

Abstract
Glucocorticoids (GCs), important therapeutic tools to treat inflammatory and immunosuppressive diseases, can also be used as part of cancer therapy. In oncology, GCs are used as anticancer drugs for lymphohematopoietic malignancies, while in solid neoplasms primarily to control the side effects of chemo/radiotherapy treatments. The molecular mechanisms underlying the effects of GCs are numerous and often overlapping, but not all have been elucidated. In normal, cancerous, and inflammatory tissues, the response to GCs differs based on the tissue type. The effects of GCs are dependent on several factors: the tumor type, the GC therapy being used, the expression level of the glucocorticoid receptor (GR), and the presence of any other stimuli such as signals from immune cells and the tumor microenvironment. Therefore, GCs may either promote or suppress tumor growth via different molecular mechanisms. Stress exposure results in dysregulation of the hypothalamic–pituitary–adrenal axis with increased levels of endogenous GCs that promote tumorigenesis, confirming the importance of GCs in tumor growth. Most of the effects of GCs are genomic and mediated by the modulation of GR gene transcription. Moreover, among the GR-induced genes, glucocorticoid-induced leucine zipper (GILZ), which was cloned and characterized primarily in our laboratory, mediates many GC anti-inflammatory effects. In this review, we analyzed the possible role for GILZ in the effects GCs have on tumors cells. We also suggest that GILZ, by affecting the immune system, tumor microenvironment, and directly cancer cell biology, has a tumor-promoting function. However, it may also induce apoptosis or decrease the proliferation of cancer cells, thus inhibiting tumor growth. The potential therapeutic implications of GILZ activity on tumor cells are discussed here.
Facts
Glucocorticoids (GCs) are crucial therapeutic tools that induce apoptosis in lymphohematopoietic neoplasms.
In non-hematological malignancies, GCs are used as adjuvant therapy to control the side effects of radiotherapy and chemotherapy, but they can either inhibit or induce tumor growth.
Most GC-elicited effects result from the transcriptional regulation of GC receptor target genes, such as glucocorticoid-induced leucine zipper (GILZ) and its transcriptional variant long-GILZ (L-GILZ).
GILZ mediates most of anti-inflammatory/immunosuppressive GC effects.
L-GILZ inhibits tumor growth by p53 activation.
Open Questions
Could GILZ mediate GC effects on cancer cell development or inhibition?
Could GILZ, directly or indirectly, participate in neoplastic cell growth?
Does L-GILZ have different functions from GILZ in the context of tumor growth?
Introduction
Glucocorticoids (GCs) are produced by the adrenal gland and regulated by the hypothalamic–pituitary–adrenal axis (HPA). Their secretion follows a circadian rhythm, but it is also controlled by numerous stress stimuli1. Natural and synthetic GCs represent the mainstay of therapy for inflammatory and autoimmune diseases, but their role as a cancer therapy is still controversial. Endogenous GCs, together with other stress hormones and neurotransmitters, are key mediators in stress-mediated neoplastic pathologies and significantly affect tumor growth and metastasis2.
GCs have both pro- and anti-apoptotic effects. For their proapoptotic properties, GCs are used in lymphohematopoietic cancer3,4. In contrast, numerous reports suggest that GCs in non-hematological malignancies, often used as adjuvant therapy, may have tumor-promoting effects5,6. Although some therapeutic protocols include GCs (e.g., breast and prostate cancer)7, in most tumors their role is still debated and often unknown.
Most GC-elicited effects result from the transcriptional regulation of GC receptor (GR) target genes8. Some of the genes upregulated by GCs have been associated to GC sensitivity, such as glucocorticoid-induced leucine zipper (GILZ, Tsc22d3). Mouse Gilz encodes for a 137-amino acid leucine zipper protein and shares 90% sequence similarity with human Gilz9,10. Long-GILZ (L-GILZ), a transcript GILZ variant, is upregulated by GCs, but differs from GILZ for some functional features11.
GILZ is responsible for most of the anti-inflammatory and immunosuppressive GC effects because it is involved in regulating differentiation, apoptosis, and cell cycle of immune cells12–14. GILZ is involved in the modulation of transcription factor activity and signaling pathways implicated not only in immune response but also in inflammation. For example, GILZ associates with and inhibits nuclear factor-κB (NF-κB)-dependent transcription, thus mediating immunosuppressive and anti-inflammatory effects in T cells, macrophages, bone marrow mesenchymal stem cells, synovial endothelium, and bronchial and intestinal epithelium15–20. Moreover, GILZ interacts with c-Fos and c-Jun and inhibits activating protein-1 (AP-1)-dependent transcription21. It binds Raf22 and Ras23 and inhibits Ras/Raf downstream target activation, including ERK-1/2 and Akt, thus mediating GC antiproliferative effects23. In contrast, L-GILZ binds and inhibits Ras in undifferentiated spermatogonia, contributing to the regulation of spermatogenesis24, binds and activates p53 in neoplastic cells, contributing to GC anticancer effects11,25, and inhibits the proliferation of thyroid cancer cells26.
Thus, GILZ inhibits transcription factors, such as NF-κB15 and AP-121, that are implicated in tumor-promoting functions27,28 and signaling pathways, such Ras-MEK-ERK, that are involved in pro-tumorigenic effects29. L-GILZ activates p53, whose role in preventing tumor development is well established30. These observations suggest that GILZ and L-GILZ interfere with the activity of signals important not only in inflammation but also in cancer. However, while much work has been done to understand the immunosuppressive and anti-inflammatory activity of GILZ, little is known of its involvement in cancer development. Here we review the current literature about GILZ and GCs in modulating both transcription pathways deregulated in cancer cells and the immune response during the development of a neoplastic disease. We also discuss the potential biological relevance of GILZ and GCs in controlling cancer growth.
GCs and cancer
As mentioned above, GCs are included in several protocols for lymphohematopoietic malignancy treatment31 and as a monotherapy or in combination with chemotherapy for solid tumor treatments/adjuvants32. However, dysregulation of the HPA with increased GC secretion has been associated with stress- and aging-related cancer33,34. Hence, what are the effects on tumor growth when endogenous GCs are increased for stress conditions or when exogenous GCs are administered for inflammatory and non-hematological malignancies diseases? Because the immune system is a main GC target, GCs are involved in stress-induced immune dysregulation, thus affecting cancer. Alteration in GC secretion during chronic stress often results in immunosuppression and a failed immune response against the tumor2,35. In many cases, this occurs in patients receiving GC chronic therapy as, if the desired pharmacological effect of exogenous administration of GCs is the control of inflammation and the induction of the immunosuppression, the latter may become dangerous in the presence of a neoplastic disease. Therefore, GCs, by activating GR signaling, may control tumor growth (1) indirectly through modulation of immune system and cells in the tumor microenvironment, or (2) directly by controlling cancer cell survival pathways and affecting tumor cell biology (Fig. 1).
Fig. 1. Schematic illustrating the effect of GCs on cancer growth.

Increased blood levels of glucocorticoids (GCs), due to either GC anti-inflammatory therapy or acute/chronic stress, could affect tumor growth through activity on multiple levels. GCs can act on all the cells of the immune system, on tumor cells, and on the tumor microenvironment
GCs, immune cells, and cancer
GCs contribute to physiological regulation of the immune system by influencing immune cell cytokine release, apoptosis, and cell proliferation. By inhibiting proliferation and inducing apoptosis, endogenous GCs contribute to physiological development of thymus repertoire and modulation of peripheral T-cell activation35,36. The potent regulatory activity of GCs on immune system is also due to the ability of GR to interfere with the activation of many transcription factors that regulate cytokine release, adhesion molecules, receptors, and other factors affected by immune system activity35. For example, GR modulates the activity of NF-κB, AP-137, nuclear factor of activated T cells, transducer and activator of transcription (STAT), cAMP response element-binding protein, interferon regulatory factor 3, T-box expressed in T cells (T-Bet), and GATA-338,39.
By acting on one or more of the above transcription factors, GCs affect macrophage and dendritic cell (DC) functions, impairing their antigen-presenting function, decreasing costimulatory and major histocompaibility complex (MHC) class II molecule expression, downregulating proinflammatory cytokines (such as tumor necrosis factor (TNF) and interleukin (IL)-12), and inducing production of anti-inflammatory cytokines, such as IL-1040,41. Furthermore, GCs interfere with T-cell activation, inhibit activation-induced cell death, and drive T-helper differentiation35,42,43. As a result of these effects, GCs lead to immunosuppression. Under physiological conditions, this immunosuppression is counterbalanced by other factors that regulate the immune system; however, under pharmacological conditions or if chronic stress causes dysregulation of GC secretion, these immunosuppressive effects can enable a tumor to escape immune detection, favoring cancer growth (Fig. 1). This mechanism is so important for tumor survival that cancer cells have “learned” to defend themselves from the immune system through autocrine synthesis of GCs, which is a common feature of malignant cells44. For example, colon carcinoma cells synthesize and release GCs, which are implicated in immune system evasion and progression of tumor45. Furthermore, dexamethasone (DEX), a synthetic GC, upregulates an inhibitory molecule expressed by activated T cells, programmed cell death 1 (PD-1), and induces T-cell tolerance and tumor growth46. This could explain why exogenous and endogenous GCs may negatively influence the cancer response to PD-1-targeting immunotherapy, as highlighted by epidemiological studies47.
On the other hand, GC therapeutic activity mimics the physiological effects of endogenous GCs, although with greater potency and efficacy. In other words, they can induce apoptosis of lymphoid cells, which provides grounds for their inclusion in protocols for the treatment of lymphohematopoietic malignancies31,48–50 (Fig. 1), such as acute Hodgkin’s lymphoma and non-Hodgkin’s lymphoma, multiple myeloma, lymphoblastic leukemia, and chronic lymphocytic leukemia51. The most important molecular mechanisms of GC-triggered apoptosis involve induction of proapoptotic members of the B-cell lymphoma-2 (Bcl-2) family and repression of anti-apoptotic members, such as Bcl-extra-large (Bcl-xL), Bcl-2, myeloid cell leukemia-1 (Mcl-1)4. In contrast, the direct effects of GCs on non-hematological cancer cells remain a controversial issue.
GCs and non-hematological cancer cells
GCs display modest efficacy when used alone or in combination with chemotherapeutic drugs for some non-hematological malignancies, such as breast and prostate cancers7. However, in vitro and in vivo studies32,52 suggest that exogenously administered GCs, as well as high levels of natural GCs caused by stressor stimuli, may induce epithelial tumor growth (Fig. 1)7,34.
In both normal53 and cancer cells, the quantity and type of GR isoforms and the affinity of specific GCs for the GRs contribute to the direct effects of GC on tumor biology2. Clinical observations suggest that increased GR expression in patients with ovarian cancer correlates with poor prognosis54. Increased GR expression may also contribute to progression of castration-resistant prostate cancer55 or be responsible for bypassing androgen receptor blockade56.
In contrast, other studies suggest that GR functions as a tumor suppressor gene. Matthews et al.57 demonstrated that GR promotes chromosome segregation during mitosis and that the GR loss, observed in many cancers, may cause malignant transformation. GR loss or downregulation is also related to epigenetic modifications, such as hypermethylation of its promoter, as observed in a cohort sample of breast cancers58. This epigenetic alteration may confer a benefit to cancer cells. In fact, other studies have shown that GR may attenuate estrogen- and androgen-induced proliferation59,60, even though GR expression on estrogen receptor-negative breast cancer cells was related to poor prognosis61. In small cell lung cancer, GR downregulation correlates with decreased proapoptotic function, an advantage for tumor cell growth62.
Many mechanisms underlie the direct effect of GCs on epithelial cancer cell survival. For example, GCs may promote cancer progression by inhibiting the tumor suppressor protein p5332,63 or by activating p38-MAPK or Akt, signaling pathways all responsible for GC-induced cell proliferation6. In addition, cortisol can stimulate cell proliferation of human oral squamous carcinoma cells via autocrine secretion of IL-664 (Fig. 2).
Fig. 2. Glucocorticoid and epithelial cancer progression.

Glucocorticoid interaction with transcription factors and transduction pathways results in increased proliferation of epithelial cancer cells and resistance to chemotherapy
GCs can induce or protect from apoptosis under different environmental conditions, including the cell type53. In fact, GCs may mediate resistance to chemotherapy-induced apoptosis52,65–67, by inducing the expression of anti-apoptotic genes. For example, GCs induced the expression of anti-apoptotic genes such as Bcl-xL and Bak68 in a breast cancer cell line. Further, in breast, ovarian, and cervical cancers, GCs inhibit paclitaxel-induced apoptosis through inhibition of NF-κB69. Furthermore, anti-apoptotic effects of DEX on breast cancer treated with doxorubicin and/or paclitaxel have been related to DEX-induced mitogen-activated protein kinase (MAPK) phosphatase-1 or IκBα expression, which inhibit MAPK and NF-κB activation, respectively2 (Fig. 2). Of note, the inhibition of GC-induced NF-κB and/or MAPK transduction pathways plays a double role: it is critical for the GC anti-inflammatory effects resulting in inhibition of proinflammatory cytokines, but it is also important for GC-mediated cancer cell survival.
Non-genomic, GC-mediated mechanisms may also play a negative role in cancer treatment. As demonstrated in vitro in a breast cancer cell line, GCs interacting with inducible nitric oxide synthase, a key enzyme in generating nitric oxide, increases the production of reactive oxygen species and produces the oxidative damage to DNA that is responsible for a decreased response to chemotherapy70 (Fig. 2).
The conflicting observations on the effects of GCs on solid tumors is also due to the number of factors that contribute to modulate the GR response. For example, endogenous or exogenous GCs can affect the tumor microenvironment.
GCs and tumor microenvironment
The tumor microenvironment plays a key role in tumor growth71, and as such, all drug therapies affect both the neoplastic cells and the tumor microenvironment72. Hematopoietic cells (T cells, B lymphocytes, natural killer cells, macrophages, DC, and neutrophils), myeloid-derived suppressor cells, mesenchymal cells (stem cells, endothelial cells, fibroblasts, and myofibroblasts), and extracellular matrix are all components of this bidirectional dialogue between tumor and microenvironment cells73. For example, tumor-associated stroma has a gene expression profile different from that of normal stroma thus influencing tumor growth74. Although many studies have suggested a role of GCs on microenvironment-mediated tumor growth, especially in determining tumor progression, the exact contribution of GR signaling on specific microenvironment cells remains unknown. Nearly all cells in the tumor microenvironment express GR, although to varying degrees2. Immune system cells present in the tumor microenvironment are inhibited by GCs, causing a local decrease in immunosurveillance2 (Fig. 1). Moreover, tumor-associated fibroblasts, the major components of stromal cells with roles in tumor growth and metastasis, exhibit alterations in GR-induced gene transcription75. GR is overexpressed in some stroma breast carcinomas76, and increased GC signaling seems to be responsible for insulin resistance in adipocytes, a major component of the breast cancer stroma, with consequent secretion of proinflammatory lymphokines and growth factors implicated in tumor progression77. On the other hand, an in vitro experiment has revealed that DEX inhibits the proliferation of a rare cell population, stem cell-like, a side population present in breast cancer stroma (Fig. 1). The result suggests that DEX targets this population and may increase sensitivity of tumor cells to chemotherapy78. In conclusion, GCs can slow or promote tumor growth through different mechanisms depending on tumor type, microenvironment, and immune system response. Is GILZ involved in those mechanisms?
GILZ and cancer
GILZ, through regulation of cell growth and differentiation, immune cell functions, and other molecular mechanisms, mediates most of GC activities, including the immunosuppressive and anti-inflammatory effects12,13. Inflammation plays an important role in initiating malignant conversion and promoting tumor growth and metastasis79. Inflammatory cells that infiltrate the tumor are an important part of the tumor microenvironment, affecting its growth and aggressiveness. Inflammation, immunomodulation, and cancer initiation and progression occur through common and overlapping pathways80. For example, the tumor-promoting functions of NF-κB, STAT3, and AP-1 are correlated to expression of genes that stimulate cancer cell proliferation and survival81,82. STAT3 and NF-κB interfere with p53, with a potential tumor-promoting activity80,83. It could be hypothesized that GILZ inhibition of NF-κB and AP-1 transcriptional activity15,21 may prevent the development of tumor growth via inhibition of proinflammatory cytokines (Fig. 4), despite the fact that DEX-induced NF-κB inhibition may result in unresponsiveness of tumor cells to chemotherapy2. In addition, GILZ inhibition of Ras and L-GILZ activation of p53 lead to antioncogenic activity in human cancer23,30. However, concomitant GILZ-mediated immunosuppression13,14 may favor the development of tumors. Although its function in cancer has not been sufficiently studied, GILZ, much like GCs, may either promote or inhibit tumor growth depending on the context.
Fig. 4. GILZ and cancer cells.

Through its interaction with different molecules and/or pathways, GILZ has contrasting effects on cancer cells. By binding and inhibiting NF-κB and AP-1, GILZ may inhibit tumor-promoting factors (1); by binding and inhibiting the mammalian target of rapamycin complex-2 (mTORC2) and activating FOXO3Α-mediated transcription of the proapoptotic protein, Bim, GILZ increases the apoptosis induced by tyrosine kinase inhibitors (TKI) in chronic myeloid leukemia (CML) and increases apoptosis of CD34+ stem cells isolated from CML patients (2). In myeloma cells, through an unknown mechanism, GILZ mediates GC apoptotic effects (3). GILZ inhibits the proliferation of Ras-transformed-NIH-3T3 cells (through inhibition of the Ras pathway) (5). GILZ induction increased proliferation of leukemia cells (by increasing mitochondrial oxidative phosphorylation) (4), ovarian cancer cells (by activation of pAkt) (6), and murine dormant melanoma cells (via the inhibition of FOXO3A and its downstream target, the cyclin-dependent kinase inhibitor 1 [p21CIP1]) (7). In lung tumor cells, the data are conflicting: GILZ reduces the response to cyclopamine by affecting Hedgehog signaling, which is responsible for GILZ expression (8), but decreases the migration and invasion of lung cancer cells by binding and targeting hypoxia-responsive transcription factor-1α (HIF-1α) for proteasome degradation, thereby decreasing COX-2 expression (9). Red T-headed leaders indicate inhibition; green arrow-headed leaders indicate activation
GILZ and immune cells
GILZ expression negatively modulates immune cell functions84 by inhibiting transcription factors crucial for immune system activation12,13. Although there is no direct experimental evidence, it may be that GILZ mediates both endogenous stress-associated and exogenous GC effects via its activity on immune cells, causing cancer progression. For example, GILZ, which is upregulated by DEX treatment in mouse and human macrophages, interferes with macrophage and DC functions85,86. In DCs, GILZ plays a critical role in the balance between the active and tolerant DC phenotypes, and it is responsible for decreasing expression of costimulatory and MHC class II molecules and for the generation of T-regulatory (Treg) cells87. Moreover, antigen-specific, IL-10-producing Treg cells, generated by GILZ-expressing DCs, inhibit the T-lymphocyte response88 (Fig. 3). Furthermore, GILZ increases Treg cell activity by favoring transforming growth factor β (TGF-β) signaling89. In T lymphocytes, GILZ regulates T-lymphocyte differentiation, activation, and apoptosis through mechanisms involving thymic selection and Th-1 and Th-2 differentiation90,91. The role of GILZ as an immunosuppressive molecule has been validated by transgenic mouse models (GILZ-TG). For example, mice with forced GILZ expression in DC87 or in bone marrow-derived mesenchymal stem cells (BMSCs)92 exhibit an accumulation of Tregs. Furthermore, GILZ-TG mice, in which GILZ is selectively overexpressed in T cells, exhibit NF-κB inhibition and consequently lower T-cell activation; however, these mice are also less susceptible to experimental colitis18 or spinal cord injury93, indicating that GILZ similar to GCs has both immunosuppressive and anti-inflammatory activities.
Fig. 3. GILZ and immune system.
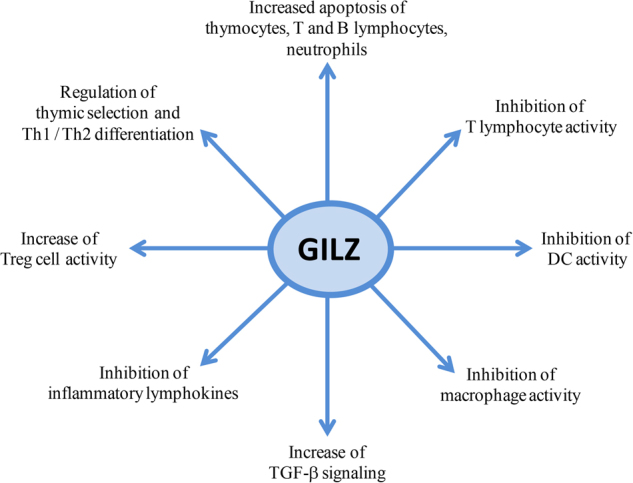
GILZ, through interactions with different transcription factors (as explained in the text), affects the cells of the immune system, thus mediating the physiological and pharmacological effects of GCs. The final outcome of GILZ expression is immunosuppression, which is the desired effect when GCs are administered for therapeutic purposes, but strongly affects tumor growth with an effect that involves both systemic and tumor microenvironment immune cells
In addition, mice lacking GILZ showed accumulation of B precursors in the bone marrow and B lymphocytes in peripheral lymphoid organs94, suggesting a role for GILZ in B-cell apoptosis. Moreover, GILZ led to apoptosis in human neutrophils via downregulation of an anti-apoptotic protein, Mcl-195. The final outcome of GILZ effect on the immune system is immunosuppression that promotes tumor growth (Fig. 3).
The effect of GILZ resembles that of GCs in its physiological control of apoptosis. In fact, thymocytes from GILZ-TG mice activate caspase-8, downregulate Bcl-xL, and undergo apoptosis, suggesting that GILZ, much like GCs50, regulates the thymic repertoire90,96,97. However, GILZ may also protect from apoptosis. GILZ overexpression rescues both the CTLL-2 cell line from IL-2 withdrawal-induced apoptosis by inhibiting Bim expression98 and mature T lymphocytes from T-cell receptor-induced apoptosis by inhibition of NF-κB activation9. This last phenomenon resembles the GC-like mechanism of mutual exclusion42. In addition, GILZ expression has been described in myeloma99, lymphoblastic leukemia, and chronic myeloid leukemia (CML)100, suggesting that GILZ has a role in the pharmacological GC-mediated apoptosis of myelodysplastic disorders. In CML, GC-induced GILZ expression abolished resistance to tyrosine kinase inhibitors, a drug family used in this syndrome, by inactivating the mammalian target of rapamycin complex-2/AKT signaling pathway and by activating FOXO3A-mediated transcription of the proapoptotic protein Bim, which plays a critical role in cell death induced by tyrosine kinase inhibitors100. By the same mechanism, GILZ may contribute to apoptosis of CD34+ stem cells isolated from CML patients100. In multiple myeloma, for which GCs are commonly used as effective therapeutics, decreasing GILZ levels by siRNA knockdown inhibited GC-induced cell death99 (Fig. 4). Accordingly, only a subset of myeloma patients responds to high-dose DEX: those showing transcriptional activity of GR and GILZ expression, whose silencing, again, reverts DEX-induced apoptosis101.
In contrast, recent studies have shown that GILZ reprograms the metabolism of the neoplastic cell, which is essentially based on glycolysis, increasing its mitochondrial oxidative phosphorylation, thus giving to leukemic cells a proliferative, metabolic advantage102 (Fig. 4). The increase of mitochondrial oxidative metabolism has been demonstrated in vitro in both cells overexpressing exogenous GILZ and DEX-treated cells overexpressing endogenous GILZ. The increased proliferation of cells overexpressing GILZ is counterbalanced by an augmented susceptibility to the cytotoxic effects of pro-oxidative, mitochondria-specific drugs (e.g., elesclomol). However, this mechanism is observed in cells expressing exogenous GILZ, but not in those treated with DEX, suggesting that DEX interferes with mitochondrial oxidative phosphorylation-mediated cell death in a GILZ-independent manner102. In addition, the increased mitochondrial oxidative phosphorylation may explain how GILZ protects cells from endoplasmic reticulum (ER) stress-mediated apoptosis, which is a consequence of genetic and epigenetic aberration in many types of cancer103, contributing to cancer cell survival under ER stress. In this case, cells overexpressing exogenous GILZ behave as those expressing DEX-induced endogenous GILZ, suggesting that GILZ mediates the effects of GCs103. Accordingly, endogenous GCs protect from ER stress in the intestinal epithelium, helping to maintain homeostasis and promoting drug effects in cases of inflammatory bowel disease104. Although an important limitation of many of these studies is that they are performed exclusively in vitro, they confirm that the effects of GILZ, much like those of GCs, depend on cell type, environmental conditions, and concomitant stimuli, producing different outcomes.
GILZ and epithelial cancer cells
Constitutive activation of the MAPK signaling pathway, which regulates physiological proliferative events, is often found in inflammation, driving proinflammatory cytokine expression and often contributing to cancer initiation or development29. An interesting involvement of GILZ in GC anti-inflammatory and immunosuppressive effects, which may also impact with cancer, is its ability to inhibit MAPK pathways through binding to some components of the MAPK cascade105. Indeed, GILZ interacts with Ras and Raf, leading to inhibition of downstream Ras-dependent signaling pathways106. By this mechanism, GILZ can mediate the DEX antiproliferative effect on activated T lymphocytes and exert antioncogenic activity in vivo and in vitro in Ras-transformed NIH-3T3 cells106 (Fig. 4). Many epithelial tumors are characterized by the hyperactivating mutations found in Ras or BRAF107. One completely unexplored field involves GILZ effect in human cancer. It is plausive that GILZ may have antioncogenic activity by, for example, binding to Ras and/or Raf. Instead, GILZ expression was observed in the cytoplasm of cancer cells, but not in the epithelium of normal ovaries and benign tumors, suggesting its potential importance for the proliferation of ovarian cancer cells108. In addition, an in vitro model with a cell line derived from ovarian cancer suggested that GILZ enhanced phosphorylated Akt expression and activity, leading to increased proliferation108 (Fig. 4). Therefore, GILZ109 and GR54 expression correlates with poor prognosis in ovarian cancer. In contrast, in estrogen-dependent breast cancer, estrogens downregulate GILZ expression, although the functional meaning of this discovery remains undetermined110.
The role of GILZ has been studied in dormant cells of murine melanoma111. These are cells in mitotic reversible quiescence, present in cancer-treated and healthy individuals alike. Their activity is regulated by dormancy-associated genes and microenvironment stimuli112. GILZ signaling, via the inhibition of FOXO3A and its downstream target, the cell cycle inhibitor, cyclin-dependent kinase inhibitor 1 (p21CIP1), antagonizes cell quiescence, induces cell cycle reactivation and tumor development in dormant murine melanoma cells. Furthermore, GILZ repression induces cellular quiescence, contributing to melanoma inactivity111 (Fig. 4).
High GILZ levels have also been associated in lung cancers with a reduced response to cyclopamine, a drug that inhibits Hedgehog signaling, which induces GILZ113, but is often deregulated in many tumors114. In contrast, it has been demonstrated that GILZ by a mechanism involving the inhibition hypoxia-responsive transcription factor-1α (HIF-1α) decreases invasiveness of epithelial lung cancer cells115. Aerobic glycolysis is a peculiar characteristic of the neoplastic cell. In hypovascularized tumor areas, hypoxia induces HIF-1α production, which intensifies anaerobic glycolysis and lactate production, suppresses the immune response, and induces invasion and metastasis. According to some studies, hypoxia causes downregulation of GR and thereby reduces the effectiveness of GCs116, while other studies suggest that hypoxia causes GR upregulation and increased sensitivity to GCs117. A recent study proposes that GCs, via GILZ expression, suppress hypoxia-induced cyclo-oxygenase of type 2 and HIF-1α activity and decrease migration and invasion of certain epithelial lung cancer cell lines115 (Fig. 4).
In summary, GILZ expression appears to have opposing effects on the neoplastic cell metabolism. It can (1) reprogram the neoplastic cell toward oxidative mitochondrial phosphorylation, as seen before with leukemic cell lines, conferring to tumor cells a proliferative advantage102, or (2) inhibit HIF-1α and decreases its signaling pathway, weakening anaerobic metabolism in lung neoplastic cells, thus hindering cancer invasiveness115. Although some of these observations have limits related to the model (exclusively in vitro), once again the effect of GILZ, just like GCs, seems to be related to the cellular type and presence of other stimuli, such as those from microenvironment.
GILZ and tumor microenvironment
GILZ has not been studied thoroughly in the tumor microenvironment, but it is a crucial mediator of GC-immunosuppressive effects. Its role in the dialogue between the tumor and the microenvironment cells is highly plausible. GILZ could, in fact, influence all the cells of the immune system that infiltrate the tumor microenvironment, as it does with the cells of the systemic immune system13,14,118. As mentioned above, GC-treated DCs express GILZ, responsible for their tolerant phenotype87,119. One recent study demonstrates in vivo and in vitro that, as a result of tumor/microenvironment interaction, GILZ is highly upregulated in tumor microenvironment DCs, resulting in the suppression of immune T-cell response against cancer, which can be restored via GILZ blockade120. Thus, in the tumor microenvironment, where GILZ could be upregulated in macrophages or DCs by immunosuppressive lymphokines such as IL-1016, the tolerogenic antigen-presenting cell phenotype may induce not only tumor-specific T-cell inhibition but also Treg cell activity. As described above, GILZ increases the TGF-β signal that not only leads to Treg cells augmentation89 but also plays an important role in tumor development, both positively and negatively, for example, by its ability to control inflammatory/immune cell and fibroblast infiltration into the tumor microenvironment121.
Other indirect evidence suggests a role of GILZ in the tumor microenvironment. BMSCs, engineered for stable GILZ expression, inhibit lymphocyte proliferation in a mixed lymphocyte reaction assay. Furthermore, GILZ expressed in BMSCs increases the production of IL-10 and decreases the production of IL-6, resulting again in an increase of Treg cell production and activity92. On the other hand, GILZ antagonizes the inhibitory effect of TNF-α on marrow mesenchymal stem cell osteogenic differentiation via the inhibition of TNF-α-induced ERK activation122.
As already seen, GILZ is involved in controlling the cellular cycle of dormant melanoma cells, especially those with stem cell characteristics111. One can hypothesis that signal exchanges between different cells in microenvironment may affect GILZ expression in dormant cells, with the end result being promotion or hindrance of cancer cell growth.
L-GILZ and cancer
L-GILZ is a transcriptional variant of the well-established and studied GILZ11. L-GILZ is upregulated by GCs, albeit at different levels and in different organs and tissues than GILZ. Due to its interaction with several different signaling proteins, L-GILZ can affect multiple outcomes. L-GILZ regulates spermatogenesis through interacting and inhibiting Ras24. L-GILZ controls myogenesis by binding and inhibiting MyoD, a crucial factor for muscle cell differentiation that regulates MyoD/HDAC1 transcriptional activity, thus mediating the anti-myogenic effect of GCs11. In addition, L-GILZ activates p53, and through that mechanism activates p53-dependent antiproliferative and proapoptotic pathways, resulting in inhibited tumor cell growth25,30.
L-GILZ’s antioncogenic activity has been demonstrated particularly in overexpression models. Indeed, L-GILZ forced expression in p53+/+, but not in p53−/−, HCT116 human colorectal carcinoma cells reduces cell proliferation and suppresses the growth of xenografts in mice30. The underlying molecular mechanism involves the ability of L-GILZ to dissociate the MDM2/p53 complex and inhibit MDM2-mediated proteasome degradation of p53, resulting in p53 activation and transcription of downstream genes p21 and PUMA30. Further strengthening the biological significance of these observations, L-GILZ was shown to mediate antiproliferative DEX activity by this mechanism. Indeed, L-GILZ, upregulated by DEX in MCF7 breast cancer cells, binds and activates p53 and induces activation of PUMA and p21, inhibiting cell proliferation. This effect is essentially due to L-GILZ as its silencing reverses the antiproliferative activity of DEX30.
Recently, L-GILZ has been shown to play a role in the proliferation of thyroid cancer cells. L-GILZ is expressed in highly differentiated thyroid cancer cells, while it is absent in less-differentiated or anaplastic cells26. Moreover, its functional role in thyroid cancer is derived from the observation that its overexpression in L-GILZ-deficient cells significantly inhibits their proliferation. In addition, L-GILZ is involved in the antiproliferative effect of kinase inhibitor drugs targeting the MAPK kinase signaling pathway, which is often constitutively activated in both the thyroid and other cancers29. Pharmacologically treating thyroid cancer cells carrying a BRAF mutation (which hyperactivates the MAPK pathway) with kinase inhibitors upregulates L-GILZ and inhibits cellular proliferation. Notably, L-GILZ silencing reverses the antiproliferative activity of the MAPK inhibitors. Moreover, a fusion protein containing L-GILZ, injected into mice transplanted with thyroid cancer cells was found to reduce the growth of xenografts26.
These are initial observations that generate many new questions. For example, could L-GILZ be essential for GC/p53 cross talk and, by this newly described mechanism, contribute to GC therapeutic efficacy? Could the upregulation of L-GILZ, induced by MAPK inhibitor drugs, and its functional significance be a phenomenon involved in multiple tumors and multiple antiproliferative drug mechanisms? And finally, could L-GILZ become an antitumor drug? Several experimental approaches could be taken to answer these intriguing questions.
CONCLUSIONS
In this review, we discussed the possible involvement of GILZ (and L-GILZ) in the effect GCs have on tumor growth. While the role of GILZ as a key mediator of anti-inflammatory and immunosuppressive GC effects is widely accepted and documented, its ability to mediate tumor cell growth inhibition or promotion has only been hypothesized. In fact, the attention of researchers has primarily been focused on the anti-inflammatory activity of GILZ, with the aim of obtaining a GILZ drug with fewer side effects than GCs, which could replace GCs. Consequently, until now, the role of GILZ on the other physiological and pharmacological effects of GCs, including its activity on cancer, remains poorly understood.
GILZ, as an immunosuppressive gene, participates in GC-mediated immunosuppressive effects on both systemic and tumor microenvironment immune cells and thus, by this mechanism, may promote tumor growth. Meanwhile, GILZ is responsible for GC-driven apoptosis in lymphohematopoietic cells, but promotes the growth of ovarian cancer cells via direct effect on tumor cells. Like GCs, GILZ can either favor or inhibit tumor development, depending on the context. Nevertheless, the ability of GILZ and L-GILZ to inhibit Ras, and that of L-GILZ to activate p53, shows their ability to modulate pathways and signals important for the development and progression of the neoplastic cell. Thus, is it plausible that GILZ and L-GILZ are involved in neoplastic pathologies regardless of their ability to mediate the effects of GCs. This observation could develop new field for investigation.
With the finding that GILZ controls the proliferation of dormant melanoma cells, especially those with stem cell characteristics, an intriguing new field of investigation has opened up that may link GCs, GILZ, the immune system, and neoplastic development. Although a role for GILZ has not been studied in the dormant cells of other malignancies, GILZ could play a general regulatory function in tumor dormancy. If this hypothesis is true, future exploration could include the signals regulating GILZ expression and function. In fact, numerous and overlapping stimuli from the microenvironment could control GILZ expression in dormant cells, either directly or indirectly through the involvement of immune cells. Moreover, stress-induced or exogenously administered GCs could act by regulating GILZ expression in dormant cells. This would confer to GILZ a decisive role in the fate of a neoplastic cell. Consequently, the pharmacological modulation of the tumor microenvironment, which may also involve GC and GILZ, becomes a critical potential therapeutic intervention.
Acknowledgements
This study was supported by Progetti di Ricerca di Rilevante Interesse Nazionale (PRIN) 2015 (2015ZT9HXY) and Fondazione Cassa di Risparmio di Perugia, Ricerca Scientifica e Tecnologica (2016.0123.021).
Conflict of interest
The authors declare that they have no conflict of interest.
Footnotes
Edited by G. Raschellà
Publisher's note: Springer Nature remains neutral with regard to jurisdictional claims in published maps and institutional affiliations.
References
- 1.Cain DW, Cidlowski JA. Specificity and sensitivity of glucocorticoid signaling in health and disease. Best. Pract. Res Clin. Endocrinol. Metab. 2015;29:545–556. doi: 10.1016/j.beem.2015.04.007. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Volden PA, Conzen SD. The influence of glucocorticoid signaling on tumor progression. Brain Behav. Immun. 2013;30:S26–S31. doi: 10.1016/j.bbi.2012.10.022. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Rhen T, Cidlowski JA. Antiinflammatory action of glucocorticoids--new mechanisms for old drugs. N. Engl. J. Med. 2005;353:1711–1723. doi: 10.1056/NEJMra050541. [DOI] [PubMed] [Google Scholar]
- 4.Schlossmacher G, Stevens A, White A. Glucocorticoid receptor-mediated apoptosis: mechanisms of resistance in cancer cells. J. Endocrinol. 2011;211:17–25. doi: 10.1530/JOE-11-0135. [DOI] [PubMed] [Google Scholar]
- 5.Coleman RE. Glucocorticoids in cancer therapy. Biotherapy. 1992;4:37–44. doi: 10.1007/BF02171708. [DOI] [PubMed] [Google Scholar]
- 6.Gundisch S, et al. Glucocorticoids augment survival and proliferation of tumor cells. Anticancer Res. 2012;32:4251–4261. [PubMed] [Google Scholar]
- 7.Lin KT, Wang LH. New dimension of glucocorticoids in cancer treatment. Steroids. 2016;111:84–88. doi: 10.1016/j.steroids.2016.02.019. [DOI] [PubMed] [Google Scholar]
- 8.Vandevyver S, Dejager L, Libert C. Comprehensive overview of the structure and regulation of the glucocorticoid receptor. Endocr. Rev. 2014;35:671–693. doi: 10.1210/er.2014-1010. [DOI] [PubMed] [Google Scholar]
- 9.D’Adamio F, et al. A new dexamethasone-induced gene of the leucine zipper family protects T lymphocytes from TCR/CD3-activated cell death. Immunity. 1997;7:803–812. doi: 10.1016/s1074-7613(00)80398-2. [DOI] [PubMed] [Google Scholar]
- 10.Cannarile L, et al. Cloning, chromosomal assignment and tissue distribution of human GILZ, a glucocorticoid hormone-induced gene. Cell Death Differ. 2001;8:201–203. doi: 10.1038/sj.cdd.4400798. [DOI] [PubMed] [Google Scholar]
- 11.Bruscoli S, et al. Glucocorticoid-induced leucine zipper (GILZ) and long GILZ inhibit myogenic differentiation and mediate anti-myogenic effects of glucocorticoids. J. Biol. Chem. 2010;285:10385–10396. doi: 10.1074/jbc.M109.070136. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Ayroldi E, Riccardi C. Glucocorticoid-induced leucine zipper (GILZ): a new important mediator of glucocorticoid action. FASEB J. 2009;23:3649–3658. doi: 10.1096/fj.09-134684. [DOI] [PubMed] [Google Scholar]
- 13.Ronchetti S, Migliorati G, Riccardi C. GILZ as a mediator of the anti-inflammatory effects of glucocorticoids. Front. Endocrinol. (Lausanne) 2015;6:170. doi: 10.3389/fendo.2015.00170. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Beaulieu E, Morand EF. Role of GILZ in immune regulation, glucocorticoid actions and rheumatoid arthritis. Nat. Rev. Rheumatol. 2011;7:340–348. doi: 10.1038/nrrheum.2011.59. [DOI] [PubMed] [Google Scholar]
- 15.Ayroldi E, et al. Modulation of T-cell activation by the glucocorticoid-induced leucine zipper factor via inhibition of nuclear factor kappaB. Blood. 2001;98:743–753. doi: 10.1182/blood.v98.3.743. [DOI] [PubMed] [Google Scholar]
- 16.Berrebi D, et al. Synthesis of glucocorticoid-induced leucine zipper (GILZ) by macrophages: an anti-inflammatory and immunosuppressive mechanism shared by glucocorticoids and IL-10. Blood. 2003;101:729–738. doi: 10.1182/blood-2002-02-0538. [DOI] [PubMed] [Google Scholar]
- 17.Eddleston J, Herschbach J, Wagelie-Steffen AL, Christiansen SC, Zuraw BL. The anti-inflammatory effect of glucocorticoids is mediated by glucocorticoid-induced leucine zipper in epithelial cells. J. Allergy Clin. Immunol. 2007;119:115–122. doi: 10.1016/j.jaci.2006.08.027. [DOI] [PubMed] [Google Scholar]
- 18.Cannarile L, et al. Glucocorticoid-induced leucine zipper is protective in Th1-mediated models of colitis. Gastroenterology. 2009;136:530–541. doi: 10.1053/j.gastro.2008.09.024. [DOI] [PubMed] [Google Scholar]
- 19.Cheng Q, et al. GILZ overexpression inhibits endothelial cell adhesive function through regulation of NF-kappaB and MAPK activity. J. Immunol. 2013;191:424–433. doi: 10.4049/jimmunol.1202662. [DOI] [PubMed] [Google Scholar]
- 20.Yang N, Zhang W, Shi XM. Glucocorticoid-induced leucine zipper (GILZ) mediates glucocorticoid action and inhibits inflammatory cytokine-induced COX-2 expression. J. Cell. Biochem. 2008;103:1760–1771. doi: 10.1002/jcb.21562. [DOI] [PubMed] [Google Scholar]
- 21.Mittelstadt PR, Ashwell JD. Inhibition of AP-1 by the glucocorticoid-inducible protein GILZ. J. Biol. Chem. 2001;276:29603–29610. doi: 10.1074/jbc.M101522200. [DOI] [PubMed] [Google Scholar]
- 22.Ayroldi E, et al. Glucocorticoid-induced leucine zipper inhibits the raf-extracellular signal-regulated kinase pathway by binding to Raf-1. Mol. Cell. Biol. 2002;22:7929–7941. doi: 10.1128/MCB.22.22.7929-7941.2002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Ayroldi E, et al. GILZ mediates the antiproliferative activity of glucocorticoids by negative regulation of Ras signaling. J. Clin. Investig. 2007;117:1605–1615. doi: 10.1172/JCI30724. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Bruscoli S, et al. Long glucocorticoid-induced leucine zipper (L-GILZ) protein interacts with ras protein pathway and contributes to spermatogenesis control. J. Biol. Chem. 2012;287:1242–1251. doi: 10.1074/jbc.M111.316372. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Ayroldi E, Marchetti C, Riccardi C. The novel partnership of L-GILZ andp53: a new affair in cancer? Mol. Cell. Oncol. 2015;2:e975087. doi: 10.4161/23723556.2014.975087. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Ayroldi E, et al. Long glucocorticoid-induced leucine zipper regulates human thyroid cancer cell proliferation. Cell Death Dis. 2018;9:305. doi: 10.1038/s41419-018-0346-y. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Tong L, Yuan Y, Wu S. Therapeutic microRNAs targeting the NF-kappa B signaling circuits of cancers. Adv. Drug Deliv. Rev. 2015;81:1–15. doi: 10.1016/j.addr.2014.09.004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Kappelmann M, Bosserhoff A, Kuphal S. AP-1/c-Jun transcription factors: regulation and function in malignant melanoma. Eur. J. Cell Biol. 2014;93:76–81. doi: 10.1016/j.ejcb.2013.10.003. [DOI] [PubMed] [Google Scholar]
- 29.De Luca A, Maiello MR, D’Alessio A, Pergameno M, Normanno N. The RAS/RAF/MEK/ERK and the PI3K/AKT signalling pathways: role in cancer pathogenesis and implications for therapeutic approaches. Expert Opin. Ther. Targets. 2012;16:S17–S27. doi: 10.1517/14728222.2011.639361. [DOI] [PubMed] [Google Scholar]
- 30.Ayroldi E, et al. L-GILZ binds p53 and MDM2 and suppresses tumor growth through p53 activation in human cancer cells. Cell Death Differ. 2015;22:118–130. doi: 10.1038/cdd.2014.129. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Kofler R. The molecular basis of glucocorticoid-induced apoptosis of lymphoblastic leukemia cells. Histochem. Cell Biol. 2000;114:1–7. doi: 10.1007/s004180000165. [DOI] [PubMed] [Google Scholar]
- 32.Azher S, et al. The non-conventional effects of glucocorticoids in cancer. J. Cell Physiol. 2016;231:2368–2373. doi: 10.1002/jcp.25408. [DOI] [PubMed] [Google Scholar]
- 33.Gassen NC, Chrousos GP, Binder EB, Zannas AS. Life stress, glucocorticoid signaling, and the aging epigenome: Implications for aging-related diseases. Neurosci. Biobehav. Rev. 2017;74:356–365. doi: 10.1016/j.neubiorev.2016.06.003. [DOI] [PubMed] [Google Scholar]
- 34.Shin KJ, et al. Molecular mechanisms underlying psychological stress and cancer. Currie. Pharm. Des. 2016;22:2389–2402. doi: 10.2174/1381612822666160226144025. [DOI] [PubMed] [Google Scholar]
- 35.Cain DW, Cidlowski JA. Immune regulation by glucocorticoids. Nat. Rev. Immunol. 2017;17:233–247. doi: 10.1038/nri.2017.1. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Weisman MH. Corticosteroids. Rheum. Dis. Clin. North Am. 2016;42:xiii. doi: 10.1016/j.rdc.2015.10.002. [DOI] [PubMed] [Google Scholar]
- 37.De Bosscher K, Vanden Berghe W, Haegeman G. The interplay between the glucocorticoid receptor and nuclear factor-kappaB or activator protein-1: molecular mechanisms for gene repression. Endocr. Rev. 2003;24:488–522. doi: 10.1210/er.2002-0006. [DOI] [PubMed] [Google Scholar]
- 38.Langlais D, Couture C, Balsalobre A, Drouin J. The Stat3/GR interaction code: predictive value of direct/indirect DNA recruitment for transcription outcome. Mol. Cell. 2012;47:38–49. doi: 10.1016/j.molcel.2012.04.021. [DOI] [PubMed] [Google Scholar]
- 39.Ratman D, et al. How glucocorticoid receptors modulate the activity of other transcription factors: a scope beyond tethering. Mol. Cell. Endocrinol. 2013;380:41–54. doi: 10.1016/j.mce.2012.12.014. [DOI] [PubMed] [Google Scholar]
- 40.Hu X, Li WP, Meng C, Ivashkiv LB. Inhibition of IFN-gamma signaling by glucocorticoids. J. Immunol. 2003;170:4833–4839. doi: 10.4049/jimmunol.170.9.4833. [DOI] [PubMed] [Google Scholar]
- 41.van den Heuvel MM, et al. Glucocorticoids modulate the development of dendritic cells from blood precursors. Clin. Exp. Immunol. 1999;115:577–583. doi: 10.1046/j.1365-2249.1999.00811.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42.Ashwell JD, Lu FW, Vacchio MS. Glucocorticoids in T cell development and function*. Annu Rev. Immunol. 2000;18:309–345. doi: 10.1146/annurev.immunol.18.1.309. [DOI] [PubMed] [Google Scholar]
- 43.Gillis S, Crabtree GR, Smith KA. Glucocorticoid-induced inhibition of T cell growth factor production. I. The effect on mitogen-induced lymphocyte proliferation. J. Immunol. 1979;123:1624–1631. [PubMed] [Google Scholar]
- 44.Cirillo N, et al. Characterisation of the cancer-associated glucocorticoid system: key role of 11beta-hydroxysteroid dehydrogenase type 2. Br. J. Cancer. 2017;117:984–993. doi: 10.1038/bjc.2017.243. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45.Sidler D, et al. Colon cancer cells produce immunoregulatory glucocorticoids. Oncogene. 2011;30:2411–2419. doi: 10.1038/onc.2010.629. [DOI] [PubMed] [Google Scholar]
- 46.Xing K, Gu B, Zhang P, Wu X. Dexamethasone enhances programmed cell death 1 (PD-1) expression during T cell activation: an insight into the optimum application of glucocorticoids in anti-cancer therapy. BMC Immunol. 2015;16:39. doi: 10.1186/s12865-015-0103-2. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47.Connell CM, et al. Cancer immunotherapy trial registrations increase exponentially but chronic immunosuppressive glucocorticoid therapy may compromise outcomes. Ann. Oncol. 2017;28:1678–1679. doi: 10.1093/annonc/mdx181. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48.Migliorati G, et al. Dexamethasone induces apoptosis in mouse natural killer cells and cytotoxic T lymphocytes. Immunology. 1994;81:21–26. [PMC free article] [PubMed] [Google Scholar]
- 49.Smith LK, Cidlowski JA. Glucocorticoid-induced apoptosis of healthy and malignant lymphocytes. Prog. Brain Res. 2010;182:1–30. doi: 10.1016/S0079-6123(10)82001-1. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 50.Schmidt S, et al. Glucocorticoid-induced apoptosis and glucocorticoid resistance: molecular mechanisms and clinical relevance. Cell Death Differ. 2004;11:S45–S55. doi: 10.1038/sj.cdd.4401456. [DOI] [PubMed] [Google Scholar]
- 51.Pufall MA. Glucocorticoids and Cancer. Adv. Exp. Med. Biol. 2015;872:315–333. doi: 10.1007/978-1-4939-2895-8_14. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 52.Herr I, Pfitzenmaier J. Glucocorticoid use in prostate cancer and other solid tumours: implications for effectiveness of cytotoxic treatment and metastases. Lancet Oncol. 2006;7:425–430. doi: 10.1016/S1470-2045(06)70694-5. [DOI] [PubMed] [Google Scholar]
- 53.Oakley RH, Cidlowski JA. The biology of the glucocorticoid receptor: new signaling mechanisms in health and disease. J. Allergy Clin. Immunol. 2013;132:1033–1044. doi: 10.1016/j.jaci.2013.09.007. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 54.Veneris JT, et al. High glucocorticoid receptor expression predicts short progression-free survival in ovarian cancer. Gynecol. Oncol. 2017;146:153–160. doi: 10.1016/j.ygyno.2017.04.012. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 55.Isikbay M, et al. Glucocorticoid receptor activity contributes to resistance to androgen-targeted therapy in prostate cancer. Horm. Cancer. 2014;5:72–89. doi: 10.1007/s12672-014-0173-2. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 56.Puhr M. The glucocorticoid receptor is a key player for prostate cancer cell survival and a target for improved anti-androgen therapy. Clin. Cancer Res. 2018;24:927–938. doi: 10.1158/1078-0432.CCR-17-0989. [DOI] [PubMed] [Google Scholar]
- 57.Matthews LC, et al. Glucocorticoid receptor regulates accurate chromosome segregation and is associated with malignancy. Proc. Natl Acad. Sci. USA. 2015;112:5479–5484. doi: 10.1073/pnas.1411356112. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 58.Nesset KA, Perri AM, Mueller CR. Frequent promoter hypermethylation and expression reduction of the glucocorticoid receptor gene in breast tumors. Epigenetics. 2014;9:851–859. doi: 10.4161/epi.28484. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 59.Karmakar S, Jin Y, Nagaich AK. Interaction of glucocorticoid receptor (GR) with estrogen receptor (ER) alpha and activator protein 1 (AP1) in dexamethasone-mediated interference of ERalpha activity. J. Biol. Chem. 2013;288:24020–24034. doi: 10.1074/jbc.M113.473819. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 60.Sahu B, et al. FoxA1 specifies unique androgen and glucocorticoid receptor binding events in prostate cancer cells. Cancer Res. 2013;73:1570–1580. doi: 10.1158/0008-5472.CAN-12-2350. [DOI] [PubMed] [Google Scholar]
- 61.Pan D, Kocherginsky M, Conzen SD. Activation of the glucocorticoid receptor is associated with poor prognosis in estrogen receptor-negative breast cancer. Cancer Res. 2011;71:6360–6370. doi: 10.1158/0008-5472.CAN-11-0362. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 62.Sommer P, et al. Glucocorticoid receptor over-expression promotes human small cell lung cancer apoptosis in vivo and thereby slows tumor growth. Endocr. Relat. Cancer. 2010;17:203–213. doi: 10.1677/ERC-09-0241. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 63.Feng Z, et al. Chronic restraint stress attenuates p53 function and promotes tumorigenesis. Proc. Natl Acad. Sci. USA. 2012;109:7013–7018. doi: 10.1073/pnas.1203930109. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 64.Bernabe DG, Tamae AC, Biasoli ER, Oliveira SH. Stress hormones increase cell proliferation and regulates interleukin-6 secretion in human oral squamous cell carcinoma cells. Brain Behav. Immun. 2011;25:574–583. doi: 10.1016/j.bbi.2010.12.012. [DOI] [PubMed] [Google Scholar]
- 65.Zhang C, et al. Corticosteroid co-treatment induces resistance to chemotherapy in surgical resections, xenografts and established cell lines of pancreatic cancer. BMC Cancer. 2006;6:61. doi: 10.1186/1471-2407-6-61. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 66.Herr I, et al. Glucocorticoid cotreatment induces apoptosis resistance toward cancer therapy in carcinomas. Cancer Res. 2003;63:3112–3120. [PubMed] [Google Scholar]
- 67.Zhang C, et al. Glucocorticoid-mediated inhibition of chemotherapy in ovarian carcinomas. Int. J. Oncol. 2006;28:551–558. [PubMed] [Google Scholar]
- 68.Vilasco M, et al. Glucocorticoid receptor and breast cancer. Breast Cancer Res. Treat. 2011;130:1–10. doi: 10.1007/s10549-011-1689-6. [DOI] [PubMed] [Google Scholar]
- 69.Huang Y, Johnson KR, Norris JS, Fan W. Nuclear factor-kappaB/IkappaB signaling pathway may contribute to the mediation of paclitaxel-induced apoptosis in solid tumor cells. Cancer Res. 2000;60:4426–4432. [PubMed] [Google Scholar]
- 70.Flaherty RL, et al. Glucocorticoids induce production of reactive oxygen species/reactive nitrogen species and DNA damage through an iNOS mediated pathway in breast cancer. Breast Cancer Res. 2017;19:35. doi: 10.1186/s13058-017-0823-8. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 71.Hui L, Chen Y. Tumor microenvironment: Sanctuary of the devil. Cancer Lett. 2015;368:7–13. doi: 10.1016/j.canlet.2015.07.039. [DOI] [PubMed] [Google Scholar]
- 72.Bakker E, Qattan M, Mutti L, Demonacos C, Krstic-Demonacos M. The role of microenvironment and immunity in drug response in leukemia. Biochim. Biophys. Acta. 2016;1863:414–426. doi: 10.1016/j.bbamcr.2015.08.003. [DOI] [PubMed] [Google Scholar]
- 73.Catalano V, et al. Tumor and its microenvironment: a synergistic interplay. Semin. Cancer Biol. 2013;23:522–532. doi: 10.1016/j.semcancer.2013.08.007. [DOI] [PubMed] [Google Scholar]
- 74.Palumbo A, Jr, Da Costa NdeO, Bonamino MH, Pinto LF, Nasciutti LE. Genetic instability in the tumor microenvironment: a new look at an old neighbor. Mol. Cancer. 2015;14:145. doi: 10.1186/s12943-015-0409-y. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 75.Hidalgo AA, et al. Biochemical characterization of nuclear receptors for vitamin D3 and glucocorticoids in prostate stroma cell microenvironment. Biochem. Biophys. Res. Commun. 2011;412:13–19. doi: 10.1016/j.bbrc.2011.06.181. [DOI] [PubMed] [Google Scholar]
- 76.Smith RA, Lea RA, Weinstein SR, Griffiths LR. Progesterone, glucocorticoid, but not estrogen receptor mRNA is altered in breast cancer stroma. Cancer Lett. 2007;255:77–84. doi: 10.1016/j.canlet.2007.03.019. [DOI] [PubMed] [Google Scholar]
- 77.Park J, Euhus DM, Scherer PE. Paracrine and endocrine effects of adipose tissue on cancer development and progression. Endocr. Rev. 2011;32:550–570. doi: 10.1210/er.2010-0030. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 78.Kim JB, Hwang SE, Yoon SP. Dexamethasone reduces side population fraction through downregulation of ABCG2 transporter in MCF-7 breast cancer cells. Mol. Med. Rep. 2017;16:453–458. doi: 10.3892/mmr.2017.6566. [DOI] [PubMed] [Google Scholar]
- 79.Sethi G, Shanmugam MK, Ramachandran L, Kumar AP, Tergaonkar V. Multifaceted link between cancer and inflammation. Biosci. Rep. 2012;32:1–15. doi: 10.1042/BSR20100136. [DOI] [PubMed] [Google Scholar]
- 80.Colotta F, Allavena P, Sica A, Garlanda C, Mantovani A. Cancer-related inflammation, the seventh hallmark of cancer: links to genetic instability. Carcinogenesis. 2009;30:1073–1081. doi: 10.1093/carcin/bgp127. [DOI] [PubMed] [Google Scholar]
- 81.Grivennikov SI, Greten FR, Karin M. Immunity, inflammation, and cancer. Cell. 2010;140:883–899. doi: 10.1016/j.cell.2010.01.025. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 82.Hoesel B, Schmid JA. The complexity of NF-kappaB signaling in inflammation and cancer. Mol. Cancer. 2013;12:86. doi: 10.1186/1476-4598-12-86. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 83.Grivennikov SI, Karin M. Dangerous liaisons: STAT3 and NF-kappaB collaboration and crosstalk in cancer. Cytokine Growth Factor Rev. 2010;21:11–19. doi: 10.1016/j.cytogfr.2009.11.005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 84.Hoppstadter J, Kiemer AK. Glucocorticoid-induced leucine zipper (GILZ) in immuno suppression: master regulator or bystander? Oncotarget. 2015;6:38446–38457. doi: 10.18632/oncotarget.6197. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 85.Hoppstadter J, et al. Glucocorticoid-induced leucine zipper: a critical factor in macrophage endotoxin tolerance. J. Immunol. 2015;194:6057–6067. doi: 10.4049/jimmunol.1403207. [DOI] [PubMed] [Google Scholar]
- 86.Krzysiek R. Role of glucocorticoid-induced leucine zipper (GILZ) expression by dendritic cells in tolerance induction. Transplant. Proc. 2010;42:3331–3332. doi: 10.1016/j.transproceed.2010.07.038. [DOI] [PubMed] [Google Scholar]
- 87.Calmette J, et al. Glucocorticoid-induced leucine zipper enhanced expression in dendritic cells is sufficient to drive regulatory T cells expansion in vivo. J. Immunol. 2014;193:5863–5872. doi: 10.4049/jimmunol.1400758. [DOI] [PubMed] [Google Scholar]
- 88.Hamdi H, et al. Induction of antigen-specific regulatory T lymphocytes by human dendritic cells expressing the glucocorticoid-induced leucine zipper. Blood. 2007;110:211–219. doi: 10.1182/blood-2007-08-107896. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 89.Bereshchenko O, et al. GILZ promotes production of peripherally induced Treg cells and mediates the crosstalk between glucocorticoids and TGF-beta signaling. Cell Rep. 2014;7:464–475. doi: 10.1016/j.celrep.2014.03.004. [DOI] [PubMed] [Google Scholar]
- 90.Delfino DV, Agostini M, Spinicelli S, Vito P, Riccardi C. Decrease of Bcl-xL and augmentation of thymocyte apoptosis in GILZ overexpressing transgenic mice. Blood. 2004;104:4134–4141. doi: 10.1182/blood-2004-03-0920. [DOI] [PubMed] [Google Scholar]
- 91.Cannarile L, et al. Increased GILZ expression in transgenic mice up-regulates Th-2 lymphokines. Blood. 2006;107:1039–1047. doi: 10.1182/blood-2005-05-2183. [DOI] [PubMed] [Google Scholar]
- 92.Yang N, Baban B, Isales CM, Shi XM. Crosstalk between bone marrow-derived mesenchymal stem cells and regulatory T cells through a glucocorticoid-induced leucine zipper/developmental endothelial locus-1-dependent mechanism. FASEB J. 2015;29:3954–3963. doi: 10.1096/fj.15-273664. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 93.Esposito E, et al. Glucocorticoid-induced leucine zipper (GILZ) over-expression in T lymphocytes inhibits inflammation and tissue damage in spinal cord injury. Neurotherapeutics. 2012;9:210–225. doi: 10.1007/s13311-011-0084-7. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 94.Bruscoli S, et al. Lack of glucocorticoid-induced leucine zipper (GILZ) deregulates B-cell survival and results in B-cell lymphocytosis in mice. Blood. 2015;126:1790–1801. doi: 10.1182/blood-2015-03-631580. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 95.Espinasse MA, et al. Glucocorticoid-induced leucine zipper is expressed in human neutrophils and promotes apoptosis through Mcl-1 down-regulation. J. Innate Immun. 2016;8:81–96. doi: 10.1159/000439052. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 96.Delfino DV, et al. Glucocorticoid-induced activation of caspase-8 protects the glucocorticoid-induced protein Gilz from proteasomal degradation and induces its binding to SUMO-1 in murine thymocytes. Cell Death Differ. 2011;18:183–190. doi: 10.1038/cdd.2010.86. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 97.Delfino DV, Pozzesi N, Pierangeli S, Ayroldi E, Fierabracci A. Manipulating thymic apoptosis for future therapy of autoimmune diseases. Curr. Pharm. Des. 2011;17:3108–3119. doi: 10.2174/138161211798157621. [DOI] [PubMed] [Google Scholar]
- 98.Asselin-Labat ML, et al. GILZ, a new target for the transcription factor FoxO3, protects T lymphocytes from interleukin-2 withdrawal-induced apoptosis. Blood. 2004;104:215–223. doi: 10.1182/blood-2003-12-4295. [DOI] [PubMed] [Google Scholar]
- 99.Grugan KD, Ma C, Singhal S, Krett NL, Rosen ST. Dual regulation of glucocorticoid-induced leucine zipper (GILZ) by the glucocorticoid receptor and the PI3-kinase/AKT pathways in multiple myeloma. J. Steroid Biochem. Mol. Biol. 2008;110:244–254. doi: 10.1016/j.jsbmb.2007.11.003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 100.Joha S, et al. GILZ inhibits the mTORC2/AKT pathway in BCR-ABL(+) cells. Oncogene. 2012;31:1419–1430. doi: 10.1038/onc.2011.328. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 101.Kervoelen C, et al. Dexamethasone-induced cell death is restricted to specific molecular subgroups of multiple myeloma. Oncotarget. 2015;6:26922–26934. doi: 10.18632/oncotarget.4616. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 102.Andre F, et al. Metabolic rewiring in cancer cells overexpressing the glucocorticoid-induced leucine zipper protein (GILZ): activation of mitochondrial oxidative phosphorylation and sensitization to oxidative cell death induced by mitochondrial targeted drugs. Int. J. Biochem. Cell Biol. 2017;85:166–174. doi: 10.1016/j.biocel.2017.02.011. [DOI] [PubMed] [Google Scholar]
- 103.Andre F, et al. GILZ overexpression attenuates endoplasmic reticulum stress-mediated cell death via the activation of mitochondrial oxidative phosphorylation. Biochem. Biophys. Res. Commun. 2016;478:513–520. doi: 10.1016/j.bbrc.2016.07.053. [DOI] [PubMed] [Google Scholar]
- 104.Das I, et al. Glucocorticoids alleviate intestinal ER stress by enhancing protein folding and degradation of misfolded proteins. J. Exp. Med. 2013;210:1201–1216. doi: 10.1084/jem.20121268. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 105.Ayroldi E, et al. Mechanisms of the anti-inflammatory effects of glucocorticoids: genomic and nongenomic interference with MAPK signaling pathways. FASEB J. 2012;26:4805–4820. doi: 10.1096/fj.12-216382. [DOI] [PubMed] [Google Scholar]
- 106.Ayroldi E, et al. GILZ mediates the antiproliferative activity of glucocorticoids by negative regulation of Ras signaling. J. Clin. Invest. 2007;117:1605–1615. doi: 10.1172/JCI30724. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 107.Oikonomou E, Koustas E, Goulielmaki M, Pintzas A. BRAF vs RAS oncogenes: are mutations of the same pathway equal? Differential signalling and therapeutic implications. Oncotarget. 2014;5:11752–11777. doi: 10.18632/oncotarget.2555. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 108.Redjimi N, et al. Identification of glucocorticoid-induced leucine zipper as a key regulator of tumor cell proliferation in epithelial ovarian cancer. Mol. Cancer. 2009;8:83. doi: 10.1186/1476-4598-8-83. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 109.Gaudin F, et al. Identification of the chemokine CX3CL1 as a new regulator of malignant cell proliferation in epithelial ovarian cancer. PLoS ONE. 2011;6:e21546. doi: 10.1371/journal.pone.0021546. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 110.Tynan SH, Lundeen SG, Allan GF. Cell type-specific bidirectional regulation of the glucocorticoid-induced leucine zipper (GILZ) gene by estrogen. J. Steroid Biochem. Mol. Biol. 2004;91:225–239. doi: 10.1016/j.jsbmb.2004.05.002. [DOI] [PubMed] [Google Scholar]
- 111.Touil Y, et al. Melanoma dormancy in a mouse model is linked to GILZ/FOXO3A-dependent quiescence of disseminated stem-like cells. Sci. Rep. 2016;6:30405. doi: 10.1038/srep30405. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 112.Gomis RR, Gawrzak S. Tumor cell dormancy. Mol. Oncol. 2017;11:62–78. doi: 10.1016/j.molonc.2016.09.009. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 113.Ingram WJ, Wicking CA, Grimmond SM, Forrest AR, Wainwright BJ. Novel genes regulated by Sonic Hedgehog in pluripotent mesenchymal cells. Oncogene. 2002;21:8196–8205. doi: 10.1038/sj.onc.1205975. [DOI] [PubMed] [Google Scholar]
- 114.Galimberti F, et al. Response to inhibition of smoothened in diverse epithelial cancer cells that lack smoothened or patched 1 mutations. Int. J. Oncol. 2012;41:1751–1761. doi: 10.3892/ijo.2012.1599. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 115.Lim W, et al. Glucocorticoids suppress hypoxia-induced COX-2 and hypoxia inducible factor-1alpha expression through the induction of glucocorticoid-induced leucine zipper. Br. J. Pharmacol. 2014;171:735–745. doi: 10.1111/bph.12491. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 116.Huang Y, Zhao JJ, Lv YY, Ding PS, Liu RY. Hypoxia down-regulates glucocorticoid receptor alpha and attenuates the anti-inflammatory actions of dexamethasone in human alveolar epithelial A549 cells. Life Sci. 2009;85:107–112. doi: 10.1016/j.lfs.2009.04.026. [DOI] [PubMed] [Google Scholar]
- 117.Leonard MO, Godson C, Brady HR, Taylor CT. Potentiation of glucocorticoid activity in hypoxia through induction of the glucocorticoid receptor. J. Immunol. 2005;174:2250–2257. doi: 10.4049/jimmunol.174.4.2250. [DOI] [PubMed] [Google Scholar]
- 118.Pozzesi N, et al. IL-2 induces and altered CD4/CD8 ratio of splenic T lymphocytes from transgenic mice overexpressing the glucocorticoid-induced protein GILZ. J. Chemother. 2007;19:562–569. doi: 10.1179/joc.2007.19.5.562. [DOI] [PubMed] [Google Scholar]
- 119.Cohen N, et al. GILZ expression in human dendritic cells redirects their maturation and prevents antigen-specific T lymphocyte response. Blood. 2006;107:2037–2044. doi: 10.1182/blood-2005-07-2760. [DOI] [PubMed] [Google Scholar]
- 120.Lebson L, Wang T, Jiang Q, Whartenby KA. Induction of the glucocorticoid-induced leucine zipper gene limits the efficacy of dendritic cell vaccines. Cancer Gene Ther. 2011;18:563–570. doi: 10.1038/cgt.2011.23. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 121.Yang L, Pang Y, Moses HL. TGF-beta and immune cells: an important regulatory axis in the tumor microenvironment and progression. Trends Immunol. 2010;31:220–227. doi: 10.1016/j.it.2010.04.002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 122.He L, Yang N, Isales CM, Shi XM. Glucocorticoid-induced leucine zipper (GILZ) antagonizes TNF-alpha inhibition of mesenchymal stem cell osteogenic differentiation. PLoS ONE. 2012;7:e31717. doi: 10.1371/journal.pone.0031717. [DOI] [PMC free article] [PubMed] [Google Scholar]