

Abstract
The chelatable iron pool (CIP) is a small but chemically significant fraction of total cellular iron. While this dynamic population of iron is limited, it is redox active and capable of generating reactive oxygen species (ROS) that can lead to oxidative stress which is associated with various pathologies. Nitric oxide (•NO), is a free radical signalling molecule that regulates numerous physiological and pathological conditions. We have previously shown that macrophages exposed to endogenously generated or exogenously administered nitric oxide (•NO) results in its interaction with CIP to form dinitrosyliron complexes with thiol containing ligands (DNICs). In this study we assessed the consequences of DNIC formation in cancer cells as •NO is known to be associated with numerous malignancies. Incubation of cancer cells with •NO led to a time and dose dependent increase in formation of DNICs. The formation of DNICs results in the sequestration of the CIP which is a major source of iron for redox reactions and reactive oxygen species (ROS) generation. Therefore, we set out to test the antioxidant effect of •NO by measuring the ability of DNICs to protect cells against oxidative stress. We observed that cancer cells treated with •NO were partially protected against H2O2 mediated cytotoxicity. This correlated to a concomitant decrease in the formation of oxidants when •NO was present during H2O2 treatment. Similar protective effects were achieved by treating cells with iron chelators in the presence of H2O2. Interestingly, •NO decreased the rate of cellular metabolism of H2O2 suggesting that a proportion of H2O2 is consumed via reactions with cellular iron. When the CIP was artificially increased by supplementation of cells with iron, a significant decrease in the cytoprotective effect of •NO was observed. Notably, •NO concentrations, at which cytoprotective and antioxidant effects were observed, correlated with concentration-dependent increases in DNIC formation. Collectively, these results demonstrate that •NO has antioxidant properties by its ability to sequester cellular iron. This could play a significant role in variety of diseases involving ROS mediated toxicity like cancer and neurodegenerative disorders where •NO has been shown to be an important etiologic factor.
Keywords: Nitric oxide, dinitrosyliron complexes, chelatable iron, oxidative stress, cancer
Graphical abstract
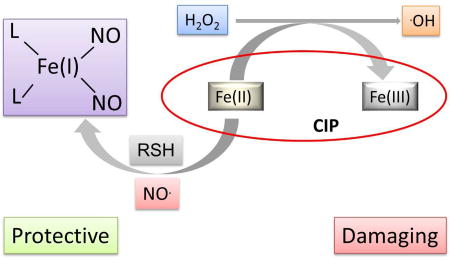
Nitric Oxide converts redox active iron into antioxidant cytoprotective DNIC.
1. INTRODUCTION
Nitric oxide (•NO) is a small free radical signalling molecule present in various biological systems (1). It is a vital effector and messenger molecule that plays role in variety of biological processes including smooth muscle tone, immune response, angiogenesis, apoptosis, and synaptic communication (1). In biological systems, nitric oxide leads to production of a complex array of reactive nitrogen oxide species (2), but it directly reacts with very small number of targets (3). These targets are either other free radicals (e.g., dioxygen, superoxide) or transition metals (e.g., Fe2+) (3). This reactivity is due to the ability of these targets to stabilize the unpaired electron on •NO (4).
Iron is an essential metal ion crutial to a variety intracellular processes such as oxygen transport, enzyme catalysis, and electron transfer reactions etc. (5). Nitric oxide is known to react with transition metals (e.g., iron) in a biological system to form various complexes (4,6). Nitric oxide is also known to react with the intracellular chelatable iron pool (CIP) which leads to the formation of dinitrosyliron complexes (DNIC) with thiol-containing ligands (7–10). The chelatable iron pool is methodologically defined because it is accessible to chemical iron chelators like desferrioxamine and accounts for minor proportion of total cellular iron (1–5%) (11,12). It is comprised by both Fe2+ and Fe3+ iron and it is associated with a diverse population of high and low molecular weight cytosolic ligands such as organic anions (phosphates and carboxylates), polypeptides, and surface component of membranes (e.g., phospholipid head groups) (12). The CIP is a dynamic pool, but under normal physiological conditions its concentration is regulated within a narrow range by homeostatic mechanisms (typically involving iron regulatory proteins, IRE-IRP pathway) (11).
Importantly the CIP forms a chemically significant portion of total intracellular iron, as it is an important source of redox active iron complexes which are known to be involved in reactions leading to the formation of reactive oxygen species e.g., the Fenton reaction (12,13). While H2O2 reacts slowly with biological molecules, the Fenton reaction results in the formation of highly oxidizing hypervalent metal-oxo complexes and hydroxyl radical (14). These species can react at diffusion controlled rates with various macromolecule in cells leading to lipid peroxidation, DNA base modifications, DNA strand breaks, and protein oxidation (15). These damaging events are considered crucial pathogenic factors for numerous diseases, like neurodegenerative disorders (Alzheimer’s and Parkinson’s disease), ischemia-reperfusion injury, cancer, etc. (11).
The biological and pathological consequences of •NO can be either deleterious or cytoprotective which depend on a variety of complex micro-environmental factors (16,17). Pro- or anti-oxidant effects of •NO depend on its local concentration, cell phenotype, and oxygen concentration as well as the presence of other reactive species in the biological milieu (16,18–20). At high local cellular concentration of •NO, it can react with a number of other reactive oxygen and nitrogen oxide species to cause deleterious effects on cellular components. In contrast, low physiological doses of •NO are shown to abate the oxidative potential of reactive oxygen species perhaps through the formation of metal-nitrosyl complexes which can lead to a decrease in the formation of Fenton related oxidants (16,21). In addition, •NO has been shown to protect cell against death caused by oxidizing agents such as hydrogen peroxide, alkylhydroperoxides (22,23).
In cancer biology, •NO plays numerous roles (24) ranging from tumor progression (25,26), DNA mutations (27), migration/invasion (28), epigenetic regulation (29–31) to even treatment (32,33). For many cancer types, however, the presence of •NO or the expression of nitric oxide synthase (NOS) is consider a negative prognostic indicator that correlates with worse patient outcomes. Although numerous cell signalling mechanisms have been reported to explain these associations we postulate that a simpler mechanism could be significant contributing factor; the ability of •NO to function as an antioxidant. Herein, we provide evidence that protection of cancer cells in the presence of oxidants results from the ability of •NO to sequester chelatable iron in the form of DNIC.
2. MATERIALS AND METHODS
2.1 Chemicals and Reagents
Diethylenetriamine nonoate (DETA/NO) and Spermine/nonoate (Sper/NO) were a generous gift from Dr Joseph A. Hrabie (National Institute of Heath, NCI). Ferric ammonium citrate (FAC), Diethylenetriamine-pentaacetic acids (DTPAC), phosphate buffer saline (PBS), desferrioxamine mesylate, CelLytic™ M cell lysis reagent, ferrous sulfate, Phenylmethanesulfonyl fluoride (PMSF), potassium iodide, 3-(2–Pyridyl)-5,6-diphenyl-1,2,4-triazine-p,p′-disulfonic acid monosodium (Ferrozine™), Neocuproine, ammonium acetate, sodium hydroxide, acetic acid, hydrochloric acid, ascorbic acid, and lipopolysacchride (LPS) were purchased from Sigma-Aldrich (St Louis, MO). Chemical iron chelator, 2,2’-dipyridal (DP), was purchased from Acros Organics (NJ). Bio-rad DC™ protein assay reagents were purchased from Bio-rad (Hercules, CA). Dihyrorhodamine 123 was purchased from Alexis Biochemicals (San Diego, CA) and alamarBlue® dye was purchased from Life Technologies (Waltham, MA). Protease inhibitor cocktail set III was purchased from Merck Millipore (Bellerica, MA). All cell culture supplies were purchased from either Invitrogen or Fischer Scientific (Pittsburgh, PA), unless otherwise specified.
2.2 Cell Culture
HCC 1806 and MDA-MB-231 breast cancer cells and HT-29 colon cancer cells were obtained from the American Type Culture Collection (ATCC, Manassas, VA). RAW 264.7 murine macrophage cells were also obtained from ATCC. All cell lines were cultured at 37°C temperature and 5% CO2 concentration in a tissue culture incubator. The cells were grown to 80-90% confluence in tissue culture plates in either DMEM (for HCC 1806 and HT-29) or RPMI (for MDA-MB-231 and RAW 264.7) growth medium containing 10% fetal bovine serum (FBS) and 1% Penicillin-Streptomycin. Prior to treatments, growth media was replaced with serum-free growth medium for 16 hours. All experiments were conducted under these culture conditions.
Iron supplementation: The cells were treated with ferric ammonium citrate (150 µg/mL) for various time points. Plates were washed with PBS containing DTPAC (100 µM) in order to remove excess extracellular iron and fresh growth medium was added to the plate prior to treatments.
2.3 Total iron determination
Total iron levels were determined using a previously established method (34) with slight modifications. Briefly, cells were lysed using lysis buffer (CelLytic™ M Cell lysis reagent with 1% protease inhibitor cocktail and 1mM PMSF). The samples were centrifuged and the supernatant was collected for further analysis. Aliquots (100 µL) of cell lysates were mixed with 100µL of 10mM HCl (the solvent of the iron standard FeCl3), and 100µL of the ironreleasing reagent (a freshly mixed solution of equal volumes of 1.4M HCl and 4.5% (w/v) KMnO4 in H2O) for 2 hour at 60 °C. After the mixtures were cooled to room temperature, 30 µL of the iron-detection reagent (6.5 mM ferrozine, 6.5 mM neocuproine, 2.5 M ammonium acetate, and 1M ascorbic acid dissolved in water) was added. After 30 minutes, 250 µL of the solution was transferred into a well of a 96-well plate and the absorbance was measured at 550 nm on a microplate reader.
Iron concentrations were estimated by comparing the absorbance of the sample to that of a known standard of FeCl3 (mixture of 100 µL of FeCl3 standards (0–300 µM) in 10 mM HCl, 100 µL lysis buffer, 100 µL releasing reagent, and 30 µL detection reagent). The intracellular iron concentrations determined were normalized to the amount of protein in the sample.
2.4 Electron Paramagnetic Resonance (EPR) Spectroscopy
EPR Spectroscopy was performed using our established method (8,28). Briefly, cells were trypsinized and re-suspended in equal volumes of PBS. DNIC signals were measured by EPR at g = 2.03 using a Bruker X-band EMX Plus EPR spectrometer, fitted with a liquid nitrogen dewar (at 77°K) with the following settings: modulation amplitude 10G, 200G scan range, 90 sec scan time, 1 scan. DNIC concentrations were estimated by comparing the signal amplitudes to that of a known standard of GSH-DNIC. For quantitative studies a microwave power saturation profile for EPR signal amplitudes was conducted to ensure we were operating below the saturation. Protein samples were collected from each experiment so that DNIC could be quantitatively compared between individual runs (pmol DNIC/mg protein).
For the CIP quantification, cells were treated with 1 mM desferrioxamine (DFO) for 4 h and then harvested for EPR analysis. The concentration of the CIP was calculated by comparing the signal amplitude to the amplitude of known concentrations of a DFO-Fe(III) standard prepared as previously described (8,24). EPR settings: g = 4.03, modulation amplitude of 10G, 200 G scan range, 30 sec scan time, 4 scans.
2.5 GSH-DNIC synthesis
The synthetic DNICs were synthesized using previously described method (35). 1 ml of 0.5 mM FeSO4 (dissolved in degassed water to avoid rapid oxidation to the ferric state) was added to 10 mL (final volume) of 0.1 M of degassed PBS buffer, pH 7.4, containing 20 mM GSH and 2 mM GSNO (25 °C) under hypoxic conditions. After 45 minutes, the reaction was almost 70% complete, and the resulting stock solution of GSH-DNIC was quantified either by UV-Vis spectroscopy (ε403=3000) or EPR spectroscopy.
2.6 Cell Viability Assay
Cell viability was assessed using an established protocol (36). Briefly, cells (10×103/well) were plated in 96-well plate 16 hour before the experiment. The cells were treated with Sper/NO for 1 h followed by H2O2 for 2 h. They were further incubated in FBS supplemented DMEM media. After 48 hours, media was replaced with media containing 10% alamarBlue® dye for 2–3 h. Absorbance was recorded at 570nm using 600nm as reference wavelength.
2.7 DHR fluorescence assay
The levels of cellular oxidants was determined using DHR fluorescence assay (37). Cells (7.5×103/well) were plated in 96-well plate 16 h before the experiment. The cells were incubated with PBS containing DHR123 (100 µM) for 1 h at 37°C. The cells were washed once with PBS and incubated with the culture medium containing Sper/NO for 1 h. H2O2 were added to the medium and incubated for 1 h. The fluorescence was measured by Spectra MAX GeminiEM plate reader (excitation: 485nm, emission: 530nm).
2.8 Cellular H2O2 metabolism
HCC 1806 cells were trypsinized and put into a reaction chamber at 6×106 cells/ml of serum-free media. Cell suspensions were constantly stirred in a sealed, water-jacketed, temperature-controlled (37 °C) chamber. The reaction chamber was equipped with a hydrogen peroxide electrode connected to an Apollo 4000 free radical analyser (World Precision Instruments, Sarasota, FL, USA). Headspace in the vessel was negligible. Reactions were initiated by bolus injection of H2O2 with a gas-tight syringe and metabolism was measured in real time.
2.9 Statistical Analysis
One way ANOVA analysis with Fisher’s LSD posthoc test was done using OriginPro8.1 software, OriginLab (Northampton, MA). The data were reported as the Mean ± Standard Error of Mean (S.E.M.).
3. RESULTS
3.1 The amounts and distributions of Cellular Iron differ between Cancer Cell types
Iron is a primary biological target for •NO in most cell types including cancers. For this reason we set out to assess the levels of total iron, CIP, and DNIC in 3 phenotypically different cancer cell types (HCC 1806 and MDA-MB-231 breast cancer cells and HT-29 colon cancer cells). Figure 1A demonstrates that the amounts of total cellular iron vary significantly between the 3 cancer cell lines with twice as much iron in the MDA-MB-231 cells compared to the HCC1806. Also, the concentration of iron in the CIP as well as the percentage total iron that made up the CIP were distinctly different between cell types (Fig. 1B,C). Interestingly, despite both breast cancer cell types (HCC 1806 and MDA-MB-231) having different levels of total cellular iron (8.6 vs. 19.2 nmol/mg protein) a similar percentage of iron was measured in the CIP (~ 1%). The HT-29 colon cancer cells had both the largest CIP (>500 pmol/mg protein) as well as 4 times the amount of total cellular iron in the CIP (>4%).
Figure 1. Comparing amounts of total Iron, chelatable Iron, and dinitrosyliron complexes in 3 cancer cell lines.

(A) Quantification of total intracellular iron. Cell lysates from three different cancer cell types (HCC 1806, HT29 and MDA-MB-231) were digested with acidic KMnO4 solution to release iron from all intracellular stores. The lysates containing free released iron were then treated with iron detection reagent (i.e., 6.5 mM ferrozine, 6.5 mM neocuproine, 2.5 M ammonium acetate, and 1M ascorbic acid dissolved in water). Absorbance was measured at 550 nm and compared to known iron standard. (B) Quantification of chelatable iron. The intracellular CIP (Fe2+/Fe3+) was measured by treating the cells with desferrioxamine (DFO, 4h) which complexes with the CIP (DFO-Fe3+) to give a distinct EPR signal at g=4.3. This was compared to standard DFO-Fe3+ complex as described previously (28). (C) Percentage CIP of total cellular iron. (D) DNIC quantification. HCC 1806, HT29 and MDA-MB-231 cell were treated with the •NO-donor Sper/NO (1 mM) for 1 h. DNICs were detected in whole cells by EPR analysis (g=2.03) and quantified by comparing to a standard curve generated with a chemically synthesized small molecular weight glutathione DNIC (GSH-DNIC), as shown previously (28). (n ≥ 3 for all experiments
3.2 DNIC formation is a function of the amount of iron in the CIP as well as •NO exposure time and concentration
We have shown previously in macrophages that DNICs are formed rapidly upon cellular exposure to •NO (8). However, the parameters of •NO exposure leading to DNIC formation in cancer cells has not be extensively studied. The three cell lines in culture were treated with the •NO-donor Sper/NO for 1 h and DNIC levels were assessed (Fig. 1D). Under these conditions significant amount of DNIC (500–900 pmoles/mg protein) were formed. Markedly higher levels of DNICs were observed in HT-29 cells compared to both HCC 1806 and MDA-MB-231 cells which paralleled differences in their respective CIP levels (Fig. 1B). In the case of the HCC 1806 cells, levels of DNIC exceeded measured amounts of the CIP (CIP 100 pmol vs. DNIC 450 pmol).
To better understand the relationship between •NO concentration and exposure time on DNIC formation we treated HCC 1806 cells with two different •NO-donors that have different •NO-release kinetics (Sper/NO and DETA/NO). Sper/NO has a relatively short half-life (39 min at 37°C) and provides a burst of •NO whereas DETA/NO has a significantly longer half-life (20 h at 37°C) and provides a continuous steady-state concentration of •NO for a prolonged period of time (38). The cells were incubated with Sper/NO (10–50 µM) for 3 h which we have determined gives a physiological steady-state •NO concentration of approximately 3–300 nM (8,38,39). Under these conditions, significant amounts DNIC were detected at Sper/NO concentrations above 20 µM (Fig. 2A). Exposure of HCC 1806 cells to DETA/NO (500–1000 µM, [•NO]SS = 25–2,000 nM) for 4 h also lead to a dose-dependent increase in the formation of DNICs (Fig. 2B). At 500 µM DETA/NO the levels of DNIC closely matched the amount of iron in the CIP, however, at higher •NO concentrations the amounts of DNIC exceeded the levels of the CIP (Fig. 1B, 2B). As the CIP is the primary source of iron for DNIC assembly this suggests that higher amounts of •NO facilitate the release of iron from other intracellular sources making it available for reaction with •NO.
Figure 2. Kinetics of DNIC assembly and degradation in Cancer Cells.

(A–C) Quantification of DNIC after NO exposure in relation to the CIP. HCC 1806 cells were treated with (A) Sper/NO (0–50 µM) for 4 h or (B) DETA/NO (0–1 mM) for 4 h or (C) DETA/NO (1 mM) for indicated time points. The cells were harvested and DNIC were measured by EPR. DNIC are expressed in terms of total cellular protein (100 pmoles/mg protein ~ 5-10 µM DNIC samples) *,p < 0.05, **,p < 0.01. (D) Stability of DNIC. HCC1806, MDA-MB-231, HT-29 cells were treated with •NO-donor Sper/NO (1 mM) for 1 h then washed and supplemented with fresh media (time = 0). The time points indicate time after removal of •NO source. ##, p < 0.01 with respect to DNIC levels at 0 h time point for HCC 1806 cells. ^, p < 0.05, ^^, p < 0.01 with respect to DNIC levels at 0 h time point for MDA-MB-231 cells. **, p < 0.01 with respect to DNIC levels at 0 h time point for HT-29 cells. 100 pmoles/mg protein ~ 5–10 µM DNIC samples. (n ≥ 3 for all experiments
To determine temporal effects of continuous •NO exposure on DNIC formation we treated HCC 1806 cells with DETA/NO (1 mM) for 12 hours and measured DNIC over time. Figure 2C demonstrates that DNICs were rapidly formed after a 1 h of •NO exposure with maximal amounts of DNICs detected by 4 hours. No increases in DNIC were observed after 4 hours but maximal amounts of DNIC were maintained throughout the duration of •NO exposure. Although the amounts of DNIC formed were 3–4 folds higher than the level of iron in the CIP a saturation level of DNIC was achieved by 4 hours even in the presence of continued •NO exposure (Fig. 1B, 2C).
3.3 Stability of Intracellular DNICs in Cancer Cells
Before we could look at downstream phenotypic effects of DNIC in cancer cells it was important to understand their stability (biological lifetime). To determine this the three cancer cell lines (HCC 1806, MDA-MB-231 and HT-29) were treated in culture with high concentrations of •NO (Sper/NO, 1 mM) to allow maximal DNIC formation. After 1 hour the •NO source was removed by washing the cells and replacing with fresh growth media. Samples were then harvested for DNIC analysis at various time points (0–4 h) after •NO removal (time = zero). Over a 4 hour period cellular DNICs decreased with a half-life of approximately 1 hour for all cell types (Fig. 2D). Taken together, these results (Fig. 2A–D) demonstrate that exposure of cancer cells to •NO results in rapid formation of DNICs and these complexes have a significant life-span in the absence of •NO.
3.4 Nitric oxide inhibits hydrogen peroxide-mediated oxidation
Although the CIP is a small fraction of total cellular iron it is redox active and pathologically significant due to its capacity to generate oxidizing reactive oxygen species (ROS) (12,13). The CIP is also the primary sources of iron for DNIC assembly. Therefore, we wanted to test whether sequestration of CIP iron via DNIC assembly would render the iron less redox active. Hydrogen peroxide (H2O2), by itself, is a relatively mild oxidant. In the presence of ferrous iron, however, Fenton chemistry converts H2O2 into highly oxidizing species (13–15,34,35). First HCC 1806 cells were pre-treated with increasing concentrations of •NO to form DNIC (Sper/NO, 0-1,000 µM, 1h). Cells were then exposed to a bolus of hydrogen peroxide (200 µM) for 2 h and intercellular oxidation was determined by measuring measuring the conversion of dihydrorhodamine 123 (DHR123) to the highly fluorescent product rhodamine. Figure 3A demonstrates a significant (p < 0.001–0.05) dose dependent decrease in H2O2-mediated oxidation in cells pre-incubated with •NO. No significant oxidation was measured in cells incubated with •NO alone (Fig. 3A). In another set of experiments, cells were incubated with increasing concentration of hydrogen peroxide (0–400 µM) in the presence or absence of low amounts of •NO (50 µM Sper/NO) (Fig. 3B). Incubation of cells with hydrogen peroxide alone leads to a dose dependent increase in formation of cellular oxidants (Fig. 3B). Importantly, in presence of •NO results in significant (p < 0.05) suppression in peroxide mediated increase in cellular oxidant levels at all concentrations of peroxide examined.
Figure 3. Nitric oxide decreases H2O2-mediated cellular oxidation.

(A) Nitric Oxide reduces cellular oxidation mediated by H2O2 in a dose dependent manner. HCC 1806 cells were loaded with fluorescent dye DHR 123 (100 µM). The cells were then treated with •NO-donor Sper/NO (10–1,000 µM) for 1 h followed by hydrogen peroxide (200 µM) for 2 h. Fluorescence was measurement and compared to untreated controls (B) Low amounts of NO diminish oxidation in the presence of increasing concentrations of H2O2. Cells were loaded with fluorescent dye, DHR 123 (100 µM) and then treated with the •NO-donor Sper/NO (50 µM, 1h) followed by hydrogen peroxide (100–400 µM, 2h). (C) Endogenous •NO decreases intracellular oxidation. RAW 264.7 cells were activated with LPS (1 µg/mL) for 16 h. The cells were then loaded with fluorescent dye, DHR 123 (100 µM) and treated with hydrogen peroxide (0.5–1 mM, 2h). Fluorescence was measurement and compared with respect to individual non-LPS treated control. (D) Cellular iron supplementation augments H2O2-mediated oxidation. HCC 1806 cells were incubated with increasing concentration of ferrous ammonium citrate (62.5–125 µg/mL) which increases the CIP. They were then loaded with DHR 123 and treated with Sper/NO (0 or 25 µM) for 1 h followed by peroxide for 2 h. with respect to individual non-iron supplemented control. (E) The effect of •NO on hydrogen peroxide metabolism in cancer cells. HCC 1806 cells in suspension (constantly stirred) were treated with NO (25 µM Sper/NO) followed by bolus addition of H2O2 (1 µM). H2O2 concentration was monitored in real time, representative electrochemical trace. H2O2 did not disappear in the absence of cells +/− •NO (data not shown). (n ≥ 3 for all experiments, *, p < 0.05; **, p < 0.01, ***, p < 0.001
Figures 3A, B demonstrate a proof of concept that •NO could decrease the amount of oxidation induced by H2O2. In general these concentrations of •NO were superphysiologic/pathologic. To model a more physiologic relevant scenario we examined the ability of endogenously produced •NO to abate H2O2-mediated oxidation. RAW 264.7 macrophage cells were activated with lipopolysaccharide (LPS; 1 µg/mL) for 16 h which we have shown induces iNOS expression and •NO production (8). These cells were then loaded with DHR 123 followed by treatment with hydrogen peroxide (500 or 1,000 µM) for 2 h. Under these conditions there was a significant decrease in intercellular oxidation in the •NOproducing cells compared to the control cells following H2O2 exposure (Fig. 3C).
Next, in order to ascertain the role of CIP in •NO-mediate anti-oxidant effects, studies were conducted with cancer cells supplemented with iron. Notably, we have previously demonstrated that supplementation of HCC 1806 cancer cells with ferric ammonium citrate (FAC) can significantly increase the CIP levels in these cells (28). Thus, the effect of CIP supplementation (via FAC) on production of oxidants was examined. HCC 1806 cells were incubated with FAC then loaded with DHR 123 dye. The cells were then treated with the •NO-donor (Sper/NO; 25 µM) for 1 h followed by hydrogen peroxide (200 µM) for 2 h. In the absence of •NO there was a significant increase in oxidant production in the cells supplemented with. However, oxidation was considerably less in cells pre-treated with Sper/NO in both the iron supplemented and non-supplemented cells (Fig. 3D). Collectively, these results (Fig. 3A–D) demonstrate that •NO can interact with the CIP to mitigate its redox ability, potentially via formation of DNICs.
3.5 Nitric Oxide decreases the rate of cellular H2O2 metabolism
It is well-known that a significant proportion of cellular hydrogen peroxide is metabolized by the enzyme catalase and we have shown that this is fairly rapid and by first-order kinetics (19). Interestingly, studies have also demonstrated that •NO can be oxidated by compound I of catalase as well as reversibly inhibit catalase activity (40). Therefore, in addition to forming DNICs, it was important to determine if the protective effects of •NO were linked to its ability to alter H2O2 metabolism. We examined the effect of •NO on hydrogen peroxide metabolism using electrochemical detection where the rate of H2O2 disappearance in the presence of cells could be monitored in real time. Notably, the rate of cellular hydrogen peroxide consumption was twice as fast in the control cells compared cells exposed to •NO (Fig. 3E). These results suggest that •NO interacts with H2O2 catabolic pathways and that although •NO is protective against oxidative damage it paradoxically increases both the half-life of H2O2 and the cellular exposure.
3.6 Cytoprotective effects of •NO
The damage caused CIP-generated oxidants can result in DNA strand breaks, base modifications, protein oxidation, and lipid peroxidation. Studies herein have demonstrated that •NO can suppress the oxidant levels in cancer cells potentially via its ability to interact with the CIP. Hence, we set out to assess the ability of •NO to alter H2O2-mediated cytotoxicity. To examine this, HCC 1806 cells were pre-incubated with increasing concentrations of the •NO-donor Sper/NO (10–100 µM) for 1 h followed by incubation with hydrogen peroxide (200 µM) for 2 h. Cells were then washed and supplemented with fresh growth media. After 48 hours, cellular viability was measured. The incubation of cells with hydrogen peroxide alone lead to a significant (p < 0.001) decrease in cell survival compared to control cells (Fig. 4A). Notably, pre-incubation with Sper/NO (25–100 µM) resulted in a significant (p < 0.001) decrease in peroxide-mediated cytotoxicity (Fig. 4A).
Figure 4. Cytoprotective effect of NO are due to its interaction with CIP in cancer cells.

Cellular viability measurments. (A) Nitric oxide suppresses peroxide mediated cytotoxicity in cancer cells. HCC 1806 cells were treated with increasing concentration of Sper/NO (10–100 µM) for 1 h followed by hydrogen peroxide (200 µM) for 2 h. Survival was measured 48 h after removal of hydrogen peroxide. ***, p < 0.01 as indicated. (B) Iron chelator suppresses H2O2 mediated cytotoxicity. HCC 1806 cells were treated with the iron chelator Dipyridal (100 or 200 µM) for 1 h followed by hydrogen peroxide (200 µM) for 2 h. Survival was measured 48 h after removal of hydrogen peroxide. ***, p < 0.01 as indicated. (C) NO suppresses H2O2 mediated cell death after iron supplementation. HCC 1806 cells were treated with FAC (150 µg/mL) for 4 h to increase the CIP. Cells were then treated with Sper/NO (0–50 µM) for 1 h followed by the addition of hydrogen peroxide (200 µM) for 2 h. Survival was measured 48 h after removal of hydrogen peroxide. ***, p < 0.001 with respect to no treatment (NT) control. ###, p < 0.001 with respect to corresponding no Sper/NO (0 µM) control. ^^^, p < 0.001 with respect to corresponding treatments with no FAC supplementation. (n ≥ 3 for all experiments
Our theory is that •NO behaves similar to chemical iron chelators by virtue of its ability to sequester iron in the form of DNICs. Thus we next sought to determine if chemical iron chelators could similarly prevent peroxide mediated cytotoxicity. HCC 1806 cells were incubated with iron chelator Dipyridal (100–200 µM) for 1 h, followed by treatment with hydrogen peroxide (200 µM) for 2 h. The cells were washed and supplemented with fresh growth media and after 48 h cell viability was measured. As in figure 4A peroxide alone resulted in a marked decrease in cellular viability. However, similar to Sper/NO, preincubation with Dipyridal lead to a significant decrease in peroxide mediated cytotoxicity (Fig. 4B). These results indicate that the protective effects of •NO and iron chelators may be due to their ability to interact with the same pool of iron (i.e., the CIP).
3.7 Nitric Oxide Mediated Cytoprotection Against Oxidative Stress is Due to its Interaction with CIP
To further elucidate the role of the CIP in •NO-mediated cytoprotection we supplemented the cells with iron to augment the CIP (FAC 150 µg/mL for 4 h). These cells were then treated with low amounts of •NO (Sper/NO 25 or 50 µM) for 1 h followed by the addition of hydrogen peroxide (200 µM) for 2 h. The cells were washed and supplemented with fresh growth medium for 48 h and cell viability was measured. Iron supplemented cells were significantly more susceptible to H2O2-mediated toxicity compared HCC 1806 cells with basal CIP levels. Although both iron supplemented and non-iron supplemented cells were partially protected from H2O2 by •NO, greater amounts of •NO were required in the cells supplemented with iron (Fig. 4C). Collectively, these results (Fig. 4A–C) demonstrate that •NO can interact with CIP and prevents oxidative stress mediated cell death, potentially via formation of DNICs.
4. DISCUSSION
DNIC were first detected in cancers as far back as the 1960s but the source of their formation and their biological functions were not known. The roles of •NO in caners are complex but in many cases it is associated with deleterious patient outcomes. In this short study, we looked into some basic parameters that contribute to DNIC formation and decay as well as potential phenotypic consequences in cancer cells. It is well-known that the CIP is a main source of iron required for the formation of these complexes (3). The major evidence for this is the observation that treatment of cells with iron chelators completely ablates DNIC assembly (8). As iron chelators display antioxidant properties due to their ability to sequester iron and render it less redox active, so to might •NO via the formation of DNIC. Thus, a potential role for •NO in cancers is to protect the tumours from oxidative host-defences mechanisms.
Although the CIP is the source of iron for DNIC assembly, we have shown that in cancer cells exposed to higher prolonged concentrations of •NO the concentrations of DNIC significantly exceed the CIP (8). This implies that •NO is capable of liberating iron from other sources such as iron storage and iron-sulphur cluster containing proteins. This indicates that a function of •NO may be to increase the availability of “free iron” (CIP) which, although not tested in this study, could potentially have pro-oxidant effects upon the removal of •NO. This would be partially dictated by the stability of DNIC upon •NO removal as well as the ability of the cells to re-establish normal iron homeostasis to bring the CIP back to basal levels. We observed that once the •NO source was removed, the DNIC signal disappeared with a half-life of approximately 60 minutes. We use the term disappear over decay because at this stage of our investigations we can only speculate on the mechanism(s) of disappearance of the EPR =2.04 DNIC signal. Potential explanations for the loss of this EPR signal could be due to multitude of reasons: conversion to non-paramagnetic •NO-iron complexes, dimeric-DNIC, decay of DNIC, and cellular export of DNIC. Turella et al showed that GST extended the half-life of DNIC in cell lysates from 2.8 minutes to 4.5-8 h (41). Another study demonstrated that Cys-DNIC was less stable under ambient air, but persists for hours under deoxygenated conditions (42). Watts et al suggested that there is an equilibrium between low and high molecular weight DNIC and the latter are exported out of the cell resulting in loss of the EPR signal (43). These results highlight the complexity •NO/iron interactions and suggest that loss of DNIC is a multifactorial process that is a function of microenvironmental conditions as well as differences in the contribution of specific thiol ligands (protein vs. non-protein).
One of the classical and probably best-studied redox reactions associated with oxidative stress the Fenton reaction. Under biological conditions, the CIP is assumed to be the major source of iron to catalyse the formation of strong oxidants by this reaction (13). Cancer cells are known to produce much higher level of H2O2 and H2O2-mediated oxidative stress is known to play an important role in carcinogenesis by inducing DNA strand breaks, mutations, and genomic instability (15). There are reports, however, demonstrating cytoprotective effects of nitric oxide against hydrogen peroxide mediated insults (22). In the current study, we tested whether •NO could abate iron-mediated H2O2 toxicity via its ability to sequester iron in the form of DNIC. It was demonstrated that cancer cells pretreated with •NO or endogenously synthesizing •NO showed both significantly less intercellular oxidation and toxicity in response to hydrogen peroxide compared to cells not exposed to •NO. These results could largely be replicated by treating cells with iron chelators instead of •NO suggesting that the mechanisms of protection are similar for both molecules (i.e., the CIP is the only target shared by both •NO and iron chelators). Further evidence for a role of DNIC in the protective effects of •NO comes from the observation that artificially increasing in the CIP via iron supplementation led to increased level of oxidant production and increased cell death. High amounts of •NO could partially overcome this excess iron-attributable toxicity.
We concluded that in cancer cells DNIC formation is an important intracellular event and their predominant function may be to protect cells from oxidative stress. One could envision a scenario where early oxidative stress induces oncogenic transformation but tumours that later acquire the ability to synthesize •NO exhibit a selective survival advantage against host-defences by being protected from iron-mediated toxicity. This compensatory mechanism could be one explanation why •NO-associated tumours correlate to increased aggressiveness and worse patient outcome.
HIGHLIGHTS.
Nitric oxide reacting with the CIP rapidly forms DNIC in cancer cells
The CIP is a major source of cellular oxidant generation upon H2O2 exposure
DNIC reduce H2O2-mediated oxidation and cell death
Acknowledgments
SS would like to thank Cancer Australia and Cure Cancer Australia Foundation for 2017 Young Investigator PdCCRs grant.
FUNDING
This work was supported in part by the National Institutes of Health R01GM085232 (DT).
ABBREVIATIONS
- DNIC
Dinitrosyliron Complexes
- NO
Nitric Oxide
- CIP
Chelatable Iron Pool
Footnotes
Publisher's Disclaimer: This is a PDF file of an unedited manuscript that has been accepted for publication. As a service to our customers we are providing this early version of the manuscript. The manuscript will undergo copyediting, typesetting, and review of the resulting proof before it is published in its final citable form. Please note that during the production process errors may be discovered which could affect the content, and all legal disclaimers that apply to the journal pertain.
References
- 1.Ignarro LJ. Nitric oxide: biology and pathobiology. Academic press; 2000. [Google Scholar]
- 2.Lancaster JR. Nitroxidative, nitrosative, and nitrative stress: kinetic predictions of reactive nitrogen species chemistry under biological conditions. Chemical research in toxicology. 2006;19:1160–1174. doi: 10.1021/tx060061w. [DOI] [PubMed] [Google Scholar]
- 3.Toledo JC, Bosworth CA, Hennon SW, Mahtani HA, Bergonia HA, Lancaster JR. Nitric oxide-induced conversion of cellular chelatable iron into macromolecule-bound paramagnetic dinitrosyliron complexes. Journal of Biological Chemistry. 2008;283:28926–28933. doi: 10.1074/jbc.M707862200. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Ford PC, Lorkovic IM. Mechanistic aspects of the reactions of nitric oxide with transition-metal complexes. Chemical reviews. 2002;102:993–1018. doi: 10.1021/cr0000271. [DOI] [PubMed] [Google Scholar]
- 5.Richardson DR, Ponka P. The molecular mechanisms of the metabolism and transport of iron in normal and neoplastic cells. Biochimica Et Biophysica Acta (BBA)-Reviews on Biomembranes. 1997;1331:1–40. doi: 10.1016/s0304-4157(96)00014-7. [DOI] [PubMed] [Google Scholar]
- 6.Thomas DD, Miranda KM, Colton CA, Citrin D, Espey MG, Wink DA. Heme proteins and nitric oxide (NO): the neglected, eloquent chemistry in NO redox signaling and regulation. Antioxidants & redox signaling. 2003;5:307–317. doi: 10.1089/152308603322110887. [DOI] [PubMed] [Google Scholar]
- 7.Bosworth CA, Toledo JC, Jr, Zmijewski JW, Li Q, Lancaster JR., Jr Dinitrosyliron complexes and the mechanism(s) of cellular protein nitrosothiol formation from nitric oxide. Proceedings of the National Academy of Sciences of the United States of America. 2009;106:4671–4676. doi: 10.1073/pnas.0710416106. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Hickok JR, Sahni S, Shen H, Arvind A, Antoniou C, Fung LW, Thomas DD. Dinitrosyliron complexes are the most abundant nitric oxide-derived cellular adduct: biological parameters of assembly and disappearance. Free radical biology & medicine. 2011;51:1558–1566. doi: 10.1016/j.freeradbiomed.2011.06.030. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Vanin AF, Sanina NA, Serezhenkov VA, Burbaev DS, Lozinsky VI, Aldoshin SM. Dinitrosyl–iron complexes with thiol-containing ligands: spatial and electronic structures. Nitric Oxide. 2007;16:82–93. doi: 10.1016/j.niox.2006.07.005. [DOI] [PubMed] [Google Scholar]
- 10.Pereira JC, Iretskii AV, Han RM, Ford PC. Dinitrosyl iron complexes with cysteine. Kinetics studies of the formation and reactions of DNICs in aqueous solution. Journal of the American Chemical Society. 2015;137:328–336. doi: 10.1021/ja510393q. [DOI] [PubMed] [Google Scholar]
- 11.Petrat F, Groot Hd, Sustmann R, Rauen U. The chelatable iron pool in living cells: a methodically defined quantity. Biological chemistry. 2002;383:489–502. doi: 10.1515/BC.2002.051. [DOI] [PubMed] [Google Scholar]
- 12.Kakhlon O, Cabantchik ZI. The labile iron pool: characterization, measurement, and participation in cellular processes. Free Radical Biology and Medicine. 2002;33:1037–1046. doi: 10.1016/s0891-5849(02)01006-7. [DOI] [PubMed] [Google Scholar]
- 13.Stohs SJ, Bagchi D. Oxidative mechanisms in the toxicity of metal ions. Free radical biology and medicine. 1995;18:321–336. doi: 10.1016/0891-5849(94)00159-h. [DOI] [PubMed] [Google Scholar]
- 14.Qian SY, Buettner GR. Iron and dioxygen chemistry is an important route to initiation of biological free radical oxidations: an electron paramagnetic resonance spin trapping study. Free radical biology and medicine. 1999;26:1447–1456. doi: 10.1016/s0891-5849(99)00002-7. [DOI] [PubMed] [Google Scholar]
- 15.Lopez-Lázaro M. Dual role of hydrogen peroxide in cancer: possible relevance to cancer chemoprevention and therapy. Cancer letters. 2007;252:1–8. doi: 10.1016/j.canlet.2006.10.029. [DOI] [PubMed] [Google Scholar]
- 16.Wink DA, Miranda KM, Espey MG, Pluta RM, Hewett SJ, Colton C, Vitek M, Feelisch M, Grisham MB. Mechanisms of the antioxidant effects of nitric oxide. Antioxidants and redox signaling. 2001;3:203–213. doi: 10.1089/152308601300185179. [DOI] [PubMed] [Google Scholar]
- 17.Thomas DD, Heinecke JL, Ridnour LA, Cheng RY, Kesarwala AH, Switzer CH, McVicar DW, Roberts DD, Glynn S, Fukuto JM, Wink DA, Miranda KM. Signaling and stress: The redox landscape in NOS2 biology. Free Radic Biol Med. 2015;87:204–225. doi: 10.1016/j.freeradbiomed.2015.06.002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Hickok JR, Vasudevan D, Jablonski K, Thomas DD. Oxygen dependence of nitric oxide-mediated signaling. Redox Biol. 2013;1:203–209. doi: 10.1016/j.redox.2012.11.002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Thomas DD, Ridnour LA, Espey MG, Donzelli S, Ambs S, Hussain SP, Harris CC, DeGraff W, Roberts DD, Mitchell JB, Wink DA. Superoxide fluxes limit nitric oxide-induced signaling. The Journal of biological chemistry. 2006;281:25984–25993. doi: 10.1074/jbc.M602242200. [DOI] [PubMed] [Google Scholar]
- 20.Thomas DD. Breathing new life into nitric oxide signaling: A brief overview of the interplay between oxygen and nitric oxide. Redox Biol. 2015;5:225–233. doi: 10.1016/j.redox.2015.05.002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Lu C, Koppenol WH. Inhibition of the Fenton reaction by nitrogen monoxide. J Biol Inorg Chem. 2005;10:732–738. doi: 10.1007/s00775-005-0019-z. [DOI] [PubMed] [Google Scholar]
- 22.Wink DA, Cook JA, Pacelli R, DeGraff W, Gamson J, Liebmann J, Krishna MC, Mitchell JB. The effect of various nitric oxide-donor agents on hydrogen peroxide-mediated toxicity: a direct correlation between nitric oxide formation and protection. Archives of biochemistry and biophysics. 1996;331:241–248. doi: 10.1006/abbi.1996.0304. [DOI] [PubMed] [Google Scholar]
- 23.Li Q, Li C, Mahtani HK, Du J, Patel AR, Lancaster JR., Jr Nitrosothiol formation and protection against Fenton chemistry by nitric oxide-induced dinitrosyliron complex formation from anoxia-initiated cellular chelatable iron increase. The Journal of biological chemistry. 2014;289:19917–19927. doi: 10.1074/jbc.M114.569764. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Hickok JR, Thomas DD. Nitric oxide and cancer therapy: the emperor has NO clothes. Current pharmaceutical design. 2010;16:381–391. doi: 10.2174/138161210790232149. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Heinecke JL, Ridnour LA, Cheng RY, Switzer CH, Lizardo MM, Khanna C, Glynn SA, Hussain SP, Young HA, Ambs S, Wink DA. Tumor microenvironment-based feed-forward regulation of NOS2 in breast cancer progression. Proceedings of the National Academy of Sciences of the United States of America. 2014;111:6323–6328. doi: 10.1073/pnas.1401799111. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Basudhar D, Somasundaram V, de Oliveira GA, Kesarwala A, Heinecke JL, Cheng RY, Glynn SA, Ambs S, Wink DA, Ridnour LA. Nitric Oxide Synthase-2-Derived Nitric Oxide Drives Multiple Pathways of Breast Cancer Progression. Antioxidants & redox signaling. 2016 doi: 10.1089/ars.2016.6813. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Wang YTDD. Nitrogen Oxides and Their Roles in Cancer Etiology. Current Pharmacology Reports. 2017;3:151–161. [Google Scholar]
- 28.Hickok JR, Sahni S, Mikhed Y, Bonini MG, Thomas DD. Nitric oxide suppresses tumor cell migration through N-Myc downstream-regulated gene-1 (NDRG1) expression: role of chelatable iron. The Journal of biological chemistry. 2011;286:41413–41424. doi: 10.1074/jbc.M111.287052. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Hickok JR, Vasudevan D, Antholine WE, Thomas DD. Nitric oxide modifies global histone methylation by inhibiting Jumonji C domain-containing demethylases. The Journal of biological chemistry. 2013;288:16004–16015. doi: 10.1074/jbc.M112.432294. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Vasudevan D, Hickok JR, Bovee RC, Pham V, Mantell LL, Bahroos N, Kanabar P, Cao XJ, Maienschein-Cline M, Garcia B, Thomas DD. Nitric oxide regulates gene expression in cancers by controlling histone posttranslational modifications. Cancer research. 2015 doi: 10.1158/0008-5472.CAN-15-1582. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Socco S. Epigenetics: The third pillar of nitric oxide signalling. 2017;121:52–58. doi: 10.1016/j.phrs.2017.04.011. [DOI] [PubMed] [Google Scholar]
- 32.Kiziltepe T, Hideshima T, Ishitsuka K, Ocio EM, Raje N, Catley L, Li CQ, Trudel LJ, Yasui H, Vallet S, Kutok JL, Chauhan D, Mitsiades CS, Saavedra JE, Wogan GN, Keefer LK, Shami PJ, Anderson KC. JS-K, a GST-activated nitric oxide generator, induces DNA double-strand breaks, activates DNA damage response pathways, and induces apoptosis in vitro and in vivo in human multiple myeloma cells. Blood. 2007;110:709–718. doi: 10.1182/blood-2006-10-052845. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Chakrapani H, Wilde TC, Citro ML, Goodblatt MM, Keefer LK, Saavedra JE. Synthesis, nitric oxide release, and anti-leukemic activity of glutathione-activated nitric oxide prodrugs: Structural analogues of PABA/NO, an anti-cancer lead compound. Bioorg Med Chem. 2008;16:2657–2664. doi: 10.1016/j.bmc.2007.11.035. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Riemer J, Hoepken HH, Czerwinska H, Robinson SR, Dringen R. Colorimetric ferrozine-based assay for the quantitation of iron in cultured cells. Analytical biochemistry. 2004;331:370–375. doi: 10.1016/j.ab.2004.03.049. [DOI] [PubMed] [Google Scholar]
- 35.Cesareo E, Parker LJ, Pedersen JZ, Nuccetelli M, Mazzetti AP, Pastore A, Federici G, Caccuri AM, Ricci G, Adams JJ. Nitrosylation of human glutathione transferase P1-1 with dinitrosyl diglutathionyl iron complex in vitro and in vivo. Journal of Biological Chemistry. 2005;280:42172–42180. doi: 10.1074/jbc.M507916200. [DOI] [PubMed] [Google Scholar]
- 36.Mikus J, Steverding D. A simple colorimetric method to screen drug cytotoxicity against Leishmania using the dye Alamar Blue®. Parasitology international. 2000;48:265–269. doi: 10.1016/s1383-5769(99)00020-3. [DOI] [PubMed] [Google Scholar]
- 37.Crow JP. Dichlorodihydrofluorescein and dihydrorhodamine 123 are sensitive indicators of peroxynitritein vitro: implications for intracellular measurement of reactive nitrogen and oxygen species. Nitric oxide. 1997;1:145–157. doi: 10.1006/niox.1996.0113. [DOI] [PubMed] [Google Scholar]
- 38.Thomas DD, Miranda KM, Espey MG, Citrin D, Jourd'heuil D, Paolocci N, Hewett SJ, Colton CA, Grisham MB, Feelisch M, Wink DA. Guide for the use of nitric oxide (NO) donors as probes of the chemistry of NO and related redox species in biological systems. Methods in enzymology. 2002;359:84–105. doi: 10.1016/s0076-6879(02)59174-6. [DOI] [PubMed] [Google Scholar]
- 39.Thomas DD, Espey MG, Ridnour LA, Hofseth LJ, Mancardi D, Harris CC, Wink DA. Hypoxic inducible factor 1alpha, extracellular signal-regulated kinase, and p53 are regulated by distinct threshold concentrations of nitric oxide. Proceedings of the National Academy of Sciences of the United States of America. 2004;101:8894–8899. doi: 10.1073/pnas.0400453101. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Bauer G. Increasing the endogenous NO level causes catalase inactivation and reactivation of intercellular apoptosis signaling specifically in tumor cells. Redox biology. 2015;6:353–371. doi: 10.1016/j.redox.2015.07.017. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.Turella P, Pedersen JZ, Caccuri AM, De Maria F, Mastroberardino P, Bello ML, Federici G, Ricci G. Glutathione transferase superfamily behaves like storage proteins for dinitrosyl-diglutathionyl-iron complex in heterogeneous systems. Journal of Biological Chemistry. 2003;278:42294–42299. doi: 10.1074/jbc.M305569200. [DOI] [PubMed] [Google Scholar]
- 42.Vanin AF, van Faassen E. DNICs: physico-chemical properties and their observations in cells and tissues. Radicals for life: the various forms of nitric oxide. 2007;19 [Google Scholar]
- 43.Watts RN, Hawkins C, Ponka P, Richardson DR. Nitrogen monoxide (NO)-mediated iron release from cells is linked to NO-induced glutathione efflux via multidrug resistance-associated protein 1. Proceedings of the National Academy of Sciences. 2006;103:7670–7675. doi: 10.1073/pnas.0602515103. [DOI] [PMC free article] [PubMed] [Google Scholar]