

ABSTRACT
CT-P10 (Truxima™) was recently approved as the world's first rituximab biosimilar product in the European Union (EU) and South Korea. To demonstrate biosimilarity of CT-P10 with the reference medicinal product (RMP), extensive 3-way similarity assessment has been conducted between CT-P10, EU-Rituximab and US-Rituximab, focusing on the physicochemical and biological quality attributes. A multitude of state-of-the-art analyses revealed that CT-P10 has identical primary and higher order structures compared to the original product. Purity/impurity profiles of CT-P10 measured by the levels of aggregates, fragments, non-glycosylated form and process-related impurities were also found to be comparable with those of RMPs. In terms of the post-translational modification, CT-P10 contains slightly less N-terminal pyro-glutamate variant, which has been known not to affect product efficacy or safety. Oligosaccharide profiling has revealed that, although CT-P10 contains the same conserved glycan species and relative proportion with the RMPs, the content of total afucosylated glycan in CT-P10 was slightly higher than in EU- or US-Rituximab. Nevertheless, the effect of the observed level of afucosylation in CT-P10 drug product on Fc receptor binding affinity or antibody-dependent cell-mediated cytotoxicity was found to be negligible based on the spiking study with highly afucosylated sample. Arrays of biological assays representative of known and putative mechanisms of action for rituximab have shown that biological activities of CT-P10 are within the quality range of RMPs. Recent results of clinical studies have further confirmed that the CT-P10 exhibits equivalent clinical efficacy and safety profiles compared to EU- and US-Rituximab. The current 3-way similarity assessment together with clinical study results confidently demonstrate that CT-P10 is highly similar with EU- and US-Rituximab in terms of physicochemical properties, biological activities, efficacy, and safety for its final approval as a biosimilar product.
KEYWORDS: rituximab, biosimilar, CT-P10, Truxima™, reference medicinal product (RMP)
Introduction
A biosimilar is a copy-version of a biological medicinal product that is developed for commercialization when the patent of the original product expires. Biosimilar products are expected to increase patients' accessibility to expensive biological medicines by promoting market competition. Despite of substantial demand, developing biosimilars is much more challenging compared to small-chemical generic drugs due to the intrinsically heterogeneous properties of biologics, which are largely dependent on manufacturing processes that biosimilar developers have no or limited knowledge about. Monoclonal antibody biosimilars are even more difficult to develop than biosimilars of smaller proteins (e.g., insulin, growth factors) because they typically contain 4 protein chains and have complex post-translational modifications. Regulatory agencies, including the US Food and Drug Administration and European Medicines Agency have established necessary guidelines so that applicants can obtain approval for their biosimilar products without conducting full clinical trials.1-5 These guidelines emphasize a “step-wise approach” for the development of biosimilars. Detailed evaluation of original products is first required to obtain information for reference product, then extensive physicochemical and biological characterization needs to be performed to demonstrate analytical similarity between biosimilar and original product. Biosimilarity is subsequently demonstrated with confirmatory non-clinical and clinical evaluation. Because extensive structural and functional characterization of both the biosimilar product and reference medicinal product (RMP) is the foundation of biosimilar development, comprehensive and robust analytical similarity assessment is essential.
Rituximab is a chimeric monoclonal antibody that selectively binds with high affinity to CD20, which is primarily found on the surface of immune system B cells.6 Rituximab destroys B cells, and thus used to treat diseases that are characterized by excessive number of B cells, overactive B cells or dysfunctional B cells. This includes many lymphomas, leukemia, transplant rejection and autoimmune disorders.7-11 CT-P10 has been developed as a similar biological medicinal product to the original rituximab products, MabThera® (EU-Rituximab) and Rituxan® (US-Rituximab). CT-P10 has identical pharmaceutical form, concentration, composition, and route of administration with the original rituximab.
As outlined in the relevant regulatory guidelines on the development of biosimilars, a step-wise approach has been taken with respect to the demonstration of similarity of CT-P10 to EU- and US-Rituximab, starting with a comprehensive physicochemical and biological characterization of CT-P10 relative to its RMP. This similarity exercise was undertaken, not only to demonstrate the similarity of CT-P10 to MabThera® and CT-P10 to Rituxan®, but also to demonstrate the comparability between Rituxan® and MabThera®, in order to support the global registration of CT-P10. The 3-way similarity assessment focused on two primary areas: 1) physicochemical similarity for detailed structural heterogeneity and purity/impurity studies, and 2) biological similarity for evaluation of functional assays, potency and binding affinity related to putative mechanisms of action.
From the extensive 3-way similarity assessment using sensitive and orthogonal methods, we have successfully demonstrated that CT-P10 has a highly similar quality profile compared to RMPs, MabThera® and Rituxan®.
Results
A wide range of state-of-the-art orthogonal methodologies was used to compare the physicochemical properties and biological activities of CT-P10, EU- and US-Rituximab. The investigated attributes include the primary structure, higher order structure, protein content, purity/impurity profiles, charged variants, glycan structures, as well as various aspects of product functionalities. A list of analytical test methods utilized for the similarity assessment is summarized in Table 1.
Table 1.
Test methods used for physicochemical and biological similarity assessment between CT-P10, EU-rituximab and US-rituximab.
| Attribute | Clinical Relevance | Test Method |
|---|---|---|
| Primary Structure | Efficacy, Safety, Immunogenicity | Peptide Mapping (HPLC) |
| Peptide Mapping (LC-MS) | ||
| Intact Mass (LC-MS) | ||
| Amino Acid Analysis | ||
| Extinction Coefficient | ||
| N-terminal Sequencing | ||
| C-terminal Sequencing | ||
| Higher Order Structure | Efficacy & Immunogenicity | Fourier Transform Infrared Spectroscopy (FTIR) |
| Differential Scanning Calorimetry (DSC) | ||
| Circular Dichroism (CD) | ||
| Free Thiol Analysis | ||
| Disulfide Bond | ||
| Content | Efficacy (PK) | Protein Concentration (UV280) |
| Purity | ||
| /Impurity | Efficacy & Immunogenicity | Size-exclusion Chromatography (SEC)-HPLC |
| Size-exclusion Chromatography (SEC)-MALS | ||
| Analytical Ultracentrifugation (AUC) | ||
| Residual Host Cell Protein | ||
| Residual Host Cell DNA | ||
| Residual rProtein A | ||
| Efficacy | Non-reduced Capillary Electrophoresis (CE)-SDS | |
| Reduced Capillary Electrophoresis (CE)-SDS | ||
| Charge Variants | Efficacy | Isoelectric Focusing (IEF) |
| Ion Exchange Chromatography (IEC)-HPLC | ||
| Glycosylation | Efficacy & Immunogenicity | Oligoprofiling by HILIC-UPLC |
| N-linked Glycan Analysis | ||
| Sialic Acid Analysis | ||
| Monosaccharide Analysis | ||
| Glycation | ||
| F(ab')2-related | Efficacy | Cell-based CD20 Binding Affinity |
| Apoptosis | ||
| Fc-F(ab')2-related | Efficacy | CDC |
| ADCC using PBMC | ||
| ADCC reporter assay | ||
| ADCP | ||
| Fc-related | Efficacy | C1q Binding Affinity |
| FcγRIIIa-V Binding Affinity | ||
| FcγRIIIa-F Binding Affinity | ||
| FcγRIIIb Binding Affinity | ||
| FcγRIIa Binding Affinity | ||
| FcγRIIb Binding Affinity | ||
| FcγRI Binding Affinity | ||
| FcRn Binding Affinity |
Primary structures
The techniques used to compare the primary structure of CT-P10, EU- and US-Rituximab include amino acid analysis, molar absorptivity, peptide mapping (HPLC and LC-MS), N/C-terminal sequencing and determination of intact mass. Amino acid analysis using HCl hydrolysis followed by RP-HPLC showed the molar ratios for the most robust amino acids (including glycine, valine and alanine) were similar to the expected ratio for all 3 product (data not shown). Molar absorptivity determined by measuring the optical density at a wavelength of 280 nm and protein molarity derived from the amino acid analysis also showed similar results for the 3 products (Table 2). The UV-based tryptic peptide map revealed a highly similar peak profile among all the samples without missing or additional new peaks with comparative retention time of each peak, suggesting identical primary sequence of the 3 products. The representative tryptic peptide maps for each product are shown in Fig. 1. The amino acid sequence was further confirmed by LC-MS peptide mapping. The detected peptides by trypsin or Asp-N of the 3 products matched the expected peptides from the amino acid sequence. In addition, the MS/MS data (data not shown) confirmed that the amino acid sequences of CT-P10, EU- and US-Rituximab matched and sequence coverage by MS/MS was 100%. The N- and C-terminal sequences of the heavy and light chain were also confirmed by peptide mapping in combination with MS/MS. The detected N- and C-terminal sequences of the light and heavy chains matched the expected sequences of rituximab for all 3 products. Peptide mapping by LC-MS was also used to identify the post-translational modifications. The results show highly similar post-translational modifications of CT-P10 compared to EU- or US-Rituximab (Table 2). The levels of asparagine deamidation and methionine oxidation were similarly very low in all 3 products. With regards to the N/C-terminal variants, the first residue was detected predominantly as pyro-glutamate in both the light and heavy chains. The majority of C-terminal sequence in the heavy chain was detected as a lysine-clipped form. Although the relative content of N/C-terminal variants was generally conserved among the 3 products, CT-P10 contains slightly lower levels (about 2%) of N-terminal pyro-glutamate in the light chain compared to EU- and US-Rituximab. Intact mass analysis yielded 4 possible masses corresponding to G0F-G0F, G0F-G1F, G1F-G1F or G0F-G2F and G2F-G2F. In all samples, the observed mass closely matches the expected mass.
Table 2.
Summary of results for physicochemical similarity assessment between CT-P10, EU-rituximab and US-rituximab.
| Min – Max Range (Mean ± SD) |
||||||
|---|---|---|---|---|---|---|
| Quality Attribute / Test Method | EU-Rituximab (N = 15) | CT-P10 Drug Product (N = 15) | US-Rituximab (N = 15) | |||
| Primary/Higher Order Structure | Molar Absorptivity | Molar Absorptivity (L∙mol−1∙cm−1) | 186,065 – 229,830 (212,656 ± 11,707) | 193,519 – 235,223 (213,636 ± 11,239) | 196,166 – 224,104 (212,717 ± 7,487) | |
| Extinction Coefficient (L∙g−1∙cm−1) | 1.29 – 1.59 (1.47 ± 0.08) | 1.34 – 1.63 (1.48 ± 0.08) | 1.36 – 1.55 (1.47 ± 0.05) | |||
| Peptide Mapping (LC-MS) | Deamidation (%) | HC Asn55 | 1.6 – 2.3 (1.9 ± 0.2) | 1.7 – 2.1 (1.9 ± 0.1) | 1.6 – 2.1 (1.9 ± 0.1) | |
| HC Asn290 | 0.1 – 0.2 (0.2 ± 0.0) | 0.1 – 0.2 (0.2 ± 0.0) | 0.1 – 0.2 (0.2 ± 0.0) | |||
| HC Asn319 | 0.4 – 0.6 (0.5 ± 0.1) | 0.5 – 0.7 (0.5 ± 0.1) | 0.4 – 0.6 (0.5 ± 0.1) | |||
| HC Asn365 | 0.1 – 0.2 (0.1 ± 0.0) | 0.1 – 0.2 (0.1 ± 0.0) | 0.1 – 0.2 (0.1 ± 0.0) | |||
| HC Asn388 | 1.8 – 2.5 (2.1 ± 0.2) | 1.5 – 2.1 (1.8 ± 0.2) | 1.8 – 2.3 (2.0 ± 0.2) | |||
| LC Asn136 | 0.1 – 0.1 (0.1 ± 0.0) | 0.1 – 0.1 (0.1 ± 0.0) | 0.1 – 0.1 (0.1 ± 0.0) | |||
| Oxidation (%) | HC Met34 | 0.1 – 0.2 (0.2 ± 0.1) | 0.1 – 0.2 (0.1 ± 0.1) | 0.1 – 0.2 (0.2 ± 0.1) | ||
| HC Met81 | 0.3 – 0.6 (0.4 ± 0.1) | 0.3 – 0.5 (0.4 ± 0.1) | 0.3 – 0.6 (0.4 ± 0.1) | |||
| HC Met256 | 1.9 – 2.9 (2.4 ± 0.3) | 2.0 – 3.6 (2.5 ± 0.5) | 1.9 – 3.4 (2.5 ± 0.4) | |||
| HC Met432 | 0.5 – 0.8 (0.6 ± 0.1) | 0.4 – 1.1 (0.7 ± 0.2) | 0.5 – 1.0 (0.7 ± 0.2) | |||
| LC Met21 | 0.1 – 0.3 (0.2 ± 0.1) | 0.1 – 0.4 (0.2 ± 0.1) | 0.1 – 0.3 (0.2 ± 0.1) | |||
| N-terminal pyro-Glu HC (%) | 99.7 – 99.9 (99.8 ± 0.1) | 99.5 – 99.8 (99.7 ± 0.1) | 99.6 – 99.9 (99.8 ± 0.1) | |||
| N-terminal pyro-Glu LC (%) | 96.1 – 98.9 (97.6 ± 0.7) | 97.1 – 98.7 (95.7 ± 0.5) | 97.4 – 98.7 (97.4 ± 0.9) | |||
| C-terminal Lys truncation HC (%) | 97.6 – 98.4 (98.1 ± 0.3) | 97.1 – 98.7 (98.1 ± 0.4) | 97.4 – 98.7 (98.2 ± 0.3) | |||
| Intact Mass (LC-MS) | G0F-G0F (Da) | 147085.9 – 147087.9 (147087.0 ± 0.6) | 147085.3 – 147086.5 (147085.8 ± 0.4) | 147085.9 – 147.088.3 (147087.0 ± 0.7) | ||
| G0F-G1F (Da) | 147246.9 – 147.249.4 (147248.3 ± 0.8) | 147246.9 – 147248.2 (147247.5 ± 0.4) | 147246.9 – 147249.3 (147248.4 ± 0.7) | |||
| G1F-G1F or G0F-G2F (Da) | 147408.2 – 147411.3 (147409.8 ± 0.8) | 147408.3 – 147410.3 (147408.9 ± 0.5) | 147408.5 – 147411.0 (147409.8 ± 0.7) | |||
| G1F-G2F (Da) | 147569.6 – 147574.1 (147571.4 ± 1.1) | 147568.8 – 147571.2 (147570.0 ± 0.7) | 147570.1 – 147573.9 (147571.6 ± 1.2) | |||
| Free Thiol Analysis | Average (free SH/IgG, µM/µM) | 0.30 – 0.35 (0.32 ± 0.02) | 0.27 – 0.31 (0.29 ± 0.01) | 0.28 – 0.36 (0.32 ± 0.02) | ||
| FTIR (cm−1) | Amide I | 1641 – 1642 (1641 ± 0.1) | 1641 – 1641 (1641 ± 0.1) | 1641 – 1641 (1641 ± 0.1) | ||
| Amide II | 1527 – 1529 (1528 ± 0.4) | 1527 – 1529 (1528 ± 0.4) | 1528 – 1528 (1528 ± 0.2) | |||
| DSC (°C) | Tm1 | 71.8 – 71.9 (71.9 ± 0.0) | 71.9 – 72.0 (71.9 ± 0.0) | 71.8 – 72.0 (71.9 ± 0.0) | ||
| Tm2 | 76.1 – 76.1 (76.1 ± 0.0) | 76.1 – 76.1 (76.1 ± 0.0) | 76.1 – 76.1 (76.1 ± 0.0 | |||
| Tm3 | 83.2 – 83.4 (83.3 ± 0.1) | 83.2 – 83.3 (83.3 ± 0.0) | 83.2 – 83.4 (83.3 ± 0.1) | |||
| Content | Protein Concentration (UV280) (mg/mL) | 10.0 – 10.6 (10.4 ± 0.2) | 9.7 – 10.6 (10.1 ± 0.3) | 10.0 – 10.5 (10.2 ± 0.2) | ||
| Purity/Impurity | SEC-HPLC | Monomer (%) | 98.75 – 99.21 (99.01 ± 0.15) | 99.17 – 99.50 (99.30 ± 0.10) | 98.60 – 99.06 (98.81 ± 0.17) | |
| HMW (%) | 0.73 – 1.13 (0.89 ± 0.13) | 0.26 – 0.82 (0.53 ± 0.19) | 0.86 – 1.27 (1.07 ± 0.14) | |||
| LMW (%) | 0.07 – 0.14 (0.10 ± 0.02) | 0.00 – 0.47 (0.17 ± 0.17) | 0.08 – 0.16 (0.12 ± 0.03) | |||
| SEC-MALS | Monomer (MW, kDa) | 137 – 139 (138 ± 1) | 137 – 139 (138 ± 1) | 137 – 139 (138 ± 1) | ||
| HMW (MW, kDa) | 247 – 344 (293 ± 25) | 166 – 307 (197 ± 40) | 237 – 364 (303 ± 35) | |||
| AUC | Monomer (%) | 97.88 – 98.48 (98.19 ± 0.23) | 97.50 – 98.52 (98.18 ± 0.30) | 97.04 – 98.45 (98.05 ± 0.40) | ||
| Dimer (%) | 1.53 – 2.12 (1.81 ± 0.23) | 1.48 – 2.50 (1.82 ± 0.30) | 1.55 – 2.96 (1.95 ± 0.40) | |||
| Monomer (s-value) | 6.03 – 6.28 (6.18 ± 0.06) | 6.10 – 6.32 (6.18 ± 0.05) | 6.09 – 6.25 (6.17 ± 0.07) | |||
| Dimer (s-value) | 8.78 – 9.12 (8.97 ± 0.11) | 8.70 – 9.19 (8.96 ± 0.12) | 8.60 – 9.09 (8.95 ± 0.12) | |||
| CE-SDS | NR | Intact IgG (%) | 90.61 – 96.16 (93.21 ± 1.44) | 94.54 – 97.18 (95.78 ± 0. 77) | 91.33 – 95.89 (93.40 ± 1.18) | |
| R | H+L (%) | 99.09 – 99.71 (99.53 ± 0.16) | 98.97 – 99.34 (99.20 ± 0.11) | 99.46 – 99.70 (99.57 ± 0.08) | ||
| Charge variants | IEC-HPLC | Acidic Group (Peak 1+2+3) (%) | 18.7 – 22.8 (20.8 ± 1.2) | 15.6 – 18.5 (16.7 ± 0.7) | 19.0 – 22.5 (20.9 ± 1.0) | |
| Main Peak (Peak 4) (%) | 66.5 – 72.8 (70.4 ± 1.8) | 65.5 – 68.4 (67.1 ± 0.9) | 67.6 – 72.0 (69.9 ± 1.5) | |||
| Basic Group (Peak 5+6+7) (%) | 7.0 – 11.0 (8.8 ± 1.0) | 15.4 – 17.4 (16.3 ± 0.5) | 8.0 – 10.6 (9.2 ± 0.9) | |||
| Glycosylation | Oligosaccharide Profile | Galactosylation (%) | 44.59 – 56.48 (51.07 ± 3.57) | 47.89 – 49.70 (48.63 ± 0.48) | 47.01 – 54.63 (50.78 ± 2.28) | |
| Afucosylation (%) | 0.77 – 2.05 (1.21 ± 0.33) | 0.36 – 0.93 (0.55 ± 0.14) | 0.66 – 1.89 (1.16 ± 0.36) | |||
| High Mannose (%) | 1.22 – 1.88 (1.43 ± 0.16) | 2.97 – 3.50 (3.29 ± 0.15) | 1.25 – 2.19 (1.63 ± 0.24) | |||
| Total Afucosylation (%) | 1.99 – 3.47 (2.64 ± 0.40) | 3.33 – 4.41 (3.84 ± 0.25) | 2.02 – 3.44 (2.79 ± 0.35) | |||
| Sialylation (%) | 0.87 – 2.98 (1.90 ± 0.63) | 1.34 – 1.50 (1.41 ± 0.05) | 0.84 – 2.93 (1.91 ± 0.64) | |||
| N-linked Glycan Analysis | Galactosylation (%) | 44.70 – 56.51 (51.17 ± 3.07) | 46.74 – 51.26 (48.89 ± 1.38) | 46.17 – 56.08 (50.97 ± 3.2) | ||
| Afucosylation (%) | 0.46 – 1.34 (0.80 ± 0.22) | 0.23 – 0.54 (0.36 ± 0.09) | 0.40 – 1.24 (0.78 ± 0.24) | |||
| High Mannose (%) | 1.46 – 2.48 (1.79 ± 0.27) | 3.13 – 4.09 (3.62 ± 0.27) | 1.54 – 2.38 (1.83 ± 0.26) | |||
| Total Afucosylation (%) | 2.10 – 3.09 (2.59 ± 0.30) | 3.46 – 4.62 (3.98 ± 0.32) | 2.22 – 3.03 (2.62 ± 0.24) | |||
| Sialylation (%) | 0.95 – 1.76 (1.44 ± 0.23) | 0.80 – 1.16 (0.92 ± 0.09) | 0.93 – 2.16 (1.54 ± 0.33) | |||
| Sialic Acid Analysis | Molar Ratio (sialic acid / protein mol / mol) | 0.07 – 0.12 (0.10 ± 0.01) | 0.07 – 0.09 (0.08 ± 0.01) | 0.07 – 0.14 (0.10 ± 0.02) | ||
| Monosaccharide Analysis (Molar Ratio) | Fuc | 1.1 – 1.3 (1.2 ± 0.1) | 1.1 – 1.3 (1.2 ± 0.1) | 1.1 – 1.4 (1.2 ± 0.1) | ||
| GlcNAc | 5.1 – 5.9 (5.6 ± 0.2) | 5.2 – 5.9 (5.5 ± 0.2) | 5.1 – 6.4 (5.6 ± 0.3) | |||
| Gal | 0.8 – 1.2 (0.9 ± 0.1) | 0.7 – 0.8 (0.8 ± 0.0) | 0.7 – 1.3 (0.9 ± 0.1) | |||
| Man | 2.7 – 3.3 (3.0 ± 0.2) | 2.8 – 3.2 (3.0 ± 0.1) | 2.8 – 3.5 (3.0 ± 0.2) | |||
| Glycation | % Glycation at LC | 2.0 – 2.7 (2.3 ± 0.2) | 1.6 – 1.8 (1.7 ± 0.1) | 2.0 – 2.5 (2.2 ± 0.1) | ||
| % Glycation at HC | 3.3 – 4.8 (3.6 ± 0.4) | 2.6 – 3.0 (2.6 ± 0.2) | 3.1 – 4.4 (3.6 ± 0.4) | |||
Figure 1.
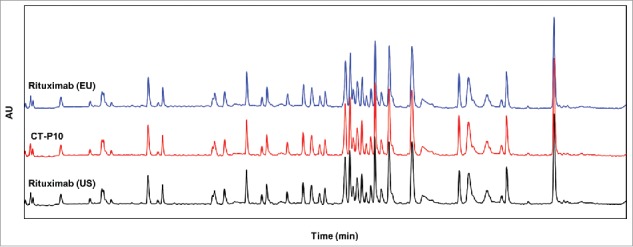
Comparison of UV chromatograms of trypsin-digested CT-P10 (red), EU-Rituximab (blue) and US-Rituximab (black). Representative tryptic peptide maps detected at 214 nm are presented for 1 batch of each product.
Higher order structures
The higher order structures of CT-P10, EU- and US-Rituximab were compared using various orthogonal methods. Ellman assay using 5,5′-dithiobis-(2-nitrobenzoic acid) (DTNB) showed that the moles of free sulfhydryl groups per mole IgG were highly similar at a low level for all 3 products (Table 2). An assessment of the disulfide bond positions by means of the native and reduced peptide mapping coupled with LC-MS revealed 8 new peaks in the native peptide map that were identified as disulfide bond-linked peptides based on the MS and MS/MS sequencing analysis. These 8 disulfide bond-linked peptides were matched in all samples, indicating the presence of the same inter and intra-chain disulfide bond linkages in all samples. The secondary or tertiary structures of the molecule were analyzed by circular dichroism (CD) and Fourier transform infrared spectroscopy (FT-IR). The far UV CD spectrum of CT-P10, EU- and US-Rituximab showed the typical shape of an antibody with a mean minimum at 217 nm and a mean maximum at 202 nm, suggesting a secondary structure dominated by β-sheet motif. No significant differences between the samples were observed in terms of maxima and minima wavelength of spectral signal and mean residue molar ellipticity (Fig. 2A). The near UV CD spectrum of CT-P10, EU- and US-Rituximab showed the typical shape of an antibody with a mean high maximum of 297 nm and the fine structure between 255 and 280 nm resulting from the aromatic amino acids and cysteines in the tertiary structure of the protein. No significant differences between the samples were observed (Fig. 2B). These data indicate that the 3 products have highly similar secondary and tertiary structures. In FT-IR spectra, all samples agreed well with respect to shape and location of the amide I and amide II bands that are the 2 most prominent vibrational bands of the protein backbone (Table 2 and Fig. 2C). The DSC thermograms of the 3 products showed highly similar thermal unfolding profiles and thermal transition midpoint temperatures, indicating that the thermal stability and conformation of CT-P10 are highly similar to those of EU- or US-Rituximab (Table 2 and Fig. 2D).
Figure 2.

Higher order structure analyzed by CD, FT-IR and DSC for CT-P10, EU-Rituximab and US-Rituximab. (A) Far-UV CD spectra, (B) Near-UV CD spectra, (C) FT-IR spectra, and (D) DSC thermograms are overlaid for 3 batches of each product.
Content
CT-P10 was manufactured at a same target concentration with EU- and US-Rituximab. The protein concentration determined by UV-Vis spectrophotometric method showed highly similar results (Table 2). The extractable volume of CT-P10 was also found to be comparable to that for the RMPs, suggesting the content of the molecule is comparable among the 3 products (data not shown).
Purity and impurity
The purity of the product was analyzed via product monomer content/molecular weight estimation (as determined by SEC-HPLC, SEC-MALS and AUC) and the amount of intact IgG and sum of heavy chain and light chain (as determined by non-reduced/reduced CE-SDS). In SEC-HPLC analysis, all 3 products showed prominent monomer peaks. CT-P10 samples have slightly higher levels (about 0.3 – 0.5%) of monomer and lower levels of high molecular weight (HMW) species in comparison to EU- and US-Rituximab (Table 2 and Fig. 3A). The level of low molecular weight (LMW) species was observed to be negligible. In SEC-MALS analysis, the molecular weight of monomer was highly similar for the 3 products; however, the calculated mass of HMW forms for CT-P10 was smaller (197 kDa) than that for EU- or US-Rituximab (293 kDa and 303 kDa, respectively). This variation was caused by the presence of different ratios of the multiple HMW species in CT-P10 versus EU- and US-Rituximab. As shown in Fig. 3B, EU- and US-Rituximab contain higher levels of peak 2 and an additional peak (peak 3) compared with CT-P10, indicating RMPs include more HMW forms than CT-P10. The molecular weight of HMW is shown as an average of all HMW species. Although the precise molecular weight of each HMW peak could not be determined due to the low resolution caused by the low levels present, when the average of these peaks are calculated for each product, the average molecular weight of HMW species in CT-P10 is smaller than that in EU- and US-Rituximab. Sedimentation velocity analytical ultracentrifugation (SV-AUC) analysis showed that all samples were composed of a single dominant monomer species at approximately 6.03 – 6.32 S with lower levels of dimer species at 8.60- 9.19 S. The relative content and sedimentation coefficients of monomer and dimer for all samples were highly similar (Table 2). CE-SDS under reduced conditions showed CT-P10 samples had slightly lower levels (about 0.3% mean difference) of H+L compared to that of EU- and US-Rituximab. This difference originated from slightly higher levels of non-glycosylated heavy chain in CT-P10 compared to EU- and US-Rituximab. CE-SDS analysis under non-reducing condition showed that the intact IgG level in CT-P10 is marginally higher (about 2% mean difference) than that observed in EU- and US-Rituximab. In addition, the levels of process-related impurities, including residual host cell protein, host cell DNA and recombinant Protein A, were found to be highly similar, with very low levels for the all 3 products (data not shown).
Figure 3.

Size-exclusion chromatogram and multi-angle light scattering chromatogram of CT-P10, EU-Rituximab and US-Rituximab. (A) SEC-HPLC chromatograms detected at 214 nm are compared for 1 batch of each product. High molecular weight (HMW) and low molecular weight (LMW) variants are separated from monomer peak. (B) SEC-MALS chromatograms detected by UV (green), RI (purple) and LS (red) are compared for 1 batch of each product. Three HMW variants (Peak 1, Peak 2 and Peak 3) are present in EU- and US-Rituximab, whereas Peak 3 is not observed in CT-P10.
Charged isoforms
Charge variants in CT-P10, EU- and US-Rituximab were analyzed by isoelectric focusing (IEF) and ion-exchange chromatography (IEC-HPLC). The results of IEF analysis showed that the calculated isoelectric point (pI) values of the 3 bands were similar and fell within a similar range for all 3 products (data not shown). In IEC-HPLC analysis, a total of 7 charge variants were separated (Fig. 4A). Although the number and distribution of IEC-HPLC peaks were conserved between CT-P10 and the RMPs, the relative proportion (peak ratio) of the seven IEC-HPLC peaks of CT-P10, particularly in the acidic and basic variants, are marginally different from ones of EU- or US-Rituximab (Table 2). The acidic peaks (sum of Peak 1, Peak 2 and Peak 3) of EU-Rituximab (18.7 – 22.8%) and US-Rituximab (19.0 – 22.5%) showed a slightly higher relative proportion compared to CT-P10 (15.6 – 18.5%), whereas the basic peak (sum of Peak 5, Peak 6 and Peak 7) of EU-Rituximab (7.0 – 11.0%) and US-Rituximab (8.0 – 10.6%) were present at a lower relative proportion than in CT-P10 lots (15.4 – 17.4%).
Figure 4.
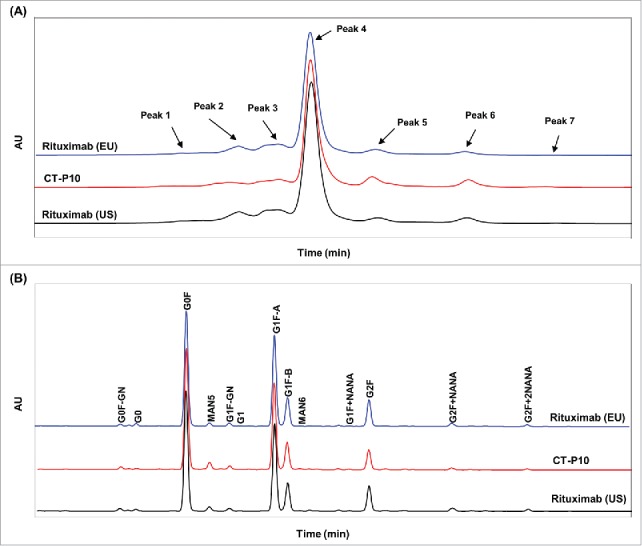
Comparison of charge variants and oligosaccharide profiles of CT-P10 (red), EU-Rituximab (blue) and US-Rituximab (black). (A) Representative ion exchange chromatograms analyzed by cation exchange-HPLC are presented for 1 batch of each product. The number and distribution of IEC-HPLC peaks are conserved between CT-P10 and RMPs. (B) Representative oligosaccharide profiles analyzed by HILIC-UPLC with 2-AB labeling are presented for 1 batch of each product. The types and proportions of the glycans are conserved among the products.
To characterize each charge variant peak, the seven charge isoforms from IEC-HPLC analysis were fractionated. Peptide mapping by LC-MS for the fractionated peaks revealed that basic peaks of CT-P10 contain slightly higher level of N-terminal glutamine variants in the light chain than those of EU- and US-Rituximab, which is in agreement with peptide mapping results by LC-MS for the unfractionated CT-P10 as described above. Nevertheless, it was confirmed that all fractionated charge variants exhibit biological activity comparable with intact samples for all 3 products (data not shown).
Glycosylation
Rituximab is a glycoprotein like other IgG1 subclass antibodies. The glycan micro-heterogeneity associated with N-glycosylation was characterized by a wide range of orthogonal methods. LC-MS analysis of the peptides generated during peptide mapping was used to identify glycosylation sites and quantify each glycan species. The results showed that Asn301 is only site for N-glycosylation in CT-P10, EU- and US-Rituximab. All 3 products were shown to contain mostly G0F and G1F structures. Minor species, including Man5, G2F and G0, were also detected. The three charged glycan species identified by this method were G1F+NANA, G2F+NANA and G2F+2NANA. The glycan micro-heterogeneity associated with CT-P10 closely reflects the heterogeneity observed in EU- and US-Rituximab. The array (type) of glycan species as well as the relative proportion (ratio) of the various glycans were shown to be highly conserved between the 3 products (Table 2).
To further characterize the glycan micro-heterogeneity associated with Asn301 as a site of N-glycosylation, PNGase F was employed for enzymatic cleavage of glycans from molecules, followed by normal phase UPLC with 2-aminobenzamide (2-AB) labeling to resolve and identify oligosaccharide structures (Fig. 4B). The oligosaccharide profiling revealed that the types and proportions of the glycans were reasonably conserved among CT-P10, EU- and US-Rituximab except for afucosylation (G0+G1) and high mannose (Man5+Man6) content (Table 2). CT-P10 samples contained slightly lower levels of afucosylated glycans (mean = 0.55%) compared to EU-Rituximab (mean = 1.21%) and US-Rituximab (mean = 1.16%). High mannose levels in CT-P10 samples were found to be slightly higher (mean = 3.29%) compared to that in EU-Rituximab (mean = 1.43%) and US-Rituximab (mean = 1.63%). Total afucosylation levels calculated by sum of afucosylation and high mannose in CT-P10 were slightly higher (mean = 3.84%) compared to that in EU-Rituximab (mean = 2.64%) and US-Rituximab (mean = 2.79%).
Because literature reports have shown that total afucosylation level has a direct relationship with increased binding affinity to FcγRIIIa and subsequently improved antibody-dependent cell-mediated cytotoxicity (ADCC),12-16 the effect of the observed difference in total afucosylation level between CT-P10, EU- and US-Rituximab was further investigated using highly afucosylated CT-P10 samples. The spiking study using variably afucosylated CT-P10 sample indicated strong correlation between afucosylated glycan level and FcγRIIIa binding affinity (R2 = 0.93, p-value < 0.0001) or between afucosylated glycan level and ADCC activity (R2 = 0.97, p-value < 0.0001) as expected (Fig. 5A). It is important to note, however, that the afucosylation levels of CT-P10 lots are below 5%, where no strong correlation exists with either FcγRIIIa binding affinity (R2 = 0.013, p-value = 0.45) or ADCC activity (R2 = 0.0032, p-value = 0.71) (Fig. 5B). Furthermore, an additional study performed with different high mannose content showed the decreased level of high mannose from 3.74% to 0.00% did not significantly affect Fab or Fc functionality based on binding affinities to FcγRIIIa, FcRn, C1q or cell-based CD20 activity. Taken together, the slightly higher level of total afucosylation level in CT-P10 in comparison with EU- or US-Rituximab is highly unlikely to have a meaningful impact on product efficacy or potency.
Figure 5.

Correlation between afucosylation level of CT-P10 with FcγRIIIa binding affinity and ADCC activity. (A) Spiking study using highly afucosylated CT-P10 (red) to check impact of variously afucosylated levels on FcγRIIIa binding affinity (left) and ADCC activity (right). Strong correlation is observed between afucosylation and biological activity when afucosylation level is increased up to 35%. (B) Relation between afucosylation level versus FcγRIIIa binding affinity (left) and ADCC activity (right) for CT-P10 (blue), EU-Rituximab (grey) and US-Rituximab (yellow) used in 3-way similarity study. Correlation between afucosylation and biological activity is not significant in the afucosylation level (< 5%) in CT-P10 drug product and RMPs.
Glycoproteins expressed by mammalian cell lines may have glycans capped by sialic acids, which are charged glycans. The sialic acid analysis results expressed as molar ratios showed that the levels of N-acetylneuraminic acid (NANA) are very low and highly similar in all 3 products (Table 2). Consistent results were obtain in oligosaccharide profiling and N-linked glycan analysis regarding sialylation level. The results of monosaccharide analysis to quantify neutral and amino sugars showed highly similar molar ratios of fucose (Fuc), N-acetyglucosamine (GlcNAc), Galactose (Gal) and Mannose (Man) between CT-P10, EU- and US-Rituximab. Finally, there was no significant difference in the glycation levels for both light chain and heavy chain in the 3 products.
Biological activities
The mechanisms of action of rituximab can be attributed to Fab and Fc functions, including the induction of apoptosis of CD20+ B-cells, complement-dependent cytotoxicity (CDC), ADCC, and antibody-dependent cellular phagocytosis (ADCP). To compare the biological activity of CT-P10, EU- and US-Rituximab, a variety of in vitro methodologies were employed. The relative binding affinities or activities were calculated relative to the CT-P10 in-house reference standard. Studies were conducted to assess the similarity of CT-P10 and EU- or US-Rituximab in binding to CD20, as well as in activities resulting from CD20 binding including CDC activity, ADCC activity, apoptosis induction by CD20 signaling, and ADCP, a putative mechanism of action. In addition, binding to C1q and Fc receptors were evaluated.
To evaluate F(ab')-related activities, CD20 binding affinity and apoptotic activity of CT-P10, EU- and US-Rituximab were measured. The results of cell-based CD20 binding affinity assay using a CHO-K1 cell line expressing recombinant CD20 antigen confirmed that the 3 products exhibit highly similar binding affinities to CD20 (Table 3 and Fig. 6A). The apoptotic activity induced by binding of CD20 was evaluated using the Raji cell line. The percentage of apoptotic cells analyzed by flow cytometry confirmed that CT-P10, EU- and US-Rituximab exhibit highly similar apoptotic activities (Table 3 and Fig. 6B).
Table 3.
Summary of results for biological similarity assessment between CT-P10, EU-rituximab and US-rituximab.
| Min – Max Range (Mean ± SD)1 |
||||
|---|---|---|---|---|
| Quality Attribute / Test Method | EU-Rituximab | CT-P10 Drug Product | US-Rituximab | |
| F(ab') related | Cell-based CD20 Binding Affinity (CELISA) (%) | 85 – 105 (96 ± 6.2) | 86 – 108 (97 ± 5.8) | 86 – 107 (96 ± 7.0) |
| Apoptosis (FACS) (%) | 95 – 114 (101 ± 5.1) | 98 – 109 (103 ± 3.1) | 87 – 106 (99 ± 5.0) | |
| Fc-F(ab') related | CDC (%) | 92 – 109 (100 ± 5.3) | 94 – 108 (100 ± 3.8) | 93 – 106 (99 ± 3.3) |
| ADCC (%) | 86 – 103 (97 ± 4.3) | 87 – 107 (97 ± 6.0) | 93 – 108 (98 ± 4.0) | |
| ADCC Reporter (%) | 81 – 117 (101 ± 9.2) | 86 – 113 (101 ± 9.3) | 82 – 113 (99 ± 10.6) | |
| ADCP (%) | 89 – 106 (99 ± 5.6) | 90 – 107 (99 ± 4.8) | 89 – 116 (101 ± 7.3) | |
| Fc related | C1q Binding Affinity (ELISA) (%) | 94 – 115 (104 ± 6.1) | 93 – 119 (104 ± 7.8) | 98 – 116 (105 ± 4.8) |
| FcγRIIIa-V Binding Affinity (SPR) (%) | 91 – 119 (104 ± 6.6) | 94 – 104 (100 ± 3.3) | 92 – 119 (103 ± 6.9) | |
| FcγRIIIa-F Binding Affinity (SPR) (%) | 95 – 123 (108 ± 8.6) | 96 – 104 (100 ± 2.5) | 93 – 120 (105 ± 8.9) | |
| FcγRIIIb Binding Affinity (SPR) (%) | 87 – 116 (105 ± 8.5) | 89 – 112 (102 ± 7.7) | 86 – 123 (101 ± 8.8) | |
| FcγRIIa Binding Affinity (SPR) (%) | 96 – 106 (100 ± 3.0) | 93 – 104 (99 ± 3.3) | 94 – 106 (100 ± 3.3) | |
| FcγRIIb Binding Affinity (SPR) (%) | 89 – 108 (97 ± 5.6) | 83 – 110 (98 ± 7.1) | 82 – 111 (94 ± 6.7) | |
| FcγRI Binding Affinity (SPR) (%) | 93 – 105 (101 ± 3.2) | 96 – 105 (100 ± 3.0) | 96 – 105 (100 ± 2.6) | |
| FcRn Binding Affinity (SPR) (%) | 96 – 103 (100 ± 2.3) | 97 – 105 (101 ± 2.4) | 96 – 104 (100 ± 2.2) | |
Relative potency (%) or binding affinity (%) in comparison to CT-P10 in-house reference standard.
Figure 6.
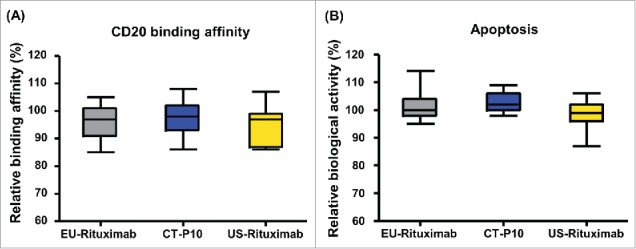
Comparison of F(ab')-related activities of CT-P10, EU-Rituximab and US-Rituximab. Box plots of relative (A) CD20 binding affinity and (B) apoptotic activity are presented for CT-P10 (blue), EU-Rituximab (grey) and US-Rituximab (yellow). Box plot shows the interquartile range (box), median (band inside of box), maximum and minimum values (whiskers).
For Fc-F(ab')-related activities of rituximab, the relative activities of CDC, ADCC and ADCP were analyzed for CT-P10, EU- and US-Rituximab. The Fc portion of IgG1 antibodies can interact with soluble C1q, resulting in lysis of the cell bound by the antibody via the classical complement pathway.17,18 CDC activity of CT-P10, EU- and US-Rituximab was assessed using the B lymphoblast cell line, Wil2-S. The relative CDC activity for the 3 products was shown to be highly similar (Table 3 and Fig. 7A). ADCC is a function of the innate immune system by which Fc-receptor-bearing effector cells kill infected cells or tumor cells through a non-phagocytic process.19,20 We performed an ADCC assay with Raji (B lymphocyte) target cells and human peripheral blood mononuclear cell (PBMC) as effector cell to compare the ADCC activity of CT-P10, EU- and US-Rituximab. An ADCC reporter assay using Raji cells and a Jurkat cell line expressing human FcγRIIIa and NFAT-induced luciferase was also employed to provide further assurance of the similarity between the 3 products with respect to initiation of ADCC activity. As shown in Table 3, Fig. 7B and Fig. 7C, CT-P10, EU- and US-Rituximab exhibited highly similar ADCC potency in both assays. ADCP results in the destruction of cells targeted by a specific antibody via monocyte or macrophage-mediated phagocytosis.19,20 Rituximab binding to CD20-expressing cells via the Fab portion of the antibody, and cross linking to Fcγ receptors (FcγRI, FcγRIIa and FcγRIIIa) expressed on the effector cells (macrophages) may result in ADCP. Whilst there is little evidence in vivo for ADCP induction by rituximab, and it is unclear whether ADCP is indeed a significant mechanism of action per se, this assay has been specifically incorporated into the similarity evaluation to provide assurance that all activities related to potential mechanisms of action have been evaluated. The ADCP activity of CT-P10, EU- and US-Rituximab was evaluated using primary monocyte-derived macrophages as effector cells and CD20-overexpressing Raji cells. The results showed the 3 products are highly similar in ADCP activity (Table 3 and Fig. 7D).
Figure 7.

Comparison of F(ab')-Fc-related activities of CT-P10, EU-Rituximab and US-Rituximab. Box plots of relative (A) CDC activity and (B) ADCC activity using PBMC, (C) ADCC activity using reporter assay and (D) ADCP activity are presented for CT-P10 (blue), EU-Rituximab (grey) and US-Rituximab (yellow). Box plot shows the interquartile range (box), median (band inside of box), maximum and minimum values (whiskers).
With regards to Fc-related activities, relative binding affinity of CT-P10, EU- and US-Rituximab to C1q as well as various types of Fc receptors were measured by ELISA and surface plasmon resonance, respectively. As summarized in Table 3 and Fig. 8, highly similar binding affinity of the 3 products to C1q, FcγRIIIa-V, FcγRIIIa-F, FcγRIIIb, FcγRIIa, FcγRIIb, FcγRI and FcRn was confirmed.
Figure 8.
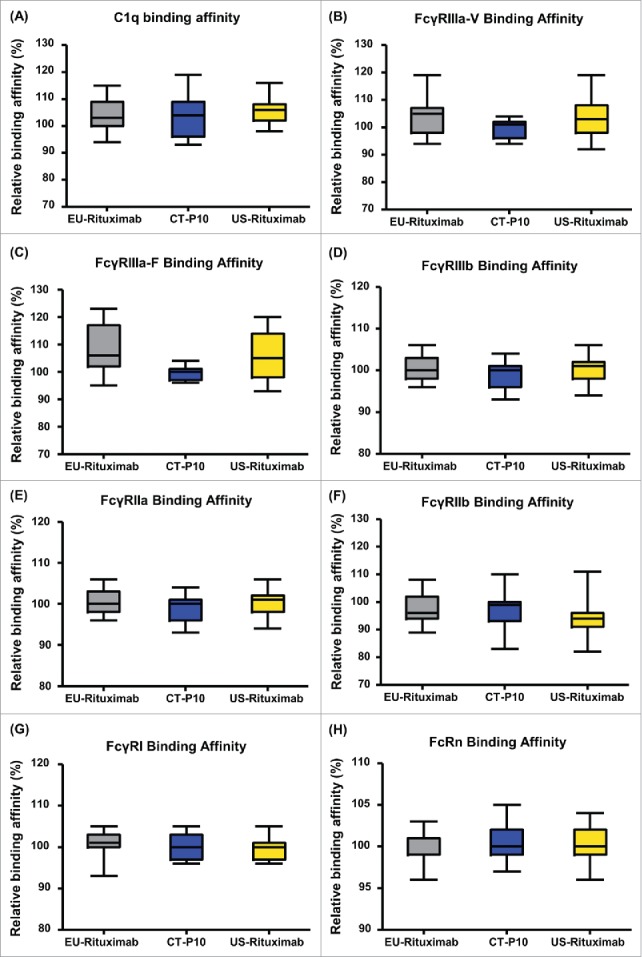
Comparison of Fc-related activities of CT-P10, EU-Rituximab and US-Rituximab. Box plots of relative (A) C1q binding affinity, (B) FcγRIIIa-V binding affinity, (C) FcγRIIIa-F binding affinity and (D) FcγRIIIb binding affinity, (E) FcγRIIa binding affinity, (F) FcγRIIb binding affinity, (G) FcγRI binding affinity and (H) FcRn binding affinity are presented for CT-P10 (blue), EU-Rituximab (grey) and US-Rituximab (yellow). Box plot shows the interquartile range (box), median (band inside of box), maximum and minimum values (whiskers).
In all biological similarity assessments, 100% of CT-P10 data points were within the quality range of EU- and US-Rituximab suggesting that the 3 products have highly similar biological activities.
Discussion
We performed a wide range of orthogonal, highly sensitive test methods to provide a meaningful set of measurements by which analytical similarity can be assessed to demonstrate biosimilarity of the proposed biosimilar product, CT-P10 with the reference medicinal products. The similarity assessments included an extensive comparative analysis of primary, secondary and tertiary structure, glycan profiles and of post-translational modifications. In addition, a large number of biological assays were used to evaluate similarity in all biological activities associated with known and putative functions and therapeutic effects.
Identical amino acid sequence with a RMP is a preemptive requirement of a biosimilar;1, 4 however, different primary structure may be caused by post-translational modifications such as N, C-terminal sequence variants. We have conclusively demonstrated that, compared to EU- and US-Rituximab, CT-P10 has an identical primary structure as shown using methods such as amino acid analysis, molar absorptivity, N/C-terminal sequencing, peptide mapping by HPLC or LC-MS, and determination of intact mass. Although slightly lower levels of N-terminal pyro-glutamate variants in the light chain were observed in CT-P10 compared to EU- and US-Rituximab, this modification has been reported to have no effect on antibody structure, antigen binding and in-vivo clearance, and is thus without safety concern.21-24 Highly similar antigen binding affinity and PK equivalence between CT-P10, EU- and US-Rituximab further confirmed that a slight difference in N-terminal pyro-glutamate variants is unlikely to be clinically meaningful. Other common post-translational modifications, including deamidation, oxidation and C-terminal lysine variants, were all observed at low levels for all 3 products.
Secondary/tertiary conformation of proteins is critical for their behavior because their functionality is determined by three-dimensional motifs. Thus, the comparison of higher order structures is essential part of similarity assessment of biosimilar products. Highly similar secondary and tertiary structure between the 3 products were confirmed by Ellman assay, disulfide positioning by LC-MS, spectroscopic and calorimetric methods, including FT-IR, CD and DSC.
Aggregation is a significant concern for biopharmaceutical products because aggregates may be associated with decreased bioactivity and increased immunogenic potential.25-27 To compare monomer/aggregate contents in CT-P10, EU- and US-Rituximab, SEC-HPLC, SEC-MALS and AUC analysis were conducted. CT-P10 contains slightly lower HMW content than EU- or US-Rituximab in SEC-HPLC analysis, whereas comparable levels of monomer and HMW were observed in SEC-MALS and AUC analysis. Although the SEC-HPLC results may be suggestive of a slight improvement in the profile of HMW species in CT-P10 relative to RMPs, the three products are considered to have no significant differences with respect to monomer and aggregate contents, taking into consideration all data from the three orthogonal methods. It should be noted that the molecular weight of HMW forms for CT-P10 was determined to be smaller than that for RMPs in SEC-MALS analysis because CT-P10 lacks some of the larger HMW species present in RMPs. Nevertheless, the HMW present in CT-P10 are highly unlikely to have an adverse effects on clinical safety and immunogenicity in comparison to EU- and US-Rituximab because the HMW species observed in CT-P10 are also present in RMPs.
Fragmentation and non-glycosylation are other critical quality attributes that can cause negative impacts on product efficacy and potency.28-31 The levels of intact IgG and non-glycosylated heavy chain were measured by CE-SDS under non-reducing and reducing conditions, respectively. The results of non-reduced CE-SDS showed that CT-P10 contains about 2% higher levels of intact IgG1 than RMPs. This may suggest that CT-P10 has a marginally better quality profile in relation to intact IgG and fragments than EU- and US-Rituximab, although this is unlikely to be clinically meaningful considering the very small difference. The reduced CE-SDS showed CT-P10 has slightly higher levels of non-glycosylated heavy chain (mean value of 0.80%) compared to that of EU-Rituximab (mean value of 0.43%) and US-Rituximab (mean value of 0.47%). Nevertheless, the magnitude of the difference is very small. This is further supported by a separate aglycosylation study, which showed no noticeable effect on FcγRIIIa and C1q binding affinities when the level of non-glycosylated heavy chain was increased to approximately 2% (data not shown). Thus, the slightly higher level of non-glycosylated heavy chain in CT-P10 has no impact on biological activities and is not clinically meaningful. Taking all of the results into account, it can be concluded that CT-P10 exhibits highly similar purity/impurity profiles with EU- and US-Rituximab.
Charge variants are one of the most common sources of heterogeneity observed in therapeutic proteins. It is reported that acidic and basic isoforms result mainly from oxidation, deamidation, isomerization, amination, cyclization, glycation, and the presence of C-terminal lysine variants,32 and could alter the mAb affinity to target and receptor molecules due to the modification of electrostatic and hydrophobic interactions with cell membranes. In addition, close monitoring of charge isoforms is critical to achieve consistent product quality because they are highly dependent on manufacturing processes.33 Similar numbers and distribution of charged variants were observed using methods IEF and IEC-HPLC. IEC-HPLC analysis demonstrates that, while the same charged variants are identified in all 3 products, the relative proportions of the charged variants are slightly different; CT-P10 contains less acidic and more basic species relative to the RMPs. Higher levels of basic variants of CT-P10 were identified due to the higher level of N-terminal glutamine compared to RMPs. However, N-terminal pyro-glutamate variant does not impact potency or safety, as discussed above. Peak characterization studies further determined that each of the charged variants were biologically active with respect to CD20 binding affinity, FcγRIIIa and FcRn binding, apoptosis, CDC and ADCC. As such the minor differences in the relative proportions of each of the charged variants are highly unlikely to influence the biological activity of the product. In addition, a literature report suggests that manufacturing process changes resulting in a reduction of basic variants from approximately 30% to 10% in EU- and US-Rituximab lots have historically been considered acceptable,33 suggesting that the slightly higher level of basic forms in CT-P10 is unlikely to have a clinical impact.
Glycosylation of monoclonal antibodies plays an important role for CDC and ADCC function through modulating the binding to the Fcγ receptor29,34 and affecting the antibody conformation and stability.31 Therefore, it is important to compare the glycosylation profile of a biosimilar with that of the original product. We found that the types and relative content of various glycan species were reasonably conserved in CT-P10, EU- and US-Rituximab. However, there were small differences in afucosylated glycans and high mannose content; CT-P10 contains a slightly lower level of afucosylated glycans and a slightly higher level of high mannose compared to those in RMPs. The spiking study with highly afucosylated CT-P10 suggests that correlation between afucosylation and biological activity is not significant in the observed afucosylation level in CT-P10 drug product (afucosylation level < 5%). Furthermore, a mannosylation study confirmed different levels of high mannose from 0.00% to 3.74% did not cause significant effects on Fab or Fc functionality based on binding affinity to FcγRIIIa, FcRn, C1q or CD20. Therefore, the minor differences in afucosylated glycans and high mannose content between CT-P10, EU- and US-Rituximab are not expected to affect product efficacy and potency, which is supported by the high similarity in biological activity between the 3 products. Mannose content can influence product half-life due to antibody clearance via the mannose receptor15; however, the global clinical study performed in RA patients confirmed the PK equivalence between CT-P10 and RMP, suggesting the observed difference in mannose content is not sufficient to significantly alter antibody clearance.35,36
Finally, highly similar biological activities were observed for CT-P10, EU- and US-Rituximab in the assays representative of the known and putative mechanisms of action of rituximab, namely, CDC, ADCC, apoptosis, C1q binding affinity, Fcγ receptors (FcγRIIIa-V, FcγRIIIa-F, FcγRIIIb, FcγRIIa, FcγRIIb and FcγRI) binding affinity and FcRn binding affinity. Statistical analysis showed 100% of the data points of CT-P10 were within the pre-determined quality range of EU- and US-Rituximab
In summary, the 3-way similarity assessment demonstrates that CT-P10, EU- and US-Rituximab are close to identical in physicochemical and structural attributes and are highly similar in biological activities. A recently reported clinical outcome further supports the conclusion that CT-P10 exhibits comparable efficacy and safety profiles with original rituximab.35-39 These results correspond with the “totality-of-evidence” concept that emphasizes comprehensive step-wise approach evaluation of multiple lines of evidence for the approval of biosimilar products.4 Taken together, we conclusively demonstrate the highly similar nature of CT-P10 with original rituximab, which enabled its approval as the world's first rituximab biosimilar product in the European Union and South Korea.40
Materials and methods
Materials
CT-P10 lots were manufactured at Celltrion Inc., Korea. EU-Rituximab (MabThera®) and US-Rituximab (Rituxan®) lots were purchased from a pharmacy located in the European Union and US, respectively. The batches of CT-P10, EU- and US-Rituximab were chosen to cover a range of expiration dates and product ages. Highly afucosylated CT-P10 used for the spiking study was generated under CT-P10 production conditions with the addition of fucosyl transferase inhibitor to inhibit fucosylation.
Amino acid analysis and molar absorptivity
Samples were first desalted using Amicon Ultra (Millipore) centrifugal filters and aliquoted in triplicate to hydrolysis tubes containing internal standards; the internal standard for this analysis was norleucine. The sample/internal standard mixtures were then hydrolyzed using 6 M HCl for 24 hr at 110°C. The hydrolysates were dried under reduced pressure, reconstituted in 80 μL of 20 mM constant-boiling HCl, and 10 μL of reconstituted sample was transferred to a microtube. Decomposed amino acids were reconstituted and derivatized by pre-column derivatization method with AQC reagent (6-aminoquinolyl-N-hydroxysuccinimidyl carbamate) so that the sample could be detected by a fluorescence detector. Retention times and fluorescence responses for each amino acid were calibrated with a standard mixture of amino acids.
Measurements of OD280 and OD320 were performed using a Beckman DU730 spectrophotometer. Using the amino acid molar ratio data from amino acid analysis, amino acid that the difference between theoretical and observed ratio is < 5% was selected as a robust amino acid. Protein concentration was determined with the concentration of robust amino acids. Molar extinction coefficient was calculated using the UV absorbance at 280 nm, concentration of protein and molecular weight of CT-P10, EU- and US-Rituximab.
HPLC peptide mapping analysis
Samples were analyzed by HPLC peptide mapping after reduction with dithiothreitol (DTT, Sigma-Aldrich), alkylation with iodoacetamide (Sigma-Aldrich), and digestion with trypsin (Promega) at 37ºC. The resulting peptides were separated by reversed phase (RP)-HPLC using a C18 column (5 µm, 250 × 4.6 mm; Vydac, Hesperia) and acetonitrile (ACN) gradient (Burdick & Jackson) containing trifluoroacetic acid (Sigma-Aldrich). Absorbance was monitored at 214 and 280 nm using a Waters-2695 Alliance HPLC system equipped with a UV detector (Waters).
LC-ESI-MS peptide mapping analysis
Samples were analysed by LC-MS peptide mapping after reduction with 10 mM DTT at 37°C for 1 hour, alkylation with 40 mM iodoacetamide (IAM), desalting with NAP5 column and digestion with trypsin (a 20:1 (sample:trypsin) treatment, at 37°C for 2 hrs), Asp-N (a 100:1 (sample:Asp-N) treatment, at 37°C for 16 hrs). The resulting peptides were separated by reversed-phase UPLC using a C18 column at a flow rate of 0.5 mL/min and following gradient conditions using 0.1% formic acid in water (mobile phase A) and 0.1% formic acid in ACN (mobile phase B); 1% B initially for 3 min, increased to 34% B in 48 min with further increase to 90% B in 0.1 min, followed by 3.9 min isocratic hold. The chromatography was performed on a Waters Acquity UPLC. An online AB SCIEX Triple TOF 5600 mass spectrometer with an electrospray source was used to collect mass spectra of the intact peptide as well as to fragment the peptides for sequencing (MS/MS analysis). The m/z (mass/charge) data were collected from 250 to 1,600 m/z.
Intact mass analysis
Intact mass by LC/MS was analyzed using an Agilent 1200 HPLC coupled online to an Agilent 6530 Q-TOF mass spectrometer. The intact protein was eluted by RP-HPLC using a C18 column at a flow rate of 0.5 mL/min and following gradient conditions using 0.1% formic acid in water (mobile phase A) and 0.1% formic acid in ACN (mobile phase B); 20% B initially for 0.5 min, increased to 90% B in 5 min, followed by 1 min isocratic hold. The m/z (mass/charge) data are collected from 900 to 4,000 m/z at a scan rate of 1 spectrum per second followed by deconvolution of the mass spectra to intensity versus molecular mass.
Free thiol analysis by Ellman assay
The free thiol (SH) groups in the samples were determined by means of the DTNB (Ellman's reagent) method. Briefly, standard and samples were mixed with DTNB in 7.5 M guanidine HCl, 125 mM sodium phosphate pH 8.0 and 1.25 mM EDTA, followed by measurement of absorbance at 412 nm. Free thiol groups could be estimated in a sample by comparison to a standard curve composed of known concentrations of a sulfhydryl-containing compound such as cysteine. The results were reported as molar ratios (free SH/IgG, μM/μM).
Disulfide bond analysis
Samples were analyzed by comparing native and reduced peptide maps. In the case of reduced peptide mapping analysis, the samples were reduced with 10 mM DTT at 37°C for 1 hour and alkylated with 40 mM IAM, whereas in the case of native peptide mapping analysis, no DTT was added to the sample. The samples were digested using trypsin after desalting with NAP-5 column. The resulting peptides were separated by RP-UPLC using a C18 column at a flow rate of 0.5 mL/min and following gradient conditions using 0.1% formic acid in water (mobile phase A) and 0.1% formic acid in ACN (mobile phase B); 1% B initially for 3 min, increased to 34% B in 48 min with further increase to 90% B in 0.1min, followed by 3.9 min isocratic hold. The chromatography was performed on a Waters Acquity UPLC. An online AB SCIEX Triple TOF 5600 mass spectrometer with an electrospray source was used to collect mass spectra of the intact peptide, as well as to fragment the peptides for sequencing (MS/MS analysis).
Fourier transform infra-red spectroscopy
FT-IR spectra were recorded on a Nicolet 6700 (Thermo-Nicolet) with a smart iTR attenuated total reflectance (ATR) accessory and diamond crystal at a resolution of 4 nm, number of scans = 32, and spectral range from 4000 – 650 cm−1. One (1) μL of sample solution was transferred onto the crystal and allowed to dry. All samples were measured in duplicate. All spectra were baseline and ATR-corrected with instrument software. In order to generate the difference spectra, buffer spectra were recorded as a blank and subsequently subtracted. Sample solutions were transferred onto the crystal and allowed to dry. FT-IR spectra were analyzed by comparing the locations and shapes of the amide I and amide II bands, as well as three other bands between 1200 and 1800 cm−1.
Circular dichroism spectroscopy
The CD experiment was performed using a Chirascan-plus CD spectrometer (Applied Photophysics) equipped with a Pelltier-controlled temperature regulator at 293 K. Cells of quartz glass along with optical path lengths of 1.0 and 0.10 cm were used for near-UV and far-UV CD measurements, respectively. Protein concentrations used for the far-UV spectra and near-UV CD spectra were 0.2 and 1.0 mg/ml, respectively. The formulation buffer was measured as a blank and subsequently subtracted. Noise reduction was applied to the baseline-corrected protein spectra using the smoothing option of the device software for the spectrometer. Conversion of the measured CD signals to mean residue molar ellipticities [(θ)MRE] was also performed using this software.
Differential scanning calorimetry
Thermal stability of the samples was evaluated by measuring their Tm values using a Microcal VP-DSC microcalorimeter (MicroCal). The thermogram was obtained with a scan rate of 1°C/min within a temperature range of 30 to 100˚C. DSC data were analyzed using a non-two state model using the Origin software package (OriginLab Corporation) to determine three thermal transition temperatures.
Size-exclusion chromatography
Samples were diluted with mobile phase buffer (20 mM sodium phosphate; 150 mM sodium chloride; pH 7.4) prior to SEC-HPLC analysis. SEC-HPLC was performed under non-denaturing conditions with the Waters-2695 Alliance HPLC system on a TSK G3000SWXL column (Tosoh, Japan) with aqueous-buffered mobile phase. The isocratic elution profile was monitored using UV detection at 214 nm.
SEC-multi angle light scattering
SEC-MALS was performed by HPLC on a TSKgel G3000SWXL column using aqueous buffered mobile phase (20 mM sodium phosphate; 150 mM sodium chloride; pH 7.4). The isocratic elution profile was monitored using MALS system, DAWN® HEREOS™II and Optilab rEX (Wyatt Technologies). Molecular weight and the monomer and dimer content were determined with MALS and RI detector.
Sedimentation velocity analytical ultracentrifugation
SV-AUC was carried out on a Beckman Coulter XLA-70 AUC instrument at 20°C. Sample was loaded into the sample channel of AUC cells having quartz windows and 12-mm double-sector Epon centerpieces. Matching buffer having the same composition and pH as the sample were loaded into the corresponding reference channel of each cell. The centrifugation was carried out at 20°C and 45,000 rpm. Radial scans of the concentration profile were collected sequentially by absorbance at 280 nm, until full sedimentation was reached. The resulting data sets were analyzed using the program SEDFIT with a continuous c(s) distribution model, yielding best-fit distributions for the number of sedimenting species and the effective molecular weights. Each sample was analyzed in two separate SV-AUC spins, resulting in two analyses per sample. The resulting c(s) distribution profile was used to calculate the percentage of each species and the estimated molecular weights.
Capillary sodium dodecyl sulfate gel electrophoresis
CE-SDS was performed under both non-reducing conditions for analysis of purity/impurities. A Beckman Coulter, PA 800 plus capillary electrophoresis system was used with a 57 cm, 50 μm I.D. bare-fused silica capillary. Reduced CE-SDS was performed for determination of purity as the corrected peak area % of the sum of heavy chain and light chain, and non-glycosylated heavy chains. Non-reduced CE-SDS was performed for determination of the corrected peak area % of intact IgG and non-assembled IgG molecules. Samples were reduced using 2-mercaptoethanol under reducing conditions and alkylated using 250 mM IAM under non-reducing condition.
Isoelectric focusing
IEF was used to determine pI values of charge variants in the samples. Electrophoresis was performed on IsoGel agarose IEF plates in the range of pH 7 – 11 using a flatbed electrophoresis system (Multiphor II, Amersham). pI values were calculated against IEF pI markers (pI range: 10.65 – 6.90) and compared to the pI values of the reference standard. The samples were focused by running the gels at 1200 V, 8 mA and 25W for 110 minutes. pI values were calculated using Quantity One software (Bio-Rad).
Ion exchange chromatography
The IEC-HPLC method was used to evaluate the distribution of charge variants by cation exchange chromatography. The HPLC system (Waters) was equipped with a Propac WCX 10 analytical column and guard column set (Dionex) at ambient temperature. Gradient NaCl elution with a mobile phase of 10 mM MES and 200 mM sodium chloride, pH 6.0 was performed, and UV signals were obtained at 214 nm. Peaks in the IEC HPLC chromatogram were integrated, and percentage peak areas of each peak were calculated.
Oligosaccharide profile analysis
For oligosaccharide profile analysis, N-linked glycans were released from the antibody using PNGase F treatment at 37°C. PNGase F-cleaved glycans were analyzed by UPLC with a fluorescence detector (Waters). Released N-linked glycans by PNGase F treatment were extracted from deglycosylated protein solution using a MassPREP HILIC µElution plate (Waters). Extracted glycans were labeled with 2-AB labeling reagent, followed by removal of excess labeling reagent using a MassPREP HILIC µElution plate. Finally, 2-AB-labeled N-linked glycans were analyzed by normal phase chromatography with a GlycoSep N column (Glyko) and fluorescence detector.
N-glycan analysis by LC-MS
For structural analysis of N-linked oligosaccharide at Asn 300 by LC-MS (AB SCIEX), trypsin-digested peptides prepared for LC-MS peptide mapping were evaluated. Extracted ion chromatograms were used to quantify each oligosaccharide species. The percentage calculation was based on each glycosylation site. For each site, all detectable oligosaccharide structures were counted.
Sialic acid analysis
Sialic acids were released from antibody by mild acid hydrolysis (0.1 M HCl) for 1 hr incubation at 80ºC. A Zorbax Extend-C18 column equipped to the HPLC system was used at a flow rate of 1.0 mL/min using 7% methanol/ 93% water (mobile phase A) and 7% methanol/93% ACN (mobile phase B). The sialic acid content was quantified based on the response of sialic acid standards (NANA) relative to an internal standard [2-keto-3-deoxy-D-glycero-D-galacto-nononic acid; deaminated neuraminic acid (KDN)]. The results were reported as molar ratios (sialic acid/protein, mole/mole).
Monosaccharide analysis
Monosaccharide analysis of neutral and amino sugars was performed by hydrolyzing the samples with 2 N trifluoroacetic acid at 100°C, followed by high performance anion exchange chromatography with the pulsed amperometric detection (HPAEC-PAD) system (Dionex) with a CarboPac PA-20 analytical column (Dionex) and isocratic elution with 200 mM NaOH. Monosaccharide content was measured based on relative response of monosaccharide standards against an internal standard (2-deoxy-D-glucose). Monosaccharide content was determined as relative mole ratios (monosaccharide/protein, mole/mole).
Glycation analysis
Samples were digested with PNGase F (1,500 U/mg treatment, at 37°C for 16 hours) to remove N-glycan, and then reduced with DTT followed by LC-ESI-MS analysis using an Agilent 1200 HPLC coupled online to an Agilent 6530 Q-TOF mass spectrometer. The m/z (mass/charge) data were collected from 900 to 4,000 m/z at a scan rate of 1 spectrum per second followed by deconvolution of the mass spectra to intensity versus molecular mass for both heavy and light chain. The percentage calculation was based on deconvoluted spectra of each chain. For the determination of % glycation in light chain, area of glycated light chain was divided by sum of both areas from native and glycated light chain. For the determination of % glycation in heavy chain, area of glycated heavy chain was divided by sum of both areas from native and glycated heavy chain.
Cell-based CD20 binding affinity
CHO-K1 cell lines expressing recombinant CD20 antigen were propagated onto the 96-well plate. After overnight incubation, cells were fixed with formaldehyde and then blocked with diluent buffer. Four-fold serial diluted sample (from 0.24 ng/mL to 4,000 ng/mL) was treated to the fixed cells. After incubation, the cells were washed and subsequently incubated with horseradish peroxide (HRP)-conjugated detection antibody (Abdserotec, Cat. No. 221–5004P). Substrate TMB (3, 3′, 5, 5′-tetramethylbenzidine) was added to each well; after the stop reaction with sulfuric acid, the plate was measured at 450 nm / 650 nm. The binding affinity (EC50) was deduced from four-parameter curve fitting of the sigmoid dose-response curves using GraphPad Prism 4 plot program. The relative cell-based CD20 binding affinity was determined from comparison with the EC50 value of CT-P10 in-house reference standard.
Complement-dependent cytotoxicity
The cell-based anti-CD20 CDC assay was performed to measure the biological activity using normal human serum as a complement source and Wil2-S cell line, a B-lymphoblast that express CD20 on the cell surface (ATCC, CRL-8885TM). Biological activity was determined by measuring Wil2-S metabolic activity as an indicator of cell viability using a colorimetric method (Cell Counting Kit-8, Dojindo). The relative potency was evaluated using EC50 value in 4-parameter logistic curve model by GraphPad PRISM software.
Antibody-dependent cell-mediated cytotoxicity
The calcein-AM release assay (ThermoFisher) was carried out to test the ADCC activity. The Raji cell line (B lymphocyte), which expresses CD20 protein on the cell surface, was used as target cell (ATCC, CRL-86TM) and human PBMCs (CTL, Cat. No. CTL-UP1) were used as effector cell. The target cell was incubated with sample and PBMCs. Then, the cell cytotoxicity was measured by fluorescence values from released calcein-AM and reported as ADCC activity. The relative ADCC activity of CT-P10, EU- and US-Rituximab was determined as mean relative ADCC activity of each cytotoxicity at 3 concentrations (10.0, 35.0 and 122.4 ng/mL) within the linear range of ADCC response compared with that of CT-P10 in-house reference standard.
Antibody-dependent cellular phagocytosis
For the analysis of ADCP, primary monocyte-derived macrophages were used as effector cells and CD20-overexpressing Raji cells (ATCC, CRL-86TM) were used as target cells. Monocytes were isolated from human PBMCs using Pan Monocyte isolation kit (MACS, Cat# 130-096-537) and were cultured with RPMI1640 medium with 10% fetal bovine serum and 25 ng/mL macrophage-colony stimulating factor for 7 – 14 days to enable differentiation into macrophages. Prior to incubation with antibodies, the target cells were labelled at 37°C for 30 minutes with PKH67 (2 × 10−7 M/1 × 106 cells), which is green fluorescent cell linker kit (Sigma-Aldrich, Cat# PKH67GL). Then, the PKH67-stained Raji cells were incubated with CT-P10, EU- and US-Rituximab for 30 minutes. The macrophages differentiated from the purified monocytes were added to incubation tubes at a 1:1 (Effector cells:Target cells) ratio. Following a 3-hrs incubation, the phagocytic activity was assessed by fluorescence-activated cell sorting analysis after staining macrophages with CD11b-APC. The % phagocytosis was calculated as the percentage of double-positive macrophages with respect to total target cells stained with PKH67. The relative phagocytic activity was determined at 3 concentrations (1.56, 6.25 and 25.0 ng/mL) within the linear range of the ADCP dose-response curve (from 0.01 ng/mL to 1,600 ng/mL) and was compared with that of CT-P10 in-house reference standard.
Apoptosis
Apoptotic activity was measured using the Raji cell line (B lymphocyte), which expresses CD20 protein on the cell surface antigen (ATCC, CRL-86TM). These cells were incubated with 0.13, 0.04 and 0.01 μg/mL of CT-P10 drug product, EU- and US-Rituximab for 48 hrs. Then, the incubated cells were harvested and labeled with an FITC Annexin-V Apoptosis Detection Kit II (BD Pharmingen™) according to the manufacturer's instructions. Flow cytometric analysis was subsequently performed and the percentage of apoptotic cell (Annexin V-FITC+/PI-) was used to quantify apoptotic cells. The relative apoptotic activity of CT-P10 drug product, EU- and US-Rituximab was determined as mean relative apoptotic activity of each apoptotic cell (%) at three concentrations (0.13, 0.04 and 0.01 μg/mL) within linear range of apoptotic response compared with that of CT-P10 in-house reference standard.
C1q binding affinity
An anti-C1q ELISA was used to evaluate the binding affinity of CT-P10 drug product, EU- and US-Rituximab to C1q complement. All samples were serially diluted and coated on a microplate surface. C1q was added and, following washing, the bound material was detected using anti-C1q-HRP conjugate (Sigma, Cat. No. A7164). The signal was measured after TMB (3, 3′, 5, 5′-tetramethylbenzidine) treatment. The optical density values for reference standard and samples were fitted using a four parameter curve fitting algorithm (GraphPad Prism® software). The relative C1q binding affinity of samples was determined from comparison of the EC50 value (effective concentration yielding a 50% response) with the CT-P10 in-house reference standard to the EC50 value of the samples.
Fc receptor binding affinity
The Fcγ receptor binding affinity assays were performed using Biacore 3000™ and Biacore T200™ with a Biacore CM5 chip coupled to the recombinant Fc receptors. Prior to the assay, each Fc receptor was diluted using 10 mM sodium acetate buffer, pH 5.0, then immobilized on the CM5 chip using an amine coupling reaction. Any unstable, immobilized Fc receptor was removed by at least 6 washes of pre-run solution. Samples were serially diluted in HBS-EP buffer, pH 7.4, (HBS-EP, pH 6.0 for FcRn) to generate binding curves and the binding affinity is evaluated using BIAevaluation software. The chips were regenerated using regeneration solution appropriate for the Fc receptor.
Statistical analysis of biological activities
Statistical analysis using a quality range approach has been conducted on data obtained from biological tests. The quality range of the RMP was set based on the range of the values obtained from EU- or US-Rituximab variation expressed as 3 times standard deviations (mean ± 3SD). The limit of mean ± 3SD is frequently used in the pharmaceutical industry for setting in-process acceptance criteria and lot release acceptance criteria.41,42 Where 90% or more of data points are within the quality range of EU- and US-Rituximab, the data are considered to be highly similar.
Spiking study with highly afucosylated CT-P10 sample
Various levels of afucosylated CT-P10 samples (4.5 – 40.0% afucosylated glycans) were generated by spiking different amounts of the highly afucosylated CT-P10 sample (48.22% afucosylated glycans) in the in-house reference (4.12% afucosylated glycan). Correlation between afucosylation level versus FcγRIIIa binding affinity or ADCC activity were subsequently investigated.
Funding Statement
Celltrion Inc.
Abbreviations
- ADCC
antibody-dependent cell-mediated cytotoxicity
- ADCP
antibody-dependent cellular phagocytosis
- CDC
complement-dependent cytotoxicity
- CD
circular dichroism
- CE-SDS
capillary sodium dodecyl sulfate gel electrophoresis
- DSC
differential scanning calorimetry
- DTT
dithiothreitol
- ELISA
enzyme linked immunosorbent assay
- FT-IR
Fourier transform infra-red
- LMW
low molecular weight
- HMW
high molecular weight
- HPLC
high performance liquid chromatography
- HILIC
hydrophilic interaction liquid chromatography
- HPAEC-PAD
high performance anion exchange chromatography with pulsed amperometric detection
- IAM
iodoacetamide
- IEF
isoelectric focusing
- IEC
ion exchange chromatography
- LC-ESI-MS
liquid chromatography electrospray ionization mass spectrometry
- MALS
multi angle light scattering
- MES
2-(N-morpholino)ethanesulfonic acid
- NANA
N-acetylneuraminic acid
- RMP
reference medicinal product
- RP
reversed phase
- SEC
size-exclusion chromatography
- SV-AUV
sedimentation velocity analytical ultracentrifugation
- TMB
3, 3′, 5, 5′-tetramethylbenzidine
- UPLC
ultra-performance liquid chromatography
- 2-AB
2-aminobenzamide
Disclosure of potential conflicts of interest
No potential conflicts of interest were disclosed.
References
- 1.European Medicines Agency, Committee for Medicinal Products for Human Use Guideline on similar biological medicinal products [Internet]. London, UK: European Medicines Agency; [Adopted on October 23, 2014; cited September 15, 2017] Available from: http://www.ema.europa.eu/docs/en_GB/document_library/Scientific_guideline/2014/10/WC500176768.pdf. [Google Scholar]
- 2.European Medicines Agency, Committee for Medicinal Products for Human Use Guideline on similar biological medicinal products containing biotechnology-derived proteins as active substance: quality issues (revision 1) [Internet]. London, UK: European Medicines Agency; [Adopted on May 22, 2014; cited September 15, 2017] Available from: http://www.ema.europa.eu/docs/en_GB/document_library/Scientific_guideline/2014/06/WC500167838.pdf. [Google Scholar]
- 3.European Medicines Agency, Committee for Medicinal Products for Human Use Guideline on similar biological medicinal products containing biotechnology - non-clinical and clinical issues [Internet]. London, UK: European Medicines Agency; [Adopted on May 30, 2012; cited September 15, 2017] Available from: http://www.ema.europa.eu/docs/en_GB/document_library/Scientific_guideline/2012/06/WC500128686.pdf. [Google Scholar]
- 4.US Food and Drug Administration Guidance for Industry: Scientific Consideration in Demonstrating Biosimilarity to a Reference Product; 2015. [accessed 2017 Sept 15]. https://www.fda.gov/downloads/drugs/guidances/ucm291128.pdf.
- 5.US Food and Drug Administration Guidance for Industry: Quality Considerations in Demonstrating Biosimilarity of a Therapeutic Protein Product to a Reference Product; 2015. [accessed 2017 Sept 15]. https://www.fda.gov/downloads/drugs/guidances/ucm291128.pdf
- 6.Reff ME, Carner K, Chambers KS, Chinn PC, Leonard JE, Raab R, Newman RA, Hanna N, Anderson DR. Depletion of B cells in vivo by a chimeric mouse human monoclonal antibody to CD20. Blood. 1994;83:435–45. PMID:7506951 [PubMed] [Google Scholar]
- 7.Gurcan HM, Keskin DB, Stern JN, Nitzberg MA, Shekhani H, Ahmed AR. A review of the current use of rituximab in autoimmune diseases. Int Immunopharmacol. 2009;9:10–25. doi: 10.1016/j.intimp.2008.10.004. PMID:19000786. [DOI] [PubMed] [Google Scholar]
- 8.Grillo-Lopez AJ, Rituximab. an insider's historical perspective. Semin Oncol. 2000;27:9–16. [PubMed] [Google Scholar]
- 9.Eisenberg R. Update on rituximab. Ann Rheum Dis 2005;64 Suppl 4:iv55−7. PMID:16239389. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Scheinfeld N. A review of rituximab in cutaneous medicine. Dermatol Online J. 2006;12:3. PMID:16638371. [PubMed] [Google Scholar]
- 11.Pescovitz MD. Rituximab, an anti-cd20 monoclonal antibody: History and mechanism of action. Am J Transplant. 2006;6:859–66. doi: 10.1111/j.1600-6143.2006.01288.x. PMID:16611321. [DOI] [PubMed] [Google Scholar]
- 12.Shields RL, Lai J, Keck R, O'Connell LY, Hong K, Meng YG, Weikert SH, Presta LG. Lack of fucose on human IgG1 N-linked oligosaccharide improves binding to human Fcgamma RIII and antibody-dependent cellular toxicity. J Biol Chem. 2002;277:26733–40. doi: 10.1074/jbc.M202069200. PMID:11986321. [DOI] [PubMed] [Google Scholar]
- 13.Shinkawa T, Nakamura K, Yamane N, Shoji-Hosaka E, Kanda Y, Sakurada M, Uchida K, Anazawa H, Satoh M, Yamasaki M, et al. . The absence of fucose but not the presence of galactose or bisecting N-acetylglucosamine of human IgG1 complex-type oligosaccharides shows the critical role of enhancing antibody-dependent cellular cytotoxicity. J Biol Chem. 2003;278:3466–73. doi: 10.1074/jbc.M210665200. PMID:12427744. [DOI] [PubMed] [Google Scholar]
- 14.Suzuki E, Niwa R, Saji S, Muta M, Hirose M, Iida S, Shiotsu Y, Satoh M, Shitara K, Kondo M, et al. . A nonfucosylated anti-HER2 antibody augments antibody-dependent cellular cytotoxicity in breast cancer patients. Clin Cancer Res. 2007;13:1875–82. doi: 10.1158/1078-0432.CCR-06-1335. PMID:17363544. [DOI] [PubMed] [Google Scholar]
- 15.Mori K, Kuni-Kamochi R, Yamane-Ohnuki N, Wakitani M, Yamano K, Imai H, Kanda Y, Niwa R, Iida S, Uchida K, et al. . Engineering Chinese hamster ovary cells to maximize effector function of produced antibodies using FUT8 siRNA. Biotechnol Bioeng. 2004;88:901–8. doi: 10.1002/bit.20326. PMID:15515168. [DOI] [PubMed] [Google Scholar]
- 16.Okazaki A, Shoji-Hosaka E, Nakamura K, Wakitani M, Uchida K, Kakita S, Tsumoto K, Kumagai I, Shitara K. Fucose depletion from human IgG1 oligosaccharide enhances binding enthalpy and association rate between IgG1 and FcgammaRIIIa. J Mol Biol. 2004;336:1239–49. doi: 10.1016/j.jmb.2004.01.007. PMID:15037082. [DOI] [PubMed] [Google Scholar]
- 17.Gaboriaud C, Thielens NM, Gregory LA, Rossi V, Fontecilla-Camps JC, Arlaud GJ. Structure and activation of the C1 complex of complement: unraveling the puzzle. Trends Immunol. 2004;25:368–73. doi: 10.1016/j.it.2004.04.008. PMID:15207504. [DOI] [PubMed] [Google Scholar]
- 18.Kishore U, Reid KB. C1q: structure, function, and receptors. Immunopharmacology. 2000;49:159–70. doi: 10.1016/S0162-3109(00)80301-X. PMID:10904115. [DOI] [PubMed] [Google Scholar]
- 19.Weiner LM, Surana R, Wang S. Monoclonal antibodies: versatile platforms for cancer immunotherapy. Nat Rev Immunol. 2010;10:317–27. doi: 10.1038/nri2744. PMID:20414205. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Adams GP, Weiner LM. Monoclonal antibody therapy of cancer. Nat Biotechnol. 2005;23:1147–57. doi: 10.1038/nbt1137. PMID:16151408. [DOI] [PubMed] [Google Scholar]
- 21.Yu L, Vizel A, Huff MB, Young M, Remmele RL Jr., He B. Investigation of N-terminal glutamate cyclization of recombinant monoclonal antibody in formulation development. J Pharm Biomed Anal. 2006;42:455–63. doi: 10.1016/j.jpba.2006.05.008. PMID:16828250. [DOI] [PubMed] [Google Scholar]
- 22.Lyubarskaya Y, Houde D, Woodard J, Murphy D, Mhatre R. Analysis of recombinant monoclonal antibody isoforms by electrospray ionization mass spectrometry as a strategy for streamlining characterization of recombinant monoclonal antibody charge heterogeneity. Anal Biochem. 2006;348:24–39. doi: 10.1016/j.ab.2005.10.003. PMID:16289440. [DOI] [PubMed] [Google Scholar]
- 23.Liu YD, Goetze AM, Bass RB, Flynn GC. N-terminal glutamate to pyroglutamate conversion in vivo for human IgG2 antibodies. J Biol Chem. 2011;286:11211–7. doi: 10.1074/jbc.M110.185041. PMID:21282104. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Hmiel LK, Brorson KA, Boyne MT 2nd. Post-translational structural modifications of immunoglobulin G and their effect on biological activity. Anal Bioanal Chem. 2015;407:79–94. doi: 10.1007/s00216-014-8108-x. PMID:25200070. [DOI] [PubMed] [Google Scholar]
- 25.Worobec A, Rosenberg AS. A Risk-Based Approach to Immunogenicity concerns of therapeutic protein products, part 1: Considering consequences of the immune response to a protein. BioPharm International. 2004;17:22–6. [Google Scholar]
- 26.Rosenberg AS. Effects of protein aggregates: an immunologic perspective. AAPS J. 2006;8:E501−7. doi: 10.1208/aapsj080359. PMID:17025268. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Hermeling S, Aranha L, Damen JM, Slijper M, Schellekens H, Crommelin DJ, Jiskoot W. Structural characterization and immunogenicity in wild-type and immune tolerant mice of degraded recombinant human interferon alpha2b. Pharm Res. 2005;22:1997–2006. doi: 10.1007/s11095-005-8177-9. PMID:16184451. [DOI] [PubMed] [Google Scholar]
- 28.Cordoba AJ, Shyong BJ, Breen D, Harris RJ. Non-enzymatic hinge region fragmentation of antibodies in solution. J Chromatogr B Analyt Technol Biomed Life Sci. 2005;818:115–21. doi: 10.1016/j.jchromb.2004.12.033. PMID:15734150. [DOI] [PubMed] [Google Scholar]
- 29.Ravetch JV, Bolland S. IgG Fc receptors. Annu Rev Immunol. 2001;19:275–90. doi: 10.1146/annurev.immunol.19.1.275. PMID:11244038. [DOI] [PubMed] [Google Scholar]
- 30.Jefferis R. Glycosylation of recombinant antibody therapeutics. Biotechnol Prog. 2005;21:11–6. doi: 10.1021/bp040016j. PMID:15903235. [DOI] [PubMed] [Google Scholar]
- 31.Zheng K, Bantog C, Bayer R. The impact of glycosylation on monoclonal antibody conformation and stability. MAbs. 2011;3:568–76. doi: 10.4161/mabs.3.6.17922. PMID:22123061. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Khawli LA, Goswami S, Hutchinson R, Kwong ZW, Yang J, Wang X, Yao Z, Sreedhara A, Cano T, Tesar D, et al. . Charge variants in IgG1: Isolation, characterization, in vitro binding properties and pharmacokinetics in rats. MAbs. 2010;2:613–24. doi: 10.4161/mabs.2.6.13333. PMID:20818176. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Schiestl M, Stangler T, Torella C, Cepeljnik T, Toll H, Grau R. Acceptable changes in quality attributes of glycosylated biopharmaceuticals. Nat Biotechnol. 2011;29:310–2. doi: 10.1038/nbt.1839. PMID:21478841. [DOI] [PubMed] [Google Scholar]
- 34.Jefferis R. Recombinant antibody therapeutics: the impact of glycosylation on mechanisms of action. Trends Pharmacol Sci. 2009;30:356–62. doi: 10.1016/j.tips.2009.04.007. PMID:19552968. [DOI] [PubMed] [Google Scholar]
- 35.Yoo DH, Suh CH, Shim SC, Jeka S, Cons-Molina FF, Hrycaj P, Lee EY, Medina-Rodriguez FG, Shesternya P, Radominski S, et al. . A multicentre randomised controlled trial to compare the pharmacokinetics, efficacy and safety of CT-P10 and innovator rituximab in patients with rheumatoid arthritis. Ann Rheum Dis. 2017;76:566–70. doi: 10.1136/annrheumdis-2016-209540. PMID:27624791. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Suh CH, Berrocal Kasay A, Chalouhi El- Khouri E, Miranda P, Bozic Majstorovic L, Jeka S, Hrycaj P, Rekalov D, Wiland P, Krause A, et al. . Pharmacokinetics and Safety of Three Formulations of Rituximab (CT-P10, US-sourced Innovator Rituximab and EU-sourced Innovator Rituximab) in Patients with Rheumatoid Arthritis: Results from Phase 3 Randomized Controlled Trial over 24 Weeks [abstract]. Arthritis Rheumatol. 2016; 68 (suppl 10). [Accessed 2017 Aug 30]. http://acrabstracts.org/abstract/pharmacokinetics-and-safetyof-three-formulations-ofrituximab-ct-p10-us-sourced-innovatorrituximab-and-eu-sourced-innovator-rituximab-inpatients- withrheumatoid-arthritis-results-from-phase-3-r/. [Google Scholar]
- 37.Park W, Suh CH, Shim SC, Molina FFC, Jeka S, Medina-Rodriguez FG, Hrycaj P, Wiland P, Lee EY, Shesternya P, et al. . Efficacy and Safety of Switching from Innovator Rituximab to Biosimilar CT-P10 Compared with Continued Treatment with CT-P10: Results of a 56-Week Open-Label Study in Patients with Rheumatoid Arthritis. BioDrugs. 2017;31:369–77. doi: 10.1007/s40259-017-0233-6. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Suh CH, Khouri ECE, Miranda P, Molina FFC, Shesternya P, Medina-Rodriguez FG, et al. . Randomised Double-Blind Study shows Comparable Long-Term Efficacy and Safety Between Rituximab Biosimilar CT-P10 and Innovator Rituximab in Patients with Rheumatoid Arthritis. Annals of the Rheumatic Diseases. 2017;76:824. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Kim WS, Buske C, Ogura M, Jurczak W, Sancho JM, Zhavrid E, Kim JS, Hernández-Rivas JÁ, Prokharau A, Vasilica M, et al. . Efficacy, pharmacokinetics, and safety of the biosimilar CT-P10 compared with rituximab in patients with previously untreated advanced-stage follicular lymphoma: A randomised, double-blind, parallel-group, non-inferiority phase 3 trial. Lancet Haematol. 2017;4:e362−e73. doi: 10.1016/S2352-3026(17)30120-5. PMID:28712940. [DOI] [PubMed] [Google Scholar]
- 40.European Medicines Agency, Committee for Medicinal Products for Human Use. Truxima: European public assessment report (EPAR) summary for the public [Internet]. London, UK: European Medicines Agency; [Updated in February 2017; cited September 15, 2017]. Available from: http://www.ema.europa.eu/docs/en_GB/document_library/EPAR_-_Summary_for_the_public/human/004112/WC500222696.pdf. [Google Scholar]
- 41.Seely RJ, Munyakazi L, Haury J. Statistical tools for setting in-process acceptance criteria. Dev Biol (Basel). 2003;113:17–25. PMID:14620848 [PubMed] [Google Scholar]
- 42.Wang X, Germansderfer A, Harms J, Rathore AS. Using statistical analysis for setting process validation acceptance criteria for biotech products. Biotechnol Prog. 2007;23:55–60. doi: 10.1021/bp060359c. PMID:17269671. [DOI] [PubMed] [Google Scholar]