

ABSTRACT
Antibody-drug conjugation strategies are continuously evolving as researchers work to improve the safety and efficacy of the molecules. However, as a part of process and product development, confirmation of the resulting innovative structures requires new, specialized mass spectrometry (MS) approaches and methods, as compared to those already established for antibody-drug conjugates (ADCs) and the heightened characterization practices used for monoclonal antibodies (mAbs), in order to accurately elucidate the resulting conjugate forms, which can sometimes have labile chemical bonds and more extreme chemical properties like hydrophobic patches. Here, we discuss practical approaches for characterization of ADCs using new methodologies and ultrahigh-resolution MS, and provide specific examples of these approaches. Denaturing conditions of typical liquid chromatography (LC)/MS analyses impede the successful detection of intact, 4-chain ADCs generated via cysteine site-directed chemistry approaches where hinge region disulfide bonds are partially reduced. However, this class of ADCs is detected intact reliably under non-denaturing size-exclusion chromatography/MS conditions, also referred to as native MS. For ADCs with acid labile linkers such as one used for conjugation of calicheamicin, careful selection of mobile phase composition is critical to the retention of intact linker-payload during LC/MS analysis. Increasing the pH of the mobile phase prevented cleavage of a labile bond in the linker moiety, and resulted in retention of the intact linker-payload. In-source fragmentation also was observed with typical electrospray ionization (ESI) source parameters during intact ADC mass analysis for a particular surface-accessible linker-payload moiety conjugated to the heavy chain C-terminal tag, LLQGA (via transglutaminase chemistry). Optimization of additional ESI source parameters such as cone voltages, gas pressures and ion transfer parameters led to minimal fragmentation and optimal sensitivity. Ultrahigh-resolution (UHR) MS, combined with reversed phase-ultrahigh performance (RP-UHP)LC and use of the FabRICATOR® enzyme, provides a highly resolving, antibody subunit-domain mapping method that allows rapid confirmation of integrity and the extent of conjugation. For some ADCs, the hydrophobic nature of the linker-payload hinders chromatographic separation of the modified subunit/domains or causes very late elution/poor recovery. As an alternative to the traditionally used C4 UHPLC column chemistry, a diphenyl column resulted in the complete recovery of modified subunit/domains. For ADCs based on maleimide chemistry, control of pH during proteolytic digestion is critical to minimize ring-opening. The optimum pH to balance digestion efficiency and one that does not cause ring opening needed to be established for successful peptide mapping.
KEYWORDS: ADC, Heightened characterization, Mass spectrometry, Intact mass, Subunit domain
Introduction
Antibody-drug conjugates (ADC) are therapeutic modalities composed of a target-specific monoclonal antibody (mAb) connected via a chemical linker to a small molecule drug (payload) that has cell-killing (cytotoxic) activity.1 The production of these therapeutic modalities relies on the chemical conjugation of a linker-payload (LP) to a specific natural or unnatural amino acid on the antibody.2 Depending on the conjugation chemistry and site of LP attachment, the molecular structure of the ADC could be very complex and heterogeneous.3 Furthermore, the linker chemistry can be “cleavable” or “non-cleavable”. ADCs with cleavable linkers rely on protease cleavage,4 acid hydrolysis5 or other mechanisms to release the unmodified payload as active species, whereas ADCs with non-cleavable linkers6 rely on the degradation of antibody to release the active species. Both the cleavability as well as the sites of conjugation have a substantial effect on the analytical strategy for ADC characterization.
Conventional conjugation chemistries typically involve cysteine or lysine residues. Cysteine conjugation is well established,7-8 and usually the hinge-region cysteines that are used for the formation of interchain disulfide bonds are targeted for conjugation. There are four interchain disulfide bonds in the IgG1 molecule and, after partial reduction, conjugation can occur randomly at any of these cysteine residue pairs ranging from zero LP additions to a maximum drug-to-antibody ratio (DAR) of 8. Typically, these hinge-region cysteine-conjugated ADCs have an even-number distribution of LP moieties with an average DAR of approximately 4. Even with missing interchain disulfide bonds, the molecule is held together by non-covalent bonds. However, the non-covalent nature of a cysteine-conjugated mAb has a direct effect on the type of liquid chromatography/mass spectrometry (LC/MS) analyses these molecules can tolerate, and in essence, native MS methods are required to maintain the 4-chain mAb structure.8
Site-specific conjugation at lysine residues can be either systematic or more random, and the complexity of the resulting ADC depends on various conjugation reaction parameters, including the chemical structure of the LP, surface accessibility of the lysine residues, reaction kinetics, and higher-order structure.9-10 The extent of conjugation reaction of lysine residues usually results in a more heterogeneous mixture of ADC species with wider DAR values than cysteine conjugates, with DAR typically ranging from 0 to 9.
A substantial effort has been dedicated to a development of site-specific ADCs with more controlled sites of conjugation and product stoichiometry in an effort to further enhance batch-to-batch consistency. Several novel approaches to generate site-specific ADCs have emerged in recent years.3 Among them, one site-specific conjugation approach utilizes transglutaminase (TG), which catalyzes formation of a covalent bond between a glutamine side chain of either an incorporated glutamine tag (LLQGA) or existing glutamines in the primary sequence of the mAb and primary amine on the LP.11-12 Conjugation to engineered cysteines is another well-described site-specific conjugation approach that results in more homogenous ADCs with DAR values equal to the number of new cysteines incorporated into mAb sequence.13
Compared to mAbs, heightened characterization of ADCs is more challenging because of the increased molecular complexity of ADCs, in which cytotoxic linker-drug payloads are conjugated to an already diverse set of proteoforms.14 As various ADC formats have been developed, new analytical tools and approaches have been devised to elucidate ADC covalent structure and the extent of conjugation, including native MS of cysteine-conjugated ADCs with and without enzymatic cleavage of the linker,8,15 2-dimensional (2D)-LC/MS analyses based on hydrophobic interaction chromatography (HIC) coupled to reversed-phase liquid chromatography,16-17 and ion-mobility mass spectrometric analysis.18-19 Here, we discuss practical approaches to heightened characterization of ADCs using LC-ultrahigh-resolution mass spectrometry (UHR MS) that can overcome challenges and generate enhanced information for better product understanding. Details of the methods are provided in Supplemental Material.
LC/MS analysis of most intact ADCs remains challenging due to extensive conjugation with hydrophobic LPs or chemically unstable linkers. Moreover, ADCs with hinge region cysteine conjugation (after partial reduction) and non-covalent subunits require native ESI conditions to properly detect the 4-chain molecule. The linker chemistry can present unique MS challenges, in addition to the site of conjugation, whether lysine, glutamine, or cysteine. Surface accessible LPs pose additional challenges during ADC introduction into the mass spectrometer due to their fragility and amenability to in-source fragmentation. As well, low-artifact heightened characterization methods are needed to prevent additional, artificial ADC modifications. ADC chemistry and properties thus have a substantial effect on the analytical strategy used for MS characterization, and significant method development is required to establish robust, reliable and informative methods.
Liquid-chromatography mass spectrometry of Intact ADC, Associated challenges, and their mitigation
Electrospray ionization mass spectrometry (ESI MS) coupled to organic size-exclusion chromatography (oSEC) was used to examine the extent of drug conjugation for both intact and de-N-glycosylated ADCs. The accurate mass information for intact ADC serves to confirm the integrity of the primary structure following conjugation, as well as identify the major and minor product conjugate forms. The common mobile phases used for the introduction of ADCs into the mass spectrometer contain both organic (i.e., acetonitrile) and acidic (i.e., trifluoroacetic acid (TFA)) modifiers that result in ADC denaturation. Because the interchain disulfide bonds of conventional Cys-conjugation ADCs are partially reduced prior to conjugation, many of the resulting conjugated isoforms were detected in dissociated forms under the denaturing conditions. The accurate molecular masses of these dissociated species provide information about the exact interchain disulfide bond cleavages and subsequent LP conjugation sites.
For example, for a particular Cys-conjugated mAb under denaturing conditions, two predominant chromatographic peaks are observed by oSEC (Fig. 1a), and from the accurate mass determinations, the earlier eluting oSEC peak (peak 1, Fig. 1a) contains dissociated ADC isoforms that are larger than light (L) chain, including various combinations of heavy (H) chain with and without L chain (e.g., HHL, HL, HH) with a specified number of LP moieties (Fig. 1b). The most abundant species comprises the disulfide linked L and H chains with two LP moieties: labeled as HL+2LP, where H represents heavy chain, L represents light chain, and number represents the number of LP moieties. Here, the chain composition and number of LP moieties for the dissociated HL+2LP species are consistent with both H-ss-H interchain disulfide bonds being reduced and conjugated to LP, and the L-ss-H interchain disulfide bond remaining intact. Therefore, the predominant 4-chain ADC form appears to involve two non-covalent HL+2LP species for a total of four LP.
Figure 1.
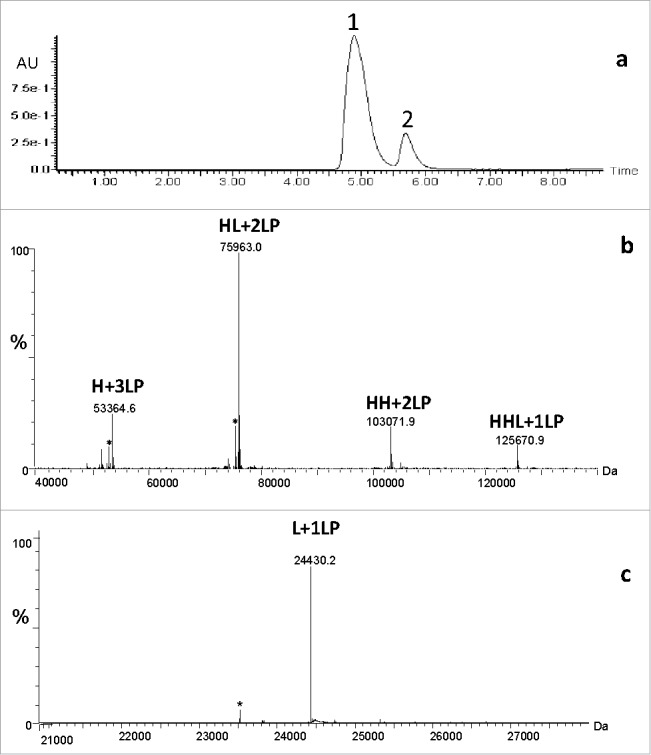
Organic (denaturing) SEC/MS of conventional cysteine chemistry ADC: a) UV profile; b) zero-charge deconvoluted mass spectrum of early eluting peak 1; c) zero-charge deconvoluted mass spectrum of later eluting peak 2. Abbreviations: H denotes heavy chain, L denotes light chain, HL denotes heavy chain linked to light chain via interchain disulfide, HH denotes two heavy chains linked via hinge-region disulfides, HHL denotes two heavy chains linked via hinge-region disulfides and to light chain via interchain disulfide, and a number LP denotes a number of LP moieties. *represent a method artifact that pertains to in-source fragmentation of LP.
Additional minor level species such as H+3LP, HH+2LP, and HHL+1LP were also observed and are consistent with cleaved interchain disulfide bonds and subsequent LP conjugation. The relative abundance of the dissociated ADC isoforms is determined by the MS signal, which is highly related to the physicochemical properties of the fragment, e.g., the L chain usually has much higher MS response than the H chain. Considering that ionization efficiency of HH+2LP and HHL+1LP might be different than that of HL+2LP, the relative abundance of these ADC isoforms might not accurately reflect their actual levels. Routine, orthogonal chromatographic or electrophoretic methods are used to assess the predominant Cys-conjugated forms, providing confirmation of the MS results. Thus, the experimental molecular masses of all conjugated H chain forms agreed well with theoretical values, which signify that the ADC has the correct molecular composition of LP, as well as the expected conjugation chemistry and specificity at hinge region cysteine residues.
The later eluting oSEC peak (peak 2, Fig. 1a) contains one LP-conjugated moiety to L chain (Fig. 1c). The experimental molecular mass is consistent with L chain plus 1 LP, in agreement with the theoretical mass, and this conjugate form results from the expected cleavage of the interchain disulfide bond between L and H chain.
In order to detect the intended, four-chain structure consistent with an IgG1 antibody and the expected drug load, non-denaturing (native) conditions needed to be developed and applied to Cys-conjugated ADCs. For this, 20 mM ammonium acetate mobile phase was used for SEC separation of the ADC prior to mass spectrometric analysis. Under native SEC-ESI MS, only one chromatographic peak was observed (Fig. 2a) that corresponded to the intact ADC molecule containing two L chains and two H chains with the correct amino acid sequences, the anticipated number of disulfide bonds, the expected G0F/G0F combination of core fucosylated, complex-type biantennary N-linked oligosaccharides in the Fc region, as well as zero, two, four, six and eight LP moieties with the intended chemical compositions. Additional minor level isoforms were also detected and consistent with the expected N-linked oligosaccharide heterogeneity in mAb. The ADC also was treated with PNGase F to remove the molecular heterogeneity resulting from N-linked glycosylation to better assess drug loading and primary structure integrity (Fig. 2c). The de-N-glycosylation of intact ADC is especially advantageous if glycosylation profile of mAb is very complex and results in overlapping m/z ions for glyco- and LP-conjugated forms, which in turn affects the deconvolution of the raw mass spectrum into zero-charge mass spectrum.
Figure 2.
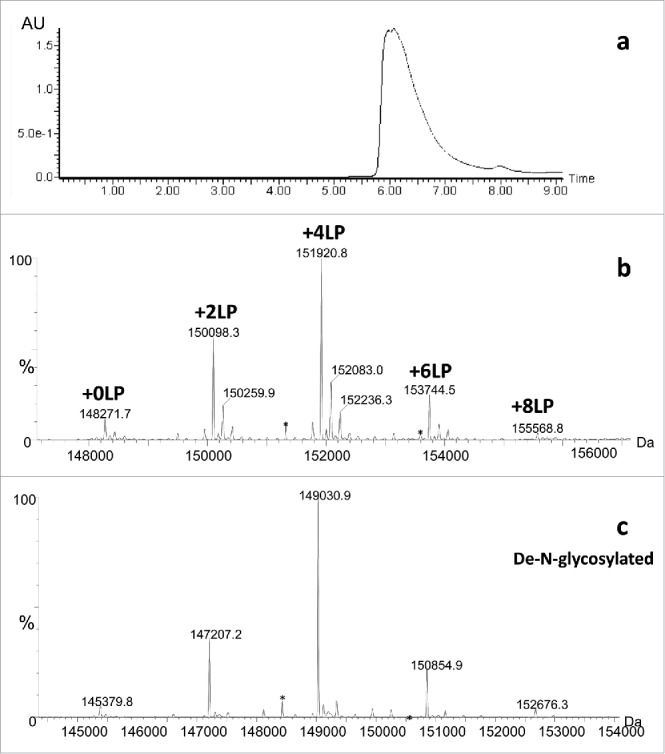
Native (non-denaturing) SEC/MS of conventional cysteine chemistry ADC: a) UV profile; b) zero-charge deconvoluted mass spectrum of intact ADC; c) zero-charge deconvoluted mass spectrum of de-N-glycosylated ADC. Abbreviations: a number LP denotes a number of LP moieties. *represent a method artifact that pertains to in-source fragmentation of LP.
For the lysine-conjugated ADCs with calicheamicin LPs (Scheme I), denaturing oSEC-ESI MS analysis using 0.1% TFA in the mobile phase resulted in rapid cleavage of calicheamicin from the ADC due to an acid labile bond in the 4-(4-acetylphenoxy)butanoic acid (AcBut) linker moiety (Fig. 3a). The resulting ESI mass spectrum of the lysine-conjugated ADC contains species with two, three, four, and five truncated linker derivatives (+204 Da) attached to the mAb molecule with the three-linker derivative being the most abundant. To detect the intact ADC with the full-length calicheamicin LP, the mobile phase containing strong acid modifier such as TFA was replaced with either a weak acid such as formic acid or ammonium acetate. This change resulted in preservation of the acid labile bond in the AcBut linker and leading to the mass spectrum of lysine-conjugated intact ADC containing species with two, three, four, and five complete calicheamicin LPs, labeled as + nCM, where CM is the abbreviation for calicheamicin and n is a number of calicheamicin moieties attached to mAb (Fig. 3b). As the calicheamicin loading increases for each conjugate form, the mass spectral signal intensities are known to progressively decrease in ESI MS analysis from ionization suppression effects due to factors such as co-elution, increased size/hydrophobicity, and decreased charge, which results in a minor underestimation of the average DAR compared to other routine orthogonal biochemical methods. The minor peaks designed with “*” represent a method artifact that pertains to the loss of the E ring from calicheamicin (-199 Da) due to in-source fragmentation or electrochemical degradation during ESI MS analysis.
Scheme 1.
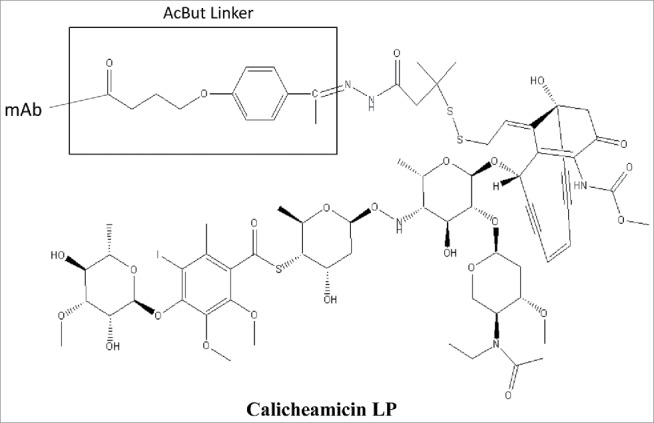
Structure of Calicheamicin Linker-Payload.
Figure 3.
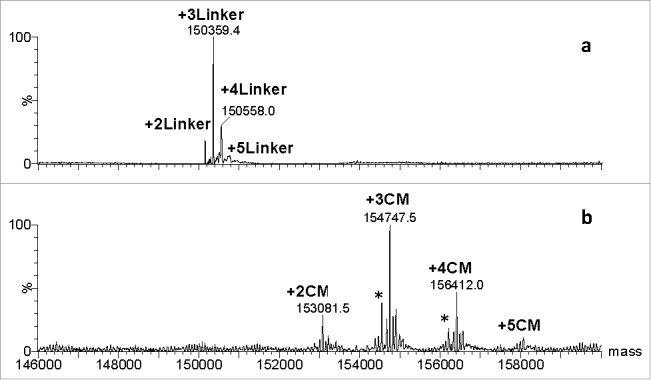
Mass spectra of conventional lysine conjugated ADC: a) in the presence of strong acid modifier, 0.1% TFA; b) in the presence of weak acid modifier, 0.1 % formic acid. Abbreviations: Linker denotes a 204-Da hydrolysis product of the AcBut linker bound to Lys; CM denotes calicheamicin, *represent a method artifact that pertains to the loss of the E ring from calicheamicin (-199 Da).
For the site-specific ADC produced by transglutaminase conjugation chemistry, the LP is known to be surface exposed. While the mass spectrum obtained under denaturing oSEC-ESI MS conditions resulted in the expected intact ADC with a four-chain covalent structure and anticipated two moles of LP derivative per antibody, there was significant in-source fragmentation (Fig. 4a). Species that correspond to loss of 786 Da (labeled with +2LP*) and 2 × 786 Da (labeled with +2LP**) are due to in-source fragmentation, where the labile bonds in the LP cleave because of the high declustering potential needed for the ion desolvation in the ESI interface and ion source, as well as from protonation of this acid labile bond given use of TFA, which is a strong acidic modifier. ESI source parameters such as cone voltage, source temperature, and source pressure were optimized to reduce in-source fragmentation. The reduction of cone voltage from 40 V to 30 V significantly reduced the in-source fragmentation (Fig. 4b). Additional reduction of cone voltage in 5 V increments to 25 and 20 V further minimized in-source fragmentation, but led to excessive salt and solvent adduct formation. Furthermore, increasing the pH of the mobile phase by using 0.1% formic acid instead of 0.1% TFA as mobile phase modifier helped to minimize the loss of 786 Da. In contrast, when milder source conditions were implemented to reduce in-source fragmentation, adduct formation was increased (Fig. 4b). Overall, 0.1% formic acid in mobile phase and 30 V cone voltage appeared to be the most optimal for reducing in-source fragmentation for the site-specific ADC with heavy chain C-terminal LP.
Figure 4.
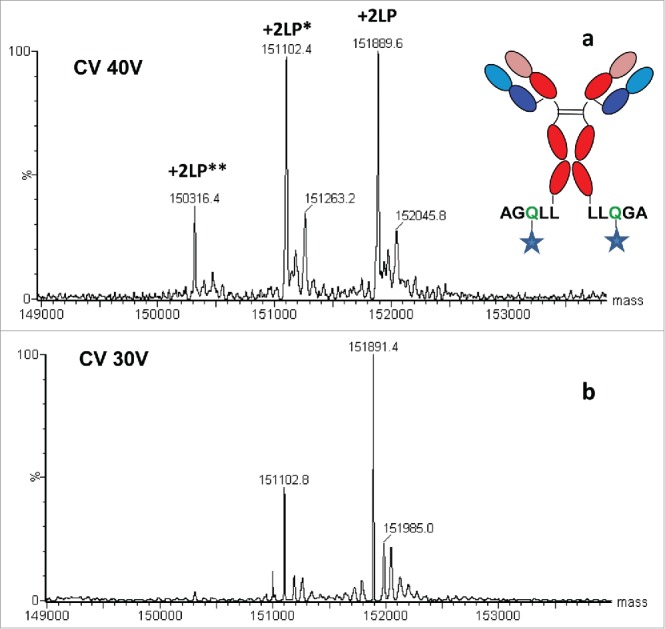
Mass spectra of site specific conjugated ADC with surface exposed LP: a) at high cone voltage of 40 V (and 0.1% TFA); b) at low cone voltage of 30 V (and 0.1% formic acid). Abbreviations: CV denotes cone voltage and a number LP denotes a number of LP moieties, *and **represents a method artifact that pertains to in-source fragmentation affecting one and two LPs, respectively.
Three-part subunit-domain analysis, associated challenges and their mitigation
To further understand the extent and fidelity of the conjugation process, three-part subunit-domain analysis is performed, which provides information about primary structure and posttranslational modifications of the ADC, and the extent of conjugation in each antibody subunit/domain. In this method, the ADC is digested with the immunoglobulin-degrading enzyme of Streptococcus pyogenes (IdeS). IdeS specifically cleaves intact IgG1 antibodies just below the hinge at a specific G-G sequence motif, yielding one Fab2 and two single-chain Fc (scFc) fragments.20-21 Upon disulfide bond reduction, the Fab2 is converted to L chain and the Fd′ part of H chain. These subunits/domains are analyzed by reversed-phase ultrahigh-performance liquid chromatography/electrospray ionization ultrahigh-resolution quadrupole time-of-flight mass spectrometry (RP-UHPLC/ESI-UHR QTOF MS). In the three-part subunit-domain assay, the L chain, and the two scFc and Fd′ H chain domains are separated chromatographically. Accurate mass determinations allow identification of product isoforms, as well as verification of the integrity of the amino acid sequence and drug conjugation. For the conventional cysteine-conjugated ADCs, the interchain cysteines sites are expected to be partially occupied with LP, and this assay can provide information about occupancy at the subunit-domain level and the integrity of the attached drug(s).
The subunit-domain mapping chromatogram for a conventional interchain cysteine-conjugation ADC is shown in Fig. 5. The scFc domain does not contain any cysteine residues associated with interchain disulfide bonds, and, as expected, no drug conjugation to the scFc domain is observed. The L chain is observed in both conjugated and un-conjugated forms. The conjugated L chain form contains the C-terminal cysteine residue that was originally associated with the interchain L to H chain disulfide bond. The Fd′ domain conjugate forms are observed with 0 to 3 drugs, corresponding to the cysteines previously associated with the L to H chain disulfide bond and the two disulfide bonds between the both H chains. Minor-level Fd′ isoforms corresponding to unconjugated Fd′ and loss of water are also observed.
Figure 5.
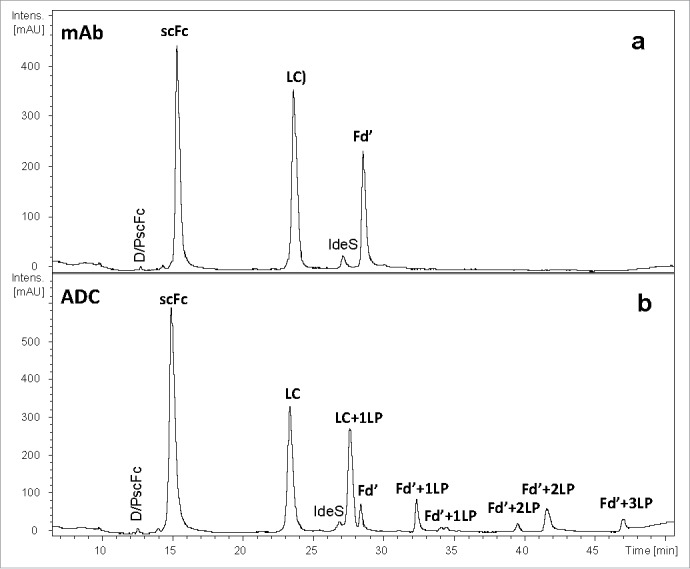
Three-part subunit/domain of cysteine-conjugated ADC obtained by using C4 column at 80 oC: a) UV 214 nm profile of mAb; b) UV 214 nm profile of ADC. IdeS denotes the immunoglobulin-degrading enzyme of Streptococcus pyogenes.
The current approach for ADC characterization by the three-part subunit-domain analysis relies on use of a C4 column with reversed-phase high performance liquid chromatography (RP-HPLC). However, partial recovery of hydrophobic subunit/domains such as Fd′ can occur, even in the mAb molecule prior to conversion to ADC (Fig. 5, top panel). Addition of the more hydrophobic LP to the already partially recovered Fd′ domain can further reduce the recovery of conjugated species (Fig. 5, bottom panel). In particular, Fd′+3LP species that contain three LP moieties conjugated via three interchain cysteines displayed very poor recovery even at a high column temperature (80 oC). The high temperature is undesirable for chromatographic separation of ADC components because it typically causes on-column method artifacts like Asp-Pro cleavages. The interaction of the ADC subunit/domains with the C4 stationery phase was too strong to be overcome with the mobile phase and high temperature. Upon switching to a diphenyl column, which is based on a different selectivity, complete recovery of both L chain and Fd′ was achieved for the mAb (Fig. 6). Use of the diphenyl column resulted in superior recovery of L chain and Fd′ conjugated to LP for the ADC (Table 1). The relative percent of column recovery was calculated for mAb and ADC subunit domain species based on the ratio of UV214 peak area for both C4 and diphenyl columns (see Table 1). The relative percent of peak area for the sum of Fd′ species for ADC increased from 14.2% to 33.0% using diphenyl instead of a C4 column. Complete recovery of conjugated subunit/domains also allowed for higher quality, more reliable mass spectra for determining the peak identities.
Figure 6.
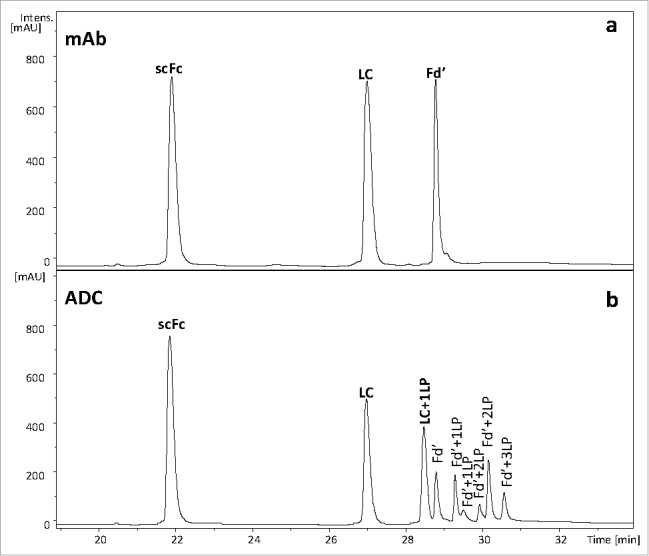
Three-part subunit/domain of cysteine-conjugated ADC obtained by using diphenyl column at 60oC: a) UV 214 nm profile of mAb; b) UV 214 nm profile of ADC.
Table 1.
Relative Abundance of mAb and ADC Subunit/Domains Using C4 and Diphenyl Columns.
| Relative Peak Area, % |
||||||
|---|---|---|---|---|---|---|
| C4 |
Diphenyl |
|||||
| Species | mAb | ADC | Total ADC Subunits | mAb | ADC | Total ADC Subunits |
| scFc | 42.5 | 45.5 | 45.5 | 39.5 | 35.4 | 35.4 |
| LC | 38.2 | 23.5 | 40.3 | 39.3 | 22.2 | 31.6 |
| LC+1LP | — | 16.8 | 9.4 | |||
| Fd′ | 19.3 | 2.7 | 14.2 | 21.2 | 0.9 | 33.0 |
| Fd′+1LP | 4.0 | 7.3 | ||||
| Fd′+1LP | 0.8 | 3.7 | ||||
| Fd′+2LP | 0.9 | 3.4 | ||||
| Fd′+2LP | 4.1 | 11.1 | ||||
| Fd′+3LP | 1.7 | 6.6 | ||||
Peptide mapping, associated challenges, and their mitigation
Analysis of the primary structure and posttranslational modifications, and confirmation of LP conjugation sites in ADCs, is typically accomplished by peptide mapping LC-MS.22 Proteolysis of the reduced and alkylated mAb and ADC with the Lys-C enzyme was optimized to be the most efficient at pH 8.2. When the same protocol was applied to the cysteine-conjugated ADCs that were manufactured using maleimide chemistry, unexpected peaks were observed in both UV 214 nm and total ion chromatograms (Fig. 7, top panel). Upon detailed examination of mass spectra of the peaks at 145 min, a new species was observed with the monoisotopic mass of 4146.1458 Da. This new species labeled as H13H14+1LP + 18 Da was 18.0028 Da higher in mass than the mass of anticipated hinge region peptide H13H14 with one LP attached (4128.1429 Da) (Fig. 7, bottom panel). The proposed mechanism for this species is based on the maleimide ring opening, which originates from the high pH conditions during sample preparation and is considered to be method artifact (Scheme II). To minimize the formation of the maleimide ring opened species, the pH of buffer used in sample preparation for enzymatic digestion was lowered to pH 7.5. As a result of lowering the enzymatic digestion pH, the +18 Da species were not observed in the UV and total ion chromatogram, or the corresponding mass spectrum (Fig. 7).
Figure 7.

Reduced and alkylated peptide mapping of cysteine conjugated ADC using maleimide chemistry: a) zoomed view of total ion current for hinge conjugated peptide at pH 8.2 (top) and 7.5 (bottom); b) mass spectra of early eluting peak at pH 8.2 that corresponds tone drug conjugate peptide with thiosuccinimide ring opening (top) and one drug conjugated peptide without thiosuccinimide ring opening (bottom). Abbreviations: H denotes heavy chain peptide.
Scheme 2.
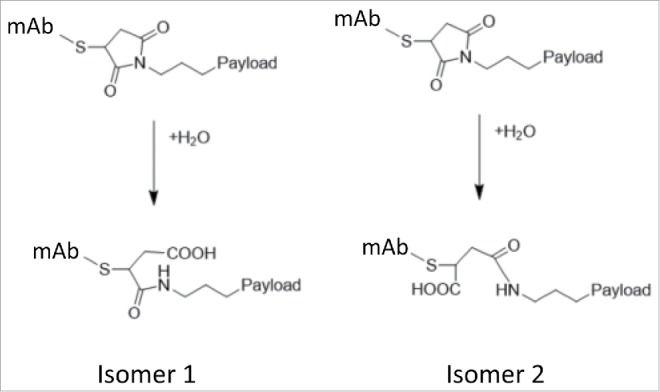
Thiosuccinimide Ring Opening.
Summary
Heightened characterization of ADCs is essential for in-depth understanding of their intended structure and, contributes overall to their successful development as biotherapeutics when combined with biological activity. Ultrahigh-resolution mass spectrometry facilitates in-depth antibody characterization, and, for ADCs, affords rapid site and occupancy determinations for drug payloads and elucidation of additional structural heterogeneity present due to particular conjugation chemistries.23 The typical ADC workflow includes accurate mass analysis of both intact and de-N-glycosylated molecular forms, two- or three-part subunit-domain analysis, and proteolytic mapping. These orthogonal MS-based analyses confirm the sequence fidelity of the ADC, as well as the extent and integrity of conjugation. The initial approach to heightened characterization of ADCs was based on subjecting them to existing characterization assays that were applied to the corresponding mAb-based intermediates. However, it was immediately recognized that many ADCs were not amenable to the LC/MS conditions developed for canonical mAbs. The difficulties with applying existing LC/MS approaches and methods to ADCs stemmed from their increased complexity and, in some cases, their non-covalent nature. Moreover, liabilities in the LP chemistries, including acid labile bonds, substantial hydrophobicity, and surface accessibility, caused additional challenges during LC/MS analysis of ADCs.
Depending on the specific assay, whether it was intact mass, subunit-domain analysis, or peptide mapping analysis, as well as the type of ADC chemistry and chemical nature of the LP, various unexpected challenges were encountered during heightened characterization. Intact mass analysis using denaturing oSEC/MS analysis was performed for confirmation of sequence fidelity, multi-chain architecture, and extent of conjugation via average mass measurement of intact and de-N-glycosylated molecule. The approach utilizes 30% acetonitrile and 0.1% TFA in the mobile phase, which denatures the ADC, but provides adequate protonation and desolvation of the molecule. oSEC/MS has been very successful in mAb characterization, but results in dissociation of cysteine-conjugated 4-chain ADCs with partially reduced disulfide bonds and non-covalent light and heavy chains. The mitigation strategy involves implementation of native (non-denaturing), MS-compatible conditions prior to introduction of the cysteine-conjugated ADC into the mass spectrometer, which results in successful detection of intact, non-covalent 4-chain ADC.
For lysine-conjugated ADCs with calicheamicin as a LP, even though the covalent nature of the ADC was preserved, the acid liable bonds within the linker moiety do not survive the harsh conditions of using 0.1% TFA in the oSEC/MS mobile phase as an ion pairing agent. The mitigation strategy for this type of ADC is to reduce the acidity of the mobile phase by either using an acid with higher pKa in the mobile phase, such as formic acid, or implement a native, ammonium acetate-based mobile phase. In our example, removing TFA from the mobile phase and replacing it with 0.1% formic acid was sufficient to minimize, if not completely eliminate, the cleavages in the calicheamicin moieties within the ADC, allowing for their complete retention on the intact ADC. Use of ammonium acetate as a mobile phase gave very similar results, but more salt adducts were seen compared to the 0.1% formic acid. Moreover, preserving intact calicheamicin on ADC during analysis allows successful monitoring of potential hydrolysis byproducts in ADC that could have happened during long-term storage conditions in the final product.
For the mass analysis of intact ADCs, ESI source parameters are often similar to those used for mAbs and primarily are optimized for adequate desolvation and subsequent ionization of large biomolecules, promoting high sensitivity but avoiding in-source fragmentation. These ESI parameters for proteins in general tend to be very different than the ones used for introduction of small organic molecules into a mass spectrometer. However, ADCs contain LP moieties that are considered to be small molecules. These small molecule moieties within ADCs, especially surface exposed ones, undergo in-source induced fragmentation under ESI conditions used for mass spectrometric analysis of ADCs. For site-specific conjugated ADC with LP conjugated to the surface-assessable C-terminus of the heavy chain, significant in-source fragmentation can prevent detection of the intact LP attached to the mAb molecule. The mitigation strategy for preserving the intact LP on the ADC in this case was based on utilizing milder ionization conditions than those typically used for mAbs. Decreasing the cone voltage by 10 V and use of 0.1% formic acid in the mobile phase were sufficient to significantly minimize the in-source fragmentation of the LP. However, complete elimination of method-induced in-source fragmentation was not achieved because further reduction of cone voltage impaired the signal-to-noise quality of raw ESI mass spectrum, making proper deconvolution to the zero-charge mass spectrum impossible. In all these examples, intact mass analysis of ADCs was used for orthogonal characterization and bioprocess development support only. Other techniques, such as HIC, RP-HPLC with UV detection, UV spectroscopy, are typically used for DAR determination for batch release and stability testing.
The three-part subunit-domain assay, which provides information on primary structure, posttranslational modifications of ADC and the extent of conjugation in each subunit, utilizes a reversed-phase liquid chromatography separation of three ∼25 kDa polypeptides (e.g., light chain, Fd′, scFc) following IdeS proteolysis and disulfide bond reduction.24 Depending on the conjugation chemistry and nature of the LP, complete chromatographic recovery of the hydrophobic domains with or without LP attached has been challenging. Due to the very hydrophobic nature of the LPs, as well as increased hydrophobicity of heavy chains and Fd′ domains, in particular, the decrease in recovery of hydrophobic subunits impairs the LC/MS analysis. Typically, low-carbon content stationary phases, such C4 or C3 columns, are used for reversed-phase separation of medium size biomolecules similar to subunit/domains. In our example, separation of three subunits on C4 column was adequate, but recoveries of the very hydrophobic Fd′ and even the less hydrophobic light chain of the mAb molecule were diminished. Upon conjugation of the hydrophobic LP to both Fd′ and light chain, the recovery of ADC subunit/domains was further reduced, making the overall analysis by LC/MS not feasible, especially, if minor and trace level species need to be characterized. However, quantitative recovery of all mAb subunit/domains was achieved by using diphenyl column, which furthermore, was not affected by addition of LP to already hydrophobic subunits. The choice of stationery phase suitable for the complete separation and quantitative recovery of the subunit/domains is driven by the amino acid sequence of the mAb and hydrophobicity of the LP. Other column chemistries could be suitable for this purpose as well.
The peptide mapping in combination with LC/MS is commonly used to address the sites of LP conjugation. Very often optimized conditions for the proteolytic digestion rely on the pH that is optimal for the enzyme activity. When applied to mAb molecules, these conditions are suitable if complete digestion takes place. Many peptide mapping protocols are available for mAbs with the most common pHs for Lys-C digestion, ranging from 8.0 to 8.2. At pH 8.2 and overnight digestion, the LP-conjugated peptides using cysteine chemistry exhibit thiosuccinimide ring opening (Scheme II). This artifactual ring opening during reduction/alkylation/digestion is undesired and was significantly minimized by reducing pH to 7.5, which was sufficient to achieve complete digestion of the protein, yet preserve the structure of the LP (Fig. 7).
In conclusion, despite many challenges presented by each type of ADC, robust, reliable and informative LC/MS methods have been developed for the sensitive and accurate characterization of these molecules at the intact, subunit, and peptide mapping level. These finely tuned methods accommodate the unique requirements of each ADC depending on conjugation chemistry, sites of conjugation, and nature of the LP. These high quality, low-artifact ADC heightened characterization methods contribute immensely to enhanced process and product understanding, which is needed ensure consistent process performance and product quality, as well as patient safety and drug efficacy.
Supplementary Material
Abbreviations
- 2D-LC/MS
2 dimensional-liquid chromatography/ mass spectro-metry
- AcBut
4-(4-acetylphenoxy)butanoic acid
- ADC
antibody-drug conjugate
- Cys
cysteine
- DAR
drug-to-antibody ratio
- ESI MS
electrospray ionization mass spectrometry
- H
heavy
- HH
heavy-heavy
- HL
heavy-light
- HHL
heavy-heavy-light
- HIC
hydrophobic interaction chromatography
- IdeS
immunoglobulin-degrading enzyme of Streptococcus pyogenes
- L
light
- LC
liquid chromatography
- LC/MS
liquid chromatography/ mass spectrometry
- LP
linker payload
- mAb
monoclonal antibody
- MS
mass spectrometry
- oSEC
organic size exclusion chr-omatography
- RP-UHPLC
reversed-phase ultrahigh performance liquid chrom-atography
- RP-UHPLC/ESI-UHR QTOF MS
reversed-phase ultrahigh-performance liquid chromatography/electrospray ionization ultrahigh-resolution quadrupole time-of-flight mass spectrometry
- SEC
size exclusion chromato-graphy
- TFA
trifluroacetic acid
- TG
transglutaminase
- UHR-MS
ultrahigh resolution mass spectrometry
- UV
ultraviolet
Disclosure of potential conflicts of interest
No potential conflicts of interest were disclosed.
Acknowledgment
The authors gratefully acknowledge the scientific assistance of Jeffry Borgmeyer, Heyi Li, and April Xu and sincerely thank the Pfizer Biotherapeutics Pharmaceutical Sciences organization for their support and critical review.
References
- 1.Sievers EL. Antibody-drug conjugates in cancer therapy. Annu Rev Med. 2013;64:15–29. doi: 10.1146/annurev-med-050311-201823. PMID:23043493 [DOI] [PubMed] [Google Scholar]
- 2.Ducry L, Stump B. Antibody−drug conjugates: Linking cytotoxic payloads to monoclonal antibodies. Bioconjug Chem. 2010, 21 (1), 5–13. doi: 10.1021/bc9002019. PMID:19769391 [DOI] [PubMed] [Google Scholar]
- 3.McCombs JR, Owen SC. Antibody drug conjugates: Design and selection of linker, payload and conjugation chemistry. AAPS J. 2015;17(2):339–351. doi: 10.1208/s12248-014-9710-8. PMID:25604608 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Burke PJ, Senter PD, Meyer DW, Miyamoto JB, Anderson M, Toki BE, Manikumar G, Wani MC, Kroll DJ, Jeffrey SC. Design, synthesis,biological evaluation of antibody−drug conjugates comprised of potent camptothecin analogues. Bioconjug Chem. 2009, 20 (6), 1242–1250. doi: 10.1021/bc9001097. PMID:19469529 [DOI] [PubMed] [Google Scholar]
- 5.Ducry L, Stump B. Antibody−drug conjugates: Linking cytotoxic payloads to monoclonal antibodies. Bioconjug Chem. 2010;21(1):5–13. doi: 10.1021/bc9002019. PMID:19769391 [DOI] [PubMed] [Google Scholar]
- 6.Lambert JM, Chari RVJ. Ado-trastuzumab Emtansine (T-DM1): An Antibody–drug conjugate (adc) for her2-positive breast cancer. J Med Chem. 2014;57(16):6949–6964. doi: 10.1021/jm500766w. PMID:24967516 [DOI] [PubMed] [Google Scholar]
- 7.Valliere-Douglass JF, Hengel SM, Pan LY. Approaches to interchain cysteine-linked ADC characterization by mass spectrometry. Mol Pharm. 2015;12(6):1774–83. doi: 10.1021/mp500614p. PMID:25474122 [DOI] [PubMed] [Google Scholar]
- 8.Valliere-Douglass JF, McFee WA, Salas-Solano O. Native intact mass determination of antibodies conjugated with monomethyl Auristatin E and F at interchain cysteine residues. Anal Chem 2012, 84 (6), 2843–9. doi: 10.1021/ac203346c. PMID:22384990 [DOI] [PubMed] [Google Scholar]
- 9.Wang L, Amphlett G, Blättler WA, Lambert JM, Zhang W. Structural characterization of the maytansinoid–monoclonal antibody immunoconjugate, huN901–DM1, by mass spectrometry. Protein Sci. 2005;14 (9):2436–2446. doi: 10.1110/ps.051478705. PMID:16081651 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Luo Q, Chung HH, Borths C, Janson M, Wen J, Joubert MK, Wypych J. Structural characterization of a monoclonal antibody-maytansinoid immunoconjugate. Anal Chem 2016;88(1):695–702. doi: 10.1021/acs.analchem.5b03709. PMID:26629796 [DOI] [PubMed] [Google Scholar]
- 11.Dennler P, Chiotellis A, Fischer E, Brégeon D, Belmant C, Gauthier L, Lhospice F, Romagne F, Schibli R. Transglutaminase-based chemo-enzymatic conjugation approach yields homogeneous antibody–drug conjugates. Bioconjug Chem. 2014;25(3):569–578. doi: 10.1021/bc400574z. PMID:24483299 [DOI] [PubMed] [Google Scholar]
- 12.Strop P, Liu SH, Dorywalska M, Delaria K, Dushin RG, Tran TT, Ho WH, Farias S, Casas MG, Abdiche Y, et al. Location matters: Site of conjugation modulates stability and pharmacokinetics of antibody drug conjugates. Chem Biol. 2013;20(2):161–167. doi: 10.1016/j.chembiol.2013.01.010. PMID:23438745 [DOI] [PubMed] [Google Scholar]
- 13.Junutula JR, Flagella KM, Graham RA, Parsons KL, Ha E, Raab H, Bhakta S, Nguyen T, Dugger DL, Li G, et al. Engineered Thio-Trastuzumab-DM1 Conjugate with an Improved Therapeutic Index to Target Human Epidermal Growth Factor Receptor 2–Positive Breast Cancer. Clin Cancer Res. 2010, 16 (19), 4769. doi: 10.1158/1078-0432.CCR-10-0987. PMID:20805300 [DOI] [PubMed] [Google Scholar]
- 14.Wakankar A, Chen Y, Gokarn Y, Jacobson FS. Analytical methods for physicochemical characterization of antibody drug conjugates. MAbs. 2011;3(2):161–72. doi: 10.4161/mabs.3.2.14960. PMID:21441786 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Firth D, Bell L, Squires M, Estdale S, McKee C. A rapid approach for characterization of thiol-conjugated antibody-drug conjugates and calculation of drug-antibody ratio by liquid chromatography mass spectrometry. Anal Biochem. 2015;485:34–42. doi: 10.1016/j.ab.2015.06.001. PMID:26070852 [DOI] [PubMed] [Google Scholar]
- 16.Sarrut M, Corgier A, Fekete S, Guillarme D, Lascoux D, Janin-Bussat MC, Beck A, Heinisch S. Analysis of antibody-drug conjugates by comprehensive on-line two-dimensional hydrophobic interaction chromatography x reversed phase liquid chromatography hyphenated to high resolution mass spectrometry. I – Optimization of separation conditions. J Chromatogr B Analyt Technol Biomed Life Sci. 2016;1032:103–11. doi: 10.1016/j.jchromb.2016.06.048. PMID:27426266 [DOI] [PubMed] [Google Scholar]
- 17.Birdsall RE, Shion H, Kotch FW, Xu A, Porter TJ, Chen W. A rapid on-line method for mass spectrometric confirmation of a cysteine-conjugated antibody-drug-conjugate structure using multidimensional chromatography. MAbs. 2015;7(6):1036–44. doi: 10.1080/19420862.2015.1083665. PMID:26305867 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Marcoux J, Champion T, Colas O, Wagner-Rousset E, Corvaia N, Van Dorsselaer A, Beck A, Cianferani S. Native mass spectrometry and ion mobility characterization of trastuzumab emtansine, a lysine-linked antibody drug conjugate. Protein Sci. 2015;24(8):1210–23. doi: 10.1002/pro.2666. PMID:25694334 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Debaene F, Boeuf A, Wagner-Rousset E, Colas O, Ayoub D, Corvaia N, Van Dorsselaer A, Beck A, Cianferani S. Innovative native MS methodologies for antibody drug conjugate characterization: High resolution native MS and IM-MS for average DAR and DAR distribution assessment. Anal Chem. 2014;86(21):10674–83. doi: 10.1021/ac502593n. PMID:25270580 [DOI] [PubMed] [Google Scholar]
- 20.Hess JL, Porsch EA, Shertz CA, Boyle MD. Immunoglobulin cleavage by the streptococcal cysteine protease IdeS can be detected using protein G capture and mass spectrometry. J Microbiol Methods. 2007;70(2):284–91. doi: 10.1016/j.mimet.2007.04.017. PMID:17543400 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Ryan MH, Petrone D, Nemeth JF, Barnathan E, Bjorck L, Jordan RE. Proteolysis of purified IgGs by human and bacterial enzymes in vitro and the detection of specific proteolytic fragments of endogenous IgG in rheumatoid synovial fluid. Mol Immunol. 2008;45(7):1837–46. doi: 10.1016/j.molimm.2007.10.043. PMID:18157932 [DOI] [PubMed] [Google Scholar]
- 22.Janin-Bussat MC, Dillenbourg M, Corvaia N, Beck A, Klinguer-Hamour C. Characterization of antibody drug conjugate positional isomers at cysteine residues by peptide mapping LC-MS analysis. J Chromatogr B Analyt Technol Biomed Life Sci. 2015;981–982:9–13. doi: 10.1016/j.jchromb.2014.12.017. PMID:25596378 [DOI] [PubMed] [Google Scholar]
- 23.Huang RY, Chen G. Characterization of antibody-drug conjugates by mass spectrometry: advances and future trends. Drug Discov Today. 2016;21(5):850–5. doi: 10.1016/j.drudis.2016.04.004. PMID:27080148 [DOI] [PubMed] [Google Scholar]
- 24.Wagner-Rousset E, Janin-Bussat MC, Colas O, Excoffier M, Ayoub D, Haeuw JF, Rilatt I, Perez M, Corvaia N, Beck A., Antibody-drug conjugate model fast characterization by LC-MS following IdeS proteolytic digestion. MAbs. 2014;6(1):173–84. doi: 10.4161/mabs.26773. PMID:25006625 [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.