

Introduction
Functional pain syndromes (FPS) occur without identifiable noxious stimulus or injury and include diverse conditions such as fibromyalgia, irritable bowel syndrome, migraine, medication overuse headache, and others [1,6,14,20,40,44,46,60,61,66,78,79]. The mechanisms underlying FPS are not well understood and while differences undoubtedly exist between these conditions it is also likely that there is shared and overlapping biology. Stress is commonly identified as a “trigger” of FPS [27,31,36,54,68,76,77]. Repeated, severe, or uncontrolled stress can elicit maladaptive dysregulation that alters brain networks to promote sensitized states (i.e. “allostatic overload”) [3,55]. Repeated stress may increase vulnerability to pain attacks and promote transformation from episodic to chronic pain [17,53]. Frequency of attacks, pain intensity, and the presence of allodynia (i.e., enhanced responses to normally innocuous stimuli) have been suggested as factors promoting increased risk of pain chronification [6,71].
An additional factor linked to pain chronification is net diminished conditioned pain modulation (CPM) [22,88,92] or diffuse noxious inhibitory controls (DNIC). The DNIC response is a pain inhibits pain phenomenon, originally discovered in rats [2]. In humans, DNIC/CPM is demonstrated by the concurrent application of noxious conditioning and test stimuli to different locations on the body. The application of the conditioning stimulus increases the pain threshold (i.e. produces analgesia) to the test stimulus in subjects with efficient DNIC/CPM reflecting a “bottom-up” modulation of pain through engagement of descending inhibition [93]. Patients with a lower CPM response, measured before scheduled surgery, were significantly more likely to develop chronic post-operative pain [43,94]. Similarly, post-operative hypersensitivity took longer to resolve in rats with a lower pre-surgery DNIC response [70]; suggesting that the DNIC/CPM response may serve as a prospective measure of vulnerability for pain chronification [93]. Critically, the CPM response has been shown to be lost or diminished in patients suffering from chronic pain including individuals with FPS [1,14,20,40,44,46,60,61,66,78,79].
While stress elicits a multitude of physiological responses, it is now understood that dynorphin, an endogenous kappa opioid receptor (KORs) agonist, is a key mediator in central stress circuits. Stress-induced activation of KORs has been shown to promote negative affective states, including depression and anxiety, as well as relapse to drug seeking in addiction [7,15,16,41,47]. Antagonism of KOR signaling with nor-binaltorphimine (nor-BNI), a KOR antagonist, blocks aversive behaviors to stress [42]. KORs are found in multiple brain regions that are relevant to pain, including the central nucleus of the amygdala (CeA) [12,41,50]. The amygdala is a limbic brain area that plays a key role in emotional responses and affective states and disorders including learned fear, anxiety, depression and pain [62]. While outputs from the CeA project to descending pain modulatory pathways including the peri-aqueductal gray (PAG), and ultimately to the spinal cord via the rostral ventromedial medulla (RVM), the potential influences of the amygdala on the DNIC response remain relatively unstudied.
We investigated whether stress-related CeA KOR signaling promotes functional pain by measuring DNIC following hyperalgesic priming with morphine. We demonstrate a lateralized KOR-dependent mechanism in the CeA that mediates a generalized loss of DNIC following stress, suggesting new strategies for therapy.
Methods
Animals
A total of 176 male, Sprague-Dawley rats (175-200 g, Envigo, Indianapolis, Indiana, USA) were used in these studies. Experiments were carried out in accordance with policies set forth by the NIH guidelines for use of laboratory animals and approval from the IACUC at the University of Arizona. Rats were kept in a climate controlled room on a 12 hour light and 12 hour dark cycle with ad libitum access to food and water. We adhered to the ARRIVE guidelines wherever possible. Animals were randomly assigned to experimental groups and the experimenters were blinded to the treatments. A post-hoc power analysis was performed using G*Power software [25] to verify that group sizes were sufficient to detect significant effects.
Morphine Priming
Anesthesia was briefly induced with 5% isoflurane and maintained with 2% isoflurane to allow subcutaneous (s.c.) implantation of osmotic minipumps (Model 2001, Alzet, Cupertino, CA, USA) (1μl/hr) delivering vehicle (0.9% saline, VetOne, Boise, Idaho, USA) or morphine sulfate (7.68 mg/kg/day) for 7 days. The experimenter was blinded to the treatment that the rats received. This dose and method of administration was chosen because it has previously been reported to cause neurons in the dorsal medullary horn to have increased receptive field size, increased activity to noxious heat stimuli, and a decreased threshold to activate to mechanical and electrical stimulation [65]. Anesthesia duration was approximately 5 minutes. Gentamycin (8 mg/kg, VetOne, Boise, Idaho, USA) was administered once immediately following surgery.
Cannulation Surgery
Stereotaxic surgeries were performed in anesthetized rats using i.p. ketamine and xylazine cocktail (80mg/kg ketamine; Western Medical Supply, Arcadia, CA, USA and 12 mg/kg xylazine; Sigma-Aldrich, St. Louis, MO, USA). For all experiments that required targeting of specific brain areas a cannula (26 gauge, PlasticsOne, Roanoke, VA, USA) was implanted on day 10 or 11 after minipump implantation. Coordinates for cannula placement were chosen from a brain atlas [69]. For CeA cannulations a single guide cannula was implanted into the CeA (±4.0 mm mediolateral, −2.0 mm anteroposterior, and −6.0 mm dorsoventral from bregma). For RVM cannulations a double guide cannula (1.2 mm spacing between cannula) was centered over the midline and implanted at 2.0 mm posterior to the intra-aural line and 8.0 mm ventral from the skull. CeA and RVM injectors extended 1 mm below the cannula. Surgeries lasted approximately 30 minutes and rats typically recovered from anesthesia around 1 hour after administration of the anesthetic. Gentamycin (8 mg/kg, VetOne, Boise, Idaho, USA) was administered once during the immediate postoperative period.
KOR Antagonist
Rats used for systemic studies received s.c. injection of the KOR antagonist, nor-binaltorphimine (nor-BNI, 3 mg/kg, 1 mL/kg, Tocris, Bristol, UK), or vehicle (0.9% saline, 1 mL/kg). Rats used for intra-amygdala or RVM studies received nor-BNI (2.5 μg in 0.5 μL per site) or vehicle (0.5 μL per site of 0.9% saline) through the cannula. All nor-BNI or vehicle injections were given 1 hour prior to each environmental bright light stress session and the experimenter was blinded to the assigned treatment group. Although nor-BNI administration has been shown to cause long-term KOR inactivation [9,24,30,32,37] that exceeds the 24 hour period between stress sessions we gave one dose one hour prior to each environmental bright light stress to ensure that KORs were maximally blocked before each stress session.
Environmental Bright Light Stress (BLS)
On days 20 and 21 post-pump implantation rats received bright light stress as described previously [29]. Briefly, rats were placed, unrestrained, in Plexiglass caging and halogen shop lamps were placed on the left and right sides of the cages to reach an illumination of approximately 1400 lux inside of the cages. Rats were exposed to the bright lights for one hour prior to transfer to chambers for testing. Days 20, and 21 post morphine priming were chosen to insure that sensory thresholds had returned to pre-morphine levels reflecting resolution of opioid-induced hyperalgesia (OIH).
Tactile Sensory Thresholds
Sensory thresholds were measured during and after administration of s.c. morphine. Rats were tested with a series of von Frey filaments (Touch Test sensory evaluators, Stoelting, Wood Dale, IL, USA) and withdrawal threshold was calculated using the Dixon up-down method [23]. Hindpaw withdrawal threshold was tested by perpendicular application of the filaments to the plantar surface of both hindpaws and periorbital withdrawal threshold was tested by applying the filaments to the center of the forehead. Left and right hindpaw withdrawal thresholds were averaged before data analysis. Cutoff was 15 g for hindpaw and 8 g for periorbital measurements.
Conditioning Stimulus
An intradermal capsaicin (Sigma-Aldrich, St. Louis, MO, USA) injection was chosen as the conditioning stimulus to induce the DNIC response. Capsaicin was prepared as previously described [26]. Briefly, capsaicin was dissolved in 1:1 tween 80 (Sigma-Aldrich, St. Louis, MO, USA) and 100% ethanol (Decon Laboratories, King of Prussia, PA, USA) to an initial concentration of 50 μg/μL. Capsaicin was then diluted to the final concentration (2.5 μg/μL) with 0.9% saline. Capsaicin solution was prepared 40 minutes prior to injection and was stored at −20°C until injection. Two hours after the second episode of BLS rats were briefly anesthetized with isoflurane (duration less than two minutes) and injected with 50 μl (125 μg) of capsaicin intradermally into the left forepaw.
Test Stimulus
The Randall–Selitto (RS) paw pressure test (Ugo Basile, Varese, Italy) was used to measure analgesia or hyperalgesia. An increasing pressure was applied to the rat hindpaw until there was a withdrawal response (cutoff at 500 g). The pressure applied when the rat withdrew its paw was recorded as the paw withdrawal threshold (PWT). In our model it serves as the test stimulus to measure the DNIC response following application of the conditioning stimulus. Prior to minipump implantation, the Randall-Selitto baseline PWT measurement was taken. On day 7 after pump implantation, a second PWT measurement was taken. After a further 14 days and before bright light stress, rats were evaluated to confirm recovery from OIH demonstrated by a return to baseline PWT. On day 21, two hours after BLS, a pre-capsaicin injection baseline measure was taken, followed by the immediate injection of capsaicin into the left forepaw and the beginning of the DNIC timecourse measurements. PWT was measured three times for each paw at each timepoint and averaged prior to data analysis.
Cannula Verification
At the end of the in vivo experiments, rats were euthanized and injected with 0.5 μl of Higgins Black Magic ink (Chartpak, Leeds, MA, USA) into the cannula for visual confirmation of injection site. Brains were harvested and fixed using 10% formalin for a period of 24 hours. Coronal sections (30 μm thick) from the brain area of interest were cut using a Microm HM 525 cryostat and mounted on Superfrost Plus microscope slides (Fisher Scientific, Pittsburgh, PA, USA). Coronal brain sections were cross-matched with a brain atlas [69] to confirm cannula placement. In total, four rats were excluded from the amygdala studies due to cannula locations being outside of the amygdala upon verification. Two rats in the left CeA experiment (one morphine/vehicle and one saline/nor-BNI) were excluded due to cannula location in the caudate putamen and two rats in the right CeA experiment (one saline/vehicle and one saline nor-BNI) were excluded for the same reason.
Dynorphin A EIA
Rats were pretreated with morphine sulfate or vehicle as described above. Rats received BLS and the DNIC response was evoked as described above. After measuring the DNIC response at the 20 minute post-capsaicin timepoint, rats were deeply anesthetized with 5% isoflurane and decapitated. Tissue punches of left and right central nucleus of the amygdala, hypothalamus, and rostral ventral medial medulla (RVM) were taken. Brains were placed upside down in a brain matrix and slices were taken at −2 to −3 mm from the optic chiasm for amygdala and hypothalamus collection and −10 to −12 mm for RVM. Tissue punches were taken from these slices (1 mm diameter for amygdala, 2 mm diameter for hypothalamus and RVM). Hypothalamus and RVM tissue was solubilized in 100 mL/g PBS that contained a peptidase and phosphatase inhibitor cocktail (Halt, Thermo Scientific, Rockford, IL, USA). Amygdala tissue samples were solubilized in 150 mL/g solution to obtain sufficient supernatant volume to run the EIA kit according to the manufacture protocol. All tissue samples were homogenized by sonication followed by heating to 95°C for 10 minutes. Samples were then centrifuged at 4°C and 10,000 rcf for 20 minutes. Dynorphin A content was measured using an EIA kit (Peninsula Laboratories, San Carlos, CA, USA). A Micro BCA (bicinchoninic acid) Protein Assay Kit (Thermo Scientific, Waltham, MA, USA), an assay for total protein, was also run to standardize the samples for varying amounts of tissue by expressing the dynorphin A content as percent of total protein. For the micro BCA assay samples were diluted 1:10 so that readings would fall in the linear range of the standard curve.
Statistics
Tactile sensory thresholds (von Frey) timecourses, 20 minute DNIC data for nor-BNI experiments, and timecourse data used for the assessment of the effects of stress on DNIC were analyzed by 2-way ANOVA followed by Tukey’s test for multiple comparisons. DNIC timecourses for the assessment of the effects of nor-BNI on the DNIC response were analyzed by 3-way ANOVA and Sidak correction for multiple comparisons was applied. Data shown as percent maximal response were calculated by subtracting the baseline value from the test point value and dividing by the cutoff value (500 g) minus the baseline value. Dynorphin A EIA results were measured by multiple t-tests without the assumption of equal variance and the DNIC responses of rats used for EIA studies were analyzed by t-test with Welch’s correction. 3-way ANOVA data was analyzed using SPSS and all other data was analyzed using GraphPad Prism 7 software.
Results
Morphine priming produces transient hypersensitivity
Delivery of morphine by s.c. minipumps produced time-related and reversible hypersensitivity to innocuous tactile stimuli (von Frey test). Morphine treated rats exhibited significant tactile periorbital (Figure 1A; p=0.0225; F(1,31)=5.765) and hindpaw (Figure 1B, p<0.0001; F(1,99)=113.4) allodynia that was apparent by day 4 (periorbital: p=0.0047, hindpaw: p<0.0001) and reached maximal levels within 7 days (periorbital and hindpaw: p<0.0001). Both periorbital and hindpaw allodynia resolved approximately 11 days after the end of morphine treatment.
Figure 1. Morphine infusion for 7 days causes hypersensitivity that resolves by day 21.
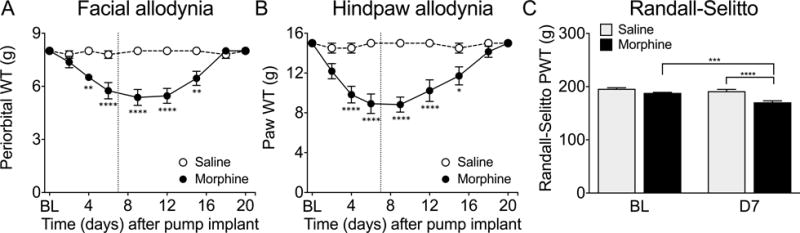
Morphine or saline was delivered by subcutaneously implanted osmotic minipump for seven days. Morphine-primed rats showed periorbital (A) and hindpaw (B) allodynia to von Frey filaments beginning on day 4 of morphine treatment. Allodynia resolved by day 18 (A and B). Morphine-treated rats also showed hindpaw hyperalgesia on the Randall-Selitto test on day 7 of drug treatment compared to morphine-treated baseline measures and compared to saline rats on day 7 (C). **** p<0.0001, *** p<0.001, ** p<0.01, * p<0.05 significant difference compared to baseline or as indicated. Allodynia experiments (A and B) n=7 per group, Randall-Selitto experiment (C) n= 98 saline, 64 morphine (data combined from rats used in all experiments in this paper). Data analyzed by two-way ANOVA followed by Tukey’s test for multiple comparisons.
Paw withdrawal threshold (PWT) to a noxious mechanical stimulus (Randall-Selitto test) was evaluated in a separate cohort of rats. Morphine, but not saline, treated rats showed a significant decrease in the Randall-Sellitto PWT in either hindpaw when tested on day 7 (Figure 1C, p<0.0001; F(1,320)=30.03). Measurements of PWT from the left and right hindpaws of each rat were averaged for each animal and the average was used to calculate the overall hyperalgesic score for the treatment group. The observed hyperalgesia resolved approximately one week following the end of morphine treatment (day 14, p=0.8135, unpaired t-test, data not shown). In rats previously implanted with cannulas, the PWT was tested on day 20 (just prior to the first stress exposure) and was not significantly different from pre-morphine levels at this timepoint (p=0.2163, unpaired t-test, data not shown).
Stress reinstates allodynia only in morphine-primed rats
In rats previously exposed to morphine, exposure to a period of environmental stress resulting from bright lights (i.e., bright light stress, BLS) reinstated tactile allodynia. Morphine-primed rats showed significant periorbital (Figure 2A, p<0.0001; F(1,77)=43.21) and hindpaw (Figure 2B, p<0.0001; F(1,77)=29.12) allodynia that was maximal by approximately 2 hours (periorbital: p<0.0001, hindaw: p=0.0022) and resolved by 5 hours after the first BLS session (day 20). After the second BLS exposure (day 21) rats again showed allodynia in both the periorbital (Figure 2C, p<0.0001; F(1,60)=55.09) and hindpaw (Figure 2D, p<0.0001; F(1,78)=199.1) regions that was greater in magnitude and longer lasting than that observed on day 20; the periorbital allodynia was significant from 2-4 hours (2: p=0.0001, 3: p=0.0009, 4: p=0.0037) and hindpaw allodynia extended beyond the 6 hour timecourse (hours 1-5: p<0.0001, 6: p=0.0016) of the measurements (Figures 2C and D). No significant changes in tactile thresholds were observed in rats previously treated with saline.
Figure 2. Bright light stress reinstates hypersensitivity in morphine-primed rats.

Morphine-primed rats showed periorbital (A and C) and hindpaw (B and D) allodynia to von Frey filaments after the first (day 20, A and B) and second (day 21, C and D) bright light session. **** p<0.0001, *** p<0.001, ** p<0.01, * p<0.05. n=7 per group. Data analyzed by two-way ANOVA followed by Tukey’s test for multiple comparisons.
Stress impaires the DNIC response in morphine-primed rats
Two hours after the second BLS exposure, the DNIC response was induced by intradermal forepaw capsaicin injection (i.e., the conditioning stimulus) and followed by the evaluation of the Randall-Selitto PWT (i.e., the test stimulus)(see Figure 3A for timecourse). DNIC was demonstrated by an increase in paw withdrawal thresholds (i.e., analgesia). Following stress, the DNIC response was significantly impaired or lost in morphine-primed animals compared to animals treated with saline (Figure 3B, p<0.0001; F(1,25)=35.35). Notably, morphine priming alone, without exposure to stress, did not alter the DNIC response (Figure 3C) suggesting a requirement for both priming and stress in the modulation of this response.
Figure 3. Morphine priming followed by stress causes a loss of DNIC that is ameliorated by systemic nor-BNI.
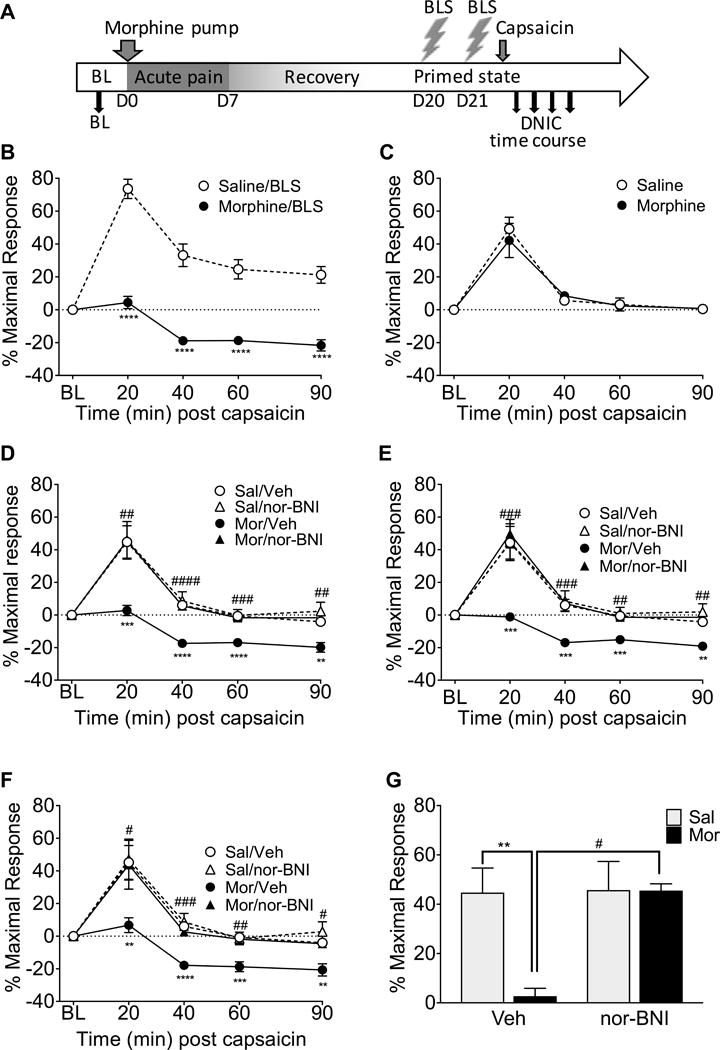
A timeline of experimental procedures is shown for clarity (A). Rats primed with morphine that received bright light stress (BLS) on day 20 and again at day 21 had a significant loss of DNIC compared to saline-primed rats (B). However, morphine-primed rats that were not exposed to BLS did not have a significantly different DNIC response compared to saline-primed rats (C). Nor-BNI administered subcutaneously prior to each BLS period significantly attenuated the loss of DNIC in morphine primed rats (D, E, F). This effect was observed in the left (D), right (E), and combined left and right (averaged together, F) hindpaw measurements. At the 20 minute DNIC timepoint (G), following BLS, the morphine-primed group treated with nor-BNI did not have a significant loss of DNIC whereas the morphine-primed rats treated with vehicle showed a significant loss of DNIC. Also, the morphine primed rats treated with nor-BNI showed significantly higher DNIC than morphine-primed vehicle-treated rats (G). **** p<0.0001, *** p<0.001, ** p<0.01, * p<0.05 significant difference from the saline/BLS group (B), saline group (C), or saline/vehicle group (D-G). #### p<0.0001, ### p<0.001, ## p<0.01, # p<0.05 significant difference between morphine/vehicle and morphine/nor-BNI rats at a timepoint (D-G). B: n= 19 saline, 8 morphine C: n=9 saline, 7 morphine, D-G: n= 10 saline/vehicle, 9 saline/nor-BNI, 10 morphine/vehicle, 6 morphine nor-BNI. Data analyzed by 2-way ANOVA followed by Tukey’s test for multiple comparisons in B, C, and G. Data in D, E, and F analyzed by 3-way ANOVA followed by Sidak’s correction for multiple comparisons.
Systemic KOR blockade prevents the stress-induced loss of DNIC in morphine-primed rats
Rats primed with morphine and exposed to BLS exhibited a significant loss of DNIC compared to saline-primed rats in the left (p=0.0049; F(1,31)=9.205), right (p=0.0028; F(1,31)=10.584), and averaged (p=0.0019; F(1,31)=11.555) hindpaw measurements. There was a significant interaction between priming and systemic nor-BNI pretreatment in the left (p=0.006; F(1,31)=8.589), right (p=0.030, F(1,31)=5.185), and averaged (p=0.008; F(1,31)=8.068) hindpaw measurements. Systemic pretreatment with nor-BNI did not alter the DNIC response in animals primed with saline in the left (Figure 3D) or right (Figure 3E) hindpaw and no effect of systemic nor-BNI was observed when both hindpaws were averaged in saline-primed rats (Figure 3F). In contrast, nor-BNI significantly prevented the loss of the DNIC response following stress that was observed in morphine-primed rats in the left (Figure 3D), right (Figure 3E), and averaged (Figure 3F) hindpaw responses. There was a significant interaction between priming and treatment (p=0.0225; F(1,31)=5.765) at the 20 minute timepoint (Figure 3G) and post-hoc analysis reveals that nor-BNI treatment significantly prevented the stress-induced loss of DNIC in morphine-primed rats (p=0.0126). These results show that KOR antagonism prevents stress-induced loss of DNIC in morphine-primed rats.
KOR blockade in the right CeA prevents stress-induced loss of DNIC in morphine-primed rats
In rats that received right CeA cannulations DNIC was significantly greater following BLS in saline- than in morphine-primed rats. This effect of priming was observed in the left (Figure 4A p<0.0001; F(1,23)=20.988), right (Figure 4B, p<0.0001; F(1,23)=25.95), and averaged (Figure 4C, p=0.0001; F(1,23)=24.792) hindpaw measurements. A significant interaction between priming and treatment was observed in the left (Figure 4A, p=0.0493; F(1,23)=4.307)), right (Figure 4B, p=0.0059; F(1,23)=9.198), and averaged (Figure 4C, p=0.0148; F(1,23)=6.942) hindpaw measurements and post-hoc analysis showed that morphine-primed rats with nor-BNI microinjected into the right CeA prior to BLS had significantly greater DNIC responses than morphine-primed rats that had vehicle microinjected prior to BLS and this effect was observed in the left (Figure 4A), right (Figure 4B), and averaged (Figure 4C) hindpaw measurements, showing that blockade of KOR signaling in the right CeA significantly prevents stress-induced loss of DNIC response bilaterally following morphine priming. Nor-BNI did not significantly alter the DNIC response in saline primed rats (Figures 4A, 4B, 4C). At the 20 min timepoint (Figure 4D) there was a significant interaction between priming and treatment (p=0.0219; F(1,23)=6.041) and post-hoc analysis shows that right CeA nor-BNI treatment significantly prevented the stress-induced loss of DNIC in morphine-primed rats (p=0.0342).
Figure 4. Nor-BNI administered into the right CeA prior to bright light stress (BLS) ameliorates the loss of DNIC.
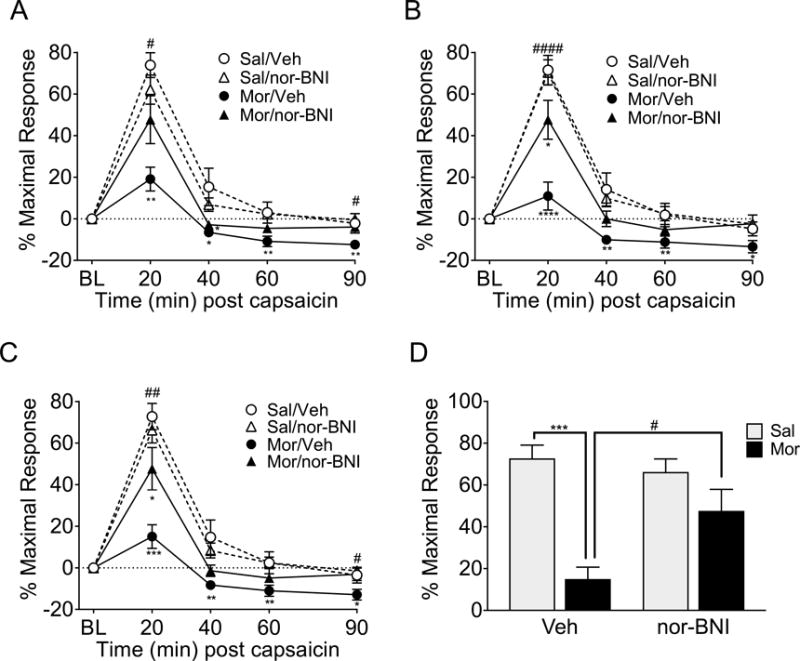
Following BLS, rats primed with morphine had a significant loss of DNIC compared to vehicle-primed rats in both the vehicle and nor-BNI treatment groups in the left (A), right (B), and averaged (C) hindpaw measurements. However, the morphine/nor-BNI treatment group had a significantly higher DNIC response in the left (A), right (B), and averaged (C) than morphine/vehicle when nor-BNI was administered into the right CeA prior to BLS. At the 20 minute DNIC timepoint (D) the morphine/vehicle group has significantly less DNIC response than the saline/vehicle group following BLS, but the morphine/nor-BNI group does not. Additionally, the morphine primed rats treated with nor-BNI have significantly higher DNIC than the morphine-primed rats treated with vehicle. **** p<0.0001, *** p<0.001, ** p<0.01, * p<0.05 significant difference between the saline/vehicle group and other treatment groups. #### p<0.0001, ## p<0.01, #p<0.05 significant difference between morphine/vehicle and morphine/nor-BNI rats at a timepoint. n= 6 saline/vehicle, 7 saline/nor-BNI, 6 morphine/vehicle, 8 morphine nor-BNI. Data analyzed by 3-way ANOVA followed by Sidak correction for multiple comparisons (A-C) or 2-way ANOVA followed by Tukey’s test for multiple comparisons (D).
KOR blockade in the left CeA does not prevent stress-induced loss of DNIC in morphine primed rats
Morphine-primed rats with left CeA cannulas had a significantly lower DNIC response than saline-primed rats in the left (Figure 5A, p<0.0001; F(1,28)=23.825), right (Figure 5B, p<0.0001; F(1,28)=23.845), and averaged (Figure 5C, p<0.0001; F(1,28)=25.600) hindpaw measurements following BLS. In contrast to the right CeA results, microinjection of nor-BNI into the left CeA one hour prior to each BLS session did not alter the stress-induced loss of DNIC in morphine-primed rats. Microinjection of nor-BNI into the left CeA also did not alter the DNIC response of saline-primed rats. These effects were observed in the left (Figure 5A), right (Figure 5B), and averaged (Figure 5C) hindpaw responses. Morphine-priming caused a significant loss of DNIC at the 20 min timepoint (Figure 5D, p<0.0001; F(1,28)=25.33). The effect of nor-BNI or vehicle pretreatment on the loss of DNIC was not significant, nor was the interaction between priming and treatment. These data suggests that KOR signaling in the right, but not the left, CeA promotes the stress-induced loss of DNIC.
Figure 5. Nor-BNI administered into the left CeA prior to bright light stress (BLS) did not ameliorate the loss of DNIC.
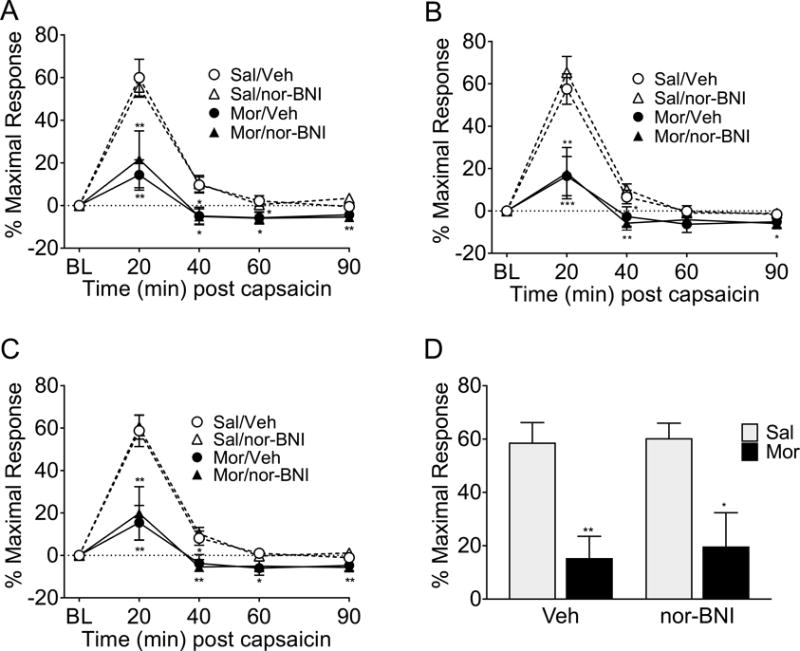
Rats primed with morphine had a significant loss of DNIC following BLS in the left (A), right (B), and averaged (C) hindpaw measurements compared to vehicle-primed rats in both the vehicle and nor-BNI treatment groups. Administration of nor-BNI into the left CeA prior to BLS did not alter the loss of DNIC in the left (A), right (B), or averaged (C) hindpaw measurements. At the 20 minute DNIC timepoint (D) both morphine-primed groups have significantly less DNIC after BLS than saline-primed controls. *** p<0.001, ** p<0.01, * p<0.05 significant difference between the saline/vehicle group and other treatment groups. n= 10 saline/vehicle, 10 saline/nor-BNI, 6 morphine/vehicle, 6 morphine nor-BNI. Data analyzed by 3-way ANOVA with Sidak correction for multiple comparisons (A-C) or 2-way ANOVA followed by Tukey’s test for multiple comparisons (D).
Rostral ventromedial medulla (RVM) nor-BNI does not restore the DNIC response in morphine primed rats
Following BLS morphine-priming caused a loss of DNIC that was not significantly modulated by bilateral administration of nor-BNI into the RVM in the left (Figure 6A p=0.0272; F(1,23) =5.566) and averaged (Figure 6C, p=0.0376, F(1,23)=4.868) hindpaw measurements following BLS. In the right hindpaw measurement (Figure 6B) the morphine-primed rats did not have a significantly different DNIC response compared to the saline-primed rats (p=0.0711, F(1,23)=3.580), but a 2-way ANOVA of left vs right hindpaw data also revealed no significant difference between the hindpaw measurements of the morphine-primed/nor-BNI injected group at any timepoint. Analysis of the 20 minute timepoint (Figure 6D) indicated a reduction of DNIC in both morphine-primed groups that was not restored by nor-BNI; a two-way ANOVA showed a significant effect of priming (p=0.0246; F(1,23)=5.787), but neither the effect of RVM pretreatment nor the interaction between priming and pretreatment was significant.
Figure 6. Nor-BNI administered into the RVM prior to bright light stress (BLS) did not ameliorate the loss of DNIC.
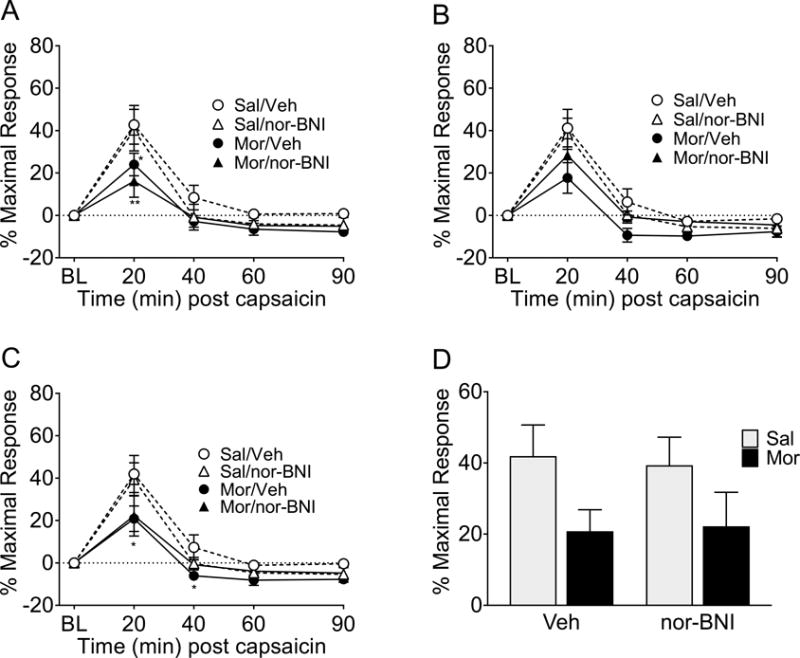
Following BLS, rats primed with morphine had a significant loss of DNIC compared to saline-primed rats in the left (A) and combined left and right (C) hindpaw measures. In the right hindpaw measurement (B) only the morphine/vehicle group had a significant loss of DNIC following BLS, however the morphine/nor-BNI treatment group did not significantly differ from the morphine/vehicle group at any timepoint (A-C). At the 20 minute DNIC timepoint (D) there was a significant effect of priming (p=0.0246), although following Tukey’s post-hoc test for multiple comparisons neither morphine-primed groups showed significantly different DNIC from saline-primed controls after BLS. ** p<0.01, * p<0.05 significant difference between the saline/vehicle group and other treatment groups. n= 6 saline/vehicle, 6 saline/nor-BNI, 9 morphine/vehicle, 6 morphine nor-BNI. Data analyzed by 3-way ANOVA with Sidak correction for multiple comparisons (A-C) or 2-way ANOVA followed by Tukey’s test for multiple comparisons (D).
Stress Increases Dynorphin A Levels in the CeA
Possible stress-induced changes in dynorphin levels were measured in a separate cohort of animals. In these rats, we verified that morphine-priming and stress produced a loss of the DNIC response measured at 20 minutes after capsaicin injection compared to rats with saline priming (Figure 7A, p<0.0001; F(14,13)=1.504). Dynorphin A content was measured at 2.5 hours after the second BLS (Figure 7B). Dynorphin A content was significantly higher in the right CeA of morphine-primed rats compared to vehicle-primed rats (p=0.0227). Dynorphin A content was increased in the left CeA, but did not reach statistical significance (p=0.1017). Significant differences in dynorphin content were not observed between saline- or morphine-primed groups in the hypothalamus or RVM (p=0.1623 and p=0.7709, respectively).
Figure 7. Dynorphin A levels are elevated in the right CeA of morphine-primed rats following stress.
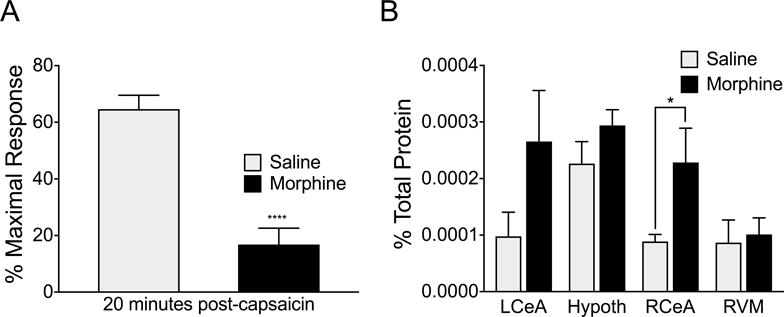
Morphine-primed rats exposed to bright light stress showed a loss of DNIC compared to saline-primed rats immediately prior to tissue collection (A). Dynorphin A levels were significantly increased (p=0.0227, df=14) in the right central nucleus of the amygdala (RCeA) of morphine-primed rats at the 20 minute timepoint of the DNIC timecourse compared to vehicle-primed rats. Quantified dynorphin A levels were not significantly different between saline and morphine-primed rats at this time point in other areas of the brain (left central nucleus of the amygdala (LCeA): p=0.1017, df=15; Hypothalamus (hypoth): p=0.1623, df=18; Rostral ventromedial medulla (RVM): p=0.7709, df=17). **** p<0.01, * p<0.05 significant difference between saline and morphine groups. Data analyzed by t-test with Welch’s correction in A and multiple t-tests assuming unequal variance in B. n-values for A=14 saline, 15 morphine. N values for B= Saline: LCeA (9), RCeA (9), RVM (9), and hypothalamus (8), Morphine: LCeA (8), RCeA (7), RVM (10), hypothalamus (12).
Discussion
Functional pain syndromes (FPS) affect more than 15% of the population worldwide and have been suggested to reflect the interplay between genetic susceptibility, gene-environment interactions, and environmental triggers to elicit a “central sensitivity syndrome” [21]. We studied the role of stress in promoting functional pain by adapting the “two-hit” hyperalgesic priming model thought to promote central sensitization relevant to pain chronification [74]. FPS are characterized by pain without obvious organic origin, and are not known to occur naturally in rodents. We treated rats with morphine to induce a hyperalgesic state similar to that seen in FPS syndromes, without use of a noxious stimulus for the initial priming event. Opioids produce long-lasting neural amplification termed opioid-induced hyperalgesia (OIH) in humans [18,19,52] and in animals [48,80]. Chronic pain patients on opiate therapy show increased sensitivity to noxious stimuli and temporal summation accompanied by decreased tolerance to maximal noxious stimuli as well as a loss of DNIC [95]. The present study investigated the role of KOR signaling by assessing the DNIC response, a dynamic assessment of pain modulatory mechanisms following morphine-priming. We report the loss of DNIC in a preclinical model without injury that requires stress. Additionally, we found that the loss of DNIC is mediated through KOR signaling from the right CeA. The involvement of the CeA or of KOR signaling in producing a loss of DNIC has not previously been reported and extends our previous findings on KOR-mediated modulation of static sensory thresholds following sumatriptan priming [91].
Morphine produced generalized and transient cephalic and extracephalic allodynia in uninjured rats that outlasted the period of drug delivery; thresholds returned to baseline levels within 11 days. OIH was also detected in either hindpaw using a noxious mechanical stimulus with thresholds returning to pre-drug baselines within 14 days of termination of the drug. Following resolution of OIH, animals with normal sensory thresholds and no tissue injury were hypersensitive to challenge with environmental stress. Uninjured morphine-primed rats showed stress-induced, delayed, and generalized allodynia that increased in magnitude and duration upon a second stress exposure, possibly consistent with increasing effects of repeated attacks in FPS [10,49,57]. Notably, our stress conditions produced mild analgesia in saline-primed animals. However, both the stress-induced analgesic and hyperalgesic effects evaluated with the noxious PWT were relatively small and stress-induced hyperalgesia in morphine-primed rats did not reach significance in this test. This bidirectional response suggests that morphine priming may increase vulnerability to stress-related pain.
The circuits underlying the DNIC response are not fully understood. Whether and how bidirectional modulatory circuits from the RVM participate in this response remains unclear. Noxious stimuli activate RVM ON cells to promote descending facilitation [35,59] while DNIC engages descending inhibition from the subnucleus reticularis dorsalis (SRD) [5]. The observed DNIC may thus reflect the combined contribution of these competing descending circuits [83]. Whether inputs from higher brain centers that are important in pain modulation, such as the cortex, hypothalamus, and amygdala, participate in DNIC remains relatively unstudied [67]. The amygdala participates in stress responses and is a part of the affective pain system [81]. We chose to target the CeA because it receives polymodal information from the basal lateral complex and direct nociceptive input from the brainstem [81] including the SRD [45], which is necessary to produce the DNIC response [5,84,86]. Additionally, the CeA sends output to multiple brainstem regions, including the PAG [82] with direct projections to the RVM. Amygdala circuits are activated by stress and we previously found that stress-induced allodynia following morphine priming was blocked by inactivation of the RVM [80].
The dynorphin/KOR system is activated by stress [7,15,16,38,47] through a hypothesized corticotropin-releasing factor (CRF) to dynorphin to KOR pathway [7,8,34,38,42]. KORs are expressed in circuits relevant to pain [12,41,50] and stress increases KOR activation in these circuits, including the amygdala [7,42]. We determined whether systemic, intra-amygdala, or intra-RVM KOR blockade would influence stress-induced DNIC in saline- or morphine-primed rats. In the absence of stress, DNIC was observed in animals previously primed with saline or with morphine. While DNIC was not influenced by stress in saline-primed rats, it was lost or greatly diminished following stress in morphine-primed animals. Systemic nor-BNI prevented the stress-induced loss of DNIC in morphine-primed rats suggesting a key role for KORs in this response.
The amygdala has increased activation during CPM assessment in humans [58,72,89]. However, whether activity in the amygdala, and specifically in the CeA, modulates DNIC has not been reported. We found that KOR antagonism in the right CeA, but not the left CeA or RVM, prevented the loss of DNIC measured in either hindpaw in morphine primed animals. A lateralized role of the amygdala has been shown in inflammatory [13,33,63,75] and neuropathic pain [28] models and mGluR5 activation in the right, but not left, CeA is sufficient to produce peripheral hypersensitivity in the absence of injury [39]. Recently, we demonstrated that microinjection of a KOR agonist into the right, but not left, CeA produced allodynia in both hindpaws following priming with sumatriptan to model medication overuse headache [91]. Collectively, these studies consistently reveal a pronociceptive role of the right, but not left, amygdala regardless of the side of the injury. Our studies extend this concept of the role of the CeA in pain modulation in the absence of injury. Interestingly, neurons in the right lateral capsular division of the central amygdala (CeLC) have much larger receptive fields for nociceptive inputs than left CeLC neurons, including whole body receptive fields even in injury free rats [33], a finding that may be relevant to DNIC that occurs independently of stimulus location. Descending pathways from the SRD to the spinal dorsal horn are, however, thought to be ipsilateral [73,85]. In morphine primed animals, DNIC was observed in both hindpaws. Nevertheless, blockade of KOR signaling in the right CeA prevented the stress-induced loss of DNIC in either hindpaw suggesting a generalized sensitized state induced by systemic morphine and amygdala lateralization in the modulation of stress-induced functional pain that corresponds with injury models.
Following morphine priming, we found that stress produced an increase in dynorphin A content in both amygdalae that reached significance in the right CeA. We had hypothesized that dynorphin A content would also be higher in the hypothalamus following stress in primed animals but this was not observed. It is possible that dynorphin A levels would be elevated in specific regions of the hypothalamus, but we did not differentiate specific nuclei and we were restricted by the use of a coronal section that did not include the entire hypothalamus. We previously reported elevated dynorphin A levels in the left and right CeA following stress in sumatriptan-primed rats [91] even though a KOR antagonist normalized stress-induced allodynia only in the right CeA [91]. Although quantification of total dynorphin A does not mean that the peptide is released our previous study also found increased pKOR in both the left and right CeA [91]. It seems likely that following priming, stress increases dynorphin A levels in the CeA bilaterally, but only the right CeA participates in pain modulation.
The diminished CPM/DNIC response observed in FPS patients is frequently interpreted as reflecting a loss of descending inhibition. An alternate, but equally plausible, interpretation is that this outcome could result from enhanced descending facilitation. A role for the RVM has not generally been supported in DNIC [4]. Recently, Meng and colleagues [64] demonstrated that RVM inactivation restored the loss of DNIC in rats primed with morphine, possibly reflecting inhibition of ON cell firing and consistent with the interpretation that enhanced facilitation may promote a loss of DNIC. The KOR is thought to be expressed on RVM OFF and NEUTRAL, rather than ON cells [90]. KORs are Gi coupled and previous work has demonstrated inhibition of OFF cell discharge with KOR agonists in the RVM [56]. Thus, blockade of RVM KORs should disinhibit OFF cells to enhance, rather than inhibit, descending inhibition. In the present study, we did not observe a stress-induced increase in RVM dynorphin A levels in morphine primed rats and RVM administration of a KOR antagonist did not alter DNIC. These data provide further support for the conclusion that the loss of DNIC results from enhanced facilitation that may mask competing descending inhibition.
Our data are consistent with the possibility that enhanced descending facilitation arising, in part, from stress-related KOR signaling in the right CeA following induction of a sensitized state due to morphine, is a key amplification mechanism that may promote functional pain conditions. We previously reported that enhanced descending facilitation is essential in maintaining chronic neuropathic pain [11]. FPS patients demonstrating a loss of CPM may therefore be especially vulnerable to chronification of pain and may demonstrate higher response outcomes to treatments that reduce descending facilitation. To date, specific mechanisms targeting descending facilitation have not been reported. Here, we demonstrate a key role of KOR signaling in a lateralized descending circuit that may be effectively targeted for the treatment of stress-related pain disorders. Importantly, novel potent, selective, and reversible KOR antagonists such as BTRX-335140 are advancing in development [51,87].
Acknowledgments
These studies were supported by R01 DA 041809, R01 NS 069575, and UH2 NS 093030 from the NIH. The authors have no conflicts of interest to disclose.
Footnotes
Disclosures: None.
References
- 1.Albu S, Gómez-Soriano J, Avila-Martin G, Taylor J. Deficient conditioned pain modulation after spinal cord injury correlates with clinical spontaneous pain measures. Pain. 2015;156:260–272. doi: 10.1097/01.j.pain.0000460306.48701.f9. [DOI] [PubMed] [Google Scholar]
- 2.Le Bars D, Dickenson AH, Besson J-M. Diffuse noxious inhibitory controls (DNIC). I. Effects on dorsal horn convergent neurones in the rat. Pain. 1979;6:283–304. doi: 10.1016/0304-3959(79)90049-6. [DOI] [PubMed] [Google Scholar]
- 3.Borsook D, Maleki N, Becerra L, McEwen B. Understanding migraine through the lens of maladaptive stress responses: a model disease of allostatic load. Neuron. 2012;73:219–34. doi: 10.1016/j.neuron.2012.01.001. [DOI] [PubMed] [Google Scholar]
- 4.Bouhassira D, Bing Z, Le Bars D. Studies of brain structures involved in diffuse noxious inhibitory controls in the rat: the rostral ventromedial medulla. J Physiol. 1993;463:667–687. doi: 10.1113/jphysiol.1993.sp019616. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Bouhassira D, Villanueva L, Bing Z, le Bars D. Involvement of the subnucleus reticularis dorsalis in diffuse noxious inhibitory controls in the rat. Brain Res. 1992;595:353–357. doi: 10.1016/0006-8993(92)91071-l. [DOI] [PubMed] [Google Scholar]
- 6.Bourke JH, Langford RM, White PD. The common link between functional somatic syndromes may be central sensitisation. J Psychosom Res. 2015;78:228–236. doi: 10.1016/j.jpsychores.2015.01.003. [DOI] [PubMed] [Google Scholar]
- 7.Bruchas MR, Land BB, Chavkin C. The dynorphin/kappa opioid system as a modulator of stress-induced and pro-addictive behaviors. Brain Res. 2010;1314:44–55. doi: 10.1016/j.brainres.2009.08.062. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Bruchas MR, Land BB, Lemos JC, Chavkin C. CRF1-R activation of the dynorphin/kappa opioid system in the mouse basolateral amygdala mediates anxiety-like behavior. PLoS One. 2009;4:17–21. doi: 10.1371/journal.pone.0008528. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Bruchas MR, Yang T, Schreiber S, Defino M, Kwan SC, Li S, Chavkin C. Long-acting kappa opioid antagonists disrupt receptor signaling and produce noncompetitive effects by activating c-Jun N-terminal kinase. J Biol Chem. 2007;282:29803–11. doi: 10.1074/jbc.M705540200. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Buchgreitz L, Lyngberg AC, Bendtsen L, Jensen R. Increased pain sensitivity is not a risk factor but a consequence of frequent headache: A population-based follow-up study. Pain. 2008;137:623–630. doi: 10.1016/j.pain.2007.10.023. [DOI] [PubMed] [Google Scholar]
- 11.Burgess SE, Gardell LR, Ossipov MH, Malan TP, Vanderah TW, Lai J, Porreca F. Time-Dependent Descending Facilitation from the Rostral Ventromedial Medulla Maintains, But Does Not Initiate, Neuropathic Pain. J Neurosci. 2002;22:5129–36. doi: 10.1523/JNEUROSCI.22-12-05129.2002. Available: http://www.jneurosci.org/content/22/12/5129.long. Accessed 2 Aug 2017. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Cahill CM, Taylor AMW, Cook C, Ong E, Morón JA, Evans CJ. Does the kappa opioid receptor system contribute to pain aversion? Front. Pharmacol. 2014;5:253. doi: 10.3389/fphar.2014.00253. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Carrasquillo Y, Gereau RW. Hemispheric lateralization of a molecular signal for pain modulation in the amygdala. Mol Pain. 2008;4:24. doi: 10.1186/1744-8069-4-24. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Chang L. Brain Responses to Visceral and Somatic Stimuli in Irritable Bowel Syndrome: a Central Nervous System Disorder? Gastroenterol. Clin North Am. 2005;34:271–279. doi: 10.1016/j.gtc.2005.02.003. [DOI] [PubMed] [Google Scholar]
- 15.Chavkin C. The therapeutic potential of κ-opioids for treatment of pain and addiction. Neuropsychopharmacology. 2011;36:369–70. doi: 10.1038/npp.2010.137. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Chavkin C, Koob GF. Dynorphin, Dysphoria, and Dependence: the Stress of Addiction. Neuropsychopharmacology. 2016;41:373–374. doi: 10.1038/npp.2015.258. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Cho S-J, Chu MK. Risk Factors of Chronic Daily Headache or Chronic Migraine. Curr Pain Headache Rep. 2015;19:465. doi: 10.1007/s11916-014-0465-9. [DOI] [PubMed] [Google Scholar]
- 18.Chu LF, Clark DJ, Angst MS, Geisslinger G, Lotsch J, Rapoport RJ, Rutstein J, Lacouture PG. Opioid tolerance and hyperalgesia in chronic pain patients after one month of oral morphine therapy: a preliminary prospective study. J Pain. 2006;7:43–8. doi: 10.1016/j.jpain.2005.08.001. [DOI] [PubMed] [Google Scholar]
- 19.Compton P, Charuvastra VC, Ling W. Pain intolerance in opioid-maintained former opiate addicts: effect of long-acting maintenance agent. Drug Alcohol Depend. 2001;63:139–46. doi: 10.1016/s0376-8716(00)00200-3. Available: http://www.ncbi.nlm.nih.gov/pubmed/11376918. Accessed 29 May 2017. [DOI] [PubMed] [Google Scholar]
- 20.Corrêa JB, Costa LOP, de Oliveira NTB, Sluka KA, Liebano RE. Central sensitization and changes in conditioned pain modulation in people with chronic nonspecific low back pain: a case–control study. Exp Brain Res. 2015;233:2391–2399. doi: 10.1007/s00221-015-4309-6. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Crabtree D, Ganty P. Common functional pain syndromes. BJA Educ. 2016;16:334–340. Available: https://oup.silverchair-cdn.com/oup/backfile/Content_public/Journal/bjaed/16/10/10.1093_bjaed_mkw010/2/mkw010.pdf?Expires=1496356365&Signature=W6CW15w6bGCkIOjNF~cIYY0pL1r7wD0KiZsGt99FZTTdtu5POLx7rnOY1aUBiqLDXr9t~PVkB-22goZqu8kxbbIjuiY-456k4q1MYV~bKZlZgZtc. Accessed 2 Jun 2017. [Google Scholar]
- 22.Davis MP. The clinical importance of conditioning pain modulation: A review and clinical implications. ACS Symp Ser. 2013;1131:9–38. [Google Scholar]
- 23.Dixon WJ. Efficient Analysis of Experimental Observations. Annu Rev Pharmacol Toxicol. 1980;20:441–462. doi: 10.1146/annurev.pa.20.040180.002301. [DOI] [PubMed] [Google Scholar]
- 24.Endoh T, Matsuura H, Tanaka C, Nagase H. Nor-binaltorphimine: a potent and selective kappa-opioid receptor antagonist with long-lasting activity in vivo. Arch Int Pharmacodyn Ther. 1992;316:30–42. Available: http://www.ncbi.nlm.nih.gov/pubmed/1326932. Accessed 2 Aug 2017. [PubMed] [Google Scholar]
- 25.Faul F, Erdfelder E, Lang A-G, Buchner A. G*Power 3: A flexible statistical power analysis program for the social, behavioral, and biomedical. Behav Res Methods. 2007;39:171–191. doi: 10.3758/bf03193146. Available: http://www.gpower.hhu.de/fileadmin/redaktion/Fakultaeten/Mathematisch-Naturwissenschaftliche_Fakultaet/Psychologie/AAP/gpower/GPower3-BRM-Paper.pdf. Accessed 31 Oct 2017. [DOI] [PubMed] [Google Scholar]
- 26.Ferrari LF, Gear RW, Levine JD. Attenuation of activity in an endogenous analgesia circuit by ongoing pain in the rat. J Neurosci. 2010;30:13699–13706. doi: 10.1523/JNEUROSCI.2867-10.2010. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Fischer S, Lemmer G, Gollwitzer M, Nater UM. Stress and resilience in functional somatic syndromes–a structural equation modeling approach. PLoS One. 2014;9:e111214. doi: 10.1371/journal.pone.0111214. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Gonçalves L, Dickenson AH. Asymmetric time-dependent activation of right central amygdala neurones in rats with peripheral neuropathy and pregabalin modulation. Eur J Neurosci. 2012;36:3204–13. doi: 10.1111/j.1460-9568.2012.08235.x. [DOI] [PubMed] [Google Scholar]
- 29.Green AL, Gu P, De Felice M, Dodick D, Ossipov MH, Porreca F. Increased susceptibility to cortical spreading depression in an animal model of medication-overuse headache. Cephalalgia. 2013;34:594–604. doi: 10.1177/0333102413515344. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Horan P, Taylor J, Yamamura HI, Porreca F. Extremely long-lasting antagonistic actions of nor-binaltorphimine (nor-BNI) in the mouse tail-flick test. J Pharmacol Exp Ther. 1992;260:1237–43. Available: http://www.ncbi.nlm.nih.gov/pubmed/1312164. Accessed 2 Aug 2017. [PubMed] [Google Scholar]
- 31.Iliopoulos P, Damigos D, Kerezoudi E, Limpitaki G, Xifaras M, Skiada D, Tsagkovits A, Skapinakis P. Trigger factors in primary headaches subtypes: a cross-sectional study from a tertiary centre in Greece. BMC Res Notes. 2015;8:393. doi: 10.1186/s13104-015-1390-7. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Jamshidi RJ, Sullivan LC, Jacobs BA, Chavera TA, Berg KA, Clarke WP. Long-term reduction of kappa opioid receptor function by the biased ligand, norbinaltorphimine, requires c-Jun N-terminal kinase activity and new protein synthesis in peripheral sensory neurons. J Pharmacol Exp Ther. 2016;359:319–328. doi: 10.1124/jpet.116.235184. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Ji G, Neugebauer V. Hemispheric lateralization of pain processing by amygdala neurons. J Neurophysiol. 2009;102:2253–64. doi: 10.1152/jn.00166.2009. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Kang-Park M, Kieffer B, Roberts AJ, Siggins GR, Moore SD. Interaction of CRF and Kappa Opioid Systems on GABAergic Neurotransmission in the Mouse Central Amygdala. J Pharmacol Exp Ther. 2015;355:206–211. doi: 10.1124/jpet.115.225870. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Kaplan H, Fields H. Hyperalgesia during acute opioid abstinence: evidence for a nociceptive facilitating function of the rostral ventromedial medulla. J Neurosci. 1991;11:1433–9. doi: 10.1523/JNEUROSCI.11-05-01433.1991. Available: http://www.jneurosci.org/content/11/5/1433. Accessed 17 Jun 2017. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Kirmayer LJ, Robbins JM. Functional Somatic Syndromes Current Concepts of Somatization: Research and Clinical Perspectives. Washington: American Psychiatric Press; 1991. pp. 79–106. [Google Scholar]
- 37.Kishioka S, Kiguchi N, Kobayashi Y, Yamamoto C, Saika F, Wakida N, Ko M-C, Woods JH. Pharmacokinetic evidence for the long-lasting effect of nor-binaltorphimine, a potent kappa opioid receptor antagonist, in mice. Neurosci Lett. 2013;552:98–102. doi: 10.1016/j.neulet.2013.07.040. [DOI] [PubMed] [Google Scholar]
- 38.Knoll AT, Carlezon WA., Jr Dynorphin, stress, and depression. Brain Res. 2010;1314:56–73. doi: 10.1016/j.brainres.2009.09.074. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Kolber BJ, Montana MC, Carrasquillo Y, Xu J, Heinemann SF, Muglia LJ, Gereau RW. Activation of metabotropic glutamate receptor 5 in the amygdala modulates pain-like behavior. J Neurosci. 2010;30:8203–13. doi: 10.1523/JNEUROSCI.1216-10.2010. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Kosek E, Hansson P. Modulatory influence on somatosensory perception from vibration and heterotopic noxious conditioning stimulation (HNCS) in fibromyalgia patients and healthy subjects. Pain. 1997;70:41–51. doi: 10.1016/s0304-3959(96)03295-2. Available: http://www.ncbi.nlm.nih.gov/pubmed/9106808. Accessed 18 Jun 2017. [DOI] [PubMed] [Google Scholar]
- 41.Lalanne L, Ayranci G, Kieffer BL, Lutz P-E. The kappa opioid receptor: from addiction to depression, and back. Front psychiatry. 2014;5:170. doi: 10.3389/fpsyt.2014.00170. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42.Land BB, Bruchas MR, Lemos JC, Xu M, Melief EJ, Chavkin C. The dysphoric component of stress is encoded by activation of the dynorphin kappa-opioid system. J Neurosci. 2008;28:407–14. doi: 10.1523/JNEUROSCI.4458-07.2008. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43.Landau R, Kraft JC, Flint LY, Carvalho B, Richebé P, Cardoso M, Lavand’homme P, Granot M, Yarnitsky D, Cahana A. An experimental paradigm for the prediction of Post-Operative Pain (PPOP) J Vis Exp. 2010;1671 doi: 10.3791/1671. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44.Lautenbacher S, Rollman GB. Possible deficiencies of pain modulation in fibromyalgia. Clin J Pain. 1997;13:189–96. doi: 10.1097/00002508-199709000-00003. Available: http://www.ncbi.nlm.nih.gov/pubmed/9303250. Accessed 18 Jun 2017. [DOI] [PubMed] [Google Scholar]
- 45.Leite-Almeida H, Valle-Fernandes A, Almeida A. Brain projections from the medullary dorsal reticular nucleus: An anterograde and retrograde tracing study in the rat. Neuroscience. 2006;140:577–595. doi: 10.1016/j.neuroscience.2006.02.022. [DOI] [PubMed] [Google Scholar]
- 46.Lewis GN, Rice DA, McNair PJ. Conditioned Pain Modulation in Populations With Chronic Pain: A Systematic Review and Meta-Analysis. J Pain. 2012;13:936–944. doi: 10.1016/j.jpain.2012.07.005. [DOI] [PubMed] [Google Scholar]
- 47.Li W, Sun H, Chen H, Yang X, Xiao L, Liu R, Shao L, Qiu Z. Major Depressive Disorder and Kappa Opioid Receptor Antagonists. Transl Perioper Pain Med. 2016;1:4–16. Available: http://www.ncbi.nlm.nih.gov/pubmed/27213169. Accessed 28 May 2017. [PMC free article] [PubMed] [Google Scholar]
- 48.Li X, Angst MS, Clark JD. Opioid-induced hyperalgesia and incisional pain. Anesth Analg. 2001;93:204–9. doi: 10.1097/00000539-200107000-00040. Available: http://www.ncbi.nlm.nih.gov/pubmed/11429366. Accessed 17 Jun 2017. [DOI] [PubMed] [Google Scholar]
- 49.Louter MA, Bosker JE, van Oosterhout WPJ, van Zwet EW, Zitman FG, Ferrari MD, Terwindt GM. Cutaneous allodynia as a predictor of migraine chronification. Brain. 2013;136:3489–3496. doi: 10.1093/brain/awt251. [DOI] [PubMed] [Google Scholar]
- 50.Mansour A, Fox CA, Akil H, Watson SJ. Opioid-receptor mRNA expression in the rat CNS: anatomical and functional implications. Trends Neurosci. 1995;18:22–29. doi: 10.1016/0166-2236(95)93946-U. [DOI] [PubMed] [Google Scholar]
- 51.Margolis E, LJ VO, Martin W. lectrophysiological characterization of BTRX-335140, a novel selective kappa opioid receptor antagonist, in ventral tegmental area dopamine neurons in rat. Kappa Theraupeutic Conf Abstr. 2017;4:72. [Google Scholar]
- 52.Marion L, SIlverman S, Hansen H, Patel V, Manchikanti L. A Comprehensive Review of Opioid-Induced Hyperalgesia. Pain Physician. 2011;14:145–161. [PubMed] [Google Scholar]
- 53.Mathew NT, Stubits E, Nigam MP. Transformation of Episodic Migraine Into Daily Headache: Analysis of Factors. Headache J Head Face Pain. 1982;22:66–68. doi: 10.1111/j.1526-4610.1982.hed2202066.x. [DOI] [PubMed] [Google Scholar]
- 54.Mayer EA. Irritable Bowel Syndrome. N Engl J Med. 2008;358:1692–1699. doi: 10.1056/NEJMcp0801447. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 55.McEwen BS. Stressed or stressed out: what is the difference? J Psychiatry Neurosci. 2005;30:315–8. Available: http://www.ncbi.nlm.nih.gov/pubmed/16151535. Accessed 17 Jun 2017. [PMC free article] [PubMed] [Google Scholar]
- 56.Meng ID, Johansen JP, Harasawa I, Fields HL. Kappa Opioids Inhibit Physiologically Identified Medullary Pain Modulating Neurons and Reduce Morphine Antinociception. J Neurophysiol. 2005;93:1138–44. doi: 10.1152/jn.00320.2004. Available: http://jn.physiology.org/content/93/3/1138.long. Accessed 17 Jun 2017. [DOI] [PubMed] [Google Scholar]
- 57.Misra UK, Kalita J, Bhoi SK. Allodynia in Migraine. Clin J Pain. 2013;29:577–582. doi: 10.1097/AJP.0b013e31826b130f. [DOI] [PubMed] [Google Scholar]
- 58.Moont R, Crispel Y, Lev R, Pud D, Yarnitsky D. Temporal changes in cortical activation during conditioned pain modulation (CPM), a LORETA study. Pain. 2011;152:1469–1477. doi: 10.1016/j.pain.2011.01.036. [DOI] [PubMed] [Google Scholar]
- 59.Morgan MM, Fields HL. Pronounced changes in the activity of nociceptive modulatory neurons in the rostral ventromedial medulla in response to prolonged thermal noxious stimuli. J Neurophysiol. 1994;72:1161–70. doi: 10.1152/jn.1994.72.3.1161. Available: http://jn.physiology.org/content/72/3/1161. Accessed 17 Jun 2017. [DOI] [PubMed] [Google Scholar]
- 60.Nahman-Averbuch H, Granovsky Y, Coghill RC, Yarnitsky D, Sprecher E, Weissman-Fogel I. Waning of “Conditioned Pain Modulation”: A Novel Expression of Subtle Pronociception in Migraine. Headache J Head Face Pain. 2013;53:1104–1115. doi: 10.1111/head.12117. [DOI] [PubMed] [Google Scholar]
- 61.Ness TJ, Lloyd LK, Fillingim RB. An Endogenous Pain Control System is Altered in Subjects with Interstitial Cystitis. J Urol. 2014;191:364–370. doi: 10.1016/j.juro.2013.08.024. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 62.Neugebauer V. Amygdala pain mechanisms. Handb Exp Pharmacol. 2015;227:261–84. doi: 10.1007/978-3-662-46450-2_13. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 63.Neugebauer V, Li W. Differential sensitization of amygdala neurons to afferent inputs in a model of arthritic pain. J Neurophysiol. 2003;89:716–27. doi: 10.1152/jn.00799.2002. [DOI] [PubMed] [Google Scholar]
- 64.Okada-Ogawa A, Porreca F, Meng ID. Sustained morphine-induced sensitization and loss of diffuse noxious inhibitory controls (DNIC) in dura-sensitive medullary dorsal horn neurons. J Neurosci. 2009;29:15828–15836. doi: 10.1523/JNEUROSCI.3623-09.2009. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 65.Okada-Ogawa A, Porreca F, Meng ID. Sustained Morphine-Induced Sensitization and Loss of Diffuse Noxious Inhibitory Controls in Dura-Sensitive Medullary Dorsal Horn Neurons. J Neurosci. 2009;29:15828–15835. doi: 10.1523/JNEUROSCI.3623-09.2009. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 66.Oono Y, Wang K, Baad-Hansen L, Futarmal S, Kohase H, Svensson P, Arendt-Nielsen L. Conditioned pain modulation in temporomandibular disorders (TMD) pain patients. Exp Brain Res. 2014;232:3111–3119. doi: 10.1007/s00221-014-3997-7. [DOI] [PubMed] [Google Scholar]
- 67.Ossipov MH, Dussor GO, Porreca F. Central modulation of pain. J Clin Invest. 2010;120:3779–3787. doi: 10.1172/JCI43766. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 68.Park J-W, Chu MK, Kim J-M, Park S-G, Cho S-J. Analysis of Trigger Factors in Episodic Migraineurs Using a Smartphone Headache Diary Applications. PLoS One. 2016;11:e0149577. doi: 10.1371/journal.pone.0149577. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 69.Paxinos G, Watson C. The rat brain in stereotaxic coordinates. 6th. Elsevier; 2007. [DOI] [PubMed] [Google Scholar]
- 70.Peters CM, Hayashida K, Suto T, Houle TT, Aschenbrenner CA, Martin TJ, Eisenach JC. Individual Differences in Acute Pain-induced Endogenous Analgesia Predict Time to Resolution of Postoperative Pain in the Rat. Anesthesiology. 2015;122:895–907. doi: 10.1097/ALN.0000000000000593. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 71.Phillips K, Clauw DJ. Central pain mechanisms in chronic pain states–maybe it is all in their head. Best Pract Res Clin Rheumatol. 2011;25:141–54. doi: 10.1016/j.berh.2011.02.005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 72.Piche M, Arsenault M, Rainville P. Cerebral and cerebrospinal processes underlying counterirritation analgesia. J Neurosci. 2009;29:14236–14246. doi: 10.1523/JNEUROSCI.2341-09.2009. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 73.Raboisson P, Dallel R, Bernard J-F, Le Bars D, Villanueva L. Organization of efferent projections from the spinal cervical enlargement to the medullary subnucleus reticularis dorsalis and the adjacent cuneate nucleus: A PHA-L study in the rat. J Comp Neurol. 1996;367:503–517. doi: 10.1002/(SICI)1096-9861(19960415)367:4<503::AID-CNE3>3.0.CO;2-8. [DOI] [PubMed] [Google Scholar]
- 74.Reichling DB, Levine JD. Critical role of nociceptor plasticity in chronic pain. Trends Neurosci. 2009;32:611–8. doi: 10.1016/j.tins.2009.07.007. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 75.Sadler KE, Gartland NM, Cavanaugh JE, Kolber BJ. Central amygdala activation of extracellular signal-regulated kinase 1 and age-dependent changes in inflammatory pain sensitivity in mice. Neurobiol Aging. 2017;56:100–107. doi: 10.1016/j.neurobiolaging.2017.04.010. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 76.Shapiro MA, Nguyen ML. Psychosocial stress and abdominal pain in adolescents. Ment Health Fam Med. 2010;7:65–9. Available: http://www.ncbi.nlm.nih.gov/pubmed/22477924. Accessed 17 Jun 2017. [PMC free article] [PubMed] [Google Scholar]
- 77.Sluka KA, Clauw DJ. Neurobiology of fibromyalgia and chronic widespread pain. Neuroscience. 2016;338:114–129. doi: 10.1016/j.neuroscience.2016.06.006. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 78.Staud R. Abnormal endogenous pain modulation is a shared characteristic of many chronic pain conditions. Expert Rev Neurother. 2012;12:577–85. doi: 10.1586/ern.12.41. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 79.Teepker M, Kunz M, Peters M, Kundermann B, Schepelmann K, Lautenbacher S. Endogenous pain inhibition during menstrual cycle in migraine. Eur J Pain (United Kingdom) 2014;18:989–998. doi: 10.1002/j.1532-2149.2013.00444.x. [DOI] [PubMed] [Google Scholar]
- 80.Vanderah TW, Suenaga NMH, Ossipov MH, Malan TP, Lai J, Porreca F. Tonic Descending Facilitation from the Rostral Ventromedial Medulla Mediates Opioid-Induced Abnormal Pain and Antinociceptive Tolerance. J Neurosci. 2001;21:279–286. doi: 10.1523/JNEUROSCI.21-01-00279.2001. Available: https://pdfs.semanticscholar.org/1e97/647f5e557ab43a5ad98b7c8ce7a5db01edde.pdf. Accessed 17 Jun 2017. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 81.Veinante P, Yalcin I, Barrot M. The amygdala between sensation and affect: a role in pain. J Mol psychiatry. 2013;1:9. doi: 10.1186/2049-9256-1-9. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 82.Vianna DML, Brandão ML. Anatomical connections of the periaqueductal gray: Specific neural substrates for different kinds of fear. Brazilian J Med Biol Res. 2003;36:557–566. doi: 10.1590/s0100-879x2003000500002. [DOI] [PubMed] [Google Scholar]
- 83.Villanueva L. Diffuse Noxious Inhibitory Control (DNIC) as a tool for exploring dysfunction of endogenous pain modulatory systems. Pain. 2009;143:161–162. doi: 10.1016/j.pain.2009.03.003. [DOI] [PubMed] [Google Scholar]
- 84.Villanueva L, Le Bars D. The activation of bulbo-spinal controls by peripheral nociceptive inputs: diffuse noxious inhibitory controls. Biol Res. 1995;28:113–25. Available: http://www.ncbi.nlm.nih.gov/pubmed/8728826. Accessed 21 Jun 2016. [PubMed] [Google Scholar]
- 85.Villanueva L, Bernard JF, Le Bars D. Distribution of spinal cord projections from the medullary subnucleus reticularis dorsalis and the adjacent cuneate nucleus: Aphaseolus vulgaris- leucoagglutinin study in the rat. J Comp Neurol. 1995;352:11–32. doi: 10.1002/cne.903520103. [DOI] [PubMed] [Google Scholar]
- 86.Villanueva L, Bouhassira D, Le Bars D. The medullary subnucleus reticularis dorsalis (SRD) as a key link in both the transmission and modulation of pain signals. Pain. 1996;67:231–240. doi: 10.1016/0304-3959(96)03121-1. [DOI] [PubMed] [Google Scholar]
- 87.Wallace T, Van Order L, Guerrero M, Riley S, Brown S, Porreca F, Rosen H, Roberts E, Martin W. Pharmacological characterization of BTRX-335140. Society for Neuroscience Abstract. 2017 Submitted. [Google Scholar]
- 88.Washington LL, Gibson SJ, Helme RD. Age-related differences in the endogenous analgesic response to repeated cold water immersion in human volunteers. Pain. 2000;89:89–96. doi: 10.1016/S0304-3959(00)00352-3. [DOI] [PubMed] [Google Scholar]
- 89.Wilder-Smith CH, Schindler D, Lovblad K, Redmond SM, Nirkko A. Brain functional magnetic resonance imaging of rectal pain and activation of endogenous inhibitory mechanisms in irritable bowel syndrome patient subgroups and healthy controls. Gut. 2004;53:1595–1601. doi: 10.1136/gut.2003.028514. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 90.Winkler CW, Hermes SM, Chavkin CI, Drake CT, Morrison SF, Aicher SA. Kappa opioid receptor (KOR) and GAD67 immunoreactivity are found in OFF and NEUTRAL cells in the rostral ventromedial medulla. J Neurophysiol. 2006;96:3465–3473. doi: 10.1152/jn.00676.2006. [DOI] [PubMed] [Google Scholar]
- 91.Xie JY, De Felice M, Kopruszinski CM, Eyde N, LaVigne J, Remeniuk B, Hernandez P, Yue X, Goshima N, Ossipov M, King T, Streicher JM, Navratilova E, Dodick D, Rosen H, Roberts E, Porreca F. Kappa opioid receptor antagonists: A possible new class of therapeutics for migraine prevention. Cephalalgia. 2017;37:780–794. doi: 10.1177/0333102417702120. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 92.Yarnitsky D. Conditioned pain modulation (the diffuse noxious inhibitory control-like effect): its relevance for acute and chronic pain states. Curr Opin Anaesthesiol. 2010;23:611–615. doi: 10.1097/ACO.0b013e32833c348b. [DOI] [PubMed] [Google Scholar]
- 93.Yarnitsky D. Role of endogenous pain modulation in chronic pain mechanisms and treatment. Pain. 2015;156(Suppl):S24–31. doi: 10.1097/01.j.pain.0000460343.46847.58. [DOI] [PubMed] [Google Scholar]
- 94.Yarnitsky D, Crispel Y, Eisenberg E, Granovsky Y, Ben-Nun A, Sprecher E, Best L-A, Granot M. Prediction of chronic post-operative pain: Pre-operative DNIC testing identifies patients at risk. Pain. 2008;138:22–28. doi: 10.1016/j.pain.2007.10.033. [DOI] [PubMed] [Google Scholar]
- 95.Zhang Y, Ahmed S, Vo T, St Hilaire K, Houghton M, Cohen AS, Mao J, Chen L. Increased Pain Sensitivity in Chronic Pain Subjects on Opioid Therapy: A Cross-Sectional Study Using Quantitative Sensory Testing. Pain Med. 2015;16:911–922. doi: 10.1111/pme.12606. [DOI] [PubMed] [Google Scholar]