

Summary
Integrins are the key mediators of cell–extracellular matrix (ECM) interaction, linking the ECM to the actin cytoskeleton. Besides localizing at the cell surface, they can be internalized and transported back to the plasma membrane (recycled) or delivered to the late endosomes/lysosomes for degradation. We and others have shown that integrin can be endocytosed together with their ECM ligands. In this short review, I will highlight how extracellular protein (including ECM) endocytosis impinges on the activation of the mechanistic target of rapamycin (mTOR) pathway, a master regulator of cell metabolism and growth. This supports the intriguing hypothesis that ECM components may be considered as nutrient sources, primarily under soluble nutrient‐depleted conditions.
Keywords: extracellular matrix, integrins, vesicular trafficking, mTOR, nutrient signalling, cell metabolism
Introduction
Integrins are a family of 24 transmembrane heterodimers, composed of a combination of 18 α and eight β subunits, that link the extracellular matrix (ECM) with the actin cytoskeleton, mediating a plethora of cell functions, including adhesion, proliferation, survival, migration and differentiation (Streuli 2016). Integrins are expressed in all cell types, except erythrocytes, and they are required for the maintenance of most tissue, as well as for adhesion‐related processes, like leucocyte activation (Manninen & Varjosalo 2017). The ECM is a complex network of secreted proteins which on the one hand provides physical support for tissues and organs and on the other hand has an active role in controlling cell behaviour, both in physiological and pathological conditions, such as in cancer (Daley & Yamada 2013). During cancer progression, tumour cells acquire the ability to detach from the primary tumour mass, invade through the surrounding tissues and form secondary tumours at distant sites, known as metastasis. This process is the main cause of mortality in patients with cancer. Several ECM components have been shown to be strongly upregulated in the stroma of invasive breast tumours compared to localized ductal carcinoma in situ (Lee et al. 2012). This observation highlights the importance of deepening our understanding of the mechanisms controlling cell‐ECM interaction, in order to elucidate how cancer cells exploit changes in the stroma to foster their invasive ability, eventually leading to metastasis formation.
Integrin‐containing adhesive structures mediate the interaction between cells and the surrounding environment. Depending on the substrate composition (presence of laminin, collagen, fibronectin or vitronectin), different integrin heterodimers promote adhesion formation. Upon initial adhesion, small focal complexes are formed, which mature into larger focal adhesions (FAs), mainly localized at the periphery of the cells. Fibrillar adhesions are longer adhesive structures predominantly located towards the middle of the cells, particularly enriched in active α5β1 integrin and tensins. Their formation has been associated with fibronectin (FN) polymerization (Clark et al. 2005). Adhesive structures are continuously remodelled during cell migration and invasion, with the formation of new adhesions at the leading edge and disassembly of mature adhesion at the rear of the cell.
Vesicular traffic of integrins
The endosomal traffic has a key role in controlling adhesion dynamics (Maritzen et al. 2015). Vesicular trafficking is composed of a complex network of vesicular and tubular compartments, whose identity is defined by specific sets of proteins present on their surface, as represented in Figure 1. These include members of the Rab and Arf family of GTPases. Besides characterizing different compartments, these GTPases also control the movement of cargos along the endosomal system. Plasma membrane receptors are internalized, through a variety of different pathways, in early endosomes (EEs), characterized by the presence of Rab5 and early endosome antigen 1 (EEA1). From here, Rab4 coordinates the fast return of cargos back to the plasma membrane, via the so‐called short‐loop recycling pathway. Alternatively, internalized receptors can be transported to the perinuclear recycling compartment (PNRC) before being targeted to the cell surface, in a Rab11‐dependent manner, via the ‘long‐loop’ recycling pathway. Proteins destined for degradation are sorted into multivesicular bodies (MVBs) and Rab7‐positive late endosomes. Fusion with the lysosomes dictates content degradation. Rather than only being the cell ‘waste compartment’, recent findings highlight the multifaceted role of the lysosomes in controlling cell behaviour. Indeed, lysosomes control secretion, plasma membrane repair, autophagy, signalling and energy metabolism (Settembre et al. 2013).
Figure 1.
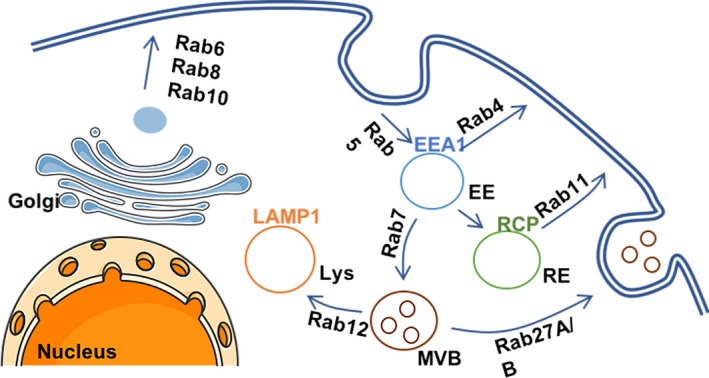
Schematic representation of endosomal trafficking and regulation by Rab small GTPases. EE, early endosome; RE, recycling endosome; MVB, multivesicular body; Lys, lysosome; EEA1, early endosome antigen 1; RCP, Rab coupling protein. [Colour figure can be viewed at http://wileyonlinelibrary.com]
Different integrin heterodimers have been shown to populate the endosomal system, including α1β1, α2β1, α3β1, α5β1, α6β1, αvβ3, αvβ6 and α6β1. Their endocytosis and recycling pathways have recently been extensively reviewed and shown to promote cancer cell proliferation, invasion and metastasis (de Franceschi et al. 2015). Interestingly, the way in which integrins are trafficked intracellularly is key in controlling how they affect cell behaviour (Caswell et al. 2009). Direct interaction between integrins and trafficking regulators dictates the route that internalized integrins will follow. For instance, protein kinase D (PKD) directly binds to the C‐terminal of integrin β3 and, through the phosphorylation of the Rab4 and Rab5 effector Rabaptin‐5, promotes the Rab4‐dependent short‐loop recycling of αvβ3. This pathway is required for fibroblast directed cell migration, as well as cancer cell invasion in the presence of αvβ3 ligands (Christoforides et al. 2012). It is important to note that, at the plasma membrane, integrins can be found in a close or inactive conformation or bound to their ligand in an active conformation. Besides controlling downstream signalling outputs, the same integrin heterodimer is trafficked through distinct compartments depending on its activation status (Arjonen et al. 2012). Different kinetics have been identified for active and inactive β1 integrin. After internalization, while inactive β1 is rapidly recycled back to the plasma membrane in a Rab4‐ and Arf6‐dependent manner, active β1 accumulates in recycling endosomes and late endosomes (Arjonen et al. 2012). Moreover, in ovarian cancer cells, inactive α5β1 is trafficked to the perinuclear recycling compartment and then recycled to the plasma membrane through a Rab11‐dependent mechanism (and not in a Rab4‐dependent manner). It is possible that other β1‐containing integrin heterodimers are responsible for the observation that inactive β1 is mainly trafficked through the Rab4 compartment in (Arjonen et al. 2012). In particular, the Rab11 effector RCP, which directly binds to β1 integrin, promotes the recycling of α5β1 to the tip of invasive protrusions when cells migrate in 3D environments, promoting their invasive ability through fibronectin‐rich matrices (Caswell et al. 2008). On the contrary, the traffic of ligand‐bound α5β1 is controlled by a member of the Rab11 family, Rab25, and the putative chloride channel CLIC3. This pathway involves the recycling of active α5β1 from the late endosomal/lysosomal compartment towards the back of cells migrating in 3D environments (Figure 2) (Dozynkiewicz et al. 2012). The functions of this are not entirely clear, and evidence supports a role for integrin recycling in controlling cell rear dynamics during migration. The kinesin Kif1C has been reported to promote α5β1 delivery to the cell tail, and this is required to maintain rear adhesion stability and directional cell migration (Theisen et al. 2012).
Figure 2.
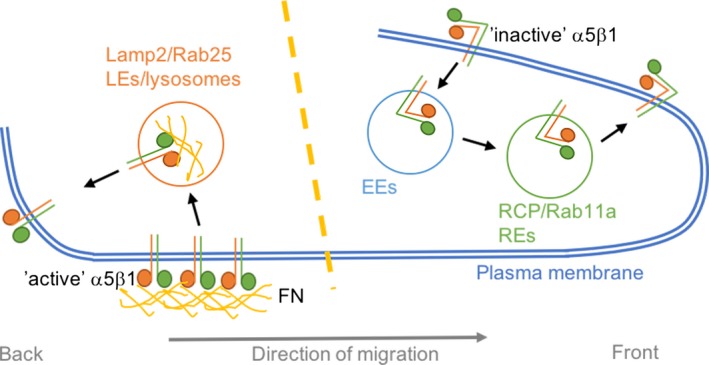
Active α5β1 and inactive α5β1 are trafficked through different endocytic routes in cells migrating through complex 3D matrices. Inactive α5β1 is internalized in early endosomes (EEs), transported to Rab coupling protein (RCP) and Rab11‐positive recycling endosomes (REs) and recycled to the plasma membrane at the tip of invasive protrusions at the front of the cell. Fibronectin (FN)‐bound α5β1 is preferentially endocytosed from subnuclear fibrillar adhesions and delivered to late endosomes (LEs)/lysosomes. From here, it is recycled to the plasma membrane at the back of migrating cells. [Colour figure can be viewed at http://wileyonlinelibrary.com]
Integrins link ECM internalization and mTOR signalling
Integrins have been shown to be internalized through a variety of endocytic mechanisms, including clathrin‐, caveolin‐, RhoA‐dependent, macropinocytosis and clathrin‐independent carriers. However, the spatial distribution of these endocytic events is not entirely clear. We aimed to characterize the mechanisms specifically controlling ligand‐bound α5β1 internalization (Rainero et al. 2015). The expression of Rab25 in ovarian cancer cells strongly induces the formation of fibrillar adhesions, mainly located underneath the nucleus, and promotes the internalization of FN‐bound α5β1, without affecting inactive integrin endocytosis. FN has been previously shown to be endocytosed in fibroblasts in a caveolin‐ and α5β1‐dependent manner (Shi & Sottile 2008). Interestingly, proteolytic digestion of the FN‐containing ECM has been shown to facilitate FN endocytosis, suggesting that FN fibrils degradation supports FN internalization (Shi & Sottile 2011). As mentioned above, tensins are a family of integrin and actin‐binding proteins particularly enriched in fibrillar adhesions. In mammals, there are four tensin isoforms: tensins 1, 2,3 and CTEN. Knock‐down of tensin 1/2/3 impairs both fibrillar adhesion formation and ligand‐bound integrin internalization. In particular, the use of the novel photoactivation‐in‐TIRF approach highlighted how ligand‐bound α5β1 is preferentially internalized from fibrillar adhesions (and not from focal adhesions) through a mechanism dependent on the small GTPase Arf4 and the SCAR/WAVE complex, a nucleator‐promoting factor that controls Arp2/3‐dependent actin polymerization. Following endocytosis, α5β1 and its ligand FN are delivered to late endosomes/lysosomes. This flux controls lysosomal positioning and the activation of the mechanistic target of rapamycin (mTOR), a key regulator of nutrient sensing. The main role of mTOR consists of controlling the fine balance between cell growth and the environmental conditions, so that cell growth is only promoted when enough energy and nutrients are available to the cells. mTOR can be activated by several growth factors, glucose and amino acids (Saxton & Sabatini 2017). mTOR exists in cells in two multiprotein complexes: mTOR complex 1 (mTORC1) and mTOR complex 2 (mTORC2). While mTORC1 is a master regulator of metabolism and cell growth, mTORC2 controls cytoskeletal dynamics and AKT activation (Saxton & Sabatini 2017). Intracellular concentration of amino acids is increased upon feeding, and it has recently been elucidated that cytosolic and intralysosomal amino acids activate mTORC1 via different pathways. In particular, an increase in amino acid concentration within the lysosome drives mTORC1 recruitment to the lysosomal membrane, thus mediating its activation through direct interaction with the Ragulator complex (Bar‐Peled et al. 2012). Interestingly, ligand‐bound integrin trafficking specifically promotes mTORC1 activation, without affecting mTORC2. Moreover, this pathway is strongly activated by [Page 9] glucose starvation (Rainero et al. 2015), suggesting a link between integrin trafficking and nutrient signalling (Figure 3). In line with this, in melanoma cells mTORC2 inhibition has been shown to promote α2 integrin expression and focal adhesion reorganization (Yoon et al. 2017). Further work is required to elucidate the mechanisms through which mTOR activation impinges on integrin expression and adhesion remodelling.
Figure 3.
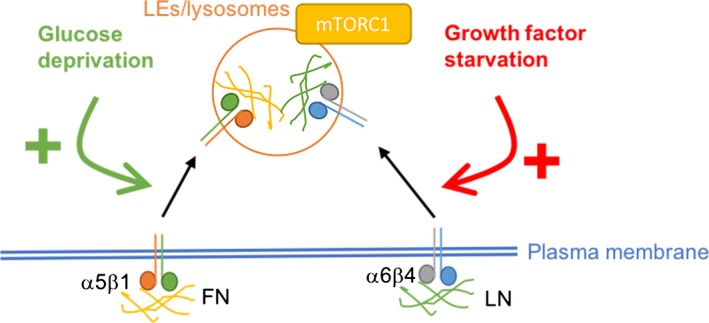
Interplay between integrin trafficking and mTORC1 activity. α5β1 integrin promotes the trafficking of fibronectin (FN) to late endosomes (LEs)/lysosomes, resulting in the recruitment of the mechanistic target of rapamycin complex 1 (mTORC1) on the lysosomal membrane and the activation of downstream signalling. This pathway is strongly promoted by glucose deprivation. α6β4 mediates the lysosomal delivery and degradation of laminin (LN), similarly leading to mTORC1 recruitment and activation. This is stimulated by growth factor starvation. [Colour figure can be viewed at http://wileyonlinelibrary.com]
In mammary epithelial cells, dietary restriction in vivo and growth factor starvation in vitro result in β4 integrin upregulation, and internalization of the ECM component laminin (LN). β4/LN complexes are delivered to late endosome/lysosomes, where LN degradation into amino acids promotes mTORC1 activation, thus preventing starvation‐induced cell death (Muranen et al. 2017) (Figure 3). It is hypothesized that mTORC1 activation is triggered by an increase in lysosomal amino acid concentration, upon digestion of ECM components, although further work is needed to specifically address the molecular mechanism(s) through which ECM internalization promotes mTOR activation.
Altogether, these findings support the idea that cells are able to use extracellular proteins as nutrient sources. In agreement with this, extracellular albumin has been shown to be internalized and degraded in lysosomes, generating carbon intermediate to support the tricarboxylic acid (TCA) cycle. Interestingly, this pathway can support pancreatic cancer cell growth in glutamine‐deprived condition and is strongly induced by the expression of oncogenic mutation of Ras (Commisso et al. 2013). More recently, Ras‐dependent extracellular protein internalization has been shown to specifically support fibroblast growth in the absence of essential amino acids (EAAs) or leucine alone, with a minimal effect in glutamine‐free conditions (Palm et al. 2015). This discrepancy could be ascribed to the use of different cell types (pancreatic cancer cells or fibroblast) in a different starvation protocol (0.1 mM or complete glutamine deprivation). Albumin uptake, in the absence of EAAs, results in mTOR lysosomal targeting and activation (Palm et al. 2015), suggesting that both ECM components and soluble proteins can lead to mTOR activation upon internalization and lysosomal degradation. Importantly, inhibition of mTORC1 (but not mTORC2) promotes extracellular protein degradation and cell proliferation in EAA‐depleted conditions, without affecting macropinocytosis‐dependent protein uptake (Palm et al. 2015). On the other end, inhibition of mTORC1, but not mTORC2, strongly induces fibrillar adhesion formation and ligand‐bound α5β1 integrin endocytosis (Rainero et al. 2015), suggesting that different endocytic/trafficking pathways are differentially controlled by mTOR signalling. Further studies are required to fully characterize the link between nutrient levels, mTOR activity and ECM component internalization.
ECM traffic and metabolism
A few examples of crosstalk between ECM trafficking and cell metabolism have been reported. Hyaluronidases (Hyal) mediate the degradation of the ECM glycosaminoglycan hyaluronan (HA). High levels of hyaluronidase 1 (Hyal1) correlate with poor prognosis in prostate cancer patients, while Hyal1 overexpression increases tumorigenesis and metastasis in mouse models. Recent evidence suggests that Hyal1 is required for clathrin‐dependent internalization of HA, promoting its delivery to Rab7‐positive late endosomes/lysosomes. HA degradation within this organelle allows increased sugar access, which, through glycolysis, can generate the energy required to sustain tumour proliferation and invasion (McAtee et al. 2015). Collagen I is the most abundant ECM protein in the human body and contains large amount of proline. It is therefore being suggested to serve as a reservoir for this amino acid. Despite data indicating a role for matrix metalloproteases (MMPs) in proline release from collagen under conditions of nutritional stress (Phang et al. 2015), further work is required to elucidate whether collagen endocytosis is required to support proline metabolism.
Energy stress promotes the activation of the metabolic sensor AMP‐activated protein kinase (AMPK), which stimulates catabolic pathways. It has recently been demonstrated that AMPK is a negative regulator of β1 integrin activity, through the regulation of tensin 1 and tensin 3 levels. Loss of AMPK promotes tensin expression, in turn leading to β1 activation and FN fibrillogenesis in fibroblasts (Georgiadou et al. 2017). It has been hypothesized that, within this FN‐rich environment, cancer cells can take up the ECM deposited by cancer‐associated fibroblasts, for the generation of the nutrients required to support cell proliferation and tumour growth (Georgiadou & Ivaska 2017). Additional work is needed to define how this process is regulated and how it contributes to tumour growth in vivo.
Conclusions
The ECM is commonly viewed as ‘dead space’ that provides a static scaffold for organ shape and acts as an obstacle for migrating cells. However, new data strengthen the idea that there is a dynamic interplay between cells and the ECM, which actively orchestrates cell behaviour. In particular, ECM receptors, including the integrin family members, promote the internalization of ECM components. Recent evidence supports the intriguing hypothesis that ECM internalization may represent a nutrient source for epithelial cells. Further research is needed to define the molecular mechanisms controlling ECM trafficking and fully elucidate how this pathway impinges on cellular metabolism.
Acknowledgements
ER was funded by the University of Sheffield and the Royal Society.
References
- Arjonen A., Alanko J., Veltel S. & Ivaska J. (2012) Distinct recycling of active and inactive beta1 integrins. Traffic 13, 610–625. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bar‐Peled L., Schweitzer L.D., Zoncu R. & Sabatini D.M. (2012) Ragulator is a GEF for the rag GTPases that signal amino acid levels to mTORC1. Cell 150, 1196–1208. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Caswell P.T., Chan M., Lindsay A.J., McCaffrey M.W., Boettiger D. & Norman J.C. (2008) Rab‐coupling protein coordinates recycling of alpha5beta1 integrin and EGFR1 to promote cell migration in 3D microenvironments. J. Cell Biol. 183, 143–155. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Caswell P.T., Vadrevu S. & Norman J.C. (2009) Integrins: masters and slaves of endocytic transport. Nat. Rev. Mol. Cell Biol. 10, 843–853. [DOI] [PubMed] [Google Scholar]
- Christoforides C., Rainero E., Brown K.K., Norman J.C. & Toker A. (2012) PKD Controls αvβ3 Integrin Recycling and Tumor Cell Invasive Migration through Its Substrate Rabaptin‐5. Dev. Cell 23, 560–572. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Clark K., Pankov R., Travis M.A. et al (2005) A specific α5β1‐integrin conformation promotes directional integrin translocation and fibronectin matrix formation. J. Cell Sci. 118, 291–300. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Commisso C., Davidson S.M., Soydaner‐Azeloglu R.G. et al (2013) Macropinocytosis of protein is an amino acid supply route in Ras‐transformed cells. Nature 497, 633–637. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Daley, W.P. , Yamada, K.M. (2013) ECM‐modulated cellular dynamics as a driving force for tissue morphogenesis. Curr. Opin. Genet. Dev. 23, 408–414. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Dozynkiewicz M.A., Jamieson N.B., Macpherson I. et al (2012) Rab25 and CLIC3 collaborate to promote integrin recycling from late endosomes/lysosomes and drive cancer progression. Dev. Cell 22, 131–145. [DOI] [PMC free article] [PubMed] [Google Scholar]
- de Franceschi N., Hamidi H., Alanko J., Sahgal P. & Ivaska J. (2015) Integrin traffic ‐ the update. J. Cell Sci. 128, 839–852. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Georgiadou M. & Ivaska J. (2017) Tensins: bridging AMP‐activated protein kinase with integrin activation. Trends Cell Biol. 27, 703–711. [DOI] [PubMed] [Google Scholar]
- Georgiadou M., Lilja J., Jacquemet G. et al (2017) AMPK negatively regulates tensin‐dependent integrin activity. J. Cell Biol. 216, 1107–1121. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lee S., Stewart S., Nagtegaal I. et al (2012) Differentially expressed genes regulating the progression of ductal carcinoma in situ to invasive breast cancer. Cancer Res. 72, 4574–4586. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Manninen A. & Varjosalo M. (2017) A proteomics view on integrin‐mediated adhesions. Proteomics 17, DOI: 10.1002/pmic.201600022. [DOI] [PubMed] [Google Scholar]
- Maritzen T., Schachtner H. & Legler D.F. (2015) On the move: endocytic trafficking in cell migration. Cell. Mol. Life Sci. 72, 2119–2134. [DOI] [PMC free article] [PubMed] [Google Scholar]
- McAtee C.O., Berkebile A.R., Elowsky C.G. et al (2015) Hyaluronidase Hyal1 Increases Tumor Cell Proliferation and Motility through Accelerated Vesicle Trafficking. J. Biol. Chem. 290, 13144–13156. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Muranen T., Iwanicki M.P., Curry N.L. et al (2017) Starved epithelial cells uptake extracellular matrix for survival. Nat. Commun. 8, 13989. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Palm W., Park Y., Wright K., Pavlova N.N., Tuveson D.A. & Thompson C.B. (2015) The utilization of extracellular proteins as nutrients is suppressed by mTORC1. Cell 162, 259–270. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Phang J.M., Liu W., Hancock C.N. & Fischer J.W. (2015) Proline metabolism and cancer: emerging links to glutamine and collagen. Curr. Opin. Clin. Nutr. Metab. Care 18, 71–77. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Rainero E., Howe J.D., Caswell P.T. et al (2015) Ligand‐occupied integrin internalization links nutrient signaling to invasive migration. Cell Rep. 2015 Jan 15. pii: S2211‐1247(14)01066‐3. [DOI] [PubMed] [Google Scholar]
- Saxton R.A. & Sabatini D.M. (2017) mTOR signaling in growth, metabolism, and disease. Cell 169, 361–371. [DOI] [PubMed] [Google Scholar]
- Settembre C., Fraldi A., Medina D.L. & Ballabio A. (2013) Signals from the lysosome: a control centre for cellular clearance and energy metabolism. Nat. Rev. Mol. Cell Biol. 14, 283–296. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Shi F. & Sottile J. (2008) Caveolin‐1‐dependent β1 integrin endocytosis is a critical regulator of fibronectin turnover. J. Cell Sci. 121, 2360–2371. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Shi F. & Sottile J. (2011) MT1‐MMP regulates the turnover and endocytosis of extracellular matrix fibronectin. J. Cell Sci. 124, 4039–4050. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Streuli C.H. (2016) Integrins as architects of cell behavior. Mol. Biol. Cell 27, 2885–2888. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Theisen, U. , Straube, E. , Straube, A. (2012) Directional persistence of migrating cells requires Kif1C‐mediated stabilization of trailing adhesions. Dev. Cell 23, 1153–1166. [DOI] [PubMed] [Google Scholar]
- Yoon S.O., Shin S., Karreth F.A. et al (2017) Focal adhesion‐ and IGF1R‐dependent survival and migratory pathways mediate tumor resistance to mTORC1/2 inhibition. Mol. Cell 67(512–527), e4. [DOI] [PMC free article] [PubMed] [Google Scholar]