

Abstract
Cell membrane engineering, including live cell membrane bioconjugation and cell membrane-derived nanomaterials is a highly promising strategy to modulate immune responses for treating diseases. Many cell membrane engineering methods have potential for translation for human clinical use in the near future. In this review, we summarize the cell membrane conjugation strategies that have been investigated for cancer immunotherapy, prevention of immune rejection to donor cells/tissues, and induction of antigen-specific tolerance in autoimmune diseases. Additionally, cell membrane-derived or membrane-coated nanomaterials are an emerging class of nanomaterials that is attracting significant attention in the field of nanomedicine. Some of these nanomaterials have been employed to elicit immune responses against cancer, toxins, and bacteria, although their application in establishing immune tolerance has not been explored. In addition to discussing potential problems, we provide our perspectives for promising future directions.
TOC image
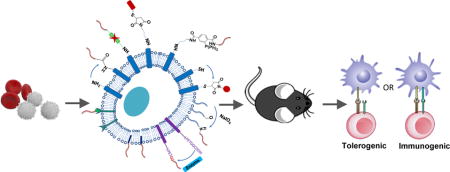
Introduction
Immunotherapy has entered the mainstream in research and clinics for treating various diseases. It functions by harnessing one’s own immune system to target and remedy the disease of interest. For example, cancer and autoimmune disorders are associated with either the suppression or overactivation, respectively, of immune responses in the body.1 To address those issues, immunotherapies can deliver therapeutics to certain immune cells for sensitizing or tolerizing them in response to specific antigens.
Cancer occurs when cells undergo uncontrolled proliferation and the immune system cannot target the aberrant cancer cells. Recently cancer immunotherapies have significantly improved the ability to treat some types of cancer. These include immune checkpoint inhibitors that block the ability of cancer cells to downregulate the T cell responses against cancer, and chimeric antigen receptor (CAR) T cell therapies that modify T cells to target specific cancer-associated antigens.2, 3 However, these treatments are still limited to specific subsets of cancer, and, in the case of CAR-T cells, require complicated and expensive manipulations of patient cells ex vivo. Therefore, strategies that complement to checkpoint inhibitors or CAR-T cells, and new anticancer immunotherapies are being actively explored.
On the other hand, autoimmune diseases are conditions in which the body’s own immune system erroneously becomes activated against self-antigens, resulting in an immune response against its own cells and tissues. Current treatment options for these conditions are limited and generally use broadly immunosuppressive drugs, which are often associated with significant side effects.4 As such, current research focuses on antigen-specific treatments, which targets only the aberrant immune cells while leaving the rest of the immune system intact to fight off normal infections.5 Recent advances in understanding how the body induces tolerance to certain antigens have highlighted the potential of cell membrane-based immunotherapies.6 Such strategies take advantage of the body’s own systems for inducing tolerance by presenting antigens in specific, non-inflammatory ways.
Since the immune system is heavily involved in virtually all types of pathologies, the ability to manipulate different components to direct immune responses is of great interest. Both synthetic and biologically-derived particles have been developed to carry drug payloads and target them to tissues of interest.7 Micro and nanomaterials-based therapeutic strategies that function at the cell and tissue level have emerged as a promising strategy for immune modulation.8, 9 On the other hand, the importance of cell membrane surfaces is increasingly recognized, as the signals they provide to other cells play major roles in directing the outcome of immune responses.10 Membrane-engineered cells and cell membrane-derived materials have been utilized to increase compatibility and interaction with targeted cells/tissues, and more importantly, to enhance therapeutic efficacy.11–17 Thus, the control of cell interactions and functions by manipulating cell surfaces via membrane conjugation methods and/or utilizing cell membrane-derived materials is an attractive area in immunotherapy (Figure 1).
Figure 1.
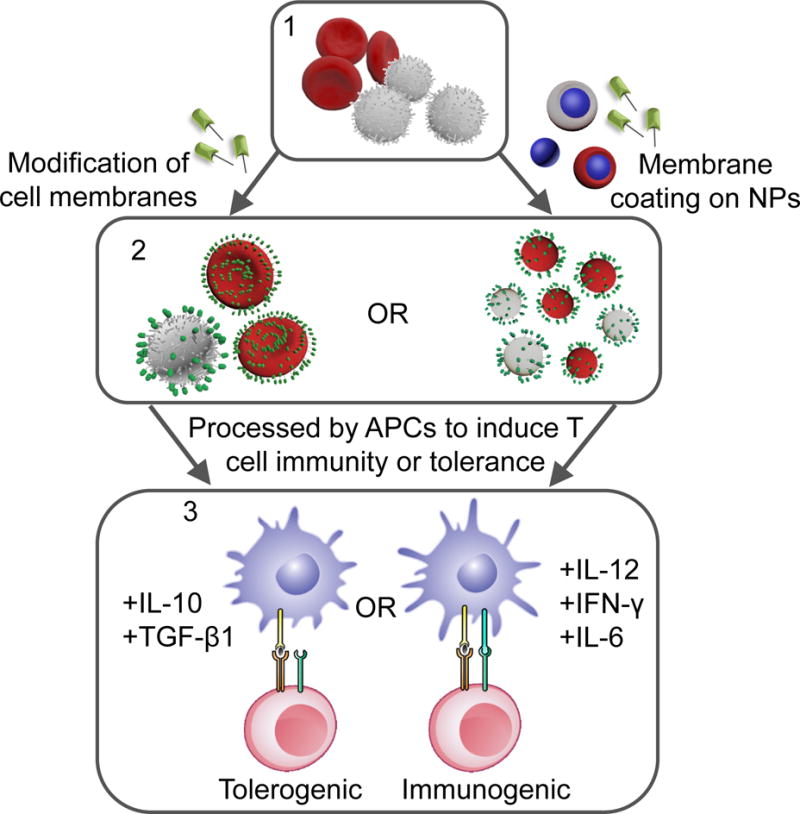
Immunotherapy strategies based on cell membrane conjugation and membrane-derived nanomaterials. Direct modification of cell membranes or membrane coating on nanoparticle surfaces generate immune-modulating therapeutics. When they are delivered to antigen-presenting cells, tolerance or immunity to effector T cells can be induced depending on the formulation.
In the first part of this review, we will address major methods in cell membrane modification with potential applications for immunotherapy. The focus will be on the chemical conjugation schemes used for engineering cell membranes that both preserve their biological properties and add functionalities. Then, a new class of nanomaterials, cell membrane-derived nanomaterials for immune system modulation will be discussed.
Cell membrane conjugation for immunotherapy
Cell membranes have been modified in various ways for immune-related therapies. These strategies include chemical conjugation, hydrophobic interactions, and cell surface-specific binding interactions (Figure 2). While some of these methods have been applied to stimulating immune responses for treating diseases like cancer, many others have been used for inducing tolerance to donor cells or for treating autoimmune diseases. This section will discuss cell membrane conjugation strategies in immune-related therapies (Figure 2).
Figure 2.
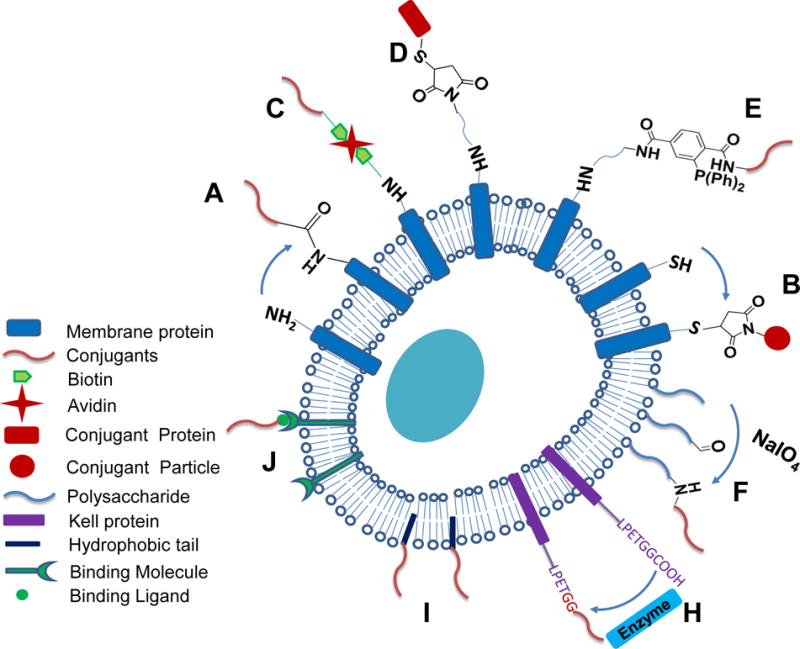
Schematic illustration of cell membrane bioconjugation strategies used for immunotherapy. Primary amines on cell membrane are used to form different linkages with conjugants, including amide bonds (A), biotin-avidin linkage (C), thiol-maleimide linkage (D), and Staudinger ligation linkage (E). Free thiols can form thiol-maleimide linkage with conjugants (B), and polysaccharides can be modified to present aldehyde groups to form secondary amine bonds (F). Biochemical methods (H), hydrophobic interactions (I), and cell surface specific binding (J) can also be used for conjugations.
Chemical conjugation
A myriad of molecules on cell membranes provide a variety of modification locations. Functional groups including primary amine, carboxylic acid, and free thiols from membrane-associated proteins and polysaccharides can be targets of various chemical reactions, offering covalent linkages to desired conjugants. Among these functional groups, primary amines are the most popular one. Conjugation can be completed in a one-step reaction or multiple reactions with the introduction of new functional groups.
One-step chemical conjugation
A simple strategy to reduce immune responses to donor cells is to prevent the presentation of stimulatory molecules to host immune cells by physically blocking access. Scott and colleagues conjugated cyanuric chloride-coupled methoxy(polyethylene glycol) (mPEG) on the membranes of red blood cells (RBCs) via an easy nucleophilic substitution of chlorides with amines on cell membranes.18 Compared to unmodified RBCs, sheep mPEG-modified RBCs were phagocytosed by human peripheral blood monocytes at much lower rates and had a higher in vivo survival rate when injected intraperitoneally into mice. Later, the same group demonstrated that PEGylation of donor peripheral blood mononuclear cells could effectively inhibit the proliferation of T cells from HLA class II disparate recipients in a mixed lymphocyte reaction.19 It was claimed that this could have applications in preventing graft-verus-host disease elicited by organ transplantation. These studies demonstrated the concept of using chemical modification to mask cells from the immune system, and facilitated the development of the concept of modifying transplanted pancreatic islets to prevent immune rejection for treating type 1 diabetes mellitus (T1DM). Since T1DM is an autoimmune disease that destroys pancreatic islets of Langerhans, which produce insulin for metabolic regulation, islet transplantation is an attractive strategy for permanently treating the disease.20 However, as with any organ transplant, the donor tissue is subject to immune rejection and current methods to protect it lead to significant suppression of the entire immune system. To reduce immune rejection of implanted donor islets by recipients, several groups have used N-hydroxysuccinimide (NHS)-functionalized polyethylene glycol (NHS-PEG) to modify islet cells via a simple one-step reaction, which links primary amines on islet cells and NHS-activated carboxyl groups to form an amide bond (Figure 2A).21–25 The reaction is faster and more biocompatible than the cyanuric chloride-based nucleophilic substitution. These studies showed significant protective effects by maintaining the ability of islets to regulate insulin production in a murine allograft model. This strategy, when combined with the short-term immunosuppressant drug anti-LFA-1 antibody, showed even greater effects, eliminating hyperglycemia in 78% of diabetic animals.25 The same membrane conjugation method has also been used to stimulate immune responses. CpG-oligonucleotides, a toll-like receptor (TLR) agonist, were modified with NHS and conjugated on mouse lung cancer cells for stimulating bone marrow-derived dendritic cells (DCs).26
Instead of utilizing pre-activated carboxyl groups, carboxylic acid-activating molecule, 1-ethyl-3-(3-dimethylaminopropyl) carbodiimide hydrochloride (EDC, which has also been named ECDI) has been used for attaching antigens on cell surfaces by simply adding to a mixture of cells and antigens. Although this is not a clean or optimal conjugation method since EDC can activate carboxyl groups of both cells and antigens, and is toxic to cells, it was later found EDC-treatment has the unique effect on antigen-presenting cells of causing them to induce tolerogenic responses when interacting with T cells.27–30 Initially, this was shown that by conjugating an antigen of interest via EDC to splenocyte membranes, the splenocytes induced T cell unresponsiveness to the antigen.27 Subsequently, Elliot et al. found that EDC treatment alone was sufficient to generate MHC-specific tolerogenic splenocytes.28 This is useful for inducing donor-specific tolerance for organ transplantation in which MHC-mismatched surface antigens are introduced to the graft recipient. Similar to autoimmune diseases, the field of organ transplantation is also focused on suppressing immune responses towards specific antigens from an allograft donor.31 When used in skin, cardiac, and islet allograft transplant models, i.v. infused EDC-conjugated donor cells significantly extended graft survival time.28–30 In optimizing the type of cells and timing of injection before graft transplantation, Miller and Luo could induce indefinite tolerance to allogeneic islet grafts in a murine model of diabetes.30 The mechanism behind this is complex and has not been fully elucidated, but one major factor is related to the fact that EDC-treated cells are much more prone to undergo apoptosis, which has been shown to exert pro-tolerogenic effects.29, 32, 33 One mechanism is that apoptotic antigen-coupled splenocytes/leukocytes accumulate in the splenic marginal zone. Spleen macrophages uptake the apoptotic cells, and then produce IL-10 and express programmed death-ligand 1 (PD-L1), both of which suppress overactive immune responses. The process also generates T regulatory cells that downregulate effector T cell responses and are essential for maintaining long-term tolerance.34 In a Phase 1 clinical trial, myelin peptide-coupled autologous apoptotic peripheral blood mononuclear cells were shown to regulate myelin specific T cell responses in multiple sclerosis patients.35
High levels of free thiols exist on T-cells, B-cells, and hematopoietic stem cells (HSCs). These thiol groups can be used to form covalent bonds with conjugants bearing sulfhydryl-reactive maleimide groups (Figure 2B). Irvine and Stephan have attached various maleimide-surfaced particles, including liposomes and lipid-enclosed polymeric nanoparticles (NPs), to antigen-specific CD8+ T cells and HSCs through reacting maleimide with pre-existing thiols on cell membranes (Figure 3A).36–39 The particles were able to stay on T cell surfaces and were nontoxic to T cells. One promising cancer treatment strategy, called adoptive T cell therapy, isolates T cells from the cancer patient, stimulates and expands them ex vivo and reinfuses them back into the patient to target tumors.40 One major challenge of this strategy is the loss of function and viability of T cells in the tumor environment. Administration of high dose IL-15 and IL-21 cytokines has been shown to stimulate T cell proliferation and effector function,41 but is limited due to dose-dependent toxicity. To address this, Stephan et al. encapsulated IL-15 and IL-21 inside liposomes and attached the liposomes onto tumor-specific T cells. The membrane conjugation method provided localized stimulation to the cells and avoided the side effects associated with systemic administration of the cytokines.37 The NP-modified CD8+ T cells with T cell receptors (TCRs) specific to melanoma antigen gp100 (Pmel-1 T cells) were able to effectively proliferate after being adoptively transferred into mice bearing melanoma tumors (Figure 3B) and extend mice survival (Figure 3C). Recently, technologies of genetically engineered CAR-T cells have achieved significant successes,3, 42–45 including being approved by the US Food and Drug Administration for treating blood cancers. However, the applications in treating solid tumors haven not been successful partly due to the loss of function of CAR-T cells in the immunosuppressive tumor environment.46, 47 Membrane-associated NPs carrying both proliferation stimulating cytokines and antibodies that overcome the immunosuppressive signaling may be promising to assist the antitumor response of CAR-T cells.
Figure 3.
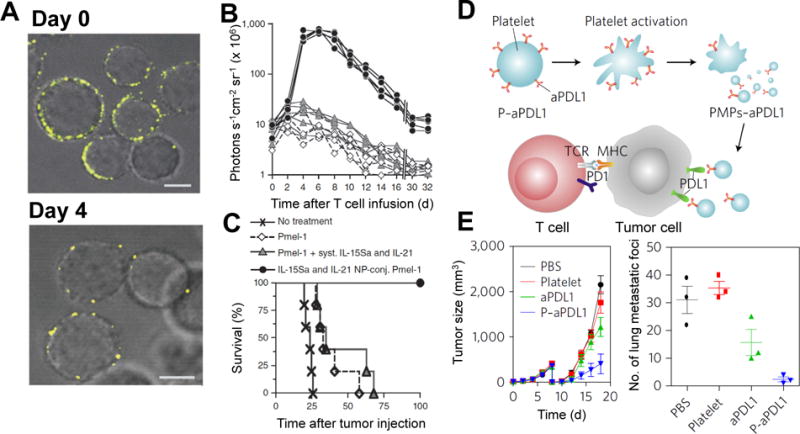
Cell membrane conjugation on T cells (A-C) and platelets (D-E) for enhanced cancer immunotherapy. Liposomes encapsulating cytokines for T cell proliferation can be attached on T cells to overcome their inhibited proliferation in tumors. (A) Confocal images of T cells conjugated with fluorescently labeled liposomes on day 0 and day 4 after stimulation. Scale bars, 2 μm (B) Bioluminescence intensity of T cells variation with time. The T cells expressed firefly luciferase, and the bioluminescence intensity was an indicator of T cell numbers in the mice. Each curve represents one mouse. (C) Survival curves of B16 melanoma-bearing mice after T cell therapy. Immune check inhibitor can be delivered by platelets to residual tumors after surgical removal of primary tumors. (D) Illustration of aPDL1 conjugated on platelets for blocking PDL1 on tumor cells to enhance T cell antitumor function. (E) Tumor growth curves of mice with an incomplete surgical resection of mouse melanoma tumors (left). Numbers of lung metastatic foci from mice treated with PDL1 antibody-modified platelets (right). (Reproduced with permission from Ref 37, Copyright© 2010, Nature Publishing Group; and Ref 54, Copyright© 2017, Nature Publishing Group)
Multi-step chemical conjugation
Many other modifications have been implemented involving multi-step modifications. For instance, biotin can bind strongly with avidin/NeutrAvidin/streptavidin, which have 4 binding sites for biotin (Figure 2C). The method is commonly used in cell membrane modification schemes. In one study, NHS-biotin was used to biotinylate the antigen protein, Tat and human erythrocytes. Tat was then anchored on the erythrocytes via the biotin-avidin interaction to induce antigen-specific T cell responses.48 Since streptavidin can bind to 4 biotins, this linkage method provides efficient and multiple attachment if necessary. Biotinylated SLeX was efficiently anchored on mesenchymal stem cell membranes (MSC) to mediate cell interaction with selectins, which are expressed on endothelial cells of inflamed blood vessels and are critical to guide immune cells to home to inflammatory tissues.49 In addition to biological molecules, NPs have been attached on MSC and endothelial cell membranes using a similar method. The cells were shown to maintain their tumoritropic properties and ability to form multicellular structures, respectively.50 A similar strategy was used to immobilize PLGA particles on islet surfaces through the biotin-streptavidin interaction.51 In the study, PLGA particles encapsulating leukemia inhibitory factor (LIF), which promotes adaptive immune tolerance, were attached on donor islets before the islets were transplanted into allogeneic diabetic mice. The modified islets helped 57% of recipient mice to maintain long-term normoglycemia, while all control mice that received unmodified allografts all became hyperglycemic.
Because of the immunogenicity of avidin and streptavidin and their “bulky” size,52 other membrane conjugation methods have become more popular. Cheng et al developed a method utilizing a thiol-maleimide reaction for cell membrane conjugation (Figure 2D). NHS-PEG2-maleimide was first used to convert cell membrane primary amine groups to maleimide, which is highly reactive with thiolated peptides and proteins under regular cell culture conditions.15, 53 Because NHS-PEG2-maleimide is not cell membrane permeable, the conjugation method was shown to not affect stem cell viability, proliferation or multipotency. Another advantage of this method is that it allows for precise control of binding sites on attached peptides or proteins by designing the location of free thiol-containing cysteine in these molecules. In a similar conjugation strategy, anti-programmed-death ligand 1 antibody (aPDL1) was conjugated onto platelets to overcome immune suppression to T cells in residual tumors after primary tumor resection (Figure 3D).54 The binding of PDL1 on cancer cells and antigen presenting cells (APCs) to the programmed-cell-death protein 1 (PD1) of T cells inactivates tumor infiltrating T lymphocytes. Although the suppression can be overcome by aPDL1,55 its efficacy is compromised by the off-targeting of aPDL1 to normal cells.56, 57 Platelets were selected as the carrier of aPDL1 because of the ability of platelets to home to surgical wounds and to capture circulating cancer cells.58, 59 Furthermore, the activation of platelets upon adhesion to injured blood vessels results in the release of platelet membrane-derived microparticles,60 facilitating membrane-associated aPDL1 blockage of PDL1 and recruitment of more immune cells. To conjugate aPDL1 on platelets, primary amines on platelet membranes were converted to sulfhydryl groups using Traut’s Reagent (2-iminothiolane), while aPDL1was modified with maleimide groups using sulfosuccinimidyl-4-(N-maleimidomethyl)-cyclohexane-1-carboxylate. The conjugation was then completed via thiol-maleimide reaction (Figure 3D). Compared to aPDL1 alone, aPDL1 conjugated on platelets accumulated more efficiently into tumor surgical sites, and reduced the recurrence of melanoma tumors and cancer cell metastasis to the lungs (Figure 3E). Since Traut’s Reagent is cell membrane permeable and toxic to cells, the conjugation method may be improved by employing cell membrane impermeable molecules instead of Traut’s Reagent in the step of functionalizing cell surfaces with thiol groups.
Staudinger ligation has also been applied in forming covalent linkages in biological systems. It is a biorthogonal reaction between azide and phosphine such as methyl-2-(diphenylphosphino) terephthalate (MDT) (Figure 2E).61 The advantage of Staudinger ligation in studying biological systems is that both chemical groups do not exist in natural biological systems and therefore react specifically, resulting in minimal noise. In one application, islet spheroids were covered with an ultra-thin conformal coating.62 NHS-PEG-N3 was used to introduce azide groups onto islet surfaces, and poly(amido amine) (PAMAM) dendrimer functionalized with MDT was attached as a second layer via Staudinger ligation. Finally, N3-terminated alginate was conformally coated on islet surfaces via the same reaction. In another application, the same group used a similar conjugation strategy to attach TGF-β1 to the surfaces of APCs in order to generate antigen-specific Tregs.63 In nature, membrane-bound TGF-β1 on Treg promotes tolerance.64 TGF-β1 was modified with MDT groups using NHS-PEG-MDT, while murine splenocytes reacted with NHS-PEG-N3 to obtain azide groups. Afterwards, TGF-β1 was immobilized onto the APC surfaces through Staudinger ligation. The modified APCs were then cocultured with naïve CD4+ T cells from OT-II mice in the presence of OVA peptide. The naïve OT-II CD4+ T cells have TCR specific to OVA and were converted to OVA-specific Tregs, which were shown to suppress the activation of OVA-specific CD4+ T cells.
Abundant polysaccharides on cell membranes have also been targeted for cell membrane conjugation. To anchor NPs on macrophages for utilizing their tumor hypoxia-tracking characteristics, sialic acid residues on macrophages were reduced by sodium periodate.65 The reaction generated aldehyde groups, which reacted with hydrazine groups on NPs to form a transient Schiff base linkage. In the presence of sodium cyanoborohydride, the linkages were converted to stable secondary amine bonds (Figure 2F). The study provided a new cell membrane conjugation method, but it did not investigate whether the method affected the tumoritropic ability of macrophages.
The strong attachment of marine mussels on substrates results from their secreted adhesive proteins containing 3,4-Dihydroxy-l-phenylalanine (DOPA), an amino acid with catechol.66 The adherence of catechol to cellular surfaces is via phenolic proton-mediated hydrogen bonding and the formation of covalent bonds through the reaction of nucleophilic groups on cell surfaces with oxidized catechols.67, 68 To reduce donor rejection of implanted islets, PEG and heparin have been conjugated to islet cell surfaces in separate studies using DOPA-modified PEG and heparin, respectively.67, 69
In contrast to the above mentioned conjugation reactions that establish linkages on cell membranes at relatively random positions, selective cell membrane conjugation can be achieved with the help of enzymes. These biochemical methods require cell membranes to present specific amino acid sequences, called acceptor peptides.70–72 For instance, the LPETGG motif can be recognized and cleaved in between threonine and glycine residues by sortase A. The produced thioester acyl-enzyme intermediate reacts with peptides with N-terminal glycine, (G)n to covalently link the motif with the peptides (Figure 2H).72 To introduce acceptor peptides on cell membranes at pre-defined positions, genetic modification of cells is required and had safety concerns until the recent establishment of targeted genome editing technology, CRISPR-Cas9 that enables the insertion of genes at pre-defined regions of the genome.73, 74 In a very recent study, CRISPR-Cas9 was used to generate RBCs presenting Kell proteins with an LPETGG motif. In the presence of sortase A, autoantigen peptides were linked on these modified RBC membranes.75 This method of conjugation was shown to be very gentle and did not damage RBCs. Furthermore, it allowed for well-controlled presentation of the antigen motif on the RBC Kell protein. To expand the clinical applicability of this strategy, a conjugation method that does not require genetic modification was also developed, taking advantage of endogenous RBC proteins with an exposed glycine at the N terminus (Figure 4A). This strategy was demonstrated to have significant potential when applied to immunotherapy for autoimmunity thanks to the tolerogenic nature of RBCs. When MOG35-55, a myelin antigen implicated in experimental autoimmune encephalomyelitis (EAE), a murine model of multiple sclerosis, was conjugated to RBC membranes in this manner and then i.v. injected in mice with EAE, the modified RBCs could both prevent and treat the disease. Similarly, conjugating an insulin peptide prevented the development of diabetes in non-obese diabetic mice. The simplified method without acceptor peptides sacrificed some degree of selectivity. It is interesting to study if other simple chemical conjugation methods discussed above can result in a similar efficacy in autoimmune disease models.
Figure 4.
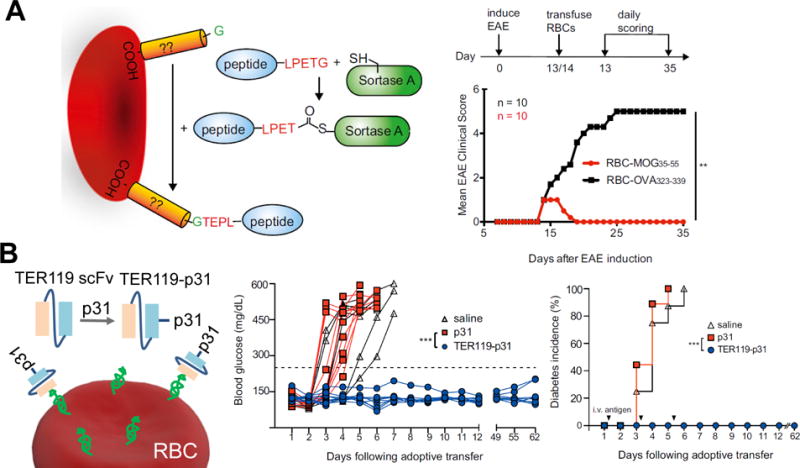
Red blood cell membrane conjugation for treating experimental autoimmune encephalomyelitis (A) and autoimmune induced diabetes (B). A) The EAE inducing peptide MOG35-55 was conjugated onto RBC membranes using the sortagging method. Around 13-14 days after induction of EAE, modified RBCs were i.v. administered into mice. Mice that received RBCs modified with the EAE peptide exhibited complete remission of disease scores approximately 5 days after treatment. Whereas the control EAE mice received RBCs modified with an irrelevant peptide, maintained disease scores. B) The anti-erythrocyte antibody TER119 was converted into a single chain Fv with an attached p31 peptide used in inducing diabetes in the BDC2.5 diabetic mouse model. In mice in which TER119-p31 was administered i.v. following adoptive transfer of diabetogenic BDC2.5 CD4+ T cells three times spaced 3 days apart, glucose levels were maintained at normal levels compared to control mice, which all developed diabetes. (Adapted from Ref 75 and Ref 79 with permission.)
Hydrophobic interactions
Cell membranes are composed of a lipid bilayer, proteins, and polysaccharides. Lipids are amphiphilic molecules and self-assemble into lipid bilayers. The hydrophobic interaction between lipid tails and protein transmembrane domains is critical to stabilize membrane proteins on cell surfaces. Similarly, hydrophobic interactions can be a facile strategy to introduce conjugants on cell membranes (Figure 2I). Compared to chemical conjugation, especially nonspecific reactions, hydrophobic interactions may cause less damage to the original physiochemical properties of cell membranes. However, its disadvantage is the instability of the inserted targets. Unlike the relatively large transmembrane domain of proteins, inserted molecules normally have a hydrophobic portion similar to lipid alkyl chains, which are prone to leave cell membranes, especially when a plethora of proteins exist in close proximity in vivo. Liu et al. have tested the conjugation efficiency of CpG-oligonucleotides terminated with various hydrophobic moieties including C18 chain, diacyllipid with or without spacer PEG, and cholesterol on B16F10 melanoma tumor cells in vitro and in vivo. Unlike free CpG-oligonucleotides, hydrophobic-tail modified CpG-oligonucleotides can insert into the tumor cells and have a better retention time in tumor tissues.76 Many hydrophobic interaction-based conjugation methods involve two steps. In one study, protein A was palmitated for inserting into leukemia cell membranes. Then fusion protein, B7-1·Fcγ1 was anchored on the cells via the binding between Fc and protein A. B7-1 is a co-stimulatory molecule for T cell activation, and its density on modified cells was shown to affect T cell proliferation in vitro.77 Some conjugants are attached through similar two-step strategies. In another study, for the conjugation of urokinase, a protein that helps dissolve blood clots, to islet cell membranes, oligo(dT)20-PEG-lipids were inserted into cell membranes though hydrophobic interactions. Hybridization between urokinase-oligo(dA)20 with oligo(dT)20 anchored the proteinase onto islet surfaces.78
Cell surface specific binding interactions
Bioconjugation can be easily achieved using fusion molecules with a ligand binding to cell membrane proteins. Building upon the discovery that apoptotic cells promote tolerance, Kontos et al. hypothesized that the large number of erythrocytes that undergo apoptosis each day can be leveraged to induce tolerance.79 This was evaluated by conjugating a protein of interest, in this case OVA, to ERY1, a 12-amino acid peptide that specifically binds to glycophorin A on erythrocyte membranes. When ERY1-OVA was injected into mice that were administered adoptively transferred OVA-specific OT-I CD8+ T cells, it was found to induce deletion of the transferred T cells. Since conjugating sufficient quantities of ERY1 on small peptides is technically challenging, an erythrocyte binding antibody, TER119, was engineered into a single-chain fragment carrying the peptide p31, an islet β-cell autoantigen implicated in diabetes pathogenesis for nonobese diabetic BDC2.5 mice (Figure 4B). When TER119-p31 was periodically administered into mice injected with activated BDC2.5 CD4+ T cells, rapid-onset diabetes was prevented compared to controls that received p31 alone. This represents an effective cell membrane conjugation method with the advantage that it does not require ex vivo cell manipulation. Double cell surface binding has also been performed. ILY4D is a binding domain of intermedilysin (ILY) on human erythrocyte membranes and some cancer cells that express CEA. Therefore, anti-CEA/ILY4D was fabricated to bind to human erythrocytes, rendering erythrocytes capable to target CEA-positive cancer cells.80
Cell membrane-derived nanomaterials for immunotherapy
Cell membrane-derived or cell membrane-coated nanomaterials (CM-NMs) are an emerging class of materials that present natural cell membranes and are highly biocompatible (Figure 1).13 In addition, CM-NMs with synthetic materials in the core own the advantages of synthetic materials in controlling drug release and imaging in vivo. The majority of CM-NMs have been utilized for harnessing the natural targeting ability of cell membrane receptors.14, 16, 17, 81 Their applications in eliciting immune protection have also gained increasing attention.
Cancer vaccine
One cancer immunotherapy under much investigation uses the concept of vaccination to induce anti-tumor immunity. This strategy typically incorporates a tumor antigen along with an immunostimulatory adjuvant to activate APCs and create an anti-tumor response. However, clinical trials of cancer vaccines have thus far shown very limited effectiveness.40 One reason for this is the limited ability of the vaccine to access DCs, which are one of the most potent APCs. In fact, one of the only FDA-approved cancer vaccines utilizes ex vivo isolated and activated DCs (PROVENGE®).82 To avoid the high costs and loss of function of ex vivo expanded DCs, Guo et al. developed a cell membrane-coated NP vaccine that targets DCs in vivo (Figure 5A).83 Their system consisted of melanoma antigen peptide-loaded PLGA-NPs coated with RBC membranes. DSPE-PEG-mannose were anchored on the coated membrane of CM-NMs for targeting DCs, while MPLA was also incorporated on the membrane as an immunostimulatory adjuvant. It was shown that the CM-NMs with both mannose and MPLA exhibited a higher degree of DC activation compared to control CM-NMs without one or both modifications, inhibited tumor growth, and suppressed tumor metastasis in a mouse melanoma tumor model.
Figure 5.
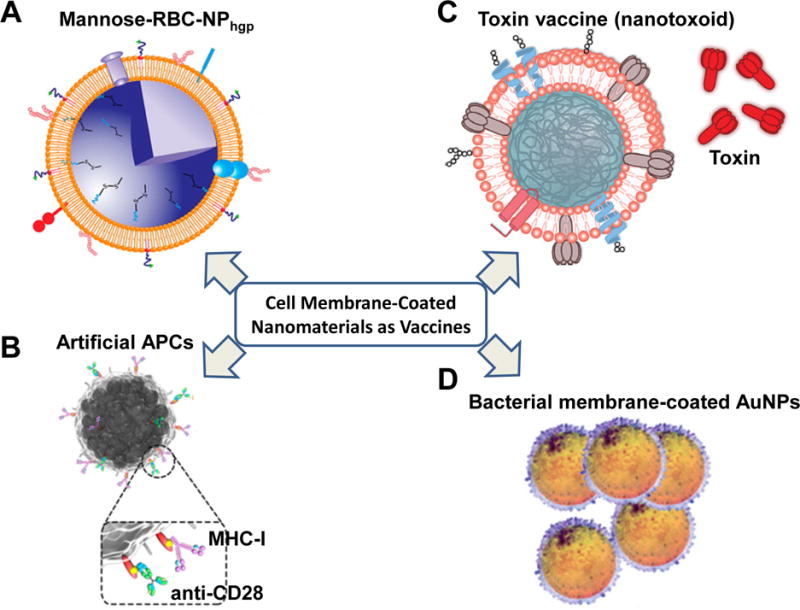
Cell membrane-derived nanomaterials for immunotherapy. (A) Mannose-RBC-NP vaccine for DC activation and tumor suppression. (B) Artificial magnetic APC vaccine for T cell stimulation and guided T cell tumor homing. The artificial APCs were magnetic nanocluster covered with leukocyte membranes and decorated with MHC-I and anti-CD28 for stimulating antigen-specific T cells. (C) Nanotoxoid for antitoxin vaccination. The Nanotoxoid was made of RBC membrane-coated NPs entrapping toxin. (D) Bacterial membrane-coated AuNPs for antibacterial immunity. (Adapted with permission from Ref 83, Copyright© 2015, ACS Publications; Ref 84, Copyright© 2017, ACS Publications; Ref 91, Copyright© 2013, Nature Publishing Group; and Ref 93, Copyright© 2015, ACS Publications.)
To circumvent the need for APC activation to elicit T cell immunity, Zhang et al. generated artificial APCs using isolated leukocyte membranes (Figure 5B).84 Leukocytes were first treated with azide-choline to present azide groups on surfaces via cell metabolism and lipid biosynthesis. After membrane isolation and coating on magnetic nanoclusters, the fabricated CM-NMs were chemically coupled with dibenzocyclooctyne-modified major histocompatibility complex class-I (MHC-I), which was loaded with OVA peptide, and dibenzocyclooctyne-modified co-stimulatory ligand anti-CD28 through the copper-free azide-alkyne cycloaddition reaction. The generated artificial APCs were highly efficient in stimulating and expanding CD8+ T cells with specific TCR against MHC-I and OVA peptide complex. After adoptive transfer of the stimulated CD8+ T cells into EG-7 tumor bearing mice, the T cells associated with the magnetic nanoclusters could by guided to tumors through magnetic control and inhibited the growth of OVA-expressing EG-7 tumors.
Although cancer vaccines targeting a single cancer cell antigen can elicit strong immune responses against tumors, the protection does not last long because of the heterogeneity of cancer cells.85 Cancer cell membranes carrying a repertoire of membrane antigens are advantageous in that they provide a broad range of antigens. B16-F10 cancer cell membrane-coated NPs (CCNPs) with MPLA attached on the surfaces were able to activate DCs and stimulate antigen-specific T cells.86 In a subsequent study, the authors subcutaneously injected CpG-encapsulated CCNPs (CpG-CCNPs) into mice either before or after challenge with B16-F10 cells.87 It was found that if the cancer vaccine was administered before cancer cell challenge, CpG-CCNPs could prevent tumor establishment in 86% of mice. In mice for which CpG-CCNPs and the checkpoint inhibitor cocktail of anti-CTLA4 and anti-PD1 were co-administered after cancer cell injection, the median survival was extended from 18 to 32 d compared to the blank control. There are two major issues that remain to be addressed in future studies. One is whether the disadvantage of relatively low amount of individual antigens can be overcome to elicit strong immune responses, especially when original T cells with specific TCRs are in low numbers. The other is to determine the importance of incorporating patient-specific intracellular neoantigens that result from tumor-specific mutations and are not in cancer cell membranes.88
Vaccines against toxin and bacteria
In the field of vaccine development, pore-forming toxins (PFTs) are highly clinically relevant since they are the main pathogenic component of many common infections.89 However, developing vaccine formulations is challenging due to the need to first inactivate them via heat or chemical methods, which can result in reduced immunogenicity and effectiveness.90 Hu et al. demonstrated that RBC membrane-coated NPs can entrap structurally intact toxins in the coating membranes and prevent the toxin from affecting normal cells, while retaining their immunogenicity to stimulate immune responses (Figure 5C).91 This nanotoxoid system using staphylococcal α-haemolysin (Hla) antigen elicited higher anti-Hla IgG titers than heat inactivated Hla alone upon administration of equivalent amounts of Hla antigen. The antibody protection extended mouse survival in response to a lethal dose of Hla, as the nanotoxoid treated mice exhibited 50% survival after just one primary immunization and 100% survival with a booster as compared to 10% and 90% survival, respectively, by treating with heat-inactivated Hla alone. They also showed that the RBC delivery vehicle did not induce any autoimmunity, which is an advantage compared to some currently used adjuvants that can result in reactogenicity and other adverse effects. To increase the antigenic breadth, the same group collected bacteria-secreted proteins from bacteria culture medium and incorporated a broad range of toxins into the RBC-coated NPs. Mice vaccinated with this new formulation of nanotoxoid could protect mice from bacterial infection.92
Another strategy used by the group is to coat small sized gold NPs with bacteria outer membranes (Figure 5D).93 After subcutaneous injection, the bacterial membrane-coated AuNPs quickly drained to lymph nodes due to their small particle size and activated DCs that elicited both bacteria-specific antibody production and T cell responses in vivo.
Perspectives and Conclusions
Cell membrane conjugation is a highly promising strategy for immunotherapy as shown from its demonstrated efficacy in animal models. To date, most studies have manipulated cell membranes to perform a single function, such as delivering an antigen to attenuate an autoimmune reaction or locally delivering a cytokine to amplify immune reactions. As these strategies become better studied, they may be combined with complementary methods to enhance their effectiveness. For instance, genetically engineered cells that express tolerogenic cytokines may increase the efficacy of cells with membrane-bound autoantigens to induce tolerance. Alternatively, to avoid gene therapy, cell membrane conjugation of NPs encapsulating tolerogenic cytokines may be used. A major limitation of many of these strategies is the difficulty in translating the treatment to broad clinical use due to the cost. Strategies that modulate targeted cells in situ without ex vivo cell engineering may pave the path for more clinical studies. CM-NMs have been mostly used for stimulating immune responses. We envision the unique nanomaterials may be modified for establishing tolerance for autoantigen and donor cell/tissues. Tolerogenic drugs encapsulated inside the synthetic core of CM-NMs may improve cell membrane-induced tolerance. During membrane coating, a small fraction of membranes will enclose the synthetic NP core in an inside-out orientation.94 This fraction of CM-NMs may need to be removed before application in order to prevent potential inflammatory responses.
Acknowledgments
This work was supported by a National Institutes of Health Grant R21AI133372.
References
- 1.Caspi RR. Immunotherapy of autoimmunity and cancer: the penalty for success. Nat Rev Immunol. 2008;8:970–976. doi: 10.1038/nri2438. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Topalian SL, Hodi FS, Brahmer JR, Gettinger SN, Smith DC, McDermott DF, Powderly JD, Carvajal RD, Sosman JA, Atkins MB, et al. Safety, Activity, and Immune Correlates of Anti-PD-1 Antibody in Cancer. N Engl J Med. 2012;366:2443–2454. doi: 10.1056/NEJMoa1200690. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Maude SL, Frey N, Shaw PA, Aplenc R, Barrett DM, Bunin NJ, Chew A, Gonzalez VE, Zheng ZH, Lacey SF, et al. Chimeric antigen receptor T cells for sustained remissions in leukemia. N Engl J Med. 2014;371:1507–1517. doi: 10.1056/NEJMoa1407222. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Wiseman AC. Immunosuppressive Medications. Clin J Am Soc Nephrol. 2016;11:332–343. doi: 10.2215/CJN.08570814. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Feldmann M, Steinman L. Design of effective immunotherapy for human autoimmunity. Nature. 2005;435:612–619. doi: 10.1038/nature03727. [DOI] [PubMed] [Google Scholar]
- 6.Miller SD, Turley DM, Podojil JR. Antigen-specific tolerance strategies for the prevention and treatment of autoimmune disease. Nat Rev Immunol. 2007;7:665–677. doi: 10.1038/nri2153. [DOI] [PubMed] [Google Scholar]
- 7.Meyer RA, Sunshine JC, Green JJ. Biomimetic particles as therapeutics. Trends Biotechnol. 2015;33:514–524. doi: 10.1016/j.tibtech.2015.07.001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Barranco C. Autoimmunity: Nanomedicine, meet autoimmune disease. Nat Rev Rheumatol. 2016;12:193. doi: 10.1038/nrrheum.2016.33. [DOI] [PubMed] [Google Scholar]
- 9.Shi J, Kantoff PW, Wooster R, Farokhzad OC. Cancer nanomedicine: progress, challenges and opportunities. Nat Rev Cancer. 2017;17:20–37. doi: 10.1038/nrc.2016.108. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Tanaka M, Sackmann E. Polymer-supported membranes as models of the cell surface. Nature. 2005;437:656–663. doi: 10.1038/nature04164. [DOI] [PubMed] [Google Scholar]
- 11.Yoo JW, Irvine DJ, Discher DE, Mitragotri S. Bio-inspired, bioengineered and biomimetic drug delivery carriers. Nat Rev Drug Discov. 2011;10:521–535. doi: 10.1038/nrd3499. [DOI] [PubMed] [Google Scholar]
- 12.Wang Q, Cheng H, Peng HS, Zhou H, Li PY, Langer R. Non-genetic engineering of cells for drug delivery and cell-based therapy. Adv Drug Deliv Rev. 2015;91:125–140. doi: 10.1016/j.addr.2014.12.003. [DOI] [PubMed] [Google Scholar]
- 13.Hu CMJ, Zhang L, Aryal S, Cheung C, Fang RH, Zhang LF. Erythrocyte membrane-camouflaged polymeric nanoparticles as a biomimetic delivery platform. Proc Natl Acad Sci U S A. 2011;108:10980–10985. doi: 10.1073/pnas.1106634108. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Hu CMJ, Fang RH, Wang KC, Luk BT, Thamphiwatana S, Dehaini D, Nguyen P, Angsantikul P, Wen CH, Kroll AV, et al. Nanoparticle biointerfacing by platelet membrane cloaking. Nature. 2015;526:118–121. doi: 10.1038/nature15373. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Zhou H, Fan ZY, Lemons PK, Cheng H. A Facile Approach to Functionalize Cell Membrane-Coated Nanoparticles. Theranostics. 2016;6:1012–1022. doi: 10.7150/thno.15095. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Hu QY, Sun WJ, Qian CG, Wang C, Bomba HN, Gu Z. Anticancer Platelet-Mimicking Nanovehicles. Adv Mater. 2015;27:7043–7050. doi: 10.1002/adma.201503323. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Parodi A, Quattrocchi N, van de Ven AL, Chiappini C, Evangelopoulos M, Martinez JO, Brown BS, Khaled SZ, Yazdi IK, Vittoria Enzo M, et al. Synthetic nanoparticles functionalized with biomimetic leukocyte membranes possess cell-like functions. Nat Nanotechnol. 2013;8:61–68. doi: 10.1038/nnano.2012.212. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Scott MD, Murad KL, Koumpouras F, Talbot M, Eaton JW. Chemical camouflage of antigenic determinants: stealth erythrocytes. Proc Natl Acad Sci U S A. 1997;94:7566–7571. doi: 10.1073/pnas.94.14.7566. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Murad KL, Gosselin EJ, Eaton JW, Scott MD. Stealth cells: prevention of major histocompatibility complex class II-mediated T-cell activation by cell surface modification. Blood. 1999;94:2135–2141. [PubMed] [Google Scholar]
- 20.Shapiro AM, Pokrywczynska M, Ricordi C. Clinical pancreatic islet transplantation. Nat Rev Endocrinol. 2017;13:268–277. doi: 10.1038/nrendo.2016.178. [DOI] [PubMed] [Google Scholar]
- 21.Contreras JL, Xie D, Mays J, Smyth CA, Eckstein C, Rahemtulla FG, Young CJ, Thompson JA, Bilbao G, Curiel DT, et al. A novel approach to xenotransplantation combining surface engineering and genetic modification of isolated adult porcine islets. Surgery. 2004;136:537–547. doi: 10.1016/j.surg.2004.05.031. [DOI] [PubMed] [Google Scholar]
- 22.Xie D, Smyth CA, Eckstein C, Bilbao G, Mays J, Eckhoff DE, Contreras JL. Cytoprotection of PEG-modified adult porcine pancreatic islets for improved xenotransplantation. Biomaterials. 2005;26:403–412. doi: 10.1016/j.biomaterials.2004.02.048. [DOI] [PubMed] [Google Scholar]
- 23.Lee DY, Park SJ, Nam JH, Byun Y. A new strategy toward improving immunoprotection in cell therapy for diabetes mellitus: Long-functioning PEGylated islets in vivo. Tissue Eng. 2006;12:615–623. doi: 10.1089/ten.2006.12.615. [DOI] [PubMed] [Google Scholar]
- 24.Lee DY, Park SJ, Lee S, Nam JH, Byun Y. Highly poly(ethylene) glycolylated islets improve long-term islet allograft survival without immunosuppressive medication. Tissue Eng. 2007;13:2133–2141. doi: 10.1089/ten.2006.0009. [DOI] [PubMed] [Google Scholar]
- 25.Giraldo JA, Molano RD, Rengifo HR, Fotino C, Gattas-Asfura KM, Pileggi A, Stabler CL. The impact of cell surface PEGylation and short-course immunotherapy on islet graft survival in an allogeneic murine model. Acta Biomater. 2017;49:272–283. doi: 10.1016/j.actbio.2016.11.060. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Tom JK, Mancini RJ, Esser-Kahn AP. Covalent modification of cell surfaces with TLR agonists improves & directs immune stimulation. ChemComm. 2013;49:9618–9620. doi: 10.1039/c3cc45468a. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Jenkins MK, Schwartz RH. Antigen presentation by chemically modified splenocytes induces antigen-specific T cell unresponsiveness in vitro and in vivo. J Exp Med. 1987;165:302–319. doi: 10.1084/jem.165.2.302. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Elliott C, Wang K, Miller S, Melvold R. Ethylcarbodiimide as an agent for induction of specific transplant tolerance. Transplantation. 1994;58:966–968. doi: 10.1097/00007890-199410270-00023. [DOI] [PubMed] [Google Scholar]
- 29.Kaneko K, Morelli AE, Wang Z, Thomson AW. Alloantigen presentation by ethylcarbodiimide-treated dendritic cells induces T cell hyporesponsiveness, and prolongs organ graft survival. Clin Immunol. 2003;108:190–198. doi: 10.1016/s1521-6616(03)00141-4. [DOI] [PubMed] [Google Scholar]
- 30.Luo X, Pothoven KL, McCarthy D, DeGutes M, Martin A, Getts DR, Xia G, He J, Zhang X, Kaufman DB, et al. ECDI-fixed allogeneic splenocytes induce donor-specific tolerance for long-term survival of islet transplants via two distinct mechanisms. Proc Natl Acad Sci U S A. 2008;105:14527–14532. doi: 10.1073/pnas.0805204105. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Scalea JR, Tomita Y, Lindholm CR, Burlingham W. Transplantation Tolerance Induction: Cell Therapies and Their Mechanisms. Front Immunol. 2016;7:87. doi: 10.3389/fimmu.2016.00087. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Ferguson TA, Herndon J, Elzey B, Griffith TS, Schoenberger S, Green DR. Uptake of apoptotic antigen-coupled cells by lymphoid dendritic cells and cross-priming of CD8(+) T cells produce active immune unresponsiveness. J Immunol. 2002;168:5589–5595. doi: 10.4049/jimmunol.168.11.5589. [DOI] [PubMed] [Google Scholar]
- 33.Turley DM, Miller SD. Peripheral tolerance induction using ethylenecarbodiimide-fixed APCs uses both direct and indirect mechanisms of antigen presentation for prevention of experimental autoimmune encephalomyelitis. J Immunol. 2007;178:2212–2220. doi: 10.4049/jimmunol.178.4.2212. [DOI] [PubMed] [Google Scholar]
- 34.Getts DR, Turley DM, Smith CE, Harp CT, McCarthy D, Feeney EM, Getts MT, Martin AJ, Luo XR, Terry RL, et al. Tolerance Induced by Apoptotic Antigen-Coupled Leukocytes Is Induced by PD-L1(+) and IL-10-Producing Splenic Macrophages and Maintained by T Regulatory Cells. J Immunol. 2011;187:2405–2417. doi: 10.4049/jimmunol.1004175. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Lutterotti A, Yousef S, Sputtek A, Sturner KH, Stellmann JP, Breiden P, Reinhardt S, Schulze C, Bester M, Heesen C, et al. Antigen-specific tolerance by autologous myelin peptide-coupled cells: A phase 1 trial in multiple sclerosis. Sci Transl Med. 2013;5:11. doi: 10.1126/scitranslmed.3006168. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Huang BN, Abraham WD, Zheng YR, Lopez SCB, Luo SS, Irvine DJ. Active targeting of chemotherapy to disseminated tumors using nanoparticle-carrying T cells. Sci Transl Med. 2015;7 doi: 10.1126/scitranslmed.aaa5447. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Stephan MT, Moon JJ, Um SH, Bershteyn A, Irvine DJ. Therapeutic cell engineering with surface-conjugated synthetic nanoparticles. Nat Med. 2010;16:1035–U135. doi: 10.1038/nm.2198. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Stephan MT, Stephan SB, Bak P, Chen JZ, Irvine DJ. Synapse-directed delivery of immunomodulators using T-cell-conjugated nanoparticles. Biomaterials. 2012;33:5776–5787. doi: 10.1016/j.biomaterials.2012.04.029. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Jones RB, Mueller S, Kumari S, Vrbanac V, Genel S, Tager AM, Allen TM, Walker BD, Irvine DJ. Antigen recognition-triggered drug delivery mediated by nanocapsule-functionalized cytotoxic T-cells. Biomaterials. 2017;117:44–53. doi: 10.1016/j.biomaterials.2016.11.048. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Farkona S, Diamandis EP, Blasutig IM. Cancer immunotherapy: the beginning of the end of cancer? BMC Med. 2016;14:73. doi: 10.1186/s12916-016-0623-5. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.Zeng R, Spolski R, Finkelstein SE, Oh SK, Kovanen PE, Hinrichs CS, Pise-Masison CA, Radonovich MF, Brady JN, Restifo NP, et al. Synergy of IL-21 and IL-15 in regulating CD8(+) T cell expansion and function. J Exp Med. 2005;201:139–148. doi: 10.1084/jem.20041057. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42.Porter DL, Levine BL, Kalos M, Bagg A, June CH. Chimeric antigen receptor-modified T cells in chronic lymphoid leukemia. N Engl J Med. 2011;365:725–733. doi: 10.1056/NEJMoa1103849. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43.Kochenderfer JN, Dudley ME, Feldman SA, Wilson WH, Spaner DE, Maric I, Stetler-Stevenson M, Phan GQ, Hughes MS, Sherry RM, et al. B-cell depletion and remissions of malignancy along with cytokine-associated toxicity in a clinical trial of anti-CD19 chimeric-antigen-receptor-transduced T cells. Blood. 2012;119:2709–2720. doi: 10.1182/blood-2011-10-384388. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44.Fedorov VD, Themeli M, Sadelain M. PD-1-and CTLA-4-based inhibitory chimeric antigen receptors (iCARs) divert off-target immunotherapy responses. Sci Transl Med. 2013;5:12. doi: 10.1126/scitranslmed.3006597. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45.Morgan RA, Yang JC, Kitano M, Dudley ME, Laurencot CM, Rosenberg SA. Case report of a serious adverse event following the administration of T cells transduced with a chimeric antigen receptor recognizing ERBB2. Mol Ther. 2010;18:843–851. doi: 10.1038/mt.2010.24. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46.Moon EK, Wang LC, Dolfi DV, Wilson CB, Ranganathan R, Sun J, Kapoor V, Scholler J, Pure E, Milone MC, et al. Multifactorial T-cell hypofunction that is reversible can limit the efficacy of chimeric antigen receptor-transduced human T cells in solid tumors. Clin Cancer Res. 2014;20:4262–4273. doi: 10.1158/1078-0432.CCR-13-2627. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47.Newick K, Moon E, Albelda SM. Chimeric antigen receptor T-cell therapy for solid tumors. Mol Ther-Oncolytics. 2016;3:7. doi: 10.1038/mto.2016.6. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48.Corinti S, Chiarantini L, Dominici S, Laguardia ME, Magnani M, Girolomoni G. Erythrocytes deliver Tat to interferon-gamma-treated human dendritic cells for efficient initiation of specific type 1 immune responses in vitro. J Leukoc Biol. 2002;71:652–658. [PubMed] [Google Scholar]
- 49.Sarkar D, Vemula PK, Teo GSL, Spelke D, Karnik R, Wee LY, Karp JM. Chemical Engineering of Mesenchymal Stem Cells to Induce a Cell Rolling Response. Bioconjugate Chem. 2008;19:2105–2109. doi: 10.1021/bc800345q. [DOI] [PubMed] [Google Scholar]
- 50.Cheng H, Kastrup CJ, Ramanathan R, Siegwart DJ, Ma ML, Bogatyrev SR, Xu QB, Whitehead KA, Langer R, Anderson DG. Nanoparticulate Cellular Patches for Cell-Mediated Tumoritropic Delivery. ACS Nano. 2010;4:625–631. doi: 10.1021/nn901319y. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 51.Dong HS, Fahmy TM, Metcalfe SM, Morton SL, Dong X, Inverardi L, Adams DB, Gao WD, Wang HJ. Immuno-Isolation of Pancreatic Islet Allografts Using Pegylated Nanotherapy Leads to Long-Term Normoglycemia in Full MHC Mismatch Recipient Mice. Plos One. 2012;7 doi: 10.1371/journal.pone.0050265. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 52.Chu TC, Twu KY, Ellington AD, Levy M. Aptamer mediated siRNA delivery. Nucleic Acids Res. 2006;34:6. doi: 10.1093/nar/gkl388. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 53.Cheng H, Byrska-Bishop M, Zhang CT, Kastrup CJ, Hwang NS, Tai AK, Lee WW, Xu XY, Nahrendorf M, Langer R, et al. Stem cell membrane engineering for cell rolling using peptide conjugation and tuning of cell-selectin interaction kinetics. Biomaterials. 2012;33:5004–5012. doi: 10.1016/j.biomaterials.2012.03.065. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 54.Wang C, Sun W, Ye Y, Hu Q, Bomba HN, Gu Z. In situ activation of platelets with checkpoint inhibitors for post-surgical cancer immunotherapy. Nat Biomed Eng. 2017;1:0011. [Google Scholar]
- 55.Zou WP, Wolchok JD, Chen LP. PD-L1 (B7-H1) and PD-1 pathway blockade for cancer therapy: Mechanisms, response biomarkers, and combinations. Sci Transl Med. 2016;8:14. doi: 10.1126/scitranslmed.aad7118. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 56.Chen LP, Han X. Anti-PD-1/PD-L1 therapy of human cancer: past, present, and future. J Clin Invest. 2015;125:3384–3391. doi: 10.1172/JCI80011. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 57.Mellati M, Eaton KD, Brooks-Worrell BM, Hagopian WA, Martins R, Palmer JP, Hirsch IB. Anti-PD-1 and anti-PDL-1 monoclonal antibodies causing type 1 diabetes. Diabetes Care. 2015;38:E137–E138. doi: 10.2337/dc15-0889. [DOI] [PubMed] [Google Scholar]
- 58.Gurtner GC, Werner S, Barrandon Y, Longaker MT. Wound repair and regeneration. Nature. 2008;453:314–321. doi: 10.1038/nature07039. [DOI] [PubMed] [Google Scholar]
- 59.Gay LJ, Felding-Habermann B. Contribution of platelets to tumour metastasis. Nat Rev Cancer. 2011;11:123–134. doi: 10.1038/nrc3004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 60.Ruggeri ZM, Mendolicchio GL. Adhesion mechanisms in platelet function. Circ Res. 2007;100:1673–1685. doi: 10.1161/01.RES.0000267878.97021.ab. [DOI] [PubMed] [Google Scholar]
- 61.Saxon E, Bertozzi CR. Cell surface engineering by a modified Staudinger reaction. Science. 2000;287:2007–2010. doi: 10.1126/science.287.5460.2007. [DOI] [PubMed] [Google Scholar]
- 62.Gattas-Asfura KM, Stabler CL. Bioorthogonal Layer-by-Layer Encapsulation of Pancreatic Islets via Hyperbranched Polymers. ACS Appl Mater Interfaces. 2013;5:9964–9974. doi: 10.1021/am401981g. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 63.Yang EY, Kronenfeld J, Gattas-Asfura K, Bayer AL, Stabler C. Engineering an Infectious Treg Biomimetic through Chemoselective Tethering of TGF-beta 1 to PEG Brush Surfaces. Tissue Eng Part A. 2015;21:S232–S232. doi: 10.1016/j.biomaterials.2015.07.009. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 64.Nakamura K, Kitani A, Strober W. Cell contact-dependent immunosuppression by CD4(+)CD25(+) regulatory T cells is mediated by cell surface-bound transforming growth factor beta. J Exp Med. 2001;194:629–644. doi: 10.1084/jem.194.5.629. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 65.Holden CA, Yuan QA, Yeudall WA, Lebman DA, Yang H. Surface engineering of macrophages with nanoparticles to generate a cell-nanoparticle hybrid vehicle for hypoxia-targeted drug delivery. Int J Nanomed. 2010;5:25–36. [PMC free article] [PubMed] [Google Scholar]
- 66.Coyne KJ, Qin XX, Waite JH. Extensible collagen in mussel byssus: A natural block copolymer. Science. 1997;277:1830–1832. doi: 10.1126/science.277.5333.1830. [DOI] [PubMed] [Google Scholar]
- 67.Brubaker CE, Kissler H, Wang LJ, Kaufman DB, Messersmith PB. Biological performance of mussel-inspired adhesive in extrahepatic islet transplantation. Biomaterials. 2010;31:420–427. doi: 10.1016/j.biomaterials.2009.09.062. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 68.Kastrup CJ, Nahrendorf M, Figueiredo JL, Lee H, Kambhampati S, Lee T, Cho SW, Gorbatov R, Iwamoto Y, Dang TT, et al. Painting blood vessels and atherosclerotic plaques with an adhesive drug depot. Proc Natl Acad Sci U S A. 2012;109:21444–21449. doi: 10.1073/pnas.1217972110. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 69.Jung YS, Jeong JH, Yook S, Im BH, Seo J, Hong SW, Park JB, Yan VC, Lee DY, Byun Y. Surface modification of pancreatic islets using heparin-DOPA conjugate and anti-CD154 mAb for the prolonged survival of intrahepatic transplanted islets in a xenograft model. Biomaterials. 2012;33:295–303. doi: 10.1016/j.biomaterials.2011.09.051. [DOI] [PubMed] [Google Scholar]
- 70.Griffin BA, Adams SR, Tsien RY. Specific covalent labeling of recombinant protein molecules inside live cells. Science. 1998;281:269–272. doi: 10.1126/science.281.5374.269. [DOI] [PubMed] [Google Scholar]
- 71.Chen I, Howarth M, Lin WY, Ting AY. Site-specific labeling of cell surface proteins with biophysical probes using biotin ligase. Nat Methods. 2005;2:99–104. doi: 10.1038/nmeth735. [DOI] [PubMed] [Google Scholar]
- 72.Popp MW, Antos JM, Grotenbreg GM, Spooner E, Ploegh HL. Sortagging: a versatile method for protein labeling. Nat Chem Biol. 2007;3:707–708. doi: 10.1038/nchembio.2007.31. [DOI] [PubMed] [Google Scholar]
- 73.Cong L, Ran FA, Cox D, Lin SL, Barretto R, Habib N, Hsu PD, Wu XB, Jiang WY, Marraffini LA, et al. Multiplex genome engineering using CRISPR/Cas systems. Science. 2013;339:819–823. doi: 10.1126/science.1231143. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 74.Jinek M, Chylinski K, Fonfara I, Hauer M, Doudna JA, Charpentier E. A programmable dual-RNA-guided DNA endonuclease in adaptive bacterial immunity. Science. 2012;337:816–821. doi: 10.1126/science.1225829. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 75.Pishesha N, Bilate AM, Wibowo MC, Huang NJ, Li ZY, Dhesycka R, Bousbaine D, Li HJ, Patterson HC, Dougan SK, et al. Engineered erythrocytes covalently linked to antigenic peptides can protect against autoimmune disease. Proc Natl Acad Sci U S A. 2017;114:3157–3162. doi: 10.1073/pnas.1701746114. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 76.Liu HP, Kwong B, Irvine DJ. Membrane Anchored Immunostimulatory Oligonucleotides for In Vivo Cell Modification and Localized Immunotherapy. Angew Chem Int Ed. 2011;50:7052–7055. doi: 10.1002/anie.201101266. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 77.Chen AS, Zheng GX, Tykocinski ML. Hierarchical costimulator thresholds for distinct immune responses: Application of a navel two-step Fc fusion protein transfer method. J Immunol. 2000;164:705–711. doi: 10.4049/jimmunol.164.2.705. [DOI] [PubMed] [Google Scholar]
- 78.Takemoto N, Teramura Y, Iwata H. Islet Surface Modification with Urokinase through DNA Hybridization. Bioconjugate Chem. 2011;22:673–678. doi: 10.1021/bc100453r. [DOI] [PubMed] [Google Scholar]
- 79.Kontos S, Kourtis IC, Dane KY, Hubbell JA. Engineering antigens for in situ erythrocyte binding induces T-cell deletion. Proc Natl Acad Sci U S A. 2013;110:E60–E68. doi: 10.1073/pnas.1216353110. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 80.Nagamune H, Ohkura K, Umezu K, Shouji H, Kourai H. A cell membrane modification technique using domain 4 of intermedilysin for immunotherapy against cancer. Anticancer Res. 2004;24:3367–3372. [PubMed] [Google Scholar]
- 81.Hu QY, Qian CG, Sun WJ, Wang JQ, Chen ZW, Bomba HN, Xin HL, Shen QD, Gu Z. Engineered nanoplatelets for enhanced treatment of multiple myeloma and thrombus. Adv Mater. 2016;28:9573–9580. doi: 10.1002/adma.201603463. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 82.Cheever MA, Higano CS. PROVENGE (Sipuleucel-T) in prostate cancer: The first FDA-approved therapeutic cancer vaccine. Clin Cancer Res. 2011;17:3520–3526. doi: 10.1158/1078-0432.CCR-10-3126. [DOI] [PubMed] [Google Scholar]
- 83.Guo Y, Wang D, Song Q, Wu T, Zhuang X, Bao Y, Kong M, Qi Y, Tan S, Zhang Z. Erythrocyte Membrane-Enveloped Polymeric Nanoparticles as Nanovaccine for Induction of Antitumor Immunity against Melanoma. ACS Nano. 2015;9:6918–6933. doi: 10.1021/acsnano.5b01042. [DOI] [PubMed] [Google Scholar]
- 84.Zhang Q, Wei W, Wang P, Zuo L, Li F, Xu J, Xi X, Gao X, Ma G, Xie HY. Biomimetic magnetosomes as versatile artificial antigen-presenting cells to potentiate T-cell-based anticancer therapy. ACS Nano. 2017;11:10724–10732. doi: 10.1021/acsnano.7b04955. [DOI] [PubMed] [Google Scholar]
- 85.van der Burg SH, Arens R, Ossendorp F, van Hall T, Melief AJM. Vaccines for established cancer: overcoming the challenges posed by immune evasion. Nat Rev Cancer. 2016;16:219–233. doi: 10.1038/nrc.2016.16. [DOI] [PubMed] [Google Scholar]
- 86.Fang RH, Hu CMJ, Luk BT, Gao WW, Copp JA, Tai YY, O’Connor DE, Zhang LF. Cancer cell membrane-coated nanoparticles for anticancer vaccination and drug delivery. Nano Lett. 2014;14:2181–2188. doi: 10.1021/nl500618u. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 87.Kroll AV, Fang RH, Jiang Y, Zhou J, Wei X, Yu CL, Gao J, Luk BT, Dehaini D, Gao W, et al. Nanoparticulate Delivery of Cancer Cell Membrane Elicits Multiantigenic Antitumor Immunity. Adv Mater. 2017;29 doi: 10.1002/adma.201703969. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 88.Schumacher TN, Schreiber RD. Neoantigens in cancer immunotherapy. Science. 2015;348:69–74. doi: 10.1126/science.aaa4971. [DOI] [PubMed] [Google Scholar]
- 89.Bubeck Wardenburg J, Schneewind O. Vaccine protection against Staphylococcus aureus pneumonia. J Exp Med. 2008;205:287–294. doi: 10.1084/jem.20072208. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 90.Cryz SJ, Jr, Furer E, Germanier R. Effect of chemical and heat inactivation on the antigenicity and immunogenicity of Vibrio cholerae. Infection and Immunity. 1982;38:21–26. doi: 10.1128/iai.38.1.21-26.1982. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 91.Hu CMJ, Fang RH, Luk BT, Zhang LF. Nanoparticle-detained toxins for safe and effective vaccination. Nat Nanotechnol. 2013;8:933–938. doi: 10.1038/nnano.2013.254. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 92.Wei XL, Gao J, Wang F, Ying M, Angsantikul P, Kroll AV, Zhou JR, Gao WW, Lu WY, Fang RH, et al. In situ capture of bacterial toxins for antivirulence vaccination. Adv Mater. 2017;29:1701644. doi: 10.1002/adma.201701644. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 93.Gao WW, Fang RH, Thamphiwatana S, Luk BT, Li JM, Angsantikul P, Zhang QZ, Hu CMJ, Zhang LF. Modulating antibacterial immunity via bacterial membrane-coated nanoparticles. Nano Lett. 2015;15:1403–1409. doi: 10.1021/nl504798g. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 94.Fan ZY, Zhou H, Li PY, Speer JE, Cheng H. Structural elucidation of cell membrane-derived nanoparticles using molecular probes. J Mat Chem B. 2014;2:8231–8238. doi: 10.1039/c4tb00980k. [DOI] [PubMed] [Google Scholar]