

Abstract
Refractory rickets is a genetic disorder that cannot be treated by vitamin D supplementation and adequate dietary calcium and phosphorus. Hereditary hypophosphatemic rickets is one of the major forms of refractory rickets in Indian children and caused due to mutations in the PHEX , FGF23 , DMP1 , ENPP1 , and SLC34A3 genes. This is the first study in India on a large number of patients reporting on mutational screening of the PHEX gene. Direct sequencing in 37 patients with refractory rickets revealed eight mutations in 13 patients of which 1 was nonsense, 2 were deletions, 1 was a deletion–insertion, and 4 were missense mutations. Of these mutations, four (c.566_567 delAG, c.651_654delACAT, c.1337delinsAATAA, and c.2048T > A) were novel mutations. This article discusses the mutations in Indian patients, collates information on the genetic causes of refractory rickets, and emphasizes the significance of genetic testing for precise diagnosis, timely treatment, and management of the condition, especially in developing countries.
Keywords: genetic disorder, hypophosphatemic rickets, PHEX, mutation, genetic testing
Introduction
Rickets is a defective mineralization of bones due to a deficiency or impaired metabolism of vitamin D, phosphorus, or calcium leading to bone deformities and stunted growth in children. Malnutrition leading to dietary insufficiency of calcium and vitamin D is the main cause of rickets in children in India and other developing countries. Treatment with calcium and vitamin D supplementation can cure this condition. Refractory rickets is a form of rickets that does not respond to treatment with vitamin D supplementation and includes entities such as hypophosphatemic rickets, Dent's disease, Fanconi syndrome, renal tubular acidosis (RTA), vitamin D-dependent rickets (VDDR), malabsorption, and chronic renal failure. 1 Hypophosphatemia is the leading cause of refractory rickets in children, which is defined as a lower than normal levels of phosphate in the blood. The normal range for serum phosphate varies with the age of an individual and ranges from 4.8 to 7.4 mg/dL in the first 3 months of life, 4.5 to 5.8 mg/dL at 1 to 2 years, 4.5 to 6.5 mg/dL up to 5 years, 3.6 to 5.8 mg/dL at 6 to 12 years, and finally decreases to adult values of 2.3 to 4.5 mg/dL by late adolescence. 2 Serum phosphate levels lower than those mentioned for the respective age groups result in hypophosphatemia caused by excessive urinary excretion of phosphate. This may be due to mutations in genes involved in maintaining phosphate homeostasis. 3 4
This article describes the regulatory mechanisms of bone mineral metabolism and provides information about the genes and mutations involved in various forms of refractory rickets and their inheritance patterns. It also presents data on mutations identified in the patients with hypophosphatemic rickets diagnosed in our hospital.
Regulation of Phosphate Homeostasis
Inorganic phosphate (Pi) is the most abundant anion in the human body and serves as a backbone for nucleic acids and cell membranes. It also plays crucial roles in cell signaling, cell metabolism, maintaining acid-base balance, and bone mineralization. Phosphate homeostasis is a complex process that involves various organs such as the intestine, kidney, bone, and parathyroid gland, and hormones such as the parathyroid hormone (PTH), vitamin D, and fibroblast growth factor-23 (FGF23). Abnormalities in any of these organs or blood levels of the hormones can result either in hypophosphatemia or hyperphosphatemia. Low blood levels of phosphate are associated with defective bone mineralization resulting in rickets and growth retardation. Among organs, the kidney is the major regulator of Pi homeostasis, because of its ability to reabsorb phosphate. Renal tubular reabsorption of phosphate occurs primarily in the proximal tubules with the help of SLC34A1 and SLC34A3 encoded sodium phosphate cotransporters NaPi-IIa and NaPi-IIc, respectively, which are expressed on the apical brush border membrane. 5 Under normal physiological conditions, 80 to 90% of phosphate is reabsorbed and the rest is excreted in the urine. The proximal tubular membrane expression of the type II NaPi cotransporter protein is regulated by hormones such as the PTH, 1,25(OH) 2 vitamin D 3 , and FGF23.
Role of Vitamin D
Vitamin D has two forms: D 2 (ergocalciferol) and D 3 (cholecalciferol). Vitamin D 2 is produced by ultraviolet B (UVB) irradiation of the ergosterol in plants and fungi. Vitamin D 3 is derived from dietary sources such as oily fish, eggs, and from conversion of 7-dehydrocholestrol on sun exposure under the influence of UVB. Both forms of vitamin D are transported by blood to the liver where they are converted to 25-hydroxy-cholecalciferol [25(OH)vitamin D 3 , calcidiol] by the hepatic microsomal enzyme vitamin D 25-hydroxylase encoded by the gene CYP2R1 . Calcidiol is the chief form of vitamin D in circulation, and its plasma concentration is the best indicator of vitamin D status. It is transported to kidney where it is hydroxylated by 25(OH)vitamin D 3 1α-hydroxylase to produce the active form of Vitamin D [1,25(OH) 2 vitamin D 3 (calcitriol)]. 6 25(OH)vitamin D 3 1α hydroxylase is encoded by CYP27B1 and is predominantly expressed in the distal convoluted tubule (DCT) under conditions of vitamin D sufficiency. 7 Calcitriol is the most potent vitamin D metabolite, but it has a very short half-life, and its plasma level is only 0.1% of the level of 25(OH)vitamin D 3 . The chief functions of calcitriol are to increase the intestinal absorption of calcium and phosphate, decrease urinary calcium losses, and increase bone resorption of calcium to maintain normal concentrations of extracellular calcium. The activity of 1α-hydroxylase is regulated by extracellular concentrations of ionized calcium, inorganic phosphate, PTH, and FGF23. When serum calcium levels are normal, feedback inhibition leads to decreased synthesis of 1,25(OH) 2 vitamin D 3 and increased conversion of 25(OH) vitamin D 3 to inactive metabolites. 8
FGF23: A Phosphate Regulating Hormone
FGF23 is recognized as an important hormone regulating phosphate reabsorption from the kidneys. Its production by bone osteocytes is increased in response to elevated serum phosphate. 9 The binding of FGF23 to the FGF receptor (FGFR) is promoted by its interaction with α-klotho, a single-pass transmembrane protein encoded by the KL gene expressed in renal tubular cells. α-klotho is an obligate coreceptor for FGF23 signaling. It binds to and activates FGFR isoforms (FGFR1c, 3c, 4), which significantly increases the affinity of these FGFRs specifically to FGF23. 10 In the kidney, full-length FGF23 suppresses the synthesis of active 1,25(OH) 2 vitamin D 3 through downregulation of renal 1α-hydroxylase, and upregulation of 24 hydroxylase, which degrades 1,25(OH) 2 vitamin D 3 . It impairs renal tubular reabsorption of phosphate by decreasing the expression of sodium phosphate cotransporters NaPi-IIa and NaPi-IIc in the proximal tubular cells. FGF23 also suppresses the production and secretion of PTH. Production of FGF23 by osteocytes is regulated by the phosphate regulating gene with homologies to endopeptidases on the X-chromosome ( PHEX ), dentin matrix protein 1 ( DMP1 ), and ectonucleotide pyrophosphatase 1 ( ENPP1 ). 11
Hypophosphatemic Rickets
Hereditary hypophosphatemic rickets is a genetic disorder and is characterized by hypophosphatemia, bone deformities in children, and osteomalacia in adults. Most cases are associated with mutations in the PHEX or FGF23 genes. While there is no absolute cure for hypophosphatemic rickets, early initiation of treatment significantly improves outcomes. Treatment is mainly phosphate supplementation at an early age, which can correct or minimize rickets and skeletal deformities and improve height velocity.
Types of Hypophosphatemic Rickets
X-Linked Hypophosphatemic Rickets
X-linked hypophosphatemic rickets (XLHR; OMIM 307800) is caused by loss-of-function mutations in the PHEX gene, which is inherited in an X-linked dominant manner. 12 It is the predominant form of hypophosphatemic rickets with a prevalence of 1 in 20,000 and accounts for approximately 80% of the familial cases of hypophosphatemic rickets. 13 The PHEX gene is located on Xp22.1, is composed of 22 exons, and encodes a transmembrane endopeptidase that belongs to the type II integral membrane zinc-dependent endopeptidase. 14 The PHEX protein is predominantly expressed on the surface of bone and teeth during osteoblast differentiation and loss of its function results in defective mineralization of bone. It plays a major role in renal phosphate metabolism but is not expressed in the kidneys. Under normal conditions, PHEX regulates the expression of hormone-like substances called phosphatonins (PTN), e.g., FGF23, FGF7, matrix extracellular phosphoglycoprotein (MEPE), and secreted frizzled-related protein 4 (sFRP-4). Phosphatonins promote phosphate excretion when blood levels of phosphate are high, thereby, maintaining phosphate homeostasis. In patients with XLHR, inactivating mutations of the PHEX gene result in a failure to regulate the expression of phosphatonins. Increased circulating levels of PTN significantly decreases renal tubular phosphate reabsorption causing renal phosphate wasting and reduced renal 1α -hydroxylase activity. 15 16 17 To date, 418 PHEX mutations have been reported (HGMD Professional 2017.2). XLH is characterized by low serum and high urine phosphate levels, inappropriately low or normal levels of serum 1,25(OH)2vitamin D3, defective calcification of cartilage and bone, lower extremity deformities, short stature, bone pain, late dentition, and frequent dental abscesses. Severity of the disease is highly variable within families. The first symptom is often bowing of the legs as children start to walk. Severity of hypophosphatemia is similar in males who are hemizygous and females who are heterozygous for a mutation in the PHEX gene, although skeletal manifestations are often more severe in males, and postpubertal males may have more severe dental disease. Treatment for all forms of hypophosphatemic rickets consists of oral administration of phosphate and active Vitamin D analogs. Phosphate supplements compensate for renal phosphate losses. Active vitamin D analogues (alfacalcidol/calcitriol) counter 1,25(OH)2vitamin D3 deficiency and prevent secondary hyperparathyroidism. Minimally invasive hemiepiphysiodesis has been used recently to correct lower limb deformities. Neutralizing antibody to FGF23 has been used in animal models, which has significantly improved skeletal abnormalities, but it is not yet commercially available. 18 19 20
Autosomal Dominant Hypophosphatemic Rickets
Autosomal dominant hypophosphatemic rickets (ADHR; OMIM 193100) is a rare condition caused by heterozygous gain-of-function mutations in the FGF23 gene, which is a member of the FGF family, located on chromosome 12p13.32, and is composed of three exons. It is transmitted in an autosomal dominant form and male-to-male transmission helps to differentiate it from XLHR. The FGF23 gene encodes a 251 amino acid protein which is a phosphate regulating hormone produced by osteocytes. It is involved in the maintenance of phosphate homeostasis and skeletogenesis. It induces phosphate excretion when blood levels of phosphate are high by decreasing the expression of sodium phosphate cotransporters NaPi-IIa and NaPi-IIc in renal proximal tubular cells. Mutations in either Arg176 or Arg179 in FGF23 give rise to ADHR. These arginine residues form a furin protease cleavage site RXXR, and mutations in either of the arginines prevent degradation of FGF23. This increases the circulating concentration of this protein consequently leading to phosphaturia. To date, there are four different FGF23 mutations (R176Q, R176W, R179W, and R179Q) resulting in a protein that is resistant to cleavage, thereby increasing its biological activity and leading to phosphaturia. 21 ADHR is inherited with variable penetrance and has a highly variable phenotype. Clinical manifestations depend on the age of onset and the severity of hypophosphatemia. When the disease occurs at an early age (1–3 years), clinical features are similar to XLHR such as severe bowing of the lower extremities, short stature, and muscle weakness. However, when the disease manifests during adulthood, clinical findings include bone pain, fatigue, muscle weakness, and repeated fractures without bowing of lower extremities. Some individuals are asymptomatic throughout life, whereas some patients alternate between being symptomatic and not. Low or normal levels of 1,25(OH) 2 vitamin D 3 are observed in patients with ADHR. Recent studies suggest that iron is one of the major factors contributing to the regulation of FGF23 expression. 22 ADHR patients with low serum iron levels have more severe hypophosphatemia due to further increase in FGF23 levels. 22 Treatment for ADHR comprises of phosphate and calcitriol supplementation to improve height, reduce bone pain, minimize skeletal deformities, and enhance mineralization of bones. 23
Autosomal Recessive Hypophosphatemic Rickets
Autosomal recessive hypophosphatemic rickets (ARHR) is a rare disease that is divided into two subtypes: ARHR type 1 (ARHR1; OMIM 241520) and type 2 (ARHR2; OMIM 613312). ARHR1 is caused by homozygous loss-of-function mutations in the DMP1 gene located on chromosome 4q22.1 and is composed of six exons. The DMP1 protein belongs to the large small integrin-binding ligand, N-linked glycoproteins (SIBLING) family of extracellular matrix proteins and is expressed in bone and teeth. It is required for normal growth and development of bone, cartilage, and dentin. It also plays an important role in phosphate homeostasis. DMP1 mutations increase FGF23 expression and stability, which causes renal phosphate wasting, hypophosphatemia, and defective bone mineralization. 24 25 26 27 28 ARHR2 is caused by homozygous loss-of-function mutations in the ENPP1 gene located on chromosome 6q23.2 and is composed of 25 exons. The ENPP1 gene encodes a protein called ectonucleotide pyrophosphatase/phosphodiesterase, which catalyzes phosphoester cleavage of adenosine triphosphate (ATP) generating pyrophosphate (PPi). PPi is an inhibitor of calcification that is hydroxyapatite crystal deposition, therefore, homozygous ENPP1 mutations also cause generalized arterial calcification of infancy. In patients with ARHR2, high circulatory levels of FGF23 have been found, which promote renal phosphate wasting and diminish renal 1α-hydroxylase activity. However, the mechanism by which ENPP1 mutations result in high serum FGF23 levels is unknown. Clinical manifestation for both ARHR1 and ARHR2 include growth retardation, lower extremity deformities, fractures, dental anomalies, and enthesopathy in some patients. To date, 9 DMP1 and 49 ENPP1 mutations have been reported (HGMD Professional 2017.2). Therapy for both ARHR1 and ARHR2 includes phosphate and calcitriol supplementation. 29 30 31
Hereditary Hypophosphatemic Rickets with Hypercalciuria
Hereditary hypophosphatemic rickets with hypercalciuria (HHRH; OMIM 241530) is an autosomal recessive disorder caused by homozygous or compound heterozygous mutations in the SLC34A3 gene, which is located on chromosome 9q34.3 and is composed of 13 exons. It encodes renal sodium phosphate cotransporter NaPi-IIc, which is expressed in the brush border membrane of proximal tubular cells. To date, 38 SLC34A3 mutations have been documented (HGMD Professional 2017.2). HHRH is characterized by renal phosphate wasting resulting in hypophosphatemia, rickets/osteomalacia, bone pain, short stature, fractures, lower limb deformities, and muscle weakness. Hypercalciuria can occur due to elevated serum 1,25(OH) 2 vitamin D 3 levels that can lead to increased intestinal absorption of calcium and phosphorus. Hypercalciuria may lead to the formation of kidney stones (nephrolithiasis) and/or result in nephrocalcinosis. It is distinguished from XLHR, ADHR, and ARHR by the presence of an elevated level of serum 1,25(OH) 2 vitamin D 3 leading to hypercalciuria and suppression of PTH secretion. Serum FGF23 is normal in HHRH in contrast to XLHR, ADHR, and ARHR. Treatment for HHRH includes phosphate supplementation alone without calcitriol supplementation. Reducing salt in the diet and the addition of thiazides can be beneficial. 32 33 34 35
Hypophosphatemic Nephrolithiasis/Osteoporosis Type 1 and 2
Hypophosphatemic nephrolithiasis/osteoporosis type 1 (NPHLOP1; OMIM 612286) is caused by heterozygous mutations in SLC34A1 gene, which is located on chromosome 5q35.3 and is composed of 14 exons. The SLC34A1 gene encodes type IIa sodium-phosphate cotransporter Na-Pi IIa, which is expressed on the apical membrane of renal proximal tubular cells. Na-Pi IIa plays a major role in phosphate reabsorption in the kidneys thereby acting as a major contributor to phosphate homeostasis. SLC34A1 mutations cause renal phosphate loss, which results in hypophosphatemia resulting in bone demineralization and nephrolithiasis. Two heterozygous missense mutations in SLC34A1 (A48F and V147M) have been reported in patients with hypophosphatemic rickets with urolithiasis/osteoporosis suggesting a dominant effect. 36 Hypophosphatemic nephrolithiasis/osteoporosis type 2 (NPHLOP2; OMIM 612287) is caused by heterozygous mutation in the SLC9A3R1 gene that is located on chromosome 17q25.1 and is composed of 6 exons. The SLC9A3R1 gene encodes a sodium-hydrogen exchanger regulatory factor 1 (NHERF1), which binds to the main renal phosphate transporter Na-Pi IIa that is essential for the membrane sorting of Na-Pi IIa. NHERF1 also binds to the PTH type 1 receptor (PTH1R). NHERF1 mutations cause impairment of renal phosphate reabsorption by increasing PTH-induced cAMP production in the renal proximal tubules. NPHLOP2 is characterized by phosphate wasting, normal PTH, and hypophosphatemia. 37 38
Hypophosphatemic Rickets with Hyperparathyroidism
Hypophosphatemic rickets with hyperparathyroidism (HRH; OMIM 612089) is characterized by hypophosphatemia, skeletal deformities, and parathyroid hyperplasia. HRH is reported to be caused by mutations in the KL gene located on chromosome 13q13.1 with 5 exons. KL encodes α-klotho protein, which is an obligate coreceptor for physiological FGF23 signaling and plays a crucial role in FGF23-mediated phosphate homeostasis. Brownstein et al identified a de novo balanced translocation t (9;13)(q21.13;q13.1) in a 23-year-old female with hypophosphatemic rickets and hyperparathyroidism. It was discovered that the disease occurs due to a chromosomal translocation with a breakpoint adjacent to α-klotho, which encodes β-glucuronidase that is involved in aging and regulation of FGF signaling. The translocation markedly increased circulating α-klotho levels, β-glucuronidase activity, and serum FGF23 levels. The novel phenotype of the patient was caused by a positional effect of the translocation that increased circulatory α-klotho and FGF23 levels. 39
Characteristic features of the different types of hypophosphatemic rickets are summarized in Table 1 .
Table 1. Characteristic features of various forms of hypophosphatemic rickets.
| XLHR | ADHR | ARHR1 ARHR2 |
HHRH | NPHLOP1 NPHLOP2 | HRH | |
|---|---|---|---|---|---|---|
| Gene involved | PHEX | FGF23 | DMP1 and ENPP1, respectively | SLC34A3 | SLC34A1 and SLC9A3R1, respectively | KL |
| Chromosomal location | Xp22.1 | 12p13 | 4q22.1 and 6q23.2 | 9q34.3 | 5q35.3 and 17q25.1 | 13q13.1 |
| Inheritance | X-linked dominant | Autosomal dominant | Autosomal recessive | Autosomal recessive | Autosomal dominant | Autosomal dominant |
| Type of mutation | Loss of function | Gain of function | Loss of function | Loss of function | Loss of function | Chromosomal translocation t (9;13)(q21.13;q13.1) |
| Pathogenesis | PHEX mutations raise FGF23 levels causing renal phosphate wasting | Mutated FGF23 becomes resistant to proteolytic inactivation, increasing FGF23 levels, causing renal phosphate wasting | DMP1 and ENPP1 mutations increase FGF23 levels causing renal phosphate wasting | SLC34A3 mutations decrease renal phosphate reabsorption causing renal phosphate wasting |
Inactivating
SLC34A1
and
SLC9A3R1
mutations decrease renal phosphate reabsorption causing renal phosphate wasting |
Chromosomal translocation breakpoint adjacent to α-Klotho results in elevated circulatory α-Klotho and FGF23 levels causing renal phosphate wasting and hyperparathyroidism |
| Serum phosphorus | Low | Low | Low | Low | Low | Low |
| Urine phosphorus | High | High | High | High | High | High |
| Serum calcium | Normal | Normal | Normal | Normal/High | Normal | Normal/High |
| Urine calcium | Low/Normal | Low/Normal | Low/Normal | High | High | Normal/High |
| Serum FGF23 level | High | High | High | Normal | Normal | High |
| 1,25 (OH) 2 vitamin D 3 level | Low/Normal | Low/Normal | Low/Normal | High | High | Low/Normal |
| Parathyroid hormone | Normal/High | Normal | Normal | Low | Low/Normal | High |
| TmP/GFR a | Low | Low | Low | Low | Low | Low |
| Muscle weakness | Minimal | Present | Minimal | Present | Present | Present |
| Hypercalciuria | No | No | No | Yes | Yes | Yes |
| Nephrolithiasis | No | No | No | Yes | Yes | Yes |
| Nephrocalcinosis | No | No | No | Yes | Yes | Yes |
Abbreviations: ADHR, autosomal dominant hypophosphatemic rickets; ARHR, autosomal recessive hypophosphatemic rickets; FGF, fibroblast growth factor; GFR, glomerular filtration rate; HRH, hypophosphatemic rickets with hyperparathyroidism; HHRH, hereditary hypophosphatemic rickets with hypercalciuria; NPHLOP, hypophosphatemic nephrolithiasis/osteoporosis type; TmP, tubular maximum reabsorption of phosphate; XLHR, X-linked hypophosphatemic rickets.
Forms of Hypophosphatemia Associated with a More Generalized Proximal Tubular Dysfunction
Dent's disease
Dent's disease is a renal tubular disorder characterized by manifestations of rickets and proximal tubule dysfunction, including hypophosphatemia, low molecular weight proteinuria, hypercalciuria, nephrolithiasis, nephrocalcinosis, and progressive renal failure. About one-third children with Dent's disease develop hypophosphatemic rickets. Recurrent vitamin A responsive night blindness has also been reported in patients with Dent's disease. These clinical features are almost exclusively manifested in males from early childhood, whereas female carriers may be asymptomatic or only mildly affected. The disease is caused by mutations in either the CLCN5 (Dent's disease 1; OMIM 300009) or OCRL (Dent's disease 2; OMIM 300555) genes located on chromosome Xp11.23 and Xq26.1, respectively, and are composed of 12 and 23 exons, respectively. Both forms are inherited in an X-linked recessive manner. The CLCN5 gene encodes a kidney-specific electrogenic Cl –/H+ exchanger ClC-5, which belongs to the CLC family of Cl – channels/transporters. The OCRL gene encodes a phosphatidylinositol-4,5-bisphosphate-5-phosphatase, and mutations are also associated with Lowe syndrome or oculocerebrorenal syndrome. 43 44 45 46 To date, 263 CLCN5 and 253 OCRL mutations have been reported (HGMD Professional 2017.2). ClC-5 and OCRL proteins are involved in the renal endocytic pathway for reabsorption of low molecular weight proteins and other molecules in the proximal tubule and mutation in any of these genes leads to proximal tubular dysfunction and causes Dent's disease. Individuals with Dent's disease 2 have additional symptoms like mild intellectual disability and hypotonia; however, they lack cataracts, unlike patients with Lowe syndrome. There is considerable intrafamilial variability in disease severity. A few patients with Dent's disease do not harbor mutations in CLCN5 and OCRL , pointing to the involvement of other genes. The care of patients with Dent's disease is supportive, focusing on the treatment of hypercalciuria with thiazides and the prevention of nephrolithiasis. 47 48 49 50 51
Fanconi syndrome
Fanconi syndrome is the impairment of proximal tubular functions characterized by phosphaturia, glucosuria, aminoaciduria, mild proteinuria, uricosuria, excessive urinary losses of sodium, potassium and bicarbonate, hypophosphatemia, and acidosis. The renal phosphate wasting results in hypophosphatemic rickets. There are various inherited as well as acquired causes of Fanconi syndrome. Cystinosis is the most common cause of Fanconi syndrome in children and are of three types: infantile nephropathic cystinosis (OMIM 219800); adult nonnephropathic cystinosis (OMIM 219750); and late-juvenile or adolescent nephropathic cystinosis (OMIM 219900). Other inherited causes include tyrosinemia type 1 (OMIM 276700), Fanconi–Bickel syndrome (OMIM 227810), Wilson's disease (OMIM 277900), Lowe syndrome (OMIM 309000), galactosemia (OMIM 230400), glycogen storage disease type 1A (OMIM 232200), and hereditary fructose intolerance (OMIM 229600) ( Table 2 ). Some of the acquired causes are the ingestion of expired tetracycline, a side effect of antiretroviral regimen containing tenofovir and didanosine, lead poisoning, and multiple myeloma. Treatment of children with Fanconi syndrome mainly consists of replacement of substances lost in the urine. 52 53 54
Table 2. Inherited causes of Fanconi syndrome.
| Disease | Inherited causes | Gene | Exons | Protein encoded | Chromosome | Inheritance pattern | Salient features |
|---|---|---|---|---|---|---|---|
| Fanconi syndrome | Cystinosis | CTNS | 12 | Cystinosin | 17p13.2 | Autosomal recessive | Infantile nephropathic cystinosis: Most severe form, occur at 3–4 mo of age with polyuria, polydipsia, failure to thrive, vomiting, constipation, growth retardation, and severe rickets |
| Adult nonnephropathic cystinosis: Photophobia is the only symptom due to deposition of cystine crystals in the cornea | |||||||
| Late-juvenile or adolescent nephropathic cystinosis: Manifests at 10–12 y of age, with proteinuria due to glomerular damage, photophobia, chronic headaches | |||||||
| Tyrosinemia type 1 | FAH | 14 | Fumarylacetoacetate hydrolase | 15q25.1 | Autosomal recessive | Failure to thrive, liver and kidney failure, rickets, increased risk of hepatocellular carcinoma | |
| Fanconi-Bickel | SLC2A2/GLUT2 | 11 | Glucose transporter GLUT2 | 3q26.2 | Autosomal recessive | Hepatorenal glycogen accumulation, proximal renal tubular dysfunction, and impaired utilization of glucose and galactose | |
| Wilson disease | ATP7B | 21 | Copper-transporting P-type ATPase | 13q14.3 | Autosomal recessive | Swelling of the legs, yellowish skin, tremors, muscle stiffness, trouble speaking, and personality changes. Vitamin D resistant rickets may occur | |
| Galactosemia | GALT | 11 | Galactose-1-phosphate uridylyltransferase | 9p13.3 | Autosomal recessive | Vomiting, jaundice, hepatosplenomegaly, food intolerance, hypoglycemia, renal tubular dysfunction, muscle hypotonia, sepsis, and cataract | |
| Lowe syndrome | OCRL | 23 | Phosphatidylinositol-4,5-bisphosphate-5-phosphatase | Xq26.1 | X-linked recessive | Renal abnormalities such as metabolic acidosis, polyuria, dehydration, hypophosphatemic rickets, neonatal hypotonia, delayed development of motor skills like sitting, standing, walking, behavioral problems, and seizures | |
| Hereditary fructose intolerance | ALDOB | 9 | Aldolase, fructose-Bisphosphate B | 13q14.3 | Autosomal recessive | Vomiting, hypoglycemia, jaundice, hemorrhage, hepatomegaly, hyperuricemia, and potentially kidney failure | |
| Glycogen storage disorder type 1A | G6PC | 5 | Glucose-6-phosphatase, catalytic | 17q21.31 | Autosomal recessive | Severe hypoglycemia and hepatomegaly due to accumulation of glycogen, growth retardation, hyperlipidemia, hyperuricemia, and hepatic adenoma in adulthood |
Hypocalcemic Rickets
Vitamin D-Dependent Rickets Type 1A
Vitamin D dependent rickets (VDDR) type 1A (VDDR1A; OMIM 264700) also known as pseudo-vitamin D deficiency rickets is caused by homozygous inactivating mutations in the CYP27B1 gene located on chromosome 12q14.1 and is composed of 9 exons. It encodes a cytochrome P450 enzyme, 1-α hydroxylase, which catalyzes the conversion of 25(OH)vitamin D 3 (Calcidiol) to its active form 1,25(OH)vitamin D 3 (calcitriol). It has an autosomal recessive inheritance and is characterized by early onset of rickets, hypotonia, seizures due to severe hypocalcemia, growth failure, motor delays, and mild to moderate hypophosphatemia. It is associated with insufficient conversion of 25(OH)vitamin D 3 to 1,25(OH) 2 vitamin D 3 resulting in normal serum levels of 25(OH) vitamin D 3 but low serum levels of 1,25(OH) 2 vitamin D 3 . Treatment is done with active vitamin D analogs like alfacalcidol or calcitriol which restores 1,25(OH) 2 vitamin D 3 levels and should be continued for life. Calcium supplementation with or without phosphorus is generally required by the patients. 55 56
Vitamin D-Dependent Rickets Type 1B
VDDR type 1B (VDDR1B; OMIM 600081) is an autosomal recessive disorder caused by homozygous loss-of-function mutations in the CYP2R1 gene, which is located on chromosome 11p15.2 and is composed of 5 exons. It encodes a cytochrome P450 enzyme, 25-hydroxylase, which catalyzes the conversion of cholecalciferol to calcidiol. It is characterized by low serum 25(OH)vitamin D 3 levels, low to normal serum calcium and phosphorus, and normal serum 1,25(OH) 2 vitamin D 3 levels. Administration of calcidiol which compensates for the enzymatic defect is the appropriate treatment for this disease to restore normal vitamin D status. 57
Vitamin D-Dependent Rickets Type 2A
VDDR type 2A (VDDR2A; OMIM 277440) is a rare genetic disorder caused by mutations in the VDR gene, which is located on chromosome 12q13.1 and is composed of 11 exons. It encodes vitamin D receptors, and VDR mutations result in the production of defective vitamin D receptors to which active vitamin D cannot bind thereby preventing its function causing end-organ resistance. It is characterized by severe hypocalcemia and rickets, usually within a few months of birth. There is marked increase in circulatory 1,25(OH) 2 vitamin D 3 levels with varying degrees of alopecia and other ectodermal defects like oligodontia, milia, and epidermal cysts in early infancy. It has an autosomal recessive pattern of inheritance. Patients with less severe mutations responds to large amounts of calcium administered orally or intravenously. Cinacalcet is a useful adjunct for therapy. 58 59
Other Forms of Refractory Rickets
Distal Renal Tubular Acidosis
The cardinal feature of distal renal tubular acidosis (dRTA) is the inability to lower urine pH. It is caused by mutations in the genes encoding the following proteins: the anion exchanger 1 (AE1) and the B1 and A4 subunits of H + -ATPase ( Table 3 ). It is characterized by severe metabolic acidosis due to insufficient secretion of H + in the distal tubule thereby resulting in inappropriately alkaline urine. Hypokalemia is a striking feature of dRTA. Hypercalciuria and phosphaturia occur due to the release of calcium and phosphate from bone for buffering excess H + during acidosis. Persistent acidosis and hypercalciuria result in nephrocalcinosis. Rickets is common in untreated cases because bones begin to serve as the buffer source. Bone resorption causes negative calcium balance resulting in calcipenic rickets. 60 61
Table 3. Genes involved in distal renal tubular acidosis.
| Type of distal RTA | Gene/ Exons |
Protein encoded by the gene | Chromosome | Inheritance pattern | Major non-nephropathic symptom |
|---|---|---|---|---|---|
| Distal RTA (OMIM 179800) |
SLC4A1/ 20 | Anion exchanger1 (AE1) protein | 17q21.31 | Autosomal dominant | Rickets |
| Distal RTA with hemolytic anemia (OMIM 611590) |
SLC4A1/ 20 | Anion exchanger1 (AE1) protein | 17q21.31 | Autosomal recessive | Hemolytic anemia, rickets |
| Distal RTA with progressive nerve deafness (OMIM 267300) | ATP6V1B1/ 14 | B1 subunits of vacuolar H + -ATPase | 2p13.3 | Autosomal recessive | Progressive nerve deafness, rickets |
| Distal RTA with late onset sensorineural hearing loss (OMIM 602722) | ATP6V0A4/ 23 | A4 subunits of vacuolar H + -ATPase | 7q34 | Autosomal recessive | Delayed sensorineural deafness, rickets |
Tumor-Induced Osteomalacia
Tumor-induced osteomalacia (TIO), also known as oncogenic osteomalacia or oncogenic hypophosphatemic osteomalacia, is an acquired cause of hypophosphatemia resulting from oversecretion of phosphatonins (PTNs) like FGF23, FGF7, MEPE, and sFRP-4 from tumors that are generally benign and of mesenchymal origin. The increased PTN production, through a feedback mechanism enhances PHEX production. However, the overproduction of PTN exceeds the capability of PHEX to regulate their inactivation. Hence, in spite of enhanced PHEX, an excessive amount of PTNs circulate to the kidney where they decrease the expression of NaPi-IIa and NaPi-IIc, thereby limiting phosphate reabsorption and causing renal phosphate wasting. Clinical features are similar to XLH and ADHR but it is rare in childhood. 62 63
Indian Scenario
Hypophosphatemic rickets, though rare, is one of the major forms of refractory rickets in Indian children. 3 64 Diagnosis of this entity is often missed due to lack of awareness about the condition, which can result in patients receiving multiple doses of calcium and vitamin D leading to toxicity. Therefore, precise diagnosis of this disorder is required to initiate phosphate supplementation at an early age minimizing the skeletal deformities. Diagnosis of this condition is not easy despite the use of current methods, which rely on clinical, biochemical, and radiographic parameters. Genetic testing could offer a molecular diagnosis by screening for PHEX or FGF23 mutations and diagnosing XLHR and ADHR based on a single blood draw at any age. Thus, genetic testing can simplify diagnosis and help to distinguish between various forms of hypophosphatemic rickets, which will allow for informed genetic counseling. At the same time, it can identify carriers in the family who are unaware or mildly affected by the disease. Only a few genetic studies on Indian patients are available. 13 45 65 66 67 68 69 Our study at a tertiary care hospital, is mainly cases of refractory rickets that were referred for diagnosis and treatment. This study aimed at identifying mutations responsible for the pathogenesis of refractory rickets.
Materials and Methods
A total of 59 clinically diagnosed refractory rickets patients from 54 unrelated nonconsanguineous families were recruited from the outpatient department of Pediatrics between 2015 and 2017 as well as their siblings and parents for genetic testing. Clinical features, biochemical profile, and family history were documented from all the patients. Of the 59 patients, 34 were males and 25 were females with mean age at onset of 31 ± 4 months. All of the patients presented with short stature and various skeletal deformities such as genu varum or genu valgum, wrist widening, rachitic rosary, and fractures. They had low serum phosphate (mean 2.1 ± 0.7 mg/dL), low TmP/GFR (mean 2.3 ± 0.2 mg/dL), normal serum calcium (mean 9.1 ± 0.4 mg/dL), elevated serum alkaline phosphatase (mean 1,317 ± 156 IU/L), and moderate to normal serum 25(OH) vitamin D 3 levels (mean 32 ± 6 ng/mL). Fifty age-matched healthy volunteers without any family history of genetic disorders were recruited who acted as controls. About 5 mL of peripheral blood was drawn in ethylenediaminetetraacetic acid (EDTA) vacutainers under aseptic condition after taking written informed consent from all the participants or their legal guardians.
PHEX Screening by Direct Sequencing and In Silico Analysis
Genomic DNA was isolated from blood samples 70 of 37 patients (32 probands and 5 affected family members), unaffected family members and 50 healthy controls, which was utilized for screening of PHEX exons by Sanger sequencing. The PHEX gene sequence was obtained from Ensembl genome browser 90 (ENSG00000102174). Sequencing results were compared with the DNA sequence of PHEX for the detection of genetic variations using in silico multiple sequence alignment tool Clustal Omega ( https://www.ebi.ac.uk/Tools/msa/clustalo/ ). The effect of the mutations on PHEX protein structure and function was assessed using various in silico pathogenicity prediction tools like Mutation Taster ( http://www.mutationtaster.org/ ), Human Splicing Finder version 3.0 (HSF 3.0, http://www.umd.be/HSF3/ ), UMD-Predictor ( http://umd-predictor.eu/ ), Sorting Intolerant From Tolerant (SIFT, http://sift.jcvi.org/ ), PolyPhen-2 ( http://genetics.bwh.harvard.edu/pph2/ ), and I-Mutant version 3.0 ( gpcr2.biocomp.unibo.it/cgi/predictors/I-Mutant3.0/I-Mutant3.0.cgi ). Crystal structures of the wild-type and the mutant PHEX protein were derived using the SWISS-MODEL online software ( https://swissmodel.expasy.org/ ). Comparison of the structure of the normal and the mutant PHEX protein was done using the TMalign online protein alignment tool ( http://zhanglab.ccmb.med.umich.edu/TM-align/ ).
Results
PHEX screening by direct sequencing identified eight mutations in 13 patients (8 probands and 5 affected family members). Of these mutations, three were missense (c. 1970A > G - Y657C, c.1601C > T - P534L, c.2188G > T- A730S), one was nonsense (c.871C > T- R291*), and four were novel (c.566_567delAG - Q189Hfs*22, c.651_654delACAT - H218Sfs*2, c.1387delinsAATAA - F446*, c.2048T > A - L683H) ( Fig. 1 ). The novel mutations were absent in the 50 healthy controls and the control databases (1000 Genomes Project, ExAC and dbSNP). In silico analysis predicted the mutations to be pathogenic and responsible for causing hypophosphatemic rickets ( Table 4 ). The amino acid residues at p.189, p.218, p.446, and p.683 in PHEX were highly conserved during evolution across different species ( Fig. 2 ). The novel deletion (Q189Hfs*22, H218Sfs*2) and deletion–insertion (F446*) mutations identified in exon 5 and 12, respectively, resulted in frameshift and premature stop codon, thereby producing truncated and nonfunctional PHEX protein ( Fig. 3 ). It was also found that the minor allele frequency (MAF) for the PHEX mutation R291* (rs866429868) was zero in various populations (African, East Asian, South Asian, Latino, Ashkenazi Jewish, Finnish European, Non-Finnish European, and others) as documented in the 1000 Genomes Project. Also, the PHEX mutation A730S (c.2188G > T) had a MAF of 0.00026 with 0.001791 in South Asian Population, 0 in East Asian, African, Finnish European, Non-Finnish European, Latino, and others as reported in the ExAC database. Therefore, these two PHEX mutations are rare and may have a deleterious effect on the phenotype of individuals in a population.
Fig. 1.
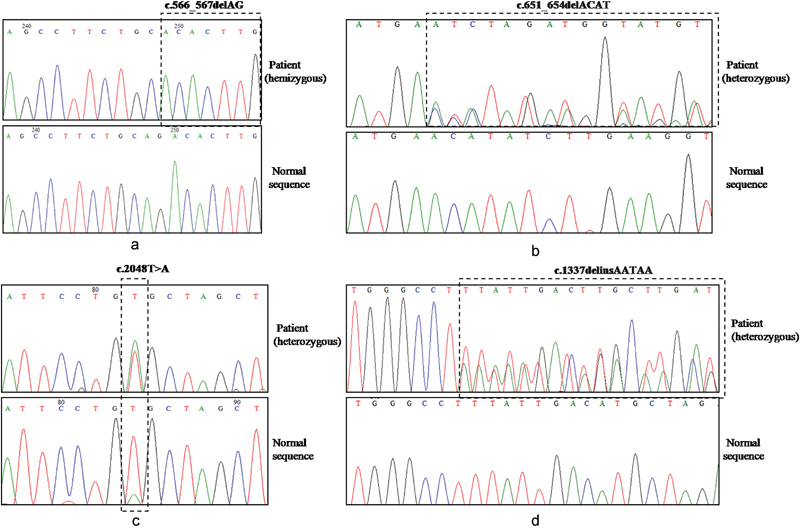
Novel PHEX mutations identified in our study. ( a – d ) Chromatograms containing novel mutations are shown in comparison to the normal sequences. ( a ) c.566_567delAG in exon 5, ( b ) c.651_654delACAT in exon 5, ( c ) c.2048T > A in exon 20, and ( d ) c.1387delinsAATAA in exon 12.
Table 4. PHEX mutations identified by Sanger sequencing in the present study .
| S. no. | PHEX mutations | Amino acid change | Type of mutation | Age (in y) /Sex of the proband |
Affected status of the family members |
Pathogenicity prediction tools | ||||||
|---|---|---|---|---|---|---|---|---|---|---|---|---|
| c.DNA position | Exon | Mutation taster ( p a ) | Human splicing finder 3.0 | UMD-predictor (Score b ) | PolyPhen-2 (Score c ) | SIFT (Score d ) | I-Mutant 2.0 (ΔΔG values e ) | |||||
| 1 | c.566_567 delAG | 5 | Q189Hfs*22 | Novel deletion, hemizygous, de novo | 17/Male | – | Disease causing ( p = 1) | Alteration of an exonic splicing enhancer (ESE) site | – | – | Causes frameshift and nonsense mediated mRNA decay (NMD) | – |
| 2 | c.651_654delACAT | 5 | H218Sfs*2 | Novel deletion, heterozygous, de novo | 14/Female | – | Disease causing ( p = 1) | Alteration of an ESE site | – | – | Causes frameshift and NMD | – |
| 3 | c.871C > T | 8 | R291* 71 72 | Reported nonsense, heterozygous | 10/Female | Affected mother and brother carry this mutation in heterozygous and hemizygous forms, respectively | Disease causing ( p = 1) | Alteration of an ESE site | Pathogenic (100) | – | Causes NMD | – |
| 4 | c.1337delinsAATAA | 12 | F446* | Novel deletion–insertion, heterozygous, de novo | 16/Female | – | Disease causing ( p = 1) | Alteration of an exonic splicing silencer (ESS) site | – | – | Causes frameshift and NMD | – |
| 5 | c.1601C > T | 15 | P534L 73 74 | Reported missense, heterozygous | 6/Female | Affected father, uncle, and sister carry this mutation in hemizygous and heterozygous forms, respectively | Disease causing ( p = 0.99) | Alteration of an ESE site | Pathogenic (93) | Probably damaging (1.00) | Damaging (0) | Does not affect protein stability (ΔΔG = –0.45 Kcal/mol) |
| 6 | c.2048T > A | 20 | L683H | Novel missense, heterozygous | 7/Female | Mother carries this mutation in heterozygous form and has short stature | Disease causing ( p = 0.99) | Does not affect splicing | Pathogenic (96) | Probably damaging (1.00) | Damaging (0) | Decreases protein stability (ΔΔG= –2.48 Kcal/mol) |
| 7 | c.1970A > G | 20 | Y657C 75 | Reported missense, heterozygous, de novo | 10/Female | – | Disease causing ( p = 0.99) | Creation of an ESS site | Pathogenic (90) | Probably damaging (1.00) | Damaging (0) | Decreases protein stability (ΔΔG = –1.15 Kcal/mol) |
| 8 | c.2188G > T | 22 | A730S | Reported missense, heterozygous, de novo | 5/Female | – | Disease causing ( p = 0.99) | Alteration of an ESE site | Pathogenic (90) | Probably damaging (0.98) | Tolerated (0.06) | Decreases protein stability (ΔΔG = –0.73 Kcal/mol) |
Mutation taster score : p a refers to the probability of prediction and a value close to 1 indicates high security of prediction. b UMD-Predictor score: Value ranges from 0 to 100 and indicates ( i ) < 50 polymorphism; ( ii ) 50–64 probable polymorphism; ( iii ) 65–74 probably pathogenic mutations; and ( iv ) > 74 pathogenic mutation. c PolyPhen-2 score: Value ranges from 0.0 (tolerated) to 1.0 (deleterious) and can be interpreted as ( i ) values close to ≤ 0.05 show benign mutation, ( ii ) > 0.05 predicts “possibly damaging” (supposed to affect protein structure or function), and ( iii ) values close to 1 “probably damaging” (high confidence of affecting protein function or structure. d SIFT score: Value ranges from 0.0 (damaging) to 1.0 (tolerated) and can be interpreted as ( i ) ≤ 0.05 damaging and ( ii ) > 0.05 (tolerated). Scores for insertions, deletions, and nonsense mutations are not generated through SIFT. e I-Mutant: ΔΔG value represents effect of the mutation on protein stability. ΔΔG < –0.5: Large decrease of stability; ΔΔG > 0.5: Large increase of stability; –0.5 ≤ ΔΔG ≥ 0.5: Neutral stability. Pathogenicity of insertions and deletions are not predicted by UMD-Predictor, PolyPhen2, and I-Mutant 2.0. *-stop codon.
Fig. 2.
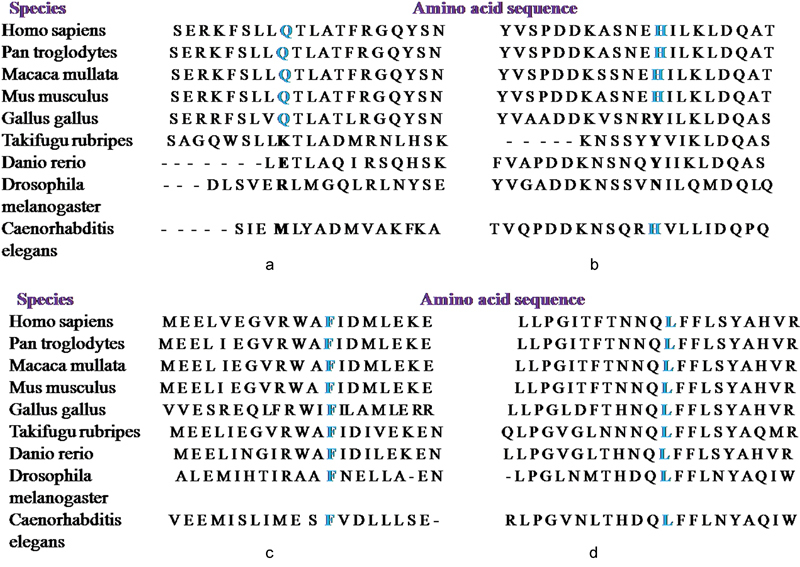
Evolutionary conservation of the amino acid residues ( a ) p.189, ( b ) p.218, ( c ) p.446, and ( d ) p.683 in PHEX. The novel PHEX mutations Q189Hfs*22, H218Sfs*2, F446*, and L683H occur at highly conserved positions in PHEX protein as shown by comparing the corresponding amino acid sequences of nine different species.
Fig. 3.
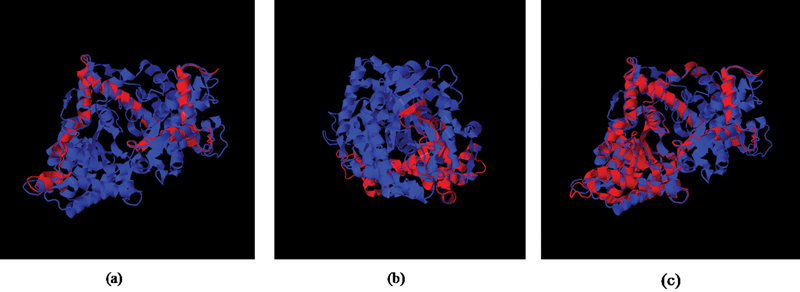
Superimposed images of normal (blue) and mutant PHEX protein (red). The novel PHEX mutations ( a ) c.566_567delAG in exon 5, ( b ) c.651_654delACAT in exon 5, and ( c ) c.1387delinsAATAA in exon 12 resulted in truncated PHEX protein (red).
The mutations identified in the patients 1, 2, 4, 7, and 8 were de novo as their parents carried wild-type alleles. The PHEX mutation L683H (c.2048T > A) was also present in the patient's mother who exhibited short stature. Of the 37 patients screened by Sanger Sequencing, PHEX mutations were detected in 13 patients while the remaining 24 patients will need to be screened for other genes like FGF23 , DMP1 , ENPP1 , etc. We have also done whole-exome sequencing in four patients to know the involvement of PHEX and other genes in the disease process. Preliminary analysis of the exome sequencing results has shown presence of a novel PHEX variation c.665_674delTGGACCAAGC (L222Qfs*7) and a reported 14 PHEX mutation c.1979G>A (W660*) in two patients. The exome sequencing results have been validated by Sanger Sequencing while the analysis in the other two patients is still in progress.
Conclusion
Rickets is one of the most common childhood disorders. Malnutrition is the major cause of rickets in children in India but inherited forms are the chief cause of rickets refractory to supplements of vitamin D and calcium. Diagnosis of this disorder is challenging as there are various forms of refractory rickets due to mutations in multiple genes. We report here four novel PHEX mutations in Indian patients of hypophosphatemic rickets. Genetic testing provides an accurate diagnosis of hypophosphatemic rickets, which allows initiation of specific therapy which can minimize skeletal deformities, and help in management of the condition, especially in developing countries.
Acknowledgments
We acknowledge the support of the Indian Council of Medical research (ICMR) Project Code: 5/7/946/2013-RCH and Department of Biotechnology (DBT)-India for this study.
Footnotes
Conflict of Interest None.
References
- 1.Sharma A. India: Jaypee Brothers; 2016. Vitamin D refractory rickets; pp. 330–345. [Google Scholar]
- 2.Eknoyan G, Levin A, Levin N W; National Kidney Foundation.K/DOQI clinical practice guidelines for bone metabolism and disease in chronic kidney disease Am J Kidney Dis 2003420403S1–S201. [PubMed] [Google Scholar]
- 3.Bajpai A, Bardia A, Mantan M, Hari P, Bagga A. Non-azotemic refractory rickets in Indian children. Indian Pediatr. 2005;42(01):23–30. [PubMed] [Google Scholar]
- 4.Christov M, Jüppner H. Insights from genetic disorders of phosphate homeostasis. Semin Nephrol. 2013;33(02):143–157. doi: 10.1016/j.semnephrol.2012.12.015. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Segawa H, Shiozaki Y, Kaneko I, Miyamoto K.The role of sodium-dependent phosphate transporter in phosphate homeostasis J Nutr Sci Vitaminol (Tokyo) 201561(Suppl):S119–S121. [DOI] [PubMed] [Google Scholar]
- 6.Bikle D D. Vitamin D metabolism, mechanism of action, and clinical applications. Chem Biol. 2014;21(03):319–329. doi: 10.1016/j.chembiol.2013.12.016. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Zehnder D, Bland R, Walker E A et al. Expression of 25-hydroxyvitamin D3-1alpha-hydroxylase in the human kidney. J Am Soc Nephrol. 1999;10(12):2465–2473. doi: 10.1681/ASN.V10122465. [DOI] [PubMed] [Google Scholar]
- 8.Perwad F, Portale A A.Vitamin D metabolism in the kidney: regulation by phosphorus and fibroblast growth factor 23 Mol Cell Endocrinol 2011347(1–2):17–24. [DOI] [PubMed] [Google Scholar]
- 9.Razzaque M S, Lanske B. The emerging role of the fibroblast growth factor-23-klotho axis in renal regulation of phosphate homeostasis. J Endocrinol. 2007;194(01):1–10. doi: 10.1677/JOE-07-0095. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Tan S J, Smith E R, Hewitson T D, Holt S G, Toussaint N D. The importance of klotho in phosphate metabolism and kidney disease. Nephrology (Carlton) 2014;19(08):439–449. doi: 10.1111/nep.12268. [DOI] [PubMed] [Google Scholar]
- 11.Rowe P S. Regulation of bone-renal mineral and energy metabolism: the PHEX, FGF23, DMP1, MEPE ASARM pathway. Crit Rev Eukaryot Gene Expr. 2012;22(01):61–86. doi: 10.1615/critreveukargeneexpr.v22.i1.50. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Francis F, Henning S, Korn B et al. A gene (PEX) with homologies to endopeptidases is mutated in patients with X-linked hypophosphatemic rickets. Nat Genet. 1995;11(02):130–136. doi: 10.1038/ng1095-130. [DOI] [PubMed] [Google Scholar]
- 13.Ma S L, Vega-Warner V, Gillies C et al. Whole exome sequencing reveals novel PHEX splice site mutations in patients with hypophosphatemic rickets. PLoS One. 2015;10(06):e0130729. doi: 10.1371/journal.pone.0130729. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Francis F, Strom T M, Hennig S et al. Genomic organization of the human PEX gene mutated in X-linked dominant hypophosphatemic rickets. Genome Res. 1997;7(06):573–585. doi: 10.1101/gr.7.6.573. [DOI] [PubMed] [Google Scholar]
- 15.Liu S, Guo R, Simpson L G, Xiao Z S, Burnham C E, Quarles L D. Regulation of fibroblastic growth factor 23 expression but not degradation by PHEX. J Biol Chem. 2003;278(39):37419–37426. doi: 10.1074/jbc.M304544200. [DOI] [PubMed] [Google Scholar]
- 16.Cheon C K, Lee H S, Kim S Y, Kwak M J, Kim G H, Yoo H W. A novel de novo mutation within PHEX gene in a young girl with hypophosphatemic rickets and review of literature. Ann Pediatr Endocrinol Metab. 2014;19(01):36–41. doi: 10.6065/apem.2014.19.1.36. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Kinoshita Y, Saito T, Shimizu Y et al. Mutational analysis of patients with FGF23-related hypophosphatemic rickets. Eur J Endocrinol. 2012;167(02):165–172. doi: 10.1530/EJE-12-0071. [DOI] [PubMed] [Google Scholar]
- 18.Aono Y, Yamazaki Y, Yasutake J et al. Therapeutic effects of anti-FGF23 antibodies in hypophosphatemic rickets/osteomalacia. J Bone Miner Res. 2009;24(11):1879–1888. doi: 10.1359/jbmr.090509. [DOI] [PubMed] [Google Scholar]
- 19.Carpenter T O, Imel E A, Ruppe M D et al. Randomized trial of the anti-FGF23 antibody KRN23 in X-linked hypophosphatemia. J Clin Invest. 2014;124(04):1587–1597. doi: 10.1172/JCI72829. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Fukumoto S. Anti-fibroblast growth factor 23 antibody therapy. Curr Opin Nephrol Hypertens. 2014;23(04):346–351. doi: 10.1097/01.mnh.0000447012.98357.da. [DOI] [PubMed] [Google Scholar]
- 21.White K E, Evans W E, Speer M C et al. Autosomal dominant hypophosphataemic rickets is associated with mutations in FGF23. Nat Genet. 2000;26(03):345–348. doi: 10.1038/81664. [DOI] [PubMed] [Google Scholar]
- 22.Clinkenbeard E L, Farrow E G, Summers L J et al. Neonatal iron deficiency causes abnormal phosphate metabolism by elevating FGF23 in normal and ADHR mice. J Bone Miner Res. 2014;29(02):361–369. doi: 10.1002/jbmr.2049. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Nanes M S. Phosphate wasting and fibroblast growth factor-23. Curr Opin Endocrinol Diabetes Obes. 2013;20(06):523–531. doi: 10.1097/01.med.0000436189.80104.80. [DOI] [PubMed] [Google Scholar]
- 24.Koshida R, Yamaguchi H, Yamasaki K, Tsuchimochi W, Yonekawa T, Nakazato M. A novel nonsense mutation in the DMP1 gene in a Japanese family with autosomal recessive hypophosphatemic rickets. J Bone Miner Metab. 2010;28(05):585–590. doi: 10.1007/s00774-010-0169-0. [DOI] [PubMed] [Google Scholar]
- 25.Mäkitie O, Pereira R C, Kaitila I et al. Long-term clinical outcome and carrier phenotype in autosomal recessive hypophosphatemia caused by a novel DMP1 mutation. J Bone Miner Res. 2010;25(10):2165–2174. doi: 10.1002/jbmr.105. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Feng J Q, Ward L M, Liu S et al. Loss of DMP1 causes rickets and osteomalacia and identifies a role for osteocytes in mineral metabolism. Nat Genet. 2006;38(11):1310–1315. doi: 10.1038/ng1905. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Turan S, Aydin C, Bereket A et al. Identification of a novel dentin matrix protein-1 (DMP-1) mutation and dental anomalies in a kindred with autosomal recessive hypophosphatemia. Bone. 2010;46(02):402–409. doi: 10.1016/j.bone.2009.09.016. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Farrow E G, Davis S I, Ward L M, White K E. The role of DMP1 in autosomal recessive hypophosphatemic rickets. J Musculoskelet Neuronal Interact. 2007;7(04):310–312. [PubMed] [Google Scholar]
- 29.Levy-Litan V, Hershkovitz E, Avizov L et al. Autosomal-recessive hypophosphatemic rickets is associated with an inactivation mutation in the ENPP1 gene. Am J Hum Genet. 2010;86(02):273–278. doi: 10.1016/j.ajhg.2010.01.010. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Lorenz-Depiereux B, Schnabel D, Tiosano D, Häusler G, Strom T M. Loss-of-function ENPP1 mutations cause both generalized arterial calcification of infancy and autosomal-recessive hypophosphatemic rickets. Am J Hum Genet. 2010;86(02):267–272. doi: 10.1016/j.ajhg.2010.01.006. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Saito T, Shimizu Y, Hori M et al. A patient with hypophosphatemic rickets and ossification of posterior longitudinal ligament caused by a novel homozygous mutation in ENPP1 gene. Bone. 2011;49(04):913–916. doi: 10.1016/j.bone.2011.06.029. [DOI] [PubMed] [Google Scholar]
- 32.Lorenz-Depiereux B, Benet-Pages A, Eckstein G et al. Hereditary hypophosphatemic rickets with hypercalciuria is caused by mutations in the sodium-phosphate cotransporter gene SLC34A3. Am J Hum Genet. 2006;78(02):193–201. doi: 10.1086/499410. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Yu Y, Sanderson S R, Reyes M et al. Novel NaPi-IIc mutations causing HHRH and idiopathic hypercalciuria in several unrelated families: long-term follow-up in one kindred. Bone. 2012;50(05):1100–1106. doi: 10.1016/j.bone.2012.02.015. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Tencza A L, Ichikawa S, Dang A et al. Hypophosphatemic rickets with hypercalciuria due to mutation in SLC34A3/type IIc sodium-phosphate cotransporter: presentation as hypercalciuria and nephrolithiasis. J Clin Endocrinol Metab. 2009;94(11):4433–4438. doi: 10.1210/jc.2009-1535. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Page K, Bergwitz C, Jaureguiberry G, Harinarayan C V, Insogna K. A patient with hypophosphatemia, a femoral fracture, and recurrent kidney stones: report of a novel mutation in SLC34A3. Endocr Pract. 2008;14(07):869–874. doi: 10.4158/EP.14.7.869. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Prié D, Huart V, Bakouh N et al. Nephrolithiasis and osteoporosis associated with hypophosphatemia caused by mutations in the type 2a sodium-phosphate cotransporter. N Engl J Med. 2002;347(13):983–991. doi: 10.1056/NEJMoa020028. [DOI] [PubMed] [Google Scholar]
- 37.Courbebaisse M, Leroy C, Bakouh N et al. A new human NHERF1 mutation decreases renal phosphate transporter NPT2a expression by a PTH-independent mechanism. PLoS One. 2012;7(04):e34764. doi: 10.1371/journal.pone.0034764. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Karim Z, Gérard B, Bakouh N et al. NHERF1 mutations and responsiveness of renal parathyroid hormone. N Engl J Med. 2008;359(11):1128–1135. doi: 10.1056/NEJMoa0802836. [DOI] [PubMed] [Google Scholar]
- 39.Brownstein C A, Adler F, Nelson-Williams C et al. A translocation causing increased alpha-klotho level results in hypophosphatemic rickets and hyperparathyroidism. Proc Natl Acad Sci U S A. 2008;105(09):3455–3460. doi: 10.1073/pnas.0712361105. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Barth J H, Jones R G, Payne R B.Calculation of renal tubular reabsorption of phosphate: the algorithm performs better than the nomogram Ann Clin Biochem 200037(Pt 1):79–81. [DOI] [PubMed] [Google Scholar]
- 41.Payne R B.Renal tubular reabsorption of phosphate (TmP/GFR): indications and interpretation Ann Clin Biochem 199835(Pt 2):201–206. [DOI] [PubMed] [Google Scholar]
- 42.Kenny A P, Glen A CA.Tests of phosphate reabsorption Lancet 19732(7821):158. [DOI] [PubMed] [Google Scholar]
- 43.Devuyst O, Thakker R V. Dent's disease. Orphanet J Rare Dis. 2010;5:28. doi: 10.1186/1750-1172-5-28. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44.Ludwig M, Utsch B, Monnens L A. Recent advances in understanding the clinical and genetic heterogeneity of Dent's disease. Nephrol Dial Transplant. 2006;21(10):2708–2717. doi: 10.1093/ndt/gfl346. [DOI] [PubMed] [Google Scholar]
- 45.Sethi S K, Ludwig M, Kabra M, Hari P, Bagga A. Vitamin A responsive night blindness in Dent's disease. Pediatr Nephrol. 2009;24(09):1765–1770. doi: 10.1007/s00467-009-1198-6. [DOI] [PubMed] [Google Scholar]
- 46.Carballo-Trujillo I, Garcia-Nieto V, Moya-Angeler F J et al. Novel truncating mutations in the ClC-5 chloride channel gene in patients with Dent's disease. Nephrol Dial Transplant. 2003;18(04):717–723. doi: 10.1093/ndt/gfg016. [DOI] [PubMed] [Google Scholar]
- 47.Lloyd S E, Pearce S H, Fisher S Eet al. A common molecular basis for three inherited kidney stone diseases Nature 1996379(6564):445–449. [DOI] [PubMed] [Google Scholar]
- 48.Morimoto T, Uchida S, Sakamoto H et al. Mutations in CLCN5 chloride channel in Japanese patients with low molecular weight proteinuria. J Am Soc Nephrol. 1998;9(05):811–818. doi: 10.1681/ASN.V95811. [DOI] [PubMed] [Google Scholar]
- 49.Hoopes R R, Jr, Hueber P A, Reid R J, Jr et al. CLCN5 chloride-channel mutations in six new North American families with X-linked nephrolithiasis. Kidney Int. 1998;54(03):698–705. doi: 10.1046/j.1523-1755.1998.00061.x. [DOI] [PubMed] [Google Scholar]
- 50.Vrljicak K, Batinić D, Milosević D, Nizić-Stancin L, Ludwig M. A boy with Dent-2 disease. Coll Antropol. 2011;35(03):925–928. [PubMed] [Google Scholar]
- 51.Claverie-Martín F, Ramos-Trujillo E, García-Nieto V. Dent's disease: clinical features and molecular basis. Pediatr Nephrol. 2011;26(05):693–704. doi: 10.1007/s00467-010-1657-0. [DOI] [PubMed] [Google Scholar]
- 52.Sinha A, Bagga A. India: Jaypee Brothers; 2016. Tubular disorders; pp. 290–329. [Google Scholar]
- 53.Bacchetta J, Greco M, Bertholet-Thomas A et al. Skeletal implications and management of cystinosis: three case reports and literature review. Bonekey Rep. 2016;5:828. doi: 10.1038/bonekey.2016.55. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 54.Selvan C, Thukral A, Chakraborthy P P et al. Refractory rickets due to Fanconi's Syndrome secondary to Wilson's disease. Indian J Endocrinol Metab. 2012;16 02:S399–S401. doi: 10.4103/2230-8210.104107. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 55.Malloy P J, Feldman D. Genetic disorders and defects in vitamin d action. Endocrinol Metab Clin North Am. 2010;39(02):333–346. doi: 10.1016/j.ecl.2010.02.004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 56.Kitanaka S, Takeyama K, Murayama A, Kato S. The molecular basis of vitamin D-dependent rickets type I. Endocr J. 2001;48(04):427–432. doi: 10.1507/endocrj.48.427. [DOI] [PubMed] [Google Scholar]
- 57.Molin A, Wiedemann A, Demers N et al. Vitamin D-dependent rickets type 1B (25-hydroxylase deficiency): a rare condition or misdiagnosed condition? J Bone Miner Res. 2017;32(09):1893–1899. doi: 10.1002/jbmr.3181. [DOI] [PubMed] [Google Scholar]
- 58.Malloy P J, Feldman D.The role of vitamin D receptor mutations in the development of alopecia Mol Cell Endocrinol 2011347(1–2):90–96. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 59.Malloy P J, Zhou Y, Wang J, Hiort O, Feldman D. Hereditary vitamin D-resistant rickets (HVDRR) owing to a heterozygous mutation in the vitamin D receptor. J Bone Miner Res. 2011;26(11):2710–2718. doi: 10.1002/jbmr.484. [DOI] [PubMed] [Google Scholar]
- 60.Lee J H, Park J H, Ha T S, Han H S. Refractory rickets caused by mild distal renal tubular acidosis. Ann Pediatr Endocrinol Metab. 2013;18(03):152–155. doi: 10.6065/apem.2013.18.3.152. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 61.Batlle D, Haque S K. Genetic causes and mechanisms of distal renal tubular acidosis. Nephrol Dial Transplant. 2012;27(10):3691–3704. doi: 10.1093/ndt/gfs442. [DOI] [PubMed] [Google Scholar]
- 62.Kumar R, Folpe A L, Mullan B P. Tumor-induced osteomalacia. Transl Endocrinol Metab. 2015;7(03):1871. [PMC free article] [PubMed] [Google Scholar]
- 63.Berndt T, Craig T A, Bowe A E et al. Secreted frizzled-related protein 4 is a potent tumor-derived phosphaturic agent. J Clin Invest. 2003;112(05):785–794. doi: 10.1172/JCI18563. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 64.Jagtap V S, Sarathi V, Lila A R, Bandgar T, Menon P, Shah N S. Hypophosphatemic rickets. Indian J Endocrinol Metab. 2012;16(02):177–182. doi: 10.4103/2230-8210.93733. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 65.Dayal D, Sharda S, Attri S V, Kumar R.Hypophosphatemic rickets caused by a novel PHEX gene mutation in an Indian girl J Pediatr Endocrinol Metab 201427(7–8):787–789. [DOI] [PubMed] [Google Scholar]
- 66.Chandran M, Chng C L, Zhao Y, Bee Y M, Phua L Y, Clarke B L. Novel PHEX gene mutation associated with X linked hypophosphatemic rickets. Nephron, Physiol. 2010;116(03):17–21. doi: 10.1159/000319318. [DOI] [PubMed] [Google Scholar]
- 67.Dayal D, Dekate P, Sharda S, Das A, Attri S. An Indian girl with Fanconi-Bickel syndrome without SLC2A2 gene mutation. J Pediatr Genet. 2013;2(02):109–112. doi: 10.3233/PGE-13056. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 68.Kanakamani J, Tomar N, Kaushal E, Tandon N, Goswami R. Presence of a deletion mutation (c.716delA) in the ligand binding domain of the vitamin D receptor in an Indian patient with vitamin D-dependent rickets type II. Calcif Tissue Int. 2010;86(01):33–41. doi: 10.1007/s00223-009-9310-2. [DOI] [PubMed] [Google Scholar]
- 69.Bhardwaj S, Thergaonkar R, Sinha A, Hari P, Hi C, Bagga A. Phenotype of Dent Disease in a cohort of Indian children. Indian Pediatr. 2016;53(11):977–982. doi: 10.1007/s13312-016-0971-4. [DOI] [PubMed] [Google Scholar]
- 70.Miller S A, Dykes D D, Polesky H F. A simple salting out procedure for extracting DNA from human nucleated cells. Nucleic Acids Res. 1988;16(03):1215. doi: 10.1093/nar/16.3.1215. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 71.Dixon P H, Christie P T, Wooding C et al. Mutational analysis of PHEX gene in X-linked hypophosphatemia. J Clin Endocrinol Metab. 1998;83(10):3615–3623. doi: 10.1210/jcem.83.10.5180. [DOI] [PubMed] [Google Scholar]
- 72.Holm I A, Huang X, Kunkel L M. Mutational analysis of the PEX gene in patients with X-linked hypophosphatemic rickets. Am J Hum Genet. 1997;60(04):790–797. [PMC free article] [PubMed] [Google Scholar]
- 73.Li S S, Gu J M, Yu W J, He J W, Fu W Z, Zhang Z L. Seven novel and six de novo PHEX gene mutations in patients with hypophosphatemic rickets. Int J Mol Med. 2016;38(06):1703–1714. doi: 10.3892/ijmm.2016.2796. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 74.Holm I A, Nelson A E, Robinson B G et al. Mutational analysis and genotype-phenotype correlation of the PHEX gene in X-linked hypophosphatemic rickets. J Clin Endocrinol Metab. 2001;86(08):3889–3899. doi: 10.1210/jcem.86.8.7761. [DOI] [PubMed] [Google Scholar]
- 75.Gaucher C, Walrant-Debray O, Nguyen T M, Esterle L, Garabédian M, Jehan F. PHEX analysis in 118 pedigrees reveals new genetic clues in hypophosphatemic rickets. Hum Genet. 2009;125(04):401–411. doi: 10.1007/s00439-009-0631-z. [DOI] [PubMed] [Google Scholar]