 mg/kg p<0.01 effect of drug and time only; 2-way RM ANOVA and Holm-Sidak post-hoc analysis. **p<0.01, n=6/group. Stimulation of soluble guanylyl cyclase causes basal hypersensitivity and acute hyperalgesia.
VL-102-associated pain is blocked by migraine medications
In order to determine if the hyperalgesia induced by VL-102 was related to migraine, we tested the effect of the acute migraine therapy sumatriptan. Sumatriptan has been shown previously to block acute NTG-induced hyperalgesia (22, 23, 25). We found that 2h after acute injection, VL-102 produced a severe mechanical hyperalgesia that was blocked by sumatriptan (Figure 3A). In addition, we tested the migraine preventatives topiramate and propranolol, which has also been shown to effectively block NTG-induced effects in mice (23, 24). Animals were treated once daily with topiramate (30 mg/kg, IP) or propranolol (20 mg/kg, IP) for 11 days. On days 3, 5, 7, 9, and 11 basal mechanical responses were determined, and mice were injected with VL-102 (1 mg/kg, IP) or vehicle and tested 2h later. Both topiramate and propranolol significantly inhibited the basal hyperalgesia induced by chronic intermittent VL-102 treatment (Figure 3B,D). Furthermore, chronic administration of either drug also prevented NTG-evoked hyperalgesia (Figure 3C, E). Together, these results indicate that VL-102 is producing a migraine-associated pain, which is very similar to that induced by NTG.
Figure 3.
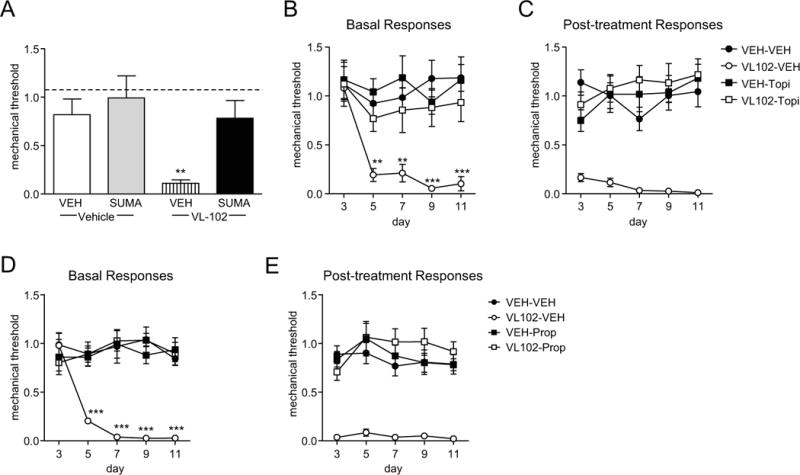
VL-102-induced hyperalgesia is blocked by acute and preventative migraine medications. (A) Sumatriptan significantly inhibited VL-102 evoked hyperalgesia. Mice were treated with vehicle (VEH) or VL-102 (1 mg/kg, IP), and 1h15min later injected with vehicle or sumatriptan (SUMA, 0.6 mg/kg, IP), and tested 45 min later. **p<0.01 as compared to VEH-VEH groups, 2-way ANOVA and Holm-Sidak post-hoc analysis. Dashed line indicates basal naïve responses. (B,C) In a separate group of animals, mice were treated with vehicle (VEH) or topiramate (30 mg/kg IP) daily for 11 days. On days 3,5,7,9, and 11 basal responses were determined (B), and mice were injected with either vehicle (VEH) or VL-102 (1 mg/kg, IP). p<0.05 effect of drug, time and interaction, 2-way RM ANOVA and Holm-Sidak post-hoc analysis. **p<0.01 and ***p<0.001 as compared to VEH-VEH controls. (C) Post-treatment responses were assessed 2h following VL-102 administration. p<0.001 effect of drug, but no significant effect of time or interaction, 2-way RM ANOVA. n=6/group. (D,E) A separate group of mice were treated with vehicle (VEH) or propranolol (20 mg/kg IP) daily for 11 days. On days 3,5,7,9, and 11 basal responses were determined (D), and mice were injected with either vehicle (VEH) or VL-102 (1 mg/kg, IP). p<0.05 effect of group, time and interaction, 2-way RM ANOVA and Holm-Sidak post-hoc analysis. **p<0.01 and ***p<0.001 as compared to VEH-VEH controls. (C) Post-treatment responses were assessed 2h following VL-102 administration. p<0.001 effect of drug, but no significant effect of time or interaction, 2-way RM ANOVA. n=9/group. Stimulation of soluble guanylyl cyclase produces a migraine-associated pain.
VL-102 induced a periorbital allodynia that was blocked by anti-migraine medications
Headache is a hallmark of migraine pain, and we tested the effect of VL-102 on head specific allodynia. Mice were treated every other day for 5 days with vehicle or VL-102 (1 mg/kg, IP) and tested for basal and post-treatment responses on days 1, 5, and 9. Similar to the hindpaw, chronic treatment with VL-102 produced a significant and sustained basal hypersensitivity in the periorbital region (Figure 4A), as well as acute allodynia 2h post-injection (Figure 4B). The administration of sumatriptan (0.6 mg/kg IP) 1h15min post-VL-102 significantly inhibited this acute periorbital allodynia. In addition, we tested animals with propranolol and VL-102 for 11 days as described above, and tested their periorbital sensitivity on day 9 (day 7 of VL-102). Chronic treatment with propranolol completely inhibited both the basal hypersensitivity and acute hyperalgesia induced by chronic intermittent VL-102 (Figure 4C and D). These results indicate that VL-102 can produce pain in the head, modeling the human migraine state.
Figure 4.
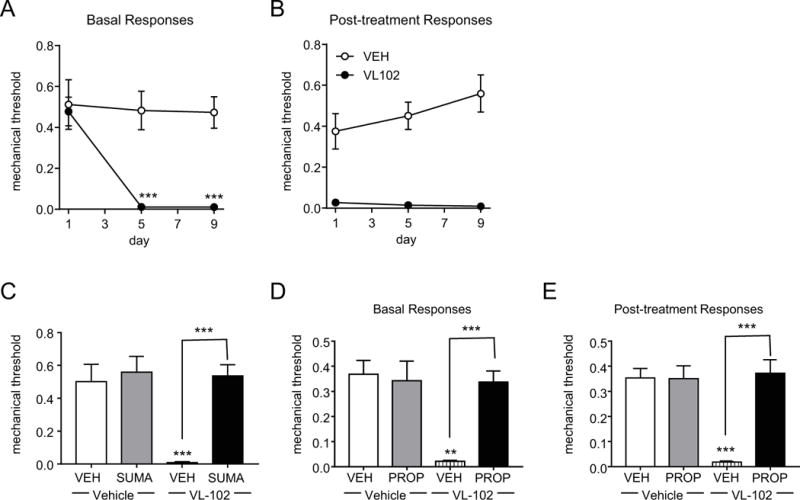
VL-102 produces periorbital allodynia that is blocked by migraine medications. C57Bl/6J mice were treated every other day with vehicle or VL-102 (1 mg/kg, IP) for 9 days. (A) Basal responses, as assessed prior to vehicle or VL-102 administration, significantly decreased in groups receiving VL-102. p<0.01 effect of group, time and interaction, 2-way RM ANOVA and Holm-Sidak post-hoc analysis. (B) VL-102 produced significant periorbital allodynia in mice tested 2 hours post-drug or vehicle administration. p<0.001 effect of group only, 2-way RM ANOVA and Holm-Sidak post-hoc analysis. n=8/group. (C) Sumatriptan (0.6 mg/kg, IP) significantly inhibited periorbital allodynia evoked by VL-102 (1 mg/kg, IP). p<0.01 group, drug, and interaction, 2-way ANOVA and Holm-Sidak post-hoc analysis. **p<0.01 as compared to VEH-VEH, ***p<0.001 as compared to VL-102-VEH, n=5-6/group. (D,E) Mice were treated with vehicle (VEH) or propranolol (20 mg/kg IP) daily for 11 days. On day 9 basal responses were determined (D), and mice were injected with either vehicle (VEH) or VL-102 (1 mg/kg, IP). p<0.05 effect of group, drug, and interaction, 2-way ANOVA and Holm-Sidak post-hoc analysis. **p<0.01 as compared to VEH-VEH, and ***p<0.001 as compared to VEH-VL-102 controls. (E) Post-treatment responses were assessed 2h following VL-102 administration. p<0.001 effect of group, but no significant effect of time or interaction, 2-way ANOVA. n=6/group.
VL-102 increases expression of c-fos and CGRP in the trigeminovascular complex
Induction of the immediate early gene, c-fos, has been used as a reliable marker of cell activation in response to stimuli. We initially tested VL-102 in the SH-SY-5Y neuroblastoma cell line. After 24h treatment, VL-102 significantly increased gene expression of c-fos, an effect that was blocked by the sGC inhibitor, ODQ (Figure 5A). We next tested the ability of acute VL-102 to induced c-fos in vivo. The trigeminal nucleus caudalis regulates head-specific pain, and NTG-based treatments that cause headache-related pain in rodents have been shown to activate cells in this brain region (25-29). In order to account for the effect of nociceptive testing, basal and post-treatment effects of VL-102 were determined, prior to tissue collection. VL-102 induced a significant increase in c-fos positive cells within the trigeminal nucleus caudalis, 2h post-treatment (Figure 5B,C), corresponding with peak hyperalgesic responses. These results indicate that VL-102 activates the immediate early gene c-fos in headache-related brain regions.
Figure 5.
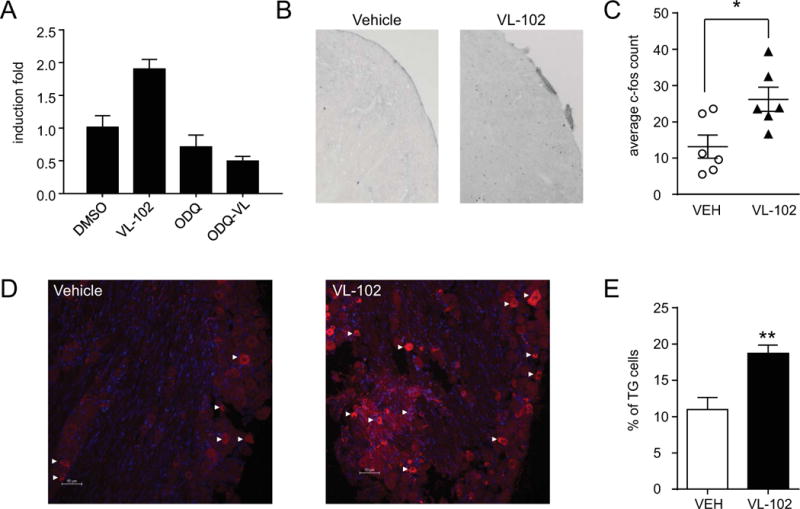
VL-102 causes an increase in c-fos and CGRP expression. (A) SH-SY-5Y cells were treated for 24h, and c-fos mRNA was quantified using qRT-PCR. p<0.01, one-way ANOVA, *p<0.05 compared to control (DMSO) group. (B) Representative images of the trigeminal nucleus caudalis from mice injected acutely with vehicle or VL-102 (1 mg/kg, IP), and sacrificed 2h later. (C) Quantification of average c-fos particles show that VL-102 significantly increased c-fos expression in the trigeminal nucleus caudalis. p<0.05, t-test, n=6 mice/group. (D) Representative images of trigeminal ganglia (TG) from mice injected chronically with vehicle or VL-102 (1 mg/kg, IP), and sacrificed 24h later. White arrow heads indicate some, but not all CGRP+ ganglia. (E) Quantification of the percentage of CGRP positive cells show that chronic VL-102 significantly increased the number of TGs expressing CGRP. p<0.01, t-test, n=4 mice/group. sGC stimulation causes increased neuronal activity, and migraine-related neuropeptide within the trigeminovascular complex.
Calcitonin gene related peptide (CGRP) is considered an endogenous migraine generator, and plays a critical role in the initiation and maintenance of migraine (30, 31). We determined if the basal hypersensitivity induced by chronic VL-102 corresponded with an increase in the number of trigeminal ganglia (TG) expressing CGRP. Dural afferents projecting from the TG are first order neurons that regulate head-specific pain. Mice were treated every other day with vehicle or VL-102 (1 mg/kg) for 9 days, which reliably induced a severe basal hypersensitivity. To avoid confounding effects of injection or testing, we determined CGRP expression 24h after the final injection of VL-102. We observed that chronic intermittent treatment with VL-102 almost doubled the number of TGs expressing CGRP (Figure 5D and E). Chronic sGC stimulation corresponds with increased CGRP expression within the trigeminal ganglia; and along with the c-fos data, our results support the notion that VL-102 is activating trigeminovascular circuits that potentiate hyperalgesia.
Inhibition of soluble guanylyl cyclase blocks NTG-induced pain
Considering that selective stimulation of the sGC pathway by VL-102 produced comparable effects to NTG, we wanted to determine if sGC inhibition would completely block NTG-induced pain. We performed a chronic experiment, in which NTG was injected every other day for 9 days (5 test-treatment days). Prior to injection of NTG (10 mg/kg IP) or vehicle, we administered the sGC inhibitor, ODQ (1 mg/kg, IP), or corresponding vehicle. We observed that ODQ completely blocked the basal hypersensitivity (Figure 6A), and the post-treatment hyperalgesia (Figure 6B) induced by NTG. These results indicate that the migraine-associated pain evoked by NTG is critically dependent on sGC activation.
Figure 6.
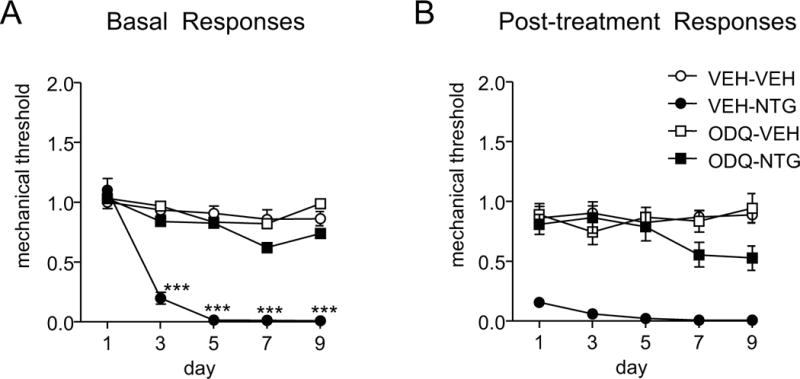
The sGC inhibitor, ODQ, inhibited NTG-induced basal and post-treatment hyperalgesia. Mice were treated chronically with vehicle (VEH) or NTG (10 mg/kg IP) every second day for 9 days. Prior to NTG/VEH administration, animals were injected with vehicle or ODQ (1 mg/kg, IP). (A) Basal responses, as assessed prior to drug treatment, were significantly decreased following NTG treatment, an effect that was completely blocked by ODQ. ***p<0.001 as compared to VEH-VEH controls. p< 0.001 effect of drug, time and interaction, 2-way RM ANOVA with Holm-Sidak post-hoc analysis. (B) Two hours following each injection, NTG-evoked a severe post-treatment hyperalgesia, which was completely prevented by ODQ treatment. p<0.001 effect of drug, no significant effect of time or interaction. n=9-15 mice/group.
Soluble guanylyl cyclase is super-activated following chronic NTG
As a final experiment, we wanted to determine if adaptations at the level of the sGC pathway could account for the maintenance of chronic migraine-associated pain. Mice received chronic intermittent treatment with NTG or vehicle, which resulted in basal hypersensitivity. On day 10, 24h following the final NTG/vehicle administration, mice that were chronically injected with NTG showed a severe basal hypersensitivity (basal responses, Figure 7), indicative of the establishment of a chronic pain state. Animals were then injected acutely with vehicle or ODQ (1 mg/kg, IP), and mechanical responses were assessed 2 and 24h later. ODQ significantly attenuated this established hypersensitivity 2h post-administration, returning mechanical responses to ~60% of control levels (post-drug responses, Figure 7A). The effect of ODQ was diminished 24h after its administration. These results indicate that the chronic pain induced by NTG is substantially mediated by aberrant sGC activation. Moreover, one injection of ODQ, 24h after the final administration of NTG, was sufficient to break out of the cycle of NO/cGMP-mediated chronic pain.
Figure 7.
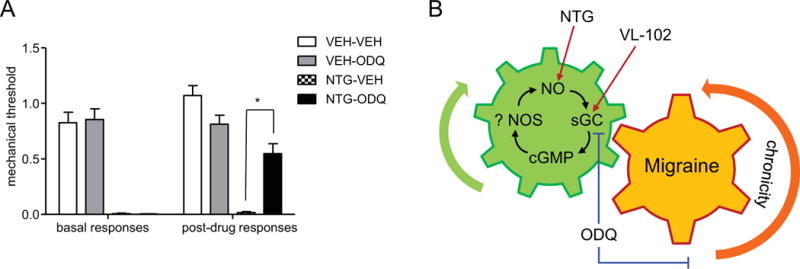
The sGC inhibitor, ODQ, inhibits already established NTG-induced basal hypersensitivity. (A) Mice were treated every second day with vehicle (VEH) or NTG (10 mg/kg, IP) for 9 days. Basal responses were significantly decreased in the NTG groups 24h after the final NTG/VEH treatment (day 10, left side). ODQ (1 mg/kg, IP) or vehicle was injected and animals were tested 2h later (post-drug responses). *p<0.05, n=7-8 mice/group. p<0.001 between groups, 1-way ANOVA with Holm-Sidak post-hoc analysis. ODQ significantly attenuated established chronic migraine-associated pain. (B) The sGC pathway is a key regulator of acute and chronic migraine. The sGC stimulator, VL-102 produces acute and chronic migraine-associated pain, comparable to NTG. In contrast, the sGC inhibitor, ODQ, completely blocks NTG-induced hyperalgesia. These results indicate that NTG-associated pain is critically mediated by the sGC pathway. ODQ also effectively blocks already established chronic migraine-associated pain, indicating that an upregulation of the sGC pathway enforces migraine chronification, potentially through adaptations of endogenous nitric oxide synthases (NOS). Our work indicates that inhibitors or negative modulators of the sGC pathway could be effective migraine treatments.
Discussion
Multiple lines of evidence implicate NO as an important component of migraine pathogenesis (5, 6), and the known human migraine trigger, NTG, is a potent NO donor. Upregulation of the sGC pathway has been implicated previously in migraine, as the phosphodiesterase 5 inhibitor, sildenafil, can increase cGMP levels and induces migraine attacks (14, 15). However, NTG is also known to directly and indirectly cause oxidative stress and has cGMP-independent actions (8, 9, 32-34), which could contribute to migraine symptomatology. In this study, we determined the precise molecular mechanism by which NTG produces migraine-related pain. We show that activation of the NO receptor, sGC, is critical for the induction and maintenance of acute and chronic migraine-associated pain. We also show that aberrant sGC signaling could be a key component regulating migraine chronification (Figure 6B), and our work indicates that sGC inhibitors may be promising migraine medications. Furthermore, through the course of this study we have also developed a novel tool for the study of migraine, the brain bioavailable sGC stimulator, VL-102, that is free of the off-target physiological effects of NTG. Our work indicates that the sGC pathway is a key signaling mechanism that regulates migraine pathophysiology.
The NO/cGMP pathway has been implicated in peripheral pain processing (35); and this pathway is of particular relevance to migraine, since NTG (36) and sildenafil (14, 15) produce headache but no other type of pain. We designed a brain bioavailable sGC stimulator, VL-102, to probe the specific role of sGC in induction of chronic migraine-associated pain. VL-102 is an analog of the sGC stimulator YC-1, and similar to this compound, VL-102 stimulated cGMP production in real-time, likely through stabilization of the sGC nitrosyl-heme complex (37). This effect was potentiated in the presence of NO, consistent with NO-independent sGC stimulation. VL-102 crosses the blood-brain barrier, and was detected in brain homogenates at similar concentrations to plasma, following systemic administration. The development of a brain-penetrant sGC stimulator provides an essential tool for interrogating the specific effect of sGC in the brain.
In behavioral assays, VL-102 produced results nearly identical to NTG. We have previously shown that NTG can produce acute hyperalgesia and basal hypersensitivity in a dose-dependent manner (23). We found that in both the head and hind paw, VL-102 also evoked acute hyperalgesia 2h post-injection, and that chronic intermittent treatment induced a prolonged basal hypersensitivity. In comparison to NTG, VL-102 was more potent and doses as low as 0.1 mg/kg produced both short and long-term hyperalgesia. In addition, VL-102-induced hyperalgesia was blocked by the migraine therapies, sumatriptan, topiramate, and propranolol. Sumatriptan is effective in other rodent (25, 38-41) and human (42) experimental models. Sumatriptan inhibited acute VL-102-induced hyperalgesia, similar to responses observed in the NTG model (22, 23, 25) which is consistent as sumatriptan is an abortive migraine therapy. The migraine preventatives, topiramate and propranolol, both inhibited VL-102-evoked acute hyperalgesia and chronic basal hypersensitivity; comparable to its effects in the NTG model (23, 24), and consistent with their actions as a preventative therapies (43, 44). Together these results indicate that direct stimulation of the sGC pathway induces the same sequelae of pain-related behaviors as NTG, and therefore the sGC pathway is an important component of migraine-related pain.
The immediate early gene, c-fos, is rapidly and transiently induced following stimulation, and has been used extensively to map neuronal activity (45). Acute sGC stimulation by VL-102 also produced increased expression of c-fos in both cells and in vivo. Studies using different preclinical models of migraine have shown that infusion of NTG (25, 28, 46, 47), capsaicin (48), facial inflammation (49), occipital afferent activation (50), dural stimulation (51), and cortical spreading depression (52) all evoked an increase in c-fos within the TNC. The stimulation of c-fos by VL-102 within the TNC establishes that this compound is activating headache-related brain regions and is also consistent with our finding that VL-102 evoked periorbital allodynia, thus supporting the notion that VL-102-induced pain is migraine-associated.
CGRP within the trigeminal ganglia (TG) plays a pivotal role in the pathophysiology of migraine (31, 53). During acute migraine attacks, CGRP serum levels are elevated (54), and sumatriptan normalizes CGRP in parallel with headache relief (55). Additionally, CGRP can induce headache (56), while CGRP receptor antagonists can abort migraine (57); thus CGRP is an active target for migraine drug development (58). In preclinical models, NTG treatment results in increased levels of CGRP in the blood (59), TNC, and dura mater (27, 46). In addition, in another rodent model of headache, the number of CGRP-expressing TGs increased in correspondence with increased pain (60). We also observed that chronic intermittent stimulation of sGC by VL-102 significantly upregulated CGRP-positive TGs. This increase corresponded with severe basal hypersensitivity, implicating CGRP as a contributing factor to VL-102-induced pain. In addition, the enhancement of CGRP in headache-associated brain regions provides further support for sGC stimulation producing a migraine-related pain. Overall, VL-102 is a novel tool to study the role of the sGC pathway in the brain, and avoids many of the caveats associated with NTG – such as oxidative stress, tolerance, dose, and solubility.
To further confirm the role of sGC activation on migraine-associated pain, we also tested the effects of the sGC inhibitor, ODQ, within the NTG model. We found that pretreatment with ODQ prior to NTG injection completely abolished the acute and chronic hyperalgesia induced by NTG. ODQ has an effect comparable to topiramate (23), and propranolol (24) in the chronic NTG model. However, ODQ was only given every other day, unlike the preventatives which were administered daily, between test days, and included a 2 day pretreatment. Importantly, sGC inhibition could also block already established migraine-associated pain. In these experiments NTG was administered chronically to produce a severe basal hypersensitivity. The acute administration of ODQ, 24h after the final injection of NTG (i.e. in the absence of exogenous sGC activation) was sufficient to reverse this hypersensitivity. These results indicate that a chronic migraine state can be established and maintained through upregulation of the sGC pathway. This increase could be due to a maladaptive enhancement of nitric oxide synthase (NOS), and the resultant endogenous NO is thus super-activating sGC. In a rat model of migraine, neuronal NOS (nNOS) was found to be upregulated in the dura mater following acute NTG infusion (27), and c-fos activation by NTG (27) or capsaicin (46) was blocked by the non-selective NOS inhibitor, L-NAME. Further, L-NAME was also found to decrease allodynia evoked by TRPM8 activation of the dura mater (61). The therapeutic benefit of sumatriptan may also be in part due to its ability to decrease NO in the brain (62, 63). In a small clinical study, the NOS inhibitor, L-NMMA, significantly improved spontaneous migraine symptoms by 60% (64, 65); showing that not only does NO activation initiate migraine, but also maintains it. Our finding that ODQ can block established migraine-related hypersensitivity supports the notion that NO dysfunction could regulate a chronic migraine state, and opens the possibility that changes may occur at both the level of the substrate (NO), and the receptor (sGC).
A better understanding of the mechanisms regulating migraine would significantly improve the prospects for novel therapies. In this study we identify the key molecular mediator through which the known human migraine trigger, NTG, evokes migraine-associated pain: soluble guanylyl cyclase. In the process we have developed a selective brain bioavailable sGC stimulator, VL-102, which can serve as a novel tool to probe sGC-mediated effects in the central nervous system. Our findings show that sGC inhibitors could be effective anti-migraine therapies, which may not only block acute migraine pain, but could also be used to help reset the chronic migraine state. To the best of our knowledge, there is no pharmaceutical program directed towards targeting centrally-active negative sGC modulators; and our work encourages the exploration of this target.
Article Highlights.
We developed a novel brain-bioavailable soluble guanylyl cyclase (sGC) stimulator VL-102; which induced migraine-related cephalic and hind-paw pain, activated brain regions involved in headache, and increased expression of calcitonin gene related peptide.
We found that inhibition of sGC completely blocked migraine-associated pain induced by the human migraine trigger nitroglycerin, and was also able to inhibit already established chronic migraine-associated pain.
These results show that sGC is a critical component for the induction and maintenance of migraine, and should be considered as a target for this disorder.
Acknowledgments
This research was supported by the NIH-NIDA Grants DA031243 and DA040688, and DoD PR141746 (AAP), and NIH-NCATS UL1TR002003 (GRT). LM and ZB are members of the UIC Graduate Program in Neuroscience.
Footnotes
The authors declare no conflicts of interests.
References
- 1.Stovner L, Hagen K, Jensen R, Katsarava Z, Lipton R, Scher A, et al. The global burden of headache: a documentation of headache prevalence and disability worldwide. Cephalalgia: an international journal of headache. 2007;27(2007):193–210. doi: 10.1111/j.1468-2982.2007.01288.x. [DOI] [PubMed] [Google Scholar]
- 2.Bigal ME, Serrano D, Reed M, Lipton RB. Chronic migraine in the population: burden, diagnosis, and satisfaction with treatment. Neurology. 2008;71(2008):559–66. doi: 10.1212/01.wnl.0000323925.29520.e7. [DOI] [PubMed] [Google Scholar]
- 3.Tfelt-Hansen P, Olesen J. Taking the negative view of current migraine treatments: the unmet needs. CNS drugs. 2012;26(2012):375–82. doi: 10.2165/11630590-000000000-00000. [DOI] [PubMed] [Google Scholar]
- 4.ICHD. The International Classification of Headache Disorders, 3rd edition (beta version) Cephalalgia: an international journal of headache. 2013;33(2013):629–808. doi: 10.1177/0333102413485658. [DOI] [PubMed] [Google Scholar]
- 5.Olesen J. The role of nitric oxide (NO) in migraine, tension-type headache and cluster headache. PharmacolTher. 2008;120(2008):157–71. doi: 10.1016/j.pharmthera.2008.08.003. [DOI] [PubMed] [Google Scholar]
- 6.Olesen J. Nitric oxide-related drug targets in headache. Neurotherapeutics: the journal of the American Society for Experimental NeuroTherapeutics. 2010;7(2010):183–90. doi: 10.1016/j.nurt.2010.03.006. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Kleschyov AL, Oelze M, Daiber A, Huang Y, Mollnau H, Schulz E, et al. Does nitric oxide mediate the vasodilator activity of nitroglycerin? Circ Res. 2003;93(2003):e104–12. doi: 10.1161/01.RES.0000100067.62876.50. [DOI] [PubMed] [Google Scholar]
- 8.Thatcher GRJ, Nicolescu AC, Bennett BM, Toader V. Nitrates and NO release: Contemporary aspects in biological and medicinal chemistry. Free Radic Biol Med. 2004;37(2004):1122–43. doi: 10.1016/j.freeradbiomed.2004.06.013. [DOI] [PubMed] [Google Scholar]
- 9.Warnholtz A, Tsilimingas N, Wendt M, Munzel T. Mechanisms underlying nitrate-induced endothelial dysfunction: insight from experimental and clinical studies. Heart Fail Rev. 2002;7(2002):335–45. doi: 10.1023/a:1020710417337. [DOI] [PubMed] [Google Scholar]
- 10.Sydow K, Daiber A, Oelze M, Chen Z, August M, Wendt M, et al. Central role of mitochondrial aldehyde dehydrogenase and reactive oxygen species in nitroglycerin tolerance and cross-tolerance. J Clin Invest. 2004;113(2004):482–9. doi: 10.1172/JCI19267. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.DiFabio J, Ji Y, Vasiliou V, Thatcher GRJ, Bennett BM. Role of mitochondrial aldehyde dehydrogenase in nitrate tolerance. Mol Pharmacol. 2003;64(2003):1109–16. doi: 10.1124/mol.64.5.1109. [DOI] [PubMed] [Google Scholar]
- 12.Borkum JM. Migraine Triggers and Oxidative Stress: A Narrative Review and Synthesis. Headache. 2016;56(2016):12–35. doi: 10.1111/head.12725. [DOI] [PubMed] [Google Scholar]
- 13.Bernecker C, Ragginer C, Fauler G, Horejsi R, Moller R, Zelzer S, et al. Oxidative stress is associated with migraine and migraine-related metabolic risk in females. Eur J Neurol. 2011;18(2011):1233–9. doi: 10.1111/j.1468-1331.2011.03414.x. [DOI] [PubMed] [Google Scholar]
- 14.Kruuse C, Thomsen LL, Jacobsen TB, Olesen J. The phosphodiesterase 5 inhibitor sildenafil has no effect on cerebral blood flow or blood velocity, but nevertheless induces headache in healthy subjects. Journal of cerebral blood flow and metabolism: official journal of the International Society of Cerebral Blood Flow and Metabolism. 2002;22(2002):1124–31. doi: 10.1097/00004647-200209000-00010. [DOI] [PubMed] [Google Scholar]
- 15.Kruuse C, Thomsen LL, Birk S, Olesen J. Migraine can be induced by sildenafil without changes in middle cerebral artery diameter. Brain: a journal of neurology. 2003;126(2003):241–7. doi: 10.1093/brain/awg009. [DOI] [PubMed] [Google Scholar]
- 16.Evgenov OV, Pacher P, Schmidt PM, Hasko G, Schmidt HH, Stasch JP. NO-independent stimulators and activators of soluble guanylate cyclase: discovery and therapeutic potential. Nature reviews Drug discovery. 2006;5(2006):755–68. doi: 10.1038/nrd2038. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Steiner AL, Parker CW, Kipnis DM. Radioimmunoassay for cyclic nucleotides. I. Preparation of antibodies and iodinated cyclic nucleotides. J Biol Chem. 1972;247(1972):1106–13. [PubMed] [Google Scholar]
- 18.Chaplan SR, Bach FW, Pogrel JW, Chung JM, Yaksh TL. Quantitative assessment of tactile allodynia in the rat paw. JNeurosciMethods. 1994;53(1994):55–63. doi: 10.1016/0165-0270(94)90144-9. [DOI] [PubMed] [Google Scholar]
- 19.Paxinos G, Franklin KBJ. The Mouse Brain in Stereotaxic Coordinates. San Diego, CA: Academic Press; 2001. [Google Scholar]
- 20.Artz JD, Toader V, Zavorin SI, Bennett BM, Thatcher GR. In vitro activation of soluble guanylyl cyclase and nitric oxide release: a comparison of NO donors and NO mimetics. Biochemistry. 2001;40(2001):9256–64. doi: 10.1021/bi002885x. [DOI] [PubMed] [Google Scholar]
- 21.Garthwaite J. From synaptically localized to volume transmission by nitric oxide. The Journal of physiology. 2016;594(2016):9–18. doi: 10.1113/JP270297. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Pradhan AA, Smith ML, Zyuzin J, Charles A. delta-Opioid receptor agonists inhibit migraine-related hyperalgesia, aversive state and cortical spreading depression in mice. British journal of pharmacology. 2014;171(2014):2375–84. doi: 10.1111/bph.12591. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Pradhan AA, Smith ML, McGuire B, Tarash I, Evans CJ, Charles A. Characterization of a novel model of chronic migraine. Pain. 2014;155(2014):269–74. doi: 10.1016/j.pain.2013.10.004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Tipton AF, Tarash I, McGuire B, Charles A, Pradhan AA. The effects of acute and preventive migraine therapies in a mouse model of chronic migraine. Cephalalgia: an international journal of headache. 2015;2015 doi: 10.1177/0333102415623070. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Bates EA, Nikai T, Brennan KC, Fu YH, Charles AC, Basbaum AI, et al. Sumatriptan alleviates nitroglycerin-induced mechanical and thermal allodynia in mice. Cephalalgia: an international journal of headache. 2010;30(2010):170–8. doi: 10.1111/j.1468-2982.2009.01864.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Ramachandran R, Bhatt DK, Ploug KB, Olesen J, Jansen-Olesen I, Hay-Schmidt A, Gupta S. A naturalistic glyceryl trinitrate infusion migraine model in the rat. Cephalalgia: an international journal of headache. 2012;32(2012):73–84. doi: 10.1177/0333102411430855. [DOI] [PubMed] [Google Scholar]
- 27.Ramachandran R, Bhatt DK, Ploug KB, Hay-Schmidt A, Jansen-Olesen I, Gupta S, Olesen J. Nitric oxide synthase, calcitonin gene-related peptide and NK-1 receptor mechanisms are involved in GTN-induced neuronal activation. Cephalalgia. 2014;34(2014):136–47. doi: 10.1177/0333102413502735. [DOI] [PubMed] [Google Scholar]
- 28.Tassorelli C, Joseph SA. NADPH-diaphorase activity and Fos expression in brain nuclei following nitroglycerin administration. Brain research. 1995;695(1995):37–44. doi: 10.1016/0006-8993(95)00732-6. [DOI] [PubMed] [Google Scholar]
- 29.Greco R, Tassorelli C, Mangione AS, Smeraldi A, Allena M, Sandrini G, et al. Effect of sex and estrogens on neuronal activation in an animal model of migraine. Headache. 2013;53(2013):288–96. doi: 10.1111/j.1526-4610.2012.02249.x. [DOI] [PubMed] [Google Scholar]
- 30.Goadsby PJ, Holland PR, Martins-Oliveira M, Hoffmann J, Schankin C, Akerman S. Pathophysiology of Migraine: A Disorder of Sensory Processing. Physiological reviews. 2017;97(2017):553–622. doi: 10.1152/physrev.00034.2015. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Russo AF. Calcitonin gene-related peptide (CGRP): a new target for migraine. Annual review of pharmacology and toxicology. 2015;55(2015):533–52. doi: 10.1146/annurev-pharmtox-010814-124701. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Dunlap T, Abdul-Hay SO, Chandrasena RE, Hagos GK, Sinha V, Wang Z, et al. Nitrates and NO-NSAIDs in cancer chemoprevention and therapy: in vitro evidence querying the NO donor functionality. Nitric Oxide. 2008;19(2008):115–24. doi: 10.1016/j.niox.2008.04.013. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Thatcher GRJ. Organic nitrates and nitrites as stores of NO. In: van Fassen E, Vanin A, editors. Radicals for Life: the various forms of nitric oxide. Amsterdam: Elsevier Science; 2007. [Google Scholar]
- 34.Daiber A, Munzel T, Gori T. Organic nitrates and nitrate tolerance–state of the art and future developments. Adv Pharmacol. 2010;60(2010):177–227. doi: 10.1016/B978-0-12-385061-4.00007-6. [DOI] [PubMed] [Google Scholar]
- 35.Schmidtko A, Tegeder I, Geisslinger G. No NO, no pain? The role of nitric oxide and cGMP in spinal pain processing. Trends Neurosci. 2009;32(2009):339–46. doi: 10.1016/j.tins.2009.01.010. [DOI] [PubMed] [Google Scholar]
- 36.Iversen HK, Olesen J, Tfelt-Hansen P. Intravenous nitroglycerin as an experimental model of vascular headache. Basic characteristics. Pain. 1989;38(1989):17–24. doi: 10.1016/0304-3959(89)90067-5. [DOI] [PubMed] [Google Scholar]
- 37.Ben Aissa M, Lee SH, Bennett BM, Thatcher GR. Targeting NO/cGMP Signaling in the CNS for Neurodegeneration and Alzheimer’s Disease. Current medicinal chemistry. 2016;23(2016):2770–88. doi: 10.2174/0929867323666160812145454. [DOI] [PubMed] [Google Scholar]
- 38.Levy D, Jakubowski M, Burstein R. Disruption of communication between peripheral and central trigeminovascular neurons mediates the antimigraine action of 5HT 1B/1D receptor agonists. ProcNatlAcadSciUSA. 2004;101(2004):4274–9. doi: 10.1073/pnas.0306147101. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Felice De M, Eyde N, Dodick D, Dussor GO, Ossipov MH, Fields HL, Porreca F. Capturing the aversive state of cephalic pain preclinically. Ann Neurol. 2013;2013 doi: 10.1002/ana.23922. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Edelmayer RM, Le LN, Yan J, Wei X, Nassini R, Materazzi S, et al. Activation of TRPA1 on dural afferents: a potential mechanism of headache pain. Pain. 2012;153(2012):1949–58. doi: 10.1016/j.pain.2012.06.012. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.Mason BN, Kaiser EA, Kuburas A, Loomis MM, Latham JA, Garcia-Martinez LF, Russo AF. Induction of Migraine-Like Photophobic Behavior in Mice by Both Peripheral and Central CGRP Mechanisms. The Journal of neuroscience: the official journal of the Society for Neuroscience. 2017;37(2017):204–16. doi: 10.1523/JNEUROSCI.2967-16.2016. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42.Iversen HK, Olesen J. Headache induced by a nitric oxide donor (nitroglycerin) responds to sumatriptan. A human model for development of migraine drugs. Cephalalgia: an international journal of headache. 1996;16(1996):412–8. doi: 10.1046/j.1468-2982.1996.1606412.x. [DOI] [PubMed] [Google Scholar]
- 43.Diener HC, Bussone G, Van Oene JC, Lahaye M, Schwalen S, Goadsby PJ. Topiramate reduces headache days in chronic migraine: a randomized, double-blind, placebo-controlled study. Cephalalgia: an international journal of headache. 2007;27(2007):814–23. doi: 10.1111/j.1468-2982.2007.01326.x. [DOI] [PubMed] [Google Scholar]
- 44.Limmroth V, Biondi D, Pfeil J, Schwalen S. Topiramate in patients with episodic migraine: reducing the risk for chronic forms of headache. Headache. 2007;47(2007):13–21. doi: 10.1111/j.1526-4610.2007.00648.x. [DOI] [PubMed] [Google Scholar]
- 45.Dragunow M, Faull R. The use of c-fos as a metabolic marker in neuronal pathway tracing. Journal of neuroscience methods. 1989;29(1989):261–5. doi: 10.1016/0165-0270(89)90150-7. [DOI] [PubMed] [Google Scholar]
- 46.Offenhauser N, Zinck T, Hoffmann J, Schiemann K, Schuh-Hofer S, Rohde W, et al. CGRP release and c-fos expression within trigeminal nucleus caudalis of the rat following glyceryltrinitrate infusion. Cephalalgia: an international journal of headache. 2005;25(2005):225–36. doi: 10.1111/j.1468-2982.2004.00845.x. [DOI] [PubMed] [Google Scholar]
- 47.Tassorelli C, Joseph SA. Systemic nitroglycerin induces Fos immunoreactivity in brainstem and forebrain structures of the rat. Brain research. 1995;682(1995):167–81. doi: 10.1016/0006-8993(95)00348-t. [DOI] [PubMed] [Google Scholar]
- 48.Sixt ML, Messlinger K, Fischer MJ. Calcitonin gene-related peptide receptor antagonist olcegepant acts in the spinal trigeminal nucleus. Brain: a journal of neurology. 2009;132(2009):3134–41. doi: 10.1093/brain/awp168. [DOI] [PubMed] [Google Scholar]
- 49.Romero-Reyes M, Akerman S, Nguyen E, Vijjeswarapu A, Hom B, Dong HW, Charles AC. Spontaneous behavioral responses in the orofacial region: a model of trigeminal pain in mouse. Headache. 2013;53(2013):137–51. doi: 10.1111/j.1526-4610.2012.02226.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 50.Le Doare K, Akerman S, Holland PR, Lasalandra MP, Bergerot A, Classey JD, et al. Occipital afferent activation of second order neurons in the trigeminocervical complex in rat. Neuroscience letters. 2006;403(2006):73–7. doi: 10.1016/j.neulet.2006.04.049. [DOI] [PubMed] [Google Scholar]
- 51.Park J, Moon H, Akerman S, Holland PR, Lasalandra MP, Andreou AP, et al. Differential trigeminovascular nociceptive responses in the thalamus in the familial hemiplegic migraine 1 knock-in mouse: a Fos protein study. Neurobiology of disease. 2014;64(2014):1–7. doi: 10.1016/j.nbd.2013.12.004. [DOI] [PubMed] [Google Scholar]
- 52.Fioravanti B, Kasasbeh A, Edelmayer R, Skinner DP, Jr, Hartings JA, Burklund RD, et al. Evaluation of cutaneous allodynia following induction of cortical spreading depression in freely moving rats. Cephalalgia: an international journal of headache. 2011;31(2011):1090–100. doi: 10.1177/0333102411410609. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 53.Ho TW, Edvinsson L, Goadsby PJ. CGRP and its receptors provide new insights into migraine pathophysiology. Nat Rev Neurol. 2010;6(2010):573–82. doi: 10.1038/nrneurol.2010.127. [DOI] [PubMed] [Google Scholar]
- 54.Goadsby PJ, Edvinsson L, Ekman R. Vasoactive peptide release in the extracerebral circulation of humans during migraine headache. Ann Neurol. 1990;28(1990):183–7. doi: 10.1002/ana.410280213. [DOI] [PubMed] [Google Scholar]
- 55.Goadsby PJ, Edvinsson L. The trigeminovascular system and migraine: studies characterizing cerebrovascular and neuropeptide changes seen in humans and cats. Ann Neurol. 1993;33(1993):48–56. doi: 10.1002/ana.410330109. [DOI] [PubMed] [Google Scholar]
- 56.Lassen LH, Haderslev PA, Jacobsen VB, Iversen HK, Sperling B, Olesen J. CGRP may play a causative role in migraine. Cephalalgia: an international journal of headache. 2002;22(2002):54–61. doi: 10.1046/j.1468-2982.2002.00310.x. [DOI] [PubMed] [Google Scholar]
- 57.Olesen J, Diener HC, Husstedt IW, Goadsby PJ, Hall D, Meier U, et al. Calcitonin gene-related peptide receptor antagonist BIBN 4096 BS for the acute treatment of migraine. NEnglJMed. 2004;350(2004):1104–10. doi: 10.1056/NEJMoa030505. [DOI] [PubMed] [Google Scholar]
- 58.Bigal ME, Walter S, Rapoport AM. Therapeutic antibodies against CGRP or its receptor. British journal of clinical pharmacology. 2015;79(2015):886–95. doi: 10.1111/bcp.12591. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 59.Di W, Zheng ZY, Xiao ZJ, Qi WW, Shi XL, Luo N, et al. Pregabalin alleviates the nitroglycerin-induced hyperalgesia in rats. Neuroscience. 2014;2014 doi: 10.1016/j.neuroscience.2014.08.056. [DOI] [PubMed] [Google Scholar]
- 60.De Felice M, Ossipov MH, Wang R, Dussor G, Lai J, Meng ID, et al. Triptan-induced enhancement of neuronal nitric oxide synthase in trigeminal ganglion dural afferents underlies increased responsiveness to potential migraine triggers. Brain: a journal of neurology. 2010;133(2010):2475–88. doi: 10.1093/brain/awq159. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 61.Burgos-Vega CC, Ahn DD, Bischoff C, Wang W, Horne D, Wang J, et al. Meningeal transient receptor potential channel M8 activation causes cutaneous facial and hindpaw allodynia in a preclinical rodent model of headache. Cephalalgia: an international journal of headache. 2016;36(2016):185–93. doi: 10.1177/0333102415584313. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 62.Read SJ, Manning P, McNeil CJ, Hunter AJ, Parsons AA. Effects of sumatriptan on nitric oxide and superoxide balance during glyceryl trinitrate infusion in the rat. Implications for antimigraine mechanisms. Brain research. 1999;847(1999):1–8. doi: 10.1016/s0006-8993(99)01985-x. [DOI] [PubMed] [Google Scholar]
- 63.Kurul SH, Demirpence S, Kiray M, Tugyan K, Yilmaz O, Kose G. Investigation of the immunoreactivities of NOS enzymes and the effect of sumatriptan in adolescent rats using an experimental model of migraine. The journal of headache and pain. 2008;9(2008):317–23. doi: 10.1007/s10194-008-0056-4. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 64.Lassen LH, Ashina M, Christiansen I, Ulrich V, Olesen J. Nitric oxide synthase inhibition in migraine. Lancet (London, England) 1997;349(1997):401–2. doi: 10.1016/s0140-6736(97)80021-9. [DOI] [PubMed] [Google Scholar]
- 65.Lassen LH, Ashina M, Christiansen I, Ulrich V, Grover R, Donaldson J, Olesen J. Nitric oxide synthase inhibition: a new principle in the treatment of migraine attacks. Cephalalgia: an international journal of headache. 1998;18(1998):27–32. doi: 10.1046/j.1468-2982.1998.1801027.x. [DOI] [PubMed] [Google Scholar]