

Abstract
The NLRP3 inflammasome is activated in response to microbial and danger signals resulting in caspase-1-dependent secretion of the proinflammatory cytokines IL-1β and IL-18. Canonical NLRP3 inflammasome activation is a two-step process requiring both priming and activation signals. During inflammasome activation, NLRP3 associates with mitochondria; however, the role for this interaction is unclear. Here we show that mouse NLRP3 and caspase-1 independently interact with the mitochondrial lipid cardiolipin (CL), which is externalized to the outer mitochondrial membrane (OMM) at priming in response to reactive oxygen species (ROS). An NLRP3 activation signal is then required for the calcium-dependent association of the adaptor molecule ASC with NLRP3 on the mitochondrial surface, resulting in inflammasome complex assembly and activation. These findings demonstrate a novel lipid interaction for caspase-1 and identify a role for mitochondria as supramolecular organizing centers (SMOCs) in the assembly and activation of the NLRP3 inflammasome.
Introduction
The concept of SMOCs proposes that individual innate receptors cluster and bind to other adaptors and effectors to form a single large structure assembled at a specific cellular location such as an organelle (1). Activation of the effector molecule is triggered, not by a traditional direct ligand-induced signaling pathway, but rather by the proximity to other effectors induced by the structure of the complex. In contrast to activation by a traditional signaling cascade, the activation via a SMOC results in a more intense and coordinated inflammatory signal, consistent with the amplification seen in initial innate immune responses (1). While oligomerization is a common feature of some nucleotide-binding domain and leucine-rich repeat containing receptor (NLR) family members, including NLRP3, the relevance of this to NLRP3 activation has been unclear (2).
NLRP3 inflammasome activation requires two steps, priming and activation, and culminates in the assembly of NLRP3, the adaptor molecule ASC, and pro-caspase-1 into the multiprotein inflammasome complex (2). Autocatalysis activates pro-caspase-1 to the active cysteine protease caspase-1 that then cleaves pro-IL-1β and pro-IL-18 into their mature secreted forms. In this study we demonstrate that caspase-1 can bind the mitochondrial lipid CL analogous to the interaction between caspase-11/4 and lipid A (3). We also show that ROS are required for the independent association of NLRP3 and caspase-1 with the OMM at priming. Furthermore, the activating NLRP3 stimulus brings the adaptor molecule ASC to the mitochondrion in a NLRP3- and calcium-dependent mechanism to complete the formation of the NLRP3 inflammasome and trigger the autocatalysis of caspase-1.
Materials and Methods
Lipids
1,2-Dioleoyl-sn-glycero-3-phosphocholine (PC) and tetralinoleoylcardiolipin (CL) were obtained from Avanti Polar Lipids. Oxidized 1-palmitoyl-2-arachidonyl-sn-glycero-3-phosphorylcholine (oxPAPC) was obtained from InvivoGen. Liposomes were prepared as previously described (4) and added at 300 μM to lysates unless otherwise stated. Unless specifically stated, liposomes used were at a 50:50 mol% PC:CL ratio.
Cell-free caspase-1 oligomerization
J774A.1 cells or BMM were primed for 4 h with 50 ng/ml ultrapure E. coli (0111:B4) LPS (Invivogen). Cells were lysed by N2 cavitation as previously described (4). Liposomes were added to lysates for 1 h at 37°C. When applicable, crosslinking was performed after 30 min of incubation with liposomes by adding 2 mM of bissulfosuccinimidyl suberate (BS3; ThermoFisher Scientific) for 30 min.
Large unilamellar vesicle (LUV) co-sedimentation
LUV were prepared by force extrusion using a miniextruder apparatus (Avanti Polar Lipids). LPS-primed J774A.1 macrophages were lysed by N2 decompression and organelle free cytosols isolated. Liposomes were added to clarified cytosols and incubated at 37°C for 30 min. LUV were then pelleted by centrifuging at 150,000g for 30 min at 4°C. Supernatants and liposome pellets were used for immunoblotting.
Mice and bone marrow-derived macrophages (BMM)
Nlrp3−/−, Asc−/−, and Casp1−/− (also deficient in caspase-11) mice have been previously described (5, 6). Wild-type C57BL/6N control mice were purchased from Charles River. The University of Iowa and Cedars-Sinai Medical Center Institutional Animal Care and Use Committee approved protocols used within this study. BMM were prepared as previously described (6).
Recombinant caspases
Plasmids expressing murine caspase-1 and caspase-3 with C-terminal Myc and DDK (FLAG) tags (pCMV6 entry) and empty vectors for expression of N-terminal Myc-DDK (pCMV6-AN-Myc-DDK) and N- and C-terminal His (pCMV6-AN-His and pCMV6-AC-His, respectively) tagged proteins were obtained from Origene Technologies. Murine caspase-11 open reading frames were cloned from LPS-primed J774A.1 mRNA and caspase-1 truncation constructs were cloned from the caspase-1 vector. Plasmids were transfected into HEK293T cells using Lipofectamine LTX PLUS (Invitrogen), and cell lines stably expressing the proteins of interest were selected with G418 (400 μg/ml). His-tagged proteins were purified using Ni-sepharose FF6 resin (GE Healthcare) according to the manufacturer’s instructions.
Lipid-coated strips
Membrane lipid strips (Echelon Biosciences) were incubated with 4 μg/ml of lysates from HEK293T cells expressing C-terminal Myc-DDK tagged murine caspase-1 or untransfected HEK293T lysates for 4 h at room temperature. Strips were washed and immunoblotted for adherent caspase-1. CL dot blot preparation was adapted from the protein lipid overlay assay described by Dowler et al. (7).
Macrophage stimulation
Unless otherwise stated, cells were primed with 100 ng/ml ultrapure E. coli (0111:B4) LPS, 100 ng/ml Pam3CSK4, or 1 μg/ml HMW poly(I:C) from Invivogen for 4 h. NLRP3 inflammasome activation was induced with 20 μM nigericin (Sigma) for 30 min. Staurosporine (Cayman) was used at 2 μM. To inhibit ROS, 25 mM N-acetyl-L-cysteine (NAC), 20 μM (2R, 4R)-4-aminopyrrolidine-2,4-dicarboxylic acid (APDC), or 500 μM mito-TEMPO (Sigma) were added 1 h prior to LPS stimulation. For inhibition of cellular calcium signaling, 50 μM BAPTA-AM (Molecular Probes) was added 30 min prior to the addition of nigericin.
Mitochondrial isolation and flow cytometry
Mitochondrial isolations were performed by differential centrifugation as previously described (4). Magnetic bead-based isolation of mitochondria was performed with anti-TOMM20 magnetic beads using a Mitochondria Isolation Kit (Miltenyi Biotec) in accordance with the manufacturer’s instructions.
Assessment of CL externalization by mitochondrial annexin V staining was previously described by Chu et al. (8). Prior to treatments, cells were incubated with 250 nM Mitotracker Green FM (Molecular Probes) for 45 min at 37°C according to the manufacturer’s instructions. Following stimulations and mitochondrial purification, resuspended mitochondria were incubated with Annexin V-Alexa Fluor 647 (Invitrogen) for 30 min on ice. Mitochondria were washed, fixed with 4% paraformaldehyde, and analyzed by flow cytometry on a BD LSR Fortessa.
Protease protection assay
Protease protection assay was performed on isolated mitochondria by preparing 0.2 mg/ml suspensions of mitochondria and digesting with 20 μg/ml proteinase K (New England Biolabs) with or without 0.5% Triton X-100 for 30 min on ice. Following incubation, proteinase K was inactivated with 2 mM phenylmethylsulfonyl fluoride.
Immunoblotting
As indicated, membranes were blotted with anti-caspase-1 p10 (sc-514, Santa Cruz Biotechnology), anti-caspase-1 p20 (Casper-1, AG-20B-0042, Adipogen), anti-NLRP3 (Cryo-2, AG-20B-0014, Adipogen), anti-ASC (AL177, AG-25B-0006, Adipogen), anti-GAPDH (6C5, CB1001, Calbiochem), anti-DDK (4C5, TA50011, Origene), anti-FLAG (M2, Sigma), anti-His (Tetra-His, Qiagen), anti-caspase-3 (8G10, 9665, Cell Signaling), anti-caspase-8 (1G12, ALX-804-447, Enzo Life Sciences), anti-caspase-11 (17D9, 14340, Cell Signaling), anti-Prohibitin (PA5-27329, Pierce), anti-VDAC (4866, Cell Signaling), anti-TSPO (9530, Cell Signaling), anti-PMP70 (ab85550, Abcam), anti-LAMP2 (ab25631, Abcam), anti-calnexin (ADI-SPA-860-F, Enzo Life Sciences), and anti-GM130 (clone 35/GM130, BD Biosciences) and probed with secondary antibodies donkey anti-rabbit-HRP (NA934V, GE Healthcare), goat anti-mouse-HRP (170-6515, BioRad), goat anti-rat-HRP (sc-2006, Santa Cruz), or rabbit anti-goat-HRP (611520, Invitrogen). Immunoblots were incubated with chemiluminescent substrates SuperSignal West Pico or Femto (Thermo Scientific) and images were captured using an Odyssey Fc (LI-COR Biosciences) imaging device.
Results and Discussion
NLRP3 and caspase-1 associate with mitochondria in response to priming
NLRP3 inflammasome activation is associated with NLRP3 migration to the mitochondria and mitochondria-associated membranes (MAM) (4, 9–12). However, the kinetics and mechanism behind this co-localization are poorly understood. To evaluate this, we isolated mitochondria and MAM after priming alone or after both priming and an activating stimulus and unexpectedly found both NLRP3 and caspase-1 associated with mitochondria and MAM following LPS priming alone (Fig. 1A). We confirmed that priming alone triggered the translocation of NLRP3 and caspase-1 to mitochondria and MAM using immunopurification of mitochondria (Supplemental Fig. 1A). To assess whether the movement of NLRP3 induced by priming involved movement to organelles other than the mitochondria and MAM, we generated mitochondrial, cytosolic, and microsomal fractions of cells. As expected, the mitochondrial fraction did reveal markers for the endoplasmic reticulum (ER) and MAM component calnexin and the golgi marker GM130 (Supplemental Fig. 1B). However, the microsomal fraction, containing ER, golgi, lysosomes, and peroxisomes, was free from mitochondrial components and did not show enhanced NLRP3 or caspase-1 association with priming (Supplemental Fig. 1B). Thus, movement of NLRP3 and caspase-1 at priming is specific to the mitochondria and MAM. The translocation of NLRP3 and caspase-1 to mitochondria is not specific to priming with LPS as TLR1/2 and TLR3 agonists, Pam3CSK4 and poly(I:C) respectively, also promoted the mitochondrial localization of caspase-1 and NLRP3 (Fig. 1B). The movement of NLRP3 and caspase-1 to the mitochondria and MAM in response to priming also occurred in the presence of cycloheximide suggesting it is a translation-independent event (Fig. 1C and Supplemental Fig. 1C). To assess whether caspase-1 and NLRP3 associated with the inner mitochondrial membrane (IMM) or the OMM in response to priming, we utilized a protease protection assay to determine their submitochondrial localization. Following priming, caspase-1 and NLRP3 associated with the OMM as both were cleaved by proteinase K in the absence of membrane permeabilization (Fig. 1D).
Figure 1.
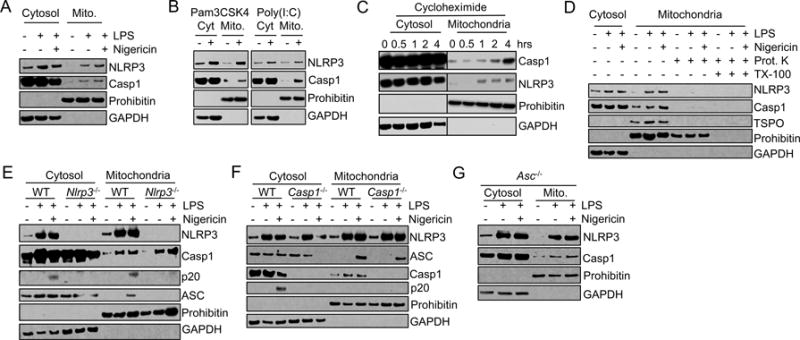
NLRP3 and caspase-1 independently associate with the mitochondria at priming. (A, B) J774A.1 cells were unstimulated, LPS-primed, or LPS-primed followed by stimulation with nigericin (A) or primed with Pam3CSK4 or HMW poly(I:C) (B). Mitochondrial and cytosolic fractions were then immunoblotted as indicated. (C) J774A.1 cells were LPS-primed for the indicated time in the presence of 10 μM cycloheximide. Mitochondrial and cytosolic fractions were then immunoblotted as indicated. (D) J774A.1 cells were unstimulated or LPS-primed followed by stimulation with nigericin as indicated. Mitochondrial and cytosolic fractions were isolated and mitochondria treated with proteinase K in the presence or absence of Triton X-100. Cytosolic and mitochondrial fractions were then immunoblotted as shown. (E-G) WT, Nlrp3−/−, Casp1 −/−, and Asc−/− BMM were unstimulated or LPS-primed and stimulated with nigericin as indicated. Mitochondrial and cytosolic fractions were subjected to immunoblot. Data shown are representative of three (A, E, G) or two (B-D, F) independent experiments.
NLRP3 and caspase-1 independently associate with mitochondria at priming
We next assessed whether NLRP3 and caspase-1 association with mitochondria was co-dependent. We found that caspase-1 association with mitochondria and MAM was triggered by priming independently of NLRP3 (Fig. 1E). We also found that ASC, unlike NLRP3 and caspase-1, was not recruited to the mitochondria at priming but instead only after the addition of nigericin (Fig. 1E). In contrast to caspase-1, the co-localization of ASC with mitochondria and MAM required the presence of NLRP3 (Fig. 1E). Utilizing Casp1−/− cells we found that caspase-1-deficiency had no impact on NLRP3 or ASC localization to mitochondria and MAM, nor did ASC-deficiency impact the recruitment of NLRP3 and caspase-1 (Fig. 1F, G). In the absence of a preceding priming step, nigericin alone did not induce translocation of ASC to mitochondria and MAM (Supplemental Fig. 1D). Hence the association of ASC with mitochondria requires the upstream event of priming, including NLRP3 recruitment to the mitochondria. Together these data show that the mitochondrial involvement in NLRP3 inflammasome activation begins at priming with the independent co-localization of NLRP3 and caspase-1 to the mitochondria and MAM. Activation of the inflammasome occurs when ASC interacts with mitochondrial NLRP3 in response to the NLRP3 activating signal.
ASC recruitment to mitochondria is calcium dependent
Increased cytosolic calcium has been shown to be required for NLRP3 inflammasome activation (13, 14). As expected chelation of cytosolic Ca2+ using BAPTA-AM blocked the activation of caspase-1 (Fig. 2A). Although chelation of Ca2+ had no effect on the movement of NLRP3 or caspase-1 to the mitochondria it did prevent the recruitment of ASC (Fig. 2A and Supplemental Fig. 1E). These data suggest the mechanism by which elevated cytosolic Ca2+ regulates NLRP3 inflammasome activation is by driving the association of ASC with mitochondria where NLRP3 and caspase-1 were previously recruited during inflammasome priming.
Figure 2.
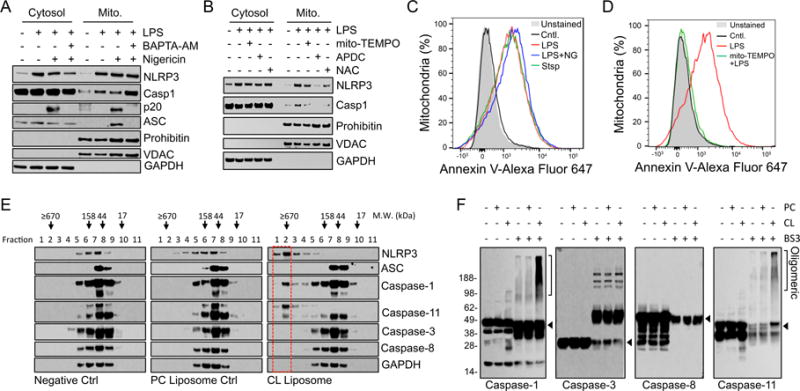
Specific roles for ROS and Ca2+ in NLRP3, caspase-1, and ASC localization to the mitochondria. (A) WT BMM were untreated or LPS-primed and treated with BAPTA-AM as indicated for 30 min prior to the addition of nigericin for a further 30 min. Mitochondrial and cytosolic fractions were isolated and immunoblotted as indicated. (B) J774A.1 were pretreated with NAC, mito-TEMPO, or APDC for 1 h and subsequently primed with LPS. Mitochondrial and cytosolic fractions were isolated and immunoblotted as indicated. (C) J774A.1 cells were left untreated, treated with staurosporine (Stsp) for 4 h or LPS-primed followed by stimulation with nigericin (NG) as indicated. Mitochondria were isolated and stained with Alexa Fluor 647-Annexin V and analyzed by flow cytometry. (D) J774A.1 cells were left untreated or LPS-primed in the presence or absence of mito-TEMPO. Mitochondria were isolated and stained with Alexa Fluor 647-Annexin V and analyzed by flow cytometry. (E) LPS-primed J774A.1 macrophages were lysed by N2 cavitation. Lysates were incubated with or without the indicated liposomes (PC, phosphatidylcholine; CL, cardiolipin). Reactions were fractionated on a sephacryl-500 column after which collected fractions were immunoblotted as indicated. The void fractions containing high molecular weight structures are highlighted with a red rectangle. (F) LPS-primed J774A.1 cells were N2 cavitated and lysates incubated with the indicated liposomes followed by BS3 crosslinking. Crosslinked lysates were then subjected to immunoblot. Data shown are representative of three (A- D, F) or two (E) independent experiments.
ROS is required for NLRP3 and caspase-1 association with mitochondria
One shared step in NLRP3 inflammasome activation is mitochondrial dysfunction associated with the generation of mitochondrial ROS (9, 10). Previous studies using the mitochondria-targeted superoxide probe mitoSOX found that TLR stimulation alone induces mitochondrial ROS (15). We also evaluated LPS-induced mitochondrial peroxide generation using mitoPY1 and found that treatment with LPS increased mitochondrial peroxide (Supplemental Fig. 1F). To determine if ROS played a role in the association of NLRP3 inflammasome components with mitochondria and MAM during priming, we used the pharmacologic inhibitors NAC, mito-TEMPO, and APDC. Inhibition of ROS prior to LPS priming with NAC, mito-TEMPO, and APDC all resulted in diminished caspase-1 activation in response to nigericin (Supplemental Fig. 1G). In addition, inhibition of ROS prior to LPS priming was also associated with a loss of priming-driven colocalization of NLRP3 and caspase-1 with mitochondria (Fig. 2B and Supplemental Fig. 1H, I). These results suggested that priming-specific mitochondrial ROS is responsible for the colocalization of both NLRP3 and caspase-1 with mitochondria and MAM.
Priming induces the movement of CL to the outer mitochondrial membrane
We have previously found that NLRP3 can interact with mitochondrial CL (4). The finding that caspase-1 and NLRP3 colocalize with the mitochondria at priming led to the hypothesis that this colocalization may in part be mediated by interactions with mitochondrial CL. As CL is found primarily on the IMM, but NLRP3 and caspase-1 interact with the OMM (Fig. 1D) we asked if CL had moved to the OMM following priming. To assess the location of CL we exploited the unique phosphatidylserine-free lipid composition of mitochondria where CL specifically binds annexin V (8). Mitochondria were isolated from untreated or LPS-primed macrophages, stained with Alexa Fluor 647-annexin V, and evaluated by flow cytometry. As expected, mitochondria from untreated cells had minimal binding of annexin V confirming the near absence of CL on the OMM of mitochondria at baseline (Fig. 2C). Mitochondria isolated from cells that had been treated with staurosporine had increased surface annexin V binding consistent with the externalization of CL in apoptosis (Fig. 2C) (16). Importantly, mitochondria from cells treated with LPS showed a similar increase in surface annexin V staining (Fig. 2C and Supplemental Fig. 1J) suggesting priming induced the translocation of CL from the IMM to the OMM. We next asked whether CL externalization to the OMM during priming was dependent upon ROS similar to NLRP3 and caspase-1 localization to mitochondria (Fig. 2B). Mitochondria were isolated from macrophages pretreated with mito-TEMPO, NAC, or APDC prior to LPS stimulation, and the presence of CL on the OMM was assessed via annexin V staining. Mito-TEMPO, NAC, and APDC pretreatment resulted in diminished mitochondrial annexin V staining (Fig. 2D and Supplemental Fig. 1K) suggesting that ROS was also required for the movement of CL to the OMM.
CL induces a molecular weight shift in NLRP3 and caspase-1
OxPAPC has been shown to interact with caspase-1 (17). In addition, caspases-4, -5, and -11 have been shown to have direct lipid A binding properties (3). Given structural similarities between lipid A and CL, we asked if CL could directly induce a molecular weight shift in caspase-1 analogous to lipid A-induced caspase-11 oligomerization (3). Utilizing gel filtration, and consistent with our previous finding showing that NLRP3 interacts with CL (4), we found that NLRP3 underwent a marked molecular weight shift in the presence of CL liposomes (Fig. 2E). Similarly, caspase-1 shifted to a high-molecular weight following the addition of CL, suggesting caspase-1 may also interact with CL (Fig. 2E). This shift was not common to all NLRP3 inflammasome components as there was no molecular weight shift in the inflammasome adaptor molecule ASC (Fig. 2E). To determine if the CL-induced shift in caspase-1 was generalizable to other caspases, we assessed the effect of CL liposomes on caspase-3, -8, and -11. Caspases-3 and -8 did not change molecular weight in the presence of CL liposomes. However, similar to the oligomerization observed in the presence of lipid A (3), caspase-11 underwent a molecular weight shift in the presence of CL (Fig. 2E). That caspase-8 was unaffected by our cardiolipin liposomes was unexpected given previous studies showing a role for cardiolipin in mediating caspase-8 association with the mitochondrial outer membrane (18, 19).
The finding that CL leads to formation of high-molecular weight protein complexes containing caspase-1 was confirmed by crosslinking assays utilizing BS3. We found caspase-1, but not caspase-3 or -8, migrated at a higher molecular weight in response to CL liposomes (Fig. 2F). BS3 crosslinking did result in a shift of caspase-3 independently of lipid addition consistent with the baseline dimerization of caspase-3 (20). We also found that caspase-11 underwent a molecular weight shift in the presence of CL (Fig. 2F). In contrast to the gel filtration findings, analysis of BS3-crosslinked samples did not reveal a CL-induced NLRP3 molecular weight shift (Supplemental Fig. 2A), suggesting NLRP3 lysines maybe insufficiently adjacent for BS3 stabilization or that an additional adaptor may be required. Consistent with the findings of Shi et al. (3) we demonstrated by BS3 crosslinking that LPS induced the oligomerization of caspase-11, but not caspase-1 (Supplemental Fig. 2B). Interestingly, we also found that oxPAPC, unlike CL, did not activate caspase-1 in our broken cell system, and also failed to shift the migration of caspase-1 but did trigger oligomerization of caspase-11 (Supplemental Fig. 2C, D). Together these data suggest that like NLRP3 (4), caspase-1 may also interact with CL in a manner analogous to the interaction of other inflammatory caspases with activating lipids (3, 17).
Caspase-1 interacts with CL
To further examine if caspase-1 can interact independently with CL, we incubated the cell lysates expressing Myc-DDK (FLAG) epitope-tagged murine caspase-1 with lipid coated membranes and found a specific binding of caspase-1 to CL (Fig. 3A). To confirm binding between caspase-1 and CL, we coated polystyrene plates with CL and again detected binding of caspase-1-Myc-DDK (Fig. 3B). In a complementary approach, we incubated CL liposomes with macrophage lysates and found that NLRP3, caspase-1, and caspase-11 pelleted with CL liposomes, while caspase-3, caspase-8 and ASC did not (Fig. 3C). Next, we generated C-terminal 6xHis-tagged constructs of murine caspase-1, -3, and -11, which we expressed in HEK293T cells, purified using Ni-sepharose and assessed binding of the purified caspases to CL coated strips. We found that caspase-1, and to a lesser extent capase-11, directly interacted with CL in a dose-dependent manner (Fig. 3D, E).
Figure 3.
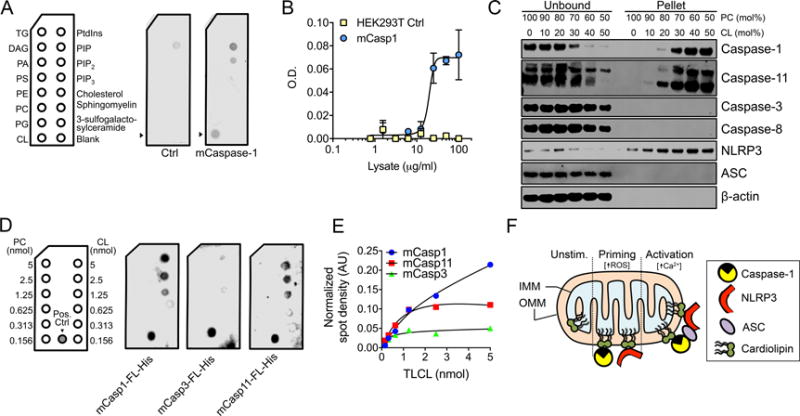
Caspase-1 directly and specifically binds to CL. (A) Lipid strips were incubated with lysates from HEK293T cells transfected with mCaspase-1-Myc-DDK or control lysates. Strips were blotted with anti-caspase-1. (B) 96-well plates coated with CL were incubated with lysates of HEK293T cells or HEK293T cells expressing mCaspase-1-Myc-DDK. Wells were washed and bound protein was detected using anti-DDK antibody followed by goat anti-mouse HRP. (C) Organelle-free cytosol from LPS-primed J774A.1 cells were incubated with large unilamellar vesicles (LUV) containing the indicated ratios of PC and CL for 30 min at 37°C followed by centrifugation to pellet the LUV. Supernatants and LUV pellets were collected and subjected to immunoblotting. (D, E) Lipid blots were prepared with increasing concentrations of CL and PC. Ni-sepharose purified protein was added as a positive control and standard for densitometric quantification. (D) Lipid blots were incubated with the indicated protein and immunoblotted with antibody to the indicated caspase. (E) Densitometric quantification of (D) normalized to positive control. (F) Model showing interactions of NLRP3 and caspase-1 with OMM in response to mitochondrial ROS at priming and the NLRP3-dependent association of ASC with OMM in response to calcium flux during activation. Data are representative of three independent experiments (A, C, D, E) or pooled from three independent experiments and expressed as the mean ± SEM (B).
The interaction of caspase-11 with lipid A is mediated primarily through the caspase activation and recruitment domain (CARD) (3). In contrast, caspase-11 interaction with oxPAPC occurred via its catalytic domain (17). To determine which domain of caspase-1 is responsible for its interaction with CL, we generated N-terminal Myc-DDK-tagged murine caspase-1 constructs (Supplemental Fig. 2E, F). We assessed binding of these constructs to PC and CL coated strips and found that binding of CL was measureable only for the full-length caspase-1 protein (Supplemental Fig. 2G, H), suggesting that caspase-1-CL interactions are distinct from other reported caspase-lipid interactions and utilize multiple domains or regions bridging domains, although it is also possible that the lack of measurable CL binding by the caspase-1 truncation constructs may represent altered tertiary structure or interference from epitope tags.
The ability of caspase-1 to directly associate with CL was not expected. However, this interaction between a lipid and caspase-1 is supported by recent findings showing similar associations for caspase-1 and -11 with oxPAPC and caspase-11 with lipid A (3, 17). Based on our current findings, we propose the requirement for mitochondria in NLRP3 inflammasome activation may be to serve as a platform, or SMOC, upon which the NLRP3 inflammasome assembles (Fig. 3F), rather than providing a specific activating ligand. We have found that NLRP3 assembly begins much earlier than previously thought, with the initial priming signal triggering the recruitment of NLRP3 and caspase-1 to the OMM in a ROS-dependent manner. The activation-associated increase in cytosolic calcium brings the adaptor molecule ASC to the mitochondria completing NLRP3 inflammasome assembly and triggering caspase-1 autocatalysis.
Supplementary Material
Acknowledgments
We thank Dr. Richard Flavell and Millennium Pharmaceuticals for providing knockout mice.
NIH grants R01 AI118719 (F.S.S.), R01 AI104706 (S.L.C.), R25 GM058939 (A.N.M.), and T32 GM007337 (E.I.E.), an American Lung Association/AAAAI Foundation grant (S.L.C.), and a grant from the Harry J. Lloyd Charitable Trust (F.S.S.) supported this work.
Footnotes
Disclosures
The authors declare no competing financial interests.
References
- 1.Kagan JC, Magupalli VG, Wu H. SMOCs: supramolecular organizing centres that control innate immunity. Nat Rev Immunol. 2014;14:821–826. doi: 10.1038/nri3757. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Elliott EI, Sutterwala FS. Initiation and perpetuation of NLRP3 inflammasome activation and assembly. Immunol Rev. 2015;265:35–52. doi: 10.1111/imr.12286. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Shi J, Zhao Y, Wang Y, Gao W, Ding J, Li P, Hu L, Shao F. Inflammatory caspases are innate immune receptors for intracellular LPS. Nature. 2014;514:187–192. doi: 10.1038/nature13683. [DOI] [PubMed] [Google Scholar]
- 4.Iyer SS, He Q, Janczy JR, Elliott EI, Zhong Z, Olivier AK, Sadler JJ, Knepper-Adrian V, Han R, Qiao L, Eisenbarth SC, Nauseef WM, Cassel SL, Sutterwala FS. Mitochondrial cardiolipin is required for Nlrp3 inflammasome activation. Immunity. 2013;39:311–323. doi: 10.1016/j.immuni.2013.08.001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Kuida K, Lippke JA, Ku G, Harding MW, Livingston DJ, Su MS, Flavell RA. Altered cytokine export and apoptosis in mice deficient in interleukin-1 beta converting enzyme. Science. 1995;267:2000–2003. doi: 10.1126/science.7535475. [DOI] [PubMed] [Google Scholar]
- 6.Sutterwala FS, Ogura Y, Szczepanik M, Lara-Tejero M, Lichtenberger GS, Grant EP, Bertin J, Coyle AJ, Galan JE, Askenase PW, Flavell RA. Critical role for NALP3/CIAS1/Cryopyrin in innate and adaptive immunity through its regulation of caspase-1. Immunity. 2006;24:317–327. doi: 10.1016/j.immuni.2006.02.004. [DOI] [PubMed] [Google Scholar]
- 7.Dowler S, Kular G, Alessi DR. Protein lipid overlay assay. Sci STKE. 2002;2002:pl6. doi: 10.1126/stke.2002.129.pl6. [DOI] [PubMed] [Google Scholar]
- 8.Chu CT, Ji J, Dagda RK, Jiang JF, Tyurina YY, Kapralov AA, Tyurin VA, Yanamala N, Shrivastava IH, Mohammadyani D, Qiang Wang KZ, Zhu J, Klein-Seetharaman J, Balasubramanian K, Amoscato AA, Borisenko G, Huang Z, Gusdon AM, Cheikhi A, Steer EK, Wang R, Baty C, Watkins S, Bahar I, Bayir H, Kagan VE. Cardiolipin externalization to the outer mitochondrial membrane acts as an elimination signal for mitophagy in neuronal cells. Nat Cell Biol. 2013;15:1197–1205. doi: 10.1038/ncb2837. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Zhou R, Yazdi AS, Menu P, Tschopp J. A role for mitochondria in NLRP3 inflammasome activation. Nature. 2011;469:221–225. doi: 10.1038/nature09663. [DOI] [PubMed] [Google Scholar]
- 10.Nakahira K, Haspel JA, Rathinam VA, Lee SJ, Dolinay T, Lam HC, Englert JA, Rabinovitch M, Cernadas M, Kim HP, Fitzgerald KA, Ryter SW, Choi AM. Autophagy proteins regulate innate immune responses by inhibiting the release of mitochondrial DNA mediated by the NALP3 inflammasome. Nat Immunol. 2011;12:222–230. doi: 10.1038/ni.1980. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Shimada K, Crother TR, Karlin J, Dagvadorj J, Chiba N, Chen S, Ramanujan VK, Wolf AJ, Vergnes L, Ojcius DM, Rentsendorj A, Vargas M, Guerrero C, Wang Y, Fitzgerald KA, Underhill DM, Town T, Arditi M. Oxidized mitochondrial DNA activates the NLRP3 inflammasome during apoptosis. Immunity. 2012;36:401–414. doi: 10.1016/j.immuni.2012.01.009. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Misawa T, Takahama M, Kozaki T, Lee H, Zou J, Saitoh T, Akira S. Microtubule-driven spatial arrangement of mitochondria promotes activation of the NLRP3 inflammasome. Nat Immunol. 2013;14:454–460. doi: 10.1038/ni.2550. [DOI] [PubMed] [Google Scholar]
- 13.Lee GS, Subramanian N, Kim AI, Aksentijevich I, Goldbach-Mansky R, Sacks DB, Germain RN, Kastner DL, Chae JJ. The calcium-sensing receptor regulates the NLRP3 inflammasome through Ca2+ and cAMP. Nature. 2012;492:123–127. doi: 10.1038/nature11588. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Rossol M, Pierer M, Raulien N, Quandt D, Meusch U, Rothe K, Schubert K, Schoneberg T, Schaefer M, Krugel U, Smajilovic S, Brauner-Osborne H, Baerwald C, Wagner U. Extracellular Ca2+ is a danger signal activating the NLRP3 inflammasome through G protein-coupled calcium sensing receptors. Nat Commun. 2012;3:1329. doi: 10.1038/ncomms2339. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.West AP, Brodsky IE, Rahner C, Woo DK, Erdjument-Bromage H, Tempst P, Walsh MC, Choi Y, Shadel GS, Ghosh S. TLR signalling augments macrophage bactericidal activity through mitochondrial ROS. Nature. 2011;472:476–480. doi: 10.1038/nature09973. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Kagan VE, Tyurina YY, Tyurin VA, Mohammadyani D, Angeli JP, Baranov SV, Klein-Seetharaman J, Friedlander RM, Mallampalli RK, Conrad M, Bayir H. Cardiolipin signaling mechanisms: collapse of asymmetry and oxidation. Antioxid Redox Signal. 2015;22:1667–1680. doi: 10.1089/ars.2014.6219. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Zanoni I, Tan Y, Di Gioia M, Broggi A, Ruan J, Shi J, Donado CA, Shao F, Wu H, Springstead JR, Kagan JC. An endogenous caspase-11 ligand elicits interleukin-1 release from living dendritic cells. Science. 2016;352:1232–1236. doi: 10.1126/science.aaf3036. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Gonzalvez F, Schug ZT, Houtkooper RH, MacKenzie ED, Brooks DG, Wanders RJ, Petit PX, Vaz FM, Gottlieb E. Cardiolipin provides an essential activating platform for caspase-8 on mitochondria. J Cell Biol. 2008;183:681–696. doi: 10.1083/jcb.200803129. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Jalmar O, Francois-Moutal L, Garcia-Saez AJ, Perry M, Granjon T, Gonzalvez F, Gottlieb E, Ayala-Sanmartin J, Klosgen B, Schwille P, Petit PX. Caspase-8 binding to cardiolipin in giant unilamellar vesicles provides a functional docking platform for bid. PLoS One. 2013;8:e55250. doi: 10.1371/journal.pone.0055250. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Boatright KM, Renatus M, Scott FL, Sperandio S, Shin H, Pedersen IM, Ricci JE, Edris WA, Sutherlin DP, Green DR, Salvesen GS. A unified model for apical caspase activation. Mol Cell. 2003;11:529–541. doi: 10.1016/s1097-2765(03)00051-0. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.