

Abstract
Juvenile polyposis (JP) syndrome is characterized by multiple hamartomatous polyps of the gastrointestinal tract. Hereditary hemorrhagic telangiectasia (HHT) is a vascular dysplasia characterized by telangiectasia in the skin, mucous membranes, and arteriovenous malformations in other organs. Individuals with JP–HHT syndrome have variable features of both rare disorders, attributed to heterozygous mutations in the SMAD4 gene. Systemic juvenile idiopathic arthritis (JIA) is a severe, chronic disease marked by arthritis and systemic inflammation for which the cause remains unknown. JIA has never been described in association with SMAD4 -related disease. We describe a case of JP–HHT syndrome with a novel SMAD4 variant, c.1052A > T (p.D351V), in which the child also had JIA manifestation.
Keywords: SMAD4, juvenile polyposis syndrome, hereditary hemorrhagic telangiectasia, juvenile idiopathic arthritis, TGF-β
Introduction
Juvenile polyposis (JP) syndrome is a disorder characterized by multiple hamartomatous polyps of the gastrointestinal (GI) tract with an associated predisposition to cancers of the GI tract. Hereditary hemorrhagic telangiectasia (HHT) is a vascular dysplasia characterized by recurrent epistaxis, telangiectasias of the skin and naso-oral mucosa, and the presence of multiple arteriovenous malformations (AVMs) affecting multiple organs. Both disorders are rare, inherited in an autosomal dominant manner, have complete, but age-related penetrance, and have variable expression leading to phenotypic heterogeneity. 1 2 3 Combined symptoms of JPS and HHT were initially reported in the 1980s, 4 5 and several patient cases have established an association between the two disorders. 6 The combined JP–HHT syndrome has since been attributed to heterozygous mutations in one gene, SMAD4 . 7
The SMAD4 gene encodes a protein in the transforming growth factor-β (TGF-β) signaling pathway, serving both as a transcription factor and as a tumor suppressor. As this pathway is involved in a wide range of cellular processes, a perturbation in the pathway lends itself to phenotypic heterogeneity. The full spectrum and extent of clinical findings have yet to be completely realized.
JPS and HHT have distinct, nonoverlapping features, and patients with the combined syndrome have the features of both but not necessarily all at once. Though hypertrophic osteoarthropathy was reported in the initial reported cases of combined JP–HHT syndrome, 8 juvenile idiopathic arthritis (JIA) has never before been described as a presenting feature or associated with individuals with a SMAD4 pathogenic variant. We describe a patient with a manifestation of JIA, in addition to features of JPS and HHT, in whom we identified a novel missense variant in the SMAD4 gene.
Clinical Report
The proband was a 6-year-old girl with a complicated medical history including JP, JIA, and symptomatic anemia. The patient was born preterm at 34 weeks to nonconsanguineous parents. The pregnancy was complicated by drug exposure (cigarette smoke, benzodiazepine, cocaine) and oligohydramnios. At birth, she required continuous positive airway pressure (CPAP) for 6 days in the neonatal intensive care unit. The newborn screening provided by the Maryland Public Health Laboratory was negative (dhmh.maryland.gov/laboratories/html/nbs.html). An additional month was spent at a subacute rehabilitation hospital for feeding and growth, and she had poor weight gain despite being on 30 kcal formula. In her first year of life, she had multiple episodes of wheezing which improved after a course of steroids. From ages 1 to 3 years, her respiratory problems progressively improved, pulmonary medications discontinued, and sweat chloride testing negative. She stopped attending pulmonology visits after 3 years old and there was a lapse in primary care for >1 year (transition of guardianship). During this time, she was seen in urgent care centers for multiple episodes of self-limited fever and sore throat.
At 4 years of age, she was hospitalized for Henoch–Schönlein purpura (abdominal pain, vomiting, fever, palpable purpura, and lower extremity pain) and acute pancreatitis and was found to be severely anemic. She had a positive stool guaiac test during this admission and was noted to have significant digital clubbing. Her family gave a history of poor weight gain despite excellent caloric intake, chronic loose stools five to six times daily, and frequent febrile illnesses. She resumed primary care appointments, and despite aggressive oral iron supplementation, hemoglobin remained below 9 g/dL and weight <10th percentile while height 50th percentile. She was also tachycardic with heart rate (HR) of 110 during well visits. At 5 years of age, she continued to have poor weight gain, frequent loose stools, and persistent anemia despite continued iron replacement therapy. A positive stool guaiac test prompted esophagogastroduodenoscopy (EGD) and colonoscopy at 5 years of age, with follow-up colonoscopy and capsule endoscopy at 6 years of age, which revealed > 50 colonic polyps ( Fig. 1 ) and no gastric or small bowel pathology. Histologic examination of biopsies revealed normal esophagus, stomach, duodenum, and polyps of the cecum and transverse colon were of the juvenile type. She was diagnosed with juvenile polyposis. Frequent stooling with intermittently bloody stools continued to be reported as part of the patient's symptomatology but epistaxis was not reported, thus her persistent anemia was attributed to GI blood loss and malabsorption. She then began experiencing knee and ankle pain and swelling and around her 6th birthday was diagnosed with polyarticular JIA by rheumatology, revised to systemic JIA. Her symptoms improved with prednisone; methotrexate was not effective. She was subsequently started on biweekly infusions of tocilizumab which have been effective in managing her systemic JIA.
Fig. 1.
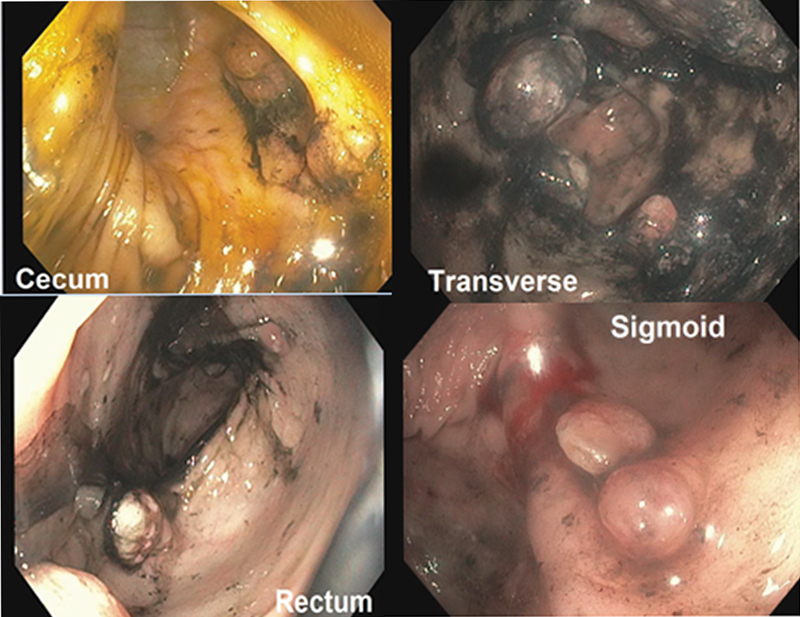
Colonoscopy (at 6 years of age). More than 50 polyps throughout the colon.
Development was normal with milestones met at appropriate ages and without difficulty in learning. The family was of Puerto Rican and Caucasian ancestry. Both parents had substance abuse issues but had no other medical conditions or features similar to those of the patient. As reported by the patient's legal guardian who is a maternal cousin, there was no family history of similar or related medical conditions. The father was deceased. The patient's brother was intellectually disabled, considered secondary to severe hypoxic ischemic encephalopathy with intracranial bleeds at birth. There was a male maternal second cousin, 14 years of age, who had recently been diagnosed with Henoch–Schönlein purpura but was otherwise healthy. The remainder of the family history was negative for polyps, GI-related conditions, rheumatologic, vascular, or cardiac conditions.
At the time of genetic consultation, the patient had presented with worsening joint pain, fevers, tachycardia, and new-onset hypoxia (SpO 2 : 88–100% on room air) without respiratory distress. Notable features included a thin female with weight at the 6th percentile, height at 44th percentile, clubbed digits, swollen knees and ankles, who was otherwise nondysmorphic and with no mucocutaneous lesions noted ( Fig. 2 ). An extensive work-up was performed to determine the etiology of her hypoxia. An infectious work-up including bronchoalveolar lavage, throat, and stool cultures was negative. An inflammatory and rheumatologic work-up, including C-reactive protein (CRP), antinuclear antibody (ANA), rheumatoid factor (RF), cyclic citrullinated peptide (CCP), Ro, La, and dsDNA tests, was negative. Chest X-ray was normal. Echocardiogram revealed mildly dilated main and left pulmonary arteries but was otherwise normal. Computed tomography (CT) scan of the chest with contrast revealed “multiple mildly enhancing lesions throughout the lungs in close proximity to the vessels” ( Fig. 3 ). This study was followed by pulmonary angiography which revealed innumerable small, medium, and large vascular pulmonary nodules some of which demonstrated shunting physiology, ultimately identified as pulmonary AVMs. She subsequently had an embolization procedure in which three pulmonary feeding arteries were embolized with improvement in hypoxemia. Magnetic resonance imaging (MRI) of the brain was negative for intracranial AVMs. CT scan of the chest and follow-up abdominal ultrasound revealed multiple small hepatic telangiectasias versus AVMs. Months following genetic evaluation, a follow-up echocardiogram revealed mild ascending aortic dilation (aortic annulus: 1.60 cm, Z = 2.47; aortic root sinus: 2.06 cm, Z = 1.59; aortic ST junction: 1.65 cm, Z = 1.34; and ascending aorta: 1.99 cm, Z = 2.29). Given the pulmonary vascular findings and history of juvenile polyposis, a genetic testing panel was performed to interrogate BMPR1A and SMAD4 genes.
Fig. 2.
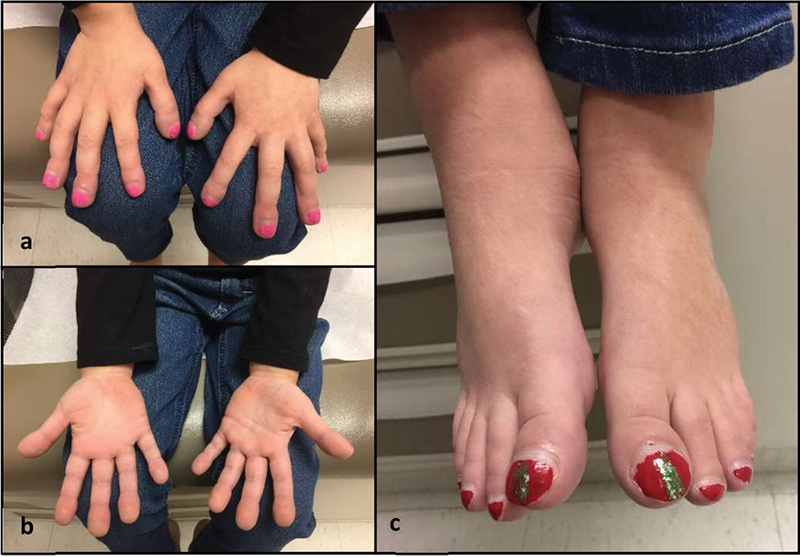
Notable physical features (at 6 years of age). ( a , b ) Digital clubbing. ( c ) Ankle swelling and digital clubbing.
Fig. 3.
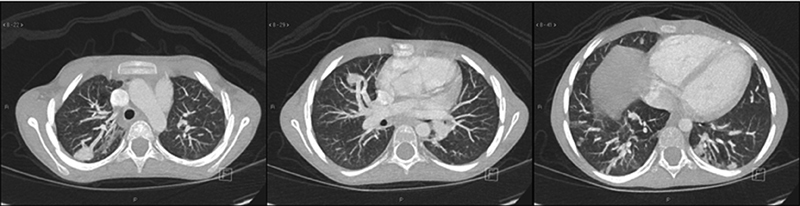
Computed tomography chest. Innumerable small, medium, and large vascular pulmonary nodules, R > L.
Methods
Informed consent was obtained from the patient's legal guardian and family. The patient was followed up at the General Pediatric, Pediatric Genetics, Gastroenterology, Rheumatology, Pulmonology, Cardiology, and Interventional Radiology Clinics at Johns Hopkins Hospital. Echocardiography was performed using an Ie33 (Phillips Medical Systems, Andover, Massachusetts, United States).
DNA Analysis
Extraction of genomic DNA and mutation analysis were performed by Invitae (San Francisco, California, United States), with reads aligned to a reference sequence (GRCh37). The analysis included the complete coding regions and 10 bp of flanking intronic sequence of the following genes: BMPR1A (NM_004329.2) and SMAD4 (NM_005359.5). Multiple databases were queried to determine the novelty of the identified SMAD4 variant. The variant was also analyzed using the missense variant pathogenicity prediction software Sorting Intolerant from Tolerant (SIFT; http://sift.bii.a-star.edu.sg/ ), 9 Polymorphism Phenotyping v2 (PolyPhen-2; http://genetics.bwh.harvard.edu/pph/ ), 10 and Align-Grantham Variation and Grantham Deviation (Align-GVGD; http://agvgd.iarc.fr/index.php ). 11
Results
Molecular genetic testing of the BMPR1A and SMAD4 genes revealed a novel, heterozygous variant of uncertain significance (VUS) in exon 9 of SMAD4 c.1052A > T (p.Asp351Val). Genetic segregation studies were not able to be performed due to the social and familial circumstances of the proband. This variant is not present in population databases (ExAC no frequency) and has not been reported in the literature in individuals with a SMAD4 -related disease. Algorithms developed to predict the effect of missense changes on protein structure and function (SIFT, PolyPhen-2, and Align-GVGD), 9 10 11 all suggest that this variant is likely to be disruptive, but these predictions have not been confirmed by published functional studies.
Discussion
Our patient has overlapping clinical features of JPS and HHT. Patients with germline mutations in SMAD4 can present with symptoms of both syndromes, having combined JP–HHT syndrome. As there are a limited number of previously reported cases of JP–HHT syndrome, the full clinical picture of this syndrome has yet to be completely elucidated. With what is known thus far about SMAD4 -related diseases, the typical phenotype of a pathogenic SMAD4 variant may include AVMs (liver, brain, and lung), GI hamartomatous polyps, mucocutaneous telangiectasias, epistaxis, rectal bleeding, anemia, hypertrophic osteoarthropathy and/or digital clubbing, and a predisposition to GI cancer. Connective tissue findings such as valvular insufficiencies and aortic dilation have also been described in SMAD4 variants. Rheumatologic conditions and systemic manifestations have not been described in the literature in association with SMAD4 variants. Our patient is the first description of a SMAD4 variant seen with systemic JIA as a presenting feature, potentially adding to the clinical picture of SMAD4 -related diseases.
Systemic JIA is characterized by the presence of arthritis with systemic symptoms, including high-spiking fevers, rash, hepatosplenomegaly, lymphadenopathy, serositis, and significantly elevated inflammatory markers. Genome-wide association studies have implicated the class II major histocompatibility complex (MHC) molecule, human leukocyte antigen—antigen D related (HLA DR), in the pathogenesis of systemic JIA, and genetic polymorphisms in several genes ( TNFA , MIF , and IL10 ) have been identified in association with systemic JIA pathogenesis. Genes associated with autoinflammatory conditions have been associated with systemic JIA ( TNFRSF1A , MVK , and MEFV genes), but the exact cause of systemic JIA is unknown and is thought to be multifactorial, including both genetic susceptibilities and environmental factors. 12 In addition to the novel SMAD4 variant found, our patient also had multiple environmental factors, including in utero exposure to cigarette smoke, benzodiazepine, and cocaine, which may have contributed to her complex medical condition.
The identified SMAD4 variant, c.1052A > T (p.D351V), is a novel missense variant and is at a conserved residue in the protein. Missense variant pathogenicity prediction software predicts the p.D351V amino acid change to be pathogenic. Heterozygous loss-of-function SMAD4 mutations are associated with both JPS and HHT. The variant identified in the patient is highly suspicious and may cause loss of function. It occurs at a highly conserved position and results in significant physicochemical consequences, with replacement of an electrically charged acid by a hydrophobic aromatic, predicted to disrupt the function of the protein.
Evidence from clinical and mouse models suggests a correlation between SMAD4 deficiency and thoracic aortic disease. 13 14 15 16 A small number of patients with SMAD4 mutations and the combined JP–HHT syndrome have been previously reported with various findings suggestive of connective tissue disorders, including aortic root dilation, thoracic aortic aneurysm, and aortic dissection. 17 Our patient has mild ascending aortic dilation, contributing to the phenotypic correlation of thoracic aortic disease to SMAD4 deficiency. Mutations in many of the genes in the TGF-β pathway are well-described causes of several connective tissue disorders. Given Smad4 is part of the TGF-β pathway, it is plausible that additional pathologic phenotypes, such as the arthritis found in our patient, may result from SMAD4 mutation. There is currently limited evidence to support this.
Xiong et al found that the TGF-β1/Smad4/7 signaling pathway may play a role in the pathological mechanism in rheumatoid arthritis, finding decreased expression of SMAD4 in cells that had suppressed synoviocyte invasiveness. 18 Mouse studies have demonstrated that blocking SMAD4 gene expression suppresses joint inflammation and the fibrosis process of synovial tissue. 19 These studies seem to suggest that altered SMAD4 expression, in part, contributes to inflammatory processes such as arthropathy. As the treatment of arthritis often involves immune suppression, patients with JP–HHT syndrome may be particularly vulnerable to increasing comorbidity given the predisposition to cancer. To what degree SMAD4 is involved in joint pain and inflammation in humans is an area that warrants further research.
Acknowledgments
We would like to thank the patient and her family for participating in this study and Invitae Corporation for performing the molecular genetic sequencing analysis on the proband.
Footnotes
Conflict of Interest None.
References
- 1.Larsen Haidle J, Howe J R. Seattle, WA: University of Washington, Seattle; 2003. Juvenile polyposis syndrome. [PubMed] [Google Scholar]
- 2.O'Malley M, LaGuardia L, Kalady M F et al. The prevalence of hereditary hemorrhagic telangiectasia in juvenile polyposis syndrome. Dis Colon Rectum. 2012;55(08):886–892. doi: 10.1097/DCR.0b013e31825aad32. [DOI] [PubMed] [Google Scholar]
- 3.Schwenter F, Faughnan M E, Gradinger A B et al. Juvenile polyposis, hereditary hemorrhagic telangiectasia, and early onset colorectal cancer in patients with SMAD4 mutation. J Gastroenterol. 2012;47(07):795–804. doi: 10.1007/s00535-012-0545-8. [DOI] [PubMed] [Google Scholar]
- 4.Cox K L, Frates R C, Jr, Wong A, Gandhi G. Hereditary generalized juvenile polyposis associated with pulmonary arteriovenous malformation. Gastroenterology. 1980;78(06):1566–1570. [PubMed] [Google Scholar]
- 5.Conte W J, Rotter J I, Schwartz A G, Congleton J E. Hereditary generalized juvenile polyposis, arteriovenous malformations and colonic carcinoma. Clin Res. 1982;30:93A. [Google Scholar]
- 6.Jelsig A M, Tørring P M, Kjeldsen A D et al. JP-HHT phenotype in Danish patients with SMAD4 mutations. Clin Genet. 2016;90(01):55–62. doi: 10.1111/cge.12693. [DOI] [PubMed] [Google Scholar]
- 7.Gallione C J, Repetto G M, Legius Eet al. A combined syndrome of juvenile polyposis and hereditary haemorrhagic telangiectasia associated with mutations in MADH4 (SMAD4) Lancet 2004363(9412):852–859. [DOI] [PubMed] [Google Scholar]
- 8.Baert A L, Casteels-Van Daele M, Broeckx J, Wijndaele L, Wilms G, Eggermont E. Generalized juvenile polyposis with pulmonary arteriovenous malformations and hypertrophic osteoarthropathy. Am J Roentgenol. 1983;141(04):661–662. doi: 10.2214/ajr.141.4.661. [DOI] [PubMed] [Google Scholar]
- 9.Ng P C, Henikoff S. Predicting deleterious amino acid substitutions. Genome Res. 2001;11(05):863–874. doi: 10.1101/gr.176601. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Adzhubei I A, Schmidt S, Peshkin L et al. A method and server for predicting damaging missense mutations. Nat Methods. 2010;7(04):248–249. doi: 10.1038/nmeth0410-248. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Tavtigian S V, Deffenbaugh A M, Yin L et al. Comprehensive statistical study of 452 BRCA1 missense substitutions with classification of eight recurrent substitutions as neutral. J Med Genet. 2006;43(04):295–305. doi: 10.1136/jmg.2005.033878. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Hersh A O, Prahalad S. Genetics of juvenile idiopathic arthritis. Rheum Dis Clin North Am. 2017;43(03):435–448. doi: 10.1016/j.rdc.2017.04.007. [DOI] [PubMed] [Google Scholar]
- 13.Teekakirikul P, Milewicz D M, Miller D T et al. Thoracic aortic disease in two patients with juvenile polyposis syndrome and SMAD4 mutations. Am J Med Genet A. 2013;161A(01):185–191. doi: 10.1002/ajmg.a.35659. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Andrabi S, Bekheirnia M R, Robbins-Furman P, Lewis R A, Prior T W, Potocki L. SMAD4 mutation segregating in a family with juvenile polyposis, aortopathy, and mitral valve dysfunction. Am J Med Genet A. 2011;155A(05):1165–1169. doi: 10.1002/ajmg.a.33968. [DOI] [PubMed] [Google Scholar]
- 15.Heald B, Rigelsky C, Moran R et al. Prevalence of thoracic aortopathy in patients with juvenile polyposis syndrome-hereditary hemorrhagic telangiectasia due to SMAD4. Am J Med Genet A. 2015;167A(08):1758–1762. doi: 10.1002/ajmg.a.37093. [DOI] [PubMed] [Google Scholar]
- 16.Holm T M, Habashi J P, Doyle J Jet al. Noncanonical TGFβ signaling contributes to aortic aneurysm progression in Marfan syndrome mice Science 2011332(6027):358–361. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Wain K E, Ellingson M S, McDonald J et al. Appreciating the broad clinical features of SMAD4 mutation carriers: a multicenter chart review. Genet Med. 2014;16(08):588–593. doi: 10.1038/gim.2014.5. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Xiong G, Huang Z, Jiang H, Pan Z, Xie J, Wang S. Inhibition of microRNA-21 decreases the invasiveness of fibroblast-like synoviocytes in rheumatoid arthritis via TGFβ/Smads signaling pathway. Iran J Basic Med Sci. 2016;19(07):787–793. [PMC free article] [PubMed] [Google Scholar]
- 19.Xue M, Gong S, Dai J, Chen G, Hu J. The treatment of fibrosis of joint synovium and frozen shoulder by SMAD4 gene silencing in rats . PLoS One. 2016;11(06):e0158093. doi: 10.1371/journal.pone.0158093. [DOI] [PMC free article] [PubMed] [Google Scholar]