

CRISPR-mediated genome engineering technologies have revolutionized genetic studies in a wide range of organisms. Here we describe new vectors and guide sequences for CRISPR mutagenesis in the important human fungal pathogens C. albicans and C. glabrata, as well as in the related yeasts S. cerevisiae and N. castellii. The design of these vectors enables efficient serial mutagenesis in each of these species by leaving few, if any, exogenous sequences in the genome. In addition, we describe strategies for the creation of unmarked deletions in each of these species and vector designs that permit the creation of vector libraries for pooled screens. These tools and strategies promise to advance genetic engineering of these medically and industrially important species.
KEYWORDS: CRISPR, Candida, Naumovozyma, Saccharomyces, albicans, castellii, cerevisiae, glabrata
ABSTRACT
We have created new vectors for clustered regularly interspaced short palindromic repeat (CRISPR) mutagenesis in Candida albicans, Saccharomyces cerevisiae, Candida glabrata, and Naumovozyma castellii. These new vectors permit a comparison of the requirements for CRISPR mutagenesis in each of these species and reveal different dependencies for repair of the Cas9 double-stranded break. Both C. albicans and S. cerevisiae rely heavily on homology-directed repair, whereas C. glabrata and N. castellii use both homology-directed and nonhomologous end-joining pathways. The high efficiency of these vectors permits the creation of unmarked deletions in each of these species and the recycling of the dominant selection marker for serial mutagenesis in prototrophs. A further refinement, represented by the "Unified" Solo vectors, incorporates Cas9, guide RNA, and repair template into a single vector, thus enabling the creation of vector libraries for pooled screens. To facilitate the design of such libraries, we have identified guide sequences for each of these species with updated guide selection algorithms.
IMPORTANCE CRISPR-mediated genome engineering technologies have revolutionized genetic studies in a wide range of organisms. Here we describe new vectors and guide sequences for CRISPR mutagenesis in the important human fungal pathogens C. albicans and C. glabrata, as well as in the related yeasts S. cerevisiae and N. castellii. The design of these vectors enables efficient serial mutagenesis in each of these species by leaving few, if any, exogenous sequences in the genome. In addition, we describe strategies for the creation of unmarked deletions in each of these species and vector designs that permit the creation of vector libraries for pooled screens. These tools and strategies promise to advance genetic engineering of these medically and industrially important species.
INTRODUCTION
Advances in clustered regularly interspaced short palindromic repeat (CRISPR) technology have greatly increased the ability to manipulate the genomes and gene expression of a wide variety of organisms (1–3). CRISPR-based mutagenesis requires a nuclease (usually Cas9) and an appropriate RNA guide targeted to the gene of interest; however, the outcome of CRISPR mutagenesis may depend on the idiosyncratic biology of the organism, especially with respect to its specific DNA repair system. The majority of systems utilize plasmid or viral expression vectors to introduce CAS9 and RNA guides into the organism, although some researchers have successfully used purified RNA/Cas9 protein complexes (4–8). The guide RNA directs the Cas9 nuclease to the complementary genomic sequence, where it makes a double-stranded break (DSB). Either the nonhomologous end-joining (NHEJ) pathway or the homology-directed repair (HDR) pathway repairs these cuts. NHEJ joins nonhomologous ends of DNA and represents the most common method of repair of CRISPR DSBs in mammals (5, 6). During NHEJ, bases are often added or deleted from the ends of DSBs, resulting in insertions or deletions (indels) in the targeted locus. When the HDR pathway is used, repairs using templates from the homologous chromosome restore the wild-type (WT) sequence. For CRISPR-mediated HDR, exogenous repair templates with homology flanking the DSBs can be used to introduce precise mutations into the genome.
Recently, we developed a CRISPR system that efficiently edits the human fungal pathogen Candida albicans (9). This system has subsequently undergone considerable optimization, and newer guide expression methods have improved the mutagenesis efficiency (10). Transient expression approaches have also permitted efficient deletion using drug resistance markers (11–13). In addition to the targeting sequence and nuclease, CRISPR mutagenesis in C. albicans and its distant relative Saccharomyces cerevisiae requires the inclusion of a repair template, as DSBs are primarily repaired by HDR (7, 9). Conservation of this requirement for a repair template among other fungi is unknown.
To explore the requirement for a repair template, we designed a number of improved vectors for CRISPR-mediated genome editing in S. cerevisiae and C. albicans as well as in two other fungi, C. glabrata and Naumovozyma castellii. Our recyclable constructs use the dominant nourseothricin resistance (Natr) drug resistance marker and are able to generate unmarked kilobase-scale deletions with a single guide sequence when coupled with a repair template. Although efficient mutagenesis in C. albicans and S. cerevisiae requires the addition of a repair template, C. glabrata and N. castellii require only CAS9 and a guide RNA. These results suggest that HDR is the predominant repair pathway in S. cerevisiae and C. albicans, in contrast to two close relatives of S. cerevisiae, N. castellii and C. glabrata, in which the NHEJ pathway dominates. Nevertheless, mutations of RAD genes that disable the HDR system bypass repair template requirements in S. cerevisiae, leading to repair via NHEJ. However, mutations in the homologous RAD genes do not bypass the repair template requirements in C. albicans, which suggests that S. cerevisiae and C. albicans have distinct repair preferences. In addition, we demonstrate that CRISPR can be used to generate loss-of-heterozygosity (LOH) events in C. albicans in the absence of exogenous repair templates.
To circumvent the requirement for cotransformation of both a Cas9/single guide RNA (sgRNA) plasmid and a repair template, we developed Unified Solo vectors, which consolidate repair templates onto the Cas9/sgRNA delivery plasmid. This consolidation enables pooled mutagenesis screening in any given background, including in diverse clinical isolates. In addition, using updated guide selection algorithms, we have designed guides for each of the aforementioned species and found that most genes (>90%) can be specifically modified by CRISPR. The combination of these improved vectors with updated guide selection algorithms sets the stage for the development of inclusive genome-wide mutant libraries in each of these species.
RESULTS
Improved vectors for mutagenesis in C. albicans.
To enable more-flexible CRISPR-based mutagenesis in C. albicans, first, we designed two improved Solo vectors for CRISPR mutagenesis (Fig. 1A, bottom). Previously, Cas9 was integrated into the ENO1 locus, resulting in a single functional copy of ENO1 (Fig. 1A, top) (9). As heterozygosity for ENO1 might affect growth under some conditions (14), our new vectors insert the CRISPR cassette into the Neut5L site, a locus whose disruption has been previously reported not to impact growth (15). Second, the previous approach removed only the guide expression module while retaining Cas9. To avoid possible complications of continued Cas9 expression, the FLP recombination target (FRT) sites were relocated to a position flanking the entire cassette such that flippase expression results in complete removal of the cassette, except for a small FRT insertion. Third, we created a variant with flippase expression under control of the inducible MAL2 gene instead of the SAP2-FLP genes because some mutant strains of C. albicans do not robustly induce SAP2-FLP expression.
FIG 1 .
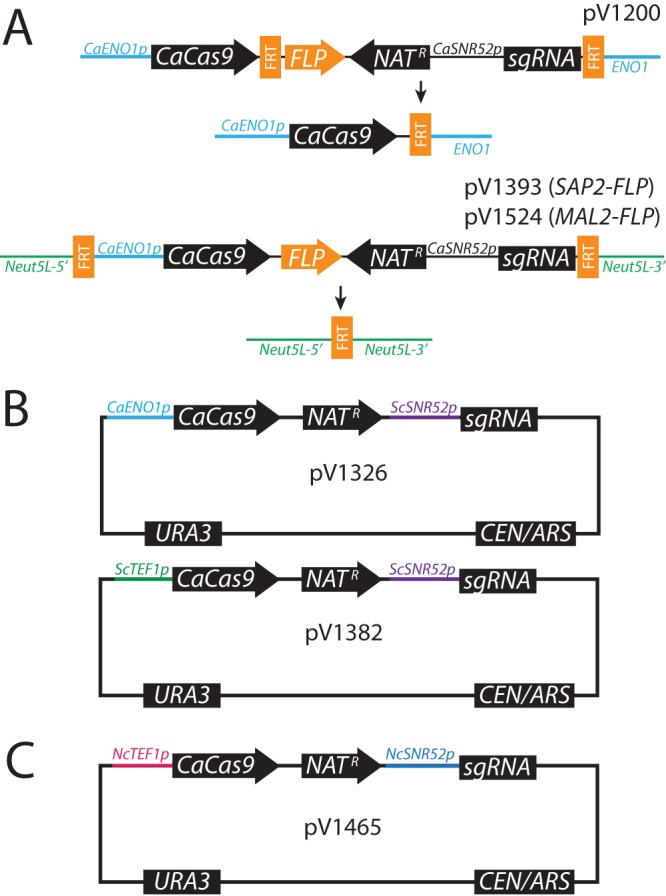
Vectors for CRISPR mutagenesis in Candida albicans, Candida glabrata, Naumovozyma castellii, and Saccharomyces cerevisiae. (A) Recyclable C. albicans CRISPR vector pV1200 (previous generation) replaces one copy of ENO1, and flipout removes NATr and sgRNA gene sequences. Current vectors pV1393 and pV1524 insert into the Neut5L locus, and flipout leaves only an FRT insertion at Neut5L. Vector pV1393 uses SAP2p to drive FLP, while pV1524 uses MAL2p. (B) Vectors pV1326 and pV1382 for CRISPR mutagenesis in C. glabrata and S. cerevisiae. (C) Vector pV1465 for CRISPR mutagenesis in N. castellii.
Markerless deletion mutants.
The limited number of dominant markers in C. albicans makes the task of creating strains with multiple mutations onerous. To determine whether one could construct unmarked deletions using a single guide sequence, we targeted the ADE2 locus using a guide that cuts within the open reading frame as previously reported (9) but utilized a repair template that juxtaposed 50 bp upstream of the open reading frame to 50 bp downstream of the ADE2 open reading frame. This repair template generated ade2/ade2 deletions of 1,652 bp at a high rate comparable to those seen with other ADE2 repair templates that created only base pair substitutions (61% for deletion versus 100% for stop codon; Fig. 2A). This result means that one can construct multiple unmarked deletion mutations in a strain without complex recycling of the limited dominant markers and without leaving FRT insertion sites at deleted loci.
FIG 2 .
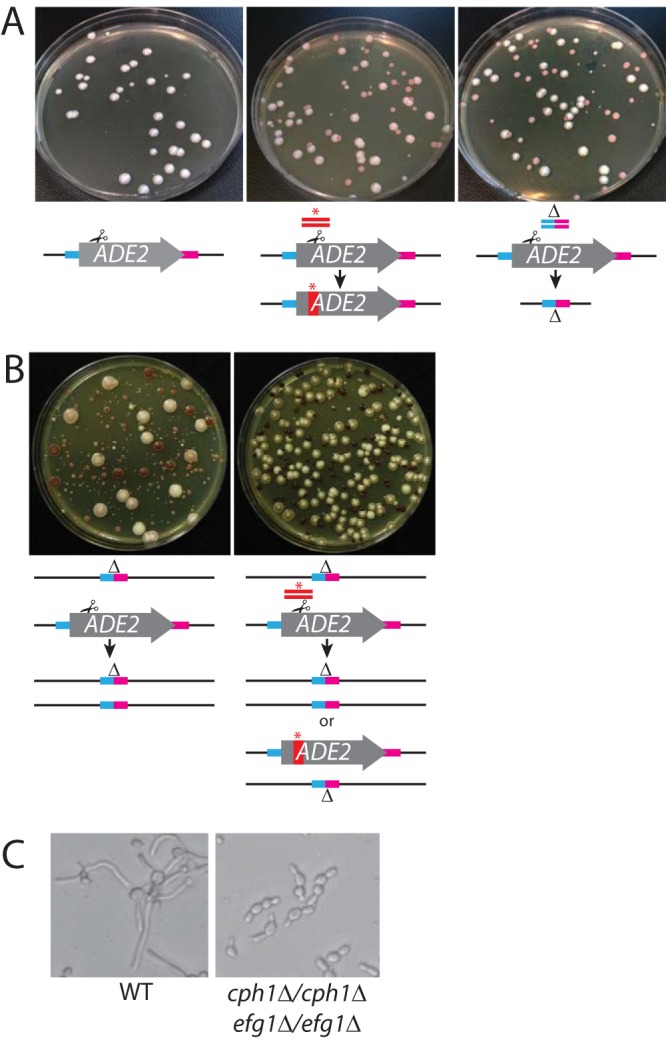
(A) CRISPR mutagenesis in C. albicans to create unmarked deletions and LOH. Strain SC5314 was transformed with ADE2-targeting plasmid pV1081 without a repair template (left), with a stop codon repair template (middle), or with a deletion repair template (right). (B) VY841 (ade2/ADE2) transformed with pV1081 without a repair template (left) or with a stop-codon repair template (right). (C) Serial mutagenesis of SC5314 with pV1393-based vectors used to create cph1/cph1 efg1/efg1 mutants. Strains of the indicated genotype were grown overnight in YPD medium and were transferred to RPMI medium–10% serum for 4 h.
Using the improved vectors (derived from pV1393 and pV1524), we demonstrated that double mutant strains of C. albicans could be produced with facility. To create a double cph1Δ/cph1Δ;efg1Δ/efg1Δ strain by sequential transformation, we first targeted the EFG1 gene with a deletion template. Transformants that were efg1Δ/efg1Δ strains were grown in medium to induce expression of flippase to remove the Cas9 machinery (see Materials and Methods), and flipout clones were then transformed with a vector containing guides and repair templates for deletion of CPH1, leading to the isolation of correct double mutant clones (cph1Δ/cph1Δ efg1Δ/efg1Δ; Fig. 2C). The frequency of deletion formation—markerless at both loci—enabled easy construction not only of morphological double mutants but also of auxotrophic double mutants (e.g., leu2Δ/leu2Δ;met15Δ/met15Δ; see Table S2 in the supplemental material) where both mutant sites were free of the marker used to select transformants.
Unified Solo vectors consolidate Cas9, guide RNA, and repair templates in a single plasmid.
A major limitation to CRISPR mutagenesis in C. albicans is the requirement to provide a separate repair template for efficient mutagenesis. This requirement makes pooled screens with a library of guides virtually impossible, as the likelihood that any given cell will get a matched guide and repair template during transformation diminishes exponentially with increasing library complexity. To circumvent this problem, we created a “Unified” Solo vector that links both guide and repair template on a single molecule.
Our first attempt utilized a stop-codon repair template for ADE2 (as in Fig. 2A) inserted immediately downstream of the sgRNA terminator (Fig. 3A). This Unified Solo vector was able to mutagenize ADE2 at a rate comparable to that achieved in our previous Solo system with a separate repair template. However, careful genotyping revealed that all of the ade2 mutants that we had recovered were the result of a translocation between the Neut5L locus (where the CRISPR system that includes the homologous repair template resides) and the ADE2 locus (Fig. 3A). We observed similar translocations in S. cerevisiae when we provided a repair template that spanned two chromosomal loci (see Text S1 and Fig. S1 in the supplemental material).
FIG 3 .

Unified Solo vectors for mutagenesis in C. albicans. (A) Unified Solo vector integrated at Neut5L directed against ADE2 results in translocations detectable by PCR with the indicated primers (red and green arrows). (B) Gene-drive-based Unified Solo vector to target CRISPR to delete ADE2. Vector pV1534 is linearized in vitro with BsmBI, while vector pV1535 is linearized in vivo by CRISPR. (C) Transformation of SC5314 with circular pV1534 (left), BsmBI-linearized pV1534 (middle), or circular pV1535 (right). (D) Maltose-induced gene drive targeting ADE2. Heterozygous mutants can be induced to create homozygous mutants by growth in maltose medium.
CRISPR mutagenesis in S. cerevisiae with multiple guides and repair templates. (A) S. cerevisiae strain L6437 was cotransformed with plasmids pV1386 and pGB9 alongside appropriate stop repair templates, as indicated. Transformants displayed either the ade2 genotype or leu2 genotype but not the ade2 leu2 genotype. (B) Strain L6437 was cotransformed with plasmids as described above except that a consolidated repair template was used that linked the stop repair templates described in the panel A legend into a single molecule. While most transformants were either ade2 or leu2, ade2 leu2 double mutants were identified. (C) PCR genotyping identified sequences consistent with translocation between the ADE2 and LEU2 chromosomes. Download FIG S1, PDF file, 0.1 MB (67.4KB, pdf) .
Copyright © 2018 Vyas et al.
This content is distributed under the terms of the Creative Commons Attribution 4.0 International license.
Targeting multiple genes in one transformation using CRISPR. Download TEXT S1, PDF file, 0.1 MB (75.8KB, pdf) .
Copyright © 2018 Vyas et al.
This content is distributed under the terms of the Creative Commons Attribution 4.0 International license.
To avoid such chromosome aberrations, which are likely to occur when homologous DNA sequences exist at two or more distinct locations in the genome, we designed a gene-drive-based Unified vector (Fig. 3B), based on a concept presented by DiCarlo et al. (16). Gene drive constructs insert the entire CRISPR system at the locus of the gene to be mutated, rendering homologous regions aligned such that all recombination events are restricted to the locus of interest. To ensure that our gene-drive construct could be inactivated after mutagenesis (for marker recycling and to safeguard against further Cas9 activity), we repositioned the FRT sequences as depicted in Fig. 3B. The repair template for this strategy differs from the previous repair templates in that its homology arms direct insertion of the entire plasmid into the target mutagenesis site, resulting in replacement of the ADE2 coding region with the Unified vector (Fig. 3B).
As episomal plasmids are not stable in C. albicans, the construct must be linearized to enable integration. We designed two strategies for vector linearization. The first strategy used the BsmBI restriction endonuclease in vitro; linearized vector generated ade2/ade2 mutants at a rate of close to 100%, whereas uncut vector failed to produce any mutants (Fig. 3C). The second strategy substituted the BsmBI sites with a target site for the sgADE2 guide and relied on Cas9/sgADE2-mediated cleavage in vivo of the circular plasmid at that target site. That strategy also produced ade2/ade2 mutants at a frequency of close to 100%, while eliminating the need to linearize the plasmid before transformation (Fig. 3B).
We reasoned that if gene-drive-mediated homozygosis requires Cas9 expression, it could be modified to create conditional knockouts. To test this, the constitutive ENO1 promoter in the Unified vector was replaced with the maltose-inducible MAL2 promoter to drive C. albicans Cas9 (CaCas9) expression. We first transformed BsmBI-digested gene-drive plasmids directed against ADE2 into a wild-type strain and plated cells onto selective medium containing glucose, where the MAL2 promoter is repressed. Heterozygous integrants (ade2Δ::FRT-MAL2p-CaCas9-FLP-Natr-sgADE2-FRT/ADE2) were grown in media containing either glucose or maltose and then plated. We found that 30% to 95% of the colonies grown in maltose media displayed an ade2 phenotype (colony color ranging from light pink to red), whereas less than 1% were red on glucose medium. The rare red colonies on glucose were likely due to leaky expression from the MAL2 promoter. For both constitutive and inducible Unified Solo vectors, we were able to remove the system by induction of the FLP gene, leaving a minimal FRT footprint in the genome and enabling serial mutagenesis.
CRISPR in C. glabrata, N. castellii, and S. cerevisiae.
The design of our Candida Solo vector CRISPR system provides advantages for engineering other fungal species, including S. cerevisiae, C. glabrata, and N. castellii. All of these species can maintain episomal plasmids, unlike C. albicans. Most published CRISPR systems for Saccharomyces use two distinct plasmids (one to express the sgRNA and the other Cas9) and require selectable auxotrophic markers. The published C. glabrata system also utilizes two plasmids and requires auxotrophic markers (17). These constraints render these systems unusable in prototrophic clinical isolates or industrial strains.
We avoided these constraints by moving our CRISPR Solo insertion into S. cerevisiae vector pRS416, which provides a CEN/ARS element for plasmid maintenance and a URA3 marker that can be used for counterselection in ura3 auxotrophs (Fig. 1B and C). We substituted species-appropriate promoter sequences for the sgRNA and CaCas9 to improve their expression. The pRS416 backbone is functional in multiple yeast species, including C. glabrata and N. castellii.
As a proof of principle, we created guides directed against ADE2 for each of these three species. For S. cerevisiae, we generated ade2 mutants efficiently only when we included a repair template for creation of either a deletion or a point mutation at ADE2 (Fig. 4A). No mutants and very few transformants were recovered when the repair template was omitted, presumably because of DSB-induced cell cycle arrest. When a repair template was included, all of the mutants that were recovered contained the sequence encoded by their respective repair templates, indicating that all repair was directed via homology-mediated mechanisms.
FIG 4 .

Efficient CRISPR mutagenesis with Solo vectors in S. cerevisiae. (A) Laboratory strain BY4741 and industrial bioethanol strain FermPro were transformed with plasmid pV1382 or plasmid pV1386 with or without the indicated repair templates. (B) FermPro was transformed with Unified Solo vector pVG1 in the presence or absence of a second repair template.
To determine if we could consolidate the CRISPR mutagenesis system in Saccharomyces as we had for C. albicans (Fig. 4B), we constructed Unified Solo vectors directed against ScADE2. Our Unified Solo plasmid efficiently mutagenized ADE2 without using an integrated gene drive mechanism. These vectors also efficiently edited the genomes of a wide variety of S. cerevisiae strains, including strains used for brewing and industrial fermentations. Addition of an exogenous repair template in the transformation greatly increased the efficiency of mutagenesis, suggesting that repair template is limiting for mutagenesis with Unified Solo vectors (Fig. 4B).
Transformation of C. glabrata and N. castellii with a plasmid containing guides for ADE2 yields ade2 mutants, both with and without a repair template (Fig. 5 and 6). Moreover, in the absence of a repair template, mutants acquire small deletions or insertions, consistent with repair via an NHEJ pathway. When a repair template was included, 62% and 71% of red ade2 C. glabrata colonies (Fig. 5, top right and bottom right, respectively) and 7% and 25% of N. castellii colonies (Fig. 6, stop repair versus deletion, respectively) contained the sequence encoded on the repair template, whereas the remaining had indels such as were seen when the repair template was omitted. For C. glabrata, we observed that the choice of CaCas9 promoter impacted the relative rates of mutagenesis in the presence or absence of a repair template (Fig. 5), similarly to previous observations (17). Mutagenesis with ENO1p-expressed CaCas9 resulted in a higher rate of NHEJ-mediated repair than of templated repair, whereas templated repair predominated for TEF1p-expressed CaCas9. The rate of mutagenesis seen with a separate repair template was notably lower in N. castellii than in the other species. Consolidation of the repair template onto a Unified Solo vector substantially improved the mutagenesis rate in N. castellii, suggesting that repair template delivery had previously been limiting (56% used a stop repair template, while 69% used a deletion repair template). In N. castellii, we found that simultaneous deletion of the N. castellii orthologues of KU70 and KU80, which are known components of the NHEJ pathway (18, 19), abolished NHEJ repair, as all of the sequenced transformants were shown to have used repair-template-mediated HDR.
FIG 5 .

Efficient CRISPR mutagenesis in C. glabrata. Strain BG2 was transformed with pV1326 (top left), pV1329 (top middle), pV1329 with a stop-codon repair template (top right), pV1435 (bottom middle), or pV1435 with a stop-codon repair template (bottom right). The yellow box (bottom left) presents a magnified view of a pV1329 with stop-codon repair transformation plate (top right). Similar results were seen with strain CLIB138. Vectors depict CaCas9 promoter differences between the two plasmids.
FIG 6 .

CRISPR mutagenesis in N. castellii. Strain DPB069 (WT) and strain DPB596 (ku70 ku80) were transformed with the indicated vectors and repair templates.
In the absence of nourseothricin selection in S. cerevisiae, C. glabrata, and N. castellii, the CRISPR plasmid was lost rapidly (without counterselection), permitting rounds of serial mutagenesis in prototrophic strains. We used this strategy for the serial creation of met15Δ mutants and then leu2 mutants in C. glabrata and ade2 and then leu2 mutants in S. cerevisiae (data not shown).
The requirement for a repair template in C. albicans.
C. albicans requires a repair template in addition to Cas9/sgRNA for mutagenesis at a given locus. We hypothesized that this was due to repair of a Cas9 double-stranded break predominantly by the HDR system. To test this directly, we measured ADE2 mutagenesis in a strain that contained a heterozygous deletion of ADE2 (ade2Δ/ADE2). We transformed both wild-type (ADE2/ADE2) and ADE2 heterozygotes with an ADE2-directed plasmid with and without a repair template containing a base pair substitution that could be distinguished from the deletion. Recovery of ade2 mutations in the ADE2/ADE2 wild-type strain required the presence of a repair template, whereas red ade2/ade2 mutants were obtained in the absence of a repair template in the ade2Δ/ADE2 heterozygote (Fig. 2B). Red mutants had only the ade2Δ/ade2Δ genotype, indicating they either had used the remaining ade2Δ allele as a repair template or had become aneuploid for the ADE2 chromosome or duplicated the ade2Δ chromosome through a 2n-1 intermediate. The increased variation in colony size that we observed without the use of an exogenous repair template was similar to results described by Feri and colleagues (20). We were unable to detect any of the indel mutations that are the hallmark of NHEJ-mediated repair. When a repair template was included in the transformation of the ade2Δ/ADE2 heterozygote, approximately 70% of the ade2 strains used the repair template, whereas the other 30% were genotyped as ade2Δ/ade2Δ.
Repair in recombination mutants.
Our data show that CRISPR mutagenesis requires a repair template in S. cerevisiae and C. albicans, suggesting that HDR is the dominant repair pathway, whereas repair can occur through both the HDR and NHEJ pathways in C. glabrata and N. castellii. We sought to determine whether mutation of the HDR machinery in either S. cerevisiae or C. albicans would increase the frequency of NHEJ-derived mutations when CRISPR targets a locus in the absence of a repair template. To this end, we transformed S. cerevisiae WT (RAD), rad51, rad52, and rad59 strains with either an untargeted Solo plasmid or an ADE2-directed Solo plasmid without a repair template (Fig. 7). As shown previously, we were unable to recover transformants of the WT strain with the ADE2 guide without the addition of a repair template. However, in strains carrying loss-of-function mutations in RAD51, RAD52, or RAD59 genes, we were able to isolate red ade2 transformants in the absence of a repair template. Sequence analysis revealed that all red ade2 transformants in the rad mutant backgrounds contained indels consistent with NHEJ-mediated repair. The few isolated white colonies contained indels in the ADE2 sequence that rendered it resistant to further CRISPR cleavage while maintaining ADE2 prototrophy.
FIG 7 .
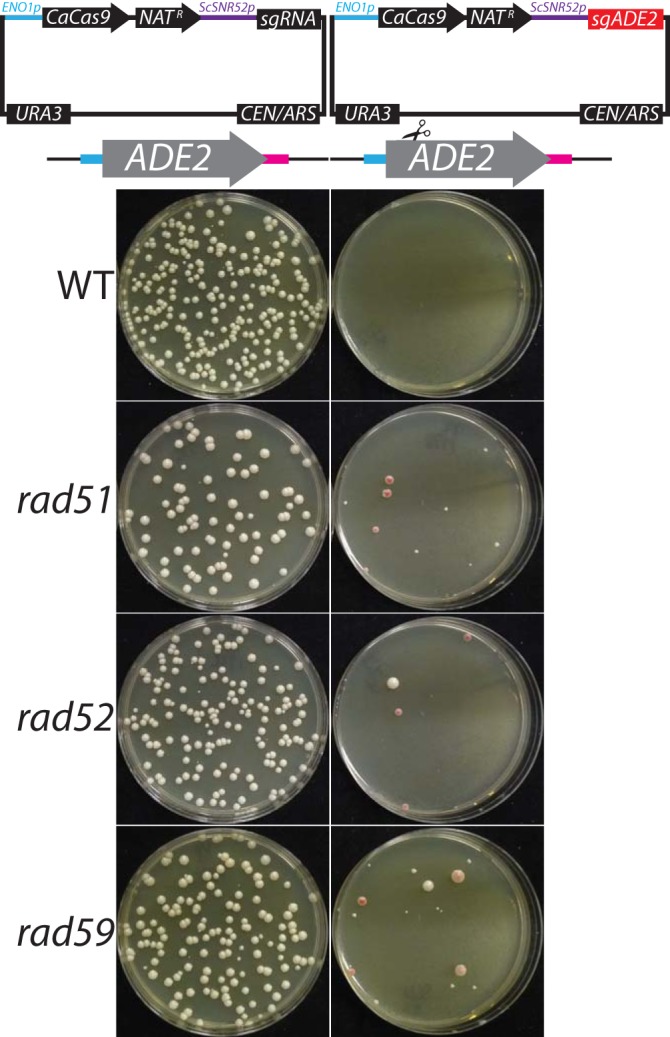
CRISPR mutagenesis in HDR mutants. S. cerevisiae strains of the indicated genotype were transformed with empty vector pV1326 (left) or ADE2-targeted vector pV1338 (right) with no repair template.
To test if the same strategy would work in C. albicans, we created homozygous mutants in the C. albicans RAD51, RAD52, and RAD59 genes (e.g., rad51/rad51) and tested mutagenesis at ADE2. We were unable to generate red ade2 colonies with our ADE2-directed vector in the absence of a repair template in these strains (data not shown).
Guide selection for C. albicans, C. glabrata, N. castellii, and S. cerevisiae.
We designed guide sequences corresponding to each annotated feature in the genomes of C. albicans, C. glabrata, N. castellii, and S. cerevisiae (see the supplemental guide table available at http://osf.io/ARDTX/), using updated guide selection and scoring methods (21), as summarized in Table 1. Most genes are uniquely targetable by multiple guide sequences, and multiple genes can be targeted with single guides (see Text S1 in the supplemental material and the supplemental guide table available at http://osf.io/ARDTX/). For C. albicans, we also designed allele-specific guides using the updated algorithm. Between 77% and 94% of diallelic genes can be uniquely targeted based on these new rules, depending on the off-target algorithm cutoff. In addition, we designed guides and repair templates for use with our (gene-drive-based) Unified Solo vector that link these sequences in cis as was done with pV1534. Targeting sequences, chosen from sequences upstream and downstream of each feature, have been screened for the presence of BsmBI sites, since they are required for vector linearization. Guides were also screened to identify those that cut the plasmid backbone. The sequences in these tables simplify cloning design for our Unified Solo vector.
TABLE 1 .
Guide availability in C. albicans, C. glabrata, N. castellii, and S. cerevisiaea
| Species | No. of unique guides |
No. (%) of uniquely targetable genes |
Avg no. of unique guides/gene |
No. of multigene guides |
No. (%) of genes targetable with other genes |
|---|---|---|---|---|---|
| C. albicans (old pipeline) | 550,045 | 6,288 of 6,466 (97) | 87 | 105,747 | 6,023 of 6,466 (93) |
| C. albicans (new pipeline) | 333,464 | 6,236 of 6,466 (96) | 53 | 12,220 | 4,647 of 6,466 (72) |
| C. albicans (allele specific, off-target cutoff of 0.2) | 13,286 | 3,112 of 4,046 (77) | 4 | NA | NA |
| C. albicans (allele specific, off-target cutoff of 0.5) | 21,765 | 3,464 of 4,046 (86) | 6 | NA | NA |
| C. albicans (allele specific, no off-target cutoff) | 39,299 | 3,805 of 4,046 (94) | 10 | NA | NA |
| C. glabrata | 197,604 | 5,285 of 5,597 (94) | 38 | 5,699 | 2,554 of 5,597 (46) |
| N. castellii | 461,932 | 5,566 of 5,594 (99) | 56 | 13,208 | 4,434 of 5,594 (79) |
| S. cerevisiae | 511,439 | 6,398 of 6,600 (97) | 80 | 14,658 | 4,522 of 6,600 (69) |
NA, not applicable (allele-specific guides by definition do not target other genes); off-target cutoff score is calculated using software developed by Doench et al. (21). A guide targeting another place in the genome with an off-target cutoff higher than the indicated score is considered unspecific.
DISCUSSION
Here we designed Solo vectors for CRISPR mutagenesis based on our original system in C. albicans (9), which has also undergone subsequent enhancements and adaptations to other species (10–13, 22). These vectors facilitate a variety of advancements in fungal genome editing. Our refined vectors enable investigators to use an episomal system (C. glabrata, N. castellii, and S. cerevisiae) or to integrate the CRISPR cassette at either a neutral site or the target mutagenesis site (C. albicans); recycle it with a minimal footprint; and/or consolidate the repair template, guide, and Cas9 onto a single molecule. Moreover, these vectors utilize a dominant selectable marker usable in prototrophic clinical isolates and industrial strains.
The redesigned repair templates allow the creation of unmarked kilobase-scale deletions without the use of repeat elements, thus enhancing the efficiency of serial mutagenesis. Though serial mutagenesis has been previously enabled by the use of FLP/FRT-based systems, residual FRT sequences in the genome create opportunities for translocations (23–25), a problem whose solution becomes exponentially more difficult with each subsequent deletion. For our new C. albicans system, we use FRT sequences at only one locus, minimizing the possibility of FLP-mediated translocations. CRISPR-based mutagenesis of members of gene families faces an additional hurdle in that DSB repair of one gene can be templated by other family members. Use of an unmarked deletion repair template addresses both of these issues. Removal of homologous family member sequences with the redesigned repair templates could decrease the likelihood of the occurrence of off-target repairs with each subsequent mutagenesis. Our vectors for C. glabrata, S. cerevisiae, and N. castellii bypass FLP/FRT requirements by virtue of being episomal.
The Solo vectors enabled us to compare results of CRISPR mutagenesis in several different fungal species with a single system based on the same design. CRISPR mutagenesis in C. albicans and S. cerevisiae requires an additional repair template, whereas CRISPR mutagenesis in C. glabrata and N. castellii requires only Cas9 and guide RNA. These results suggest that the HDR pathway is predominant over the NHEJ pathway in S. cerevisiae and C. albicans and that the NHEJ pathway predominates in N. castellii and C. glabrata. In agreement with this conclusion, mutation of HDR-enabling RAD genes bypasses the repair template requirements in S. cerevisiae, leading to repair via NHEJ. Such mutants could potentially simplify and lower the cost of pooled CRISPR screens by eliminating the need to provide a repair template, as well as providing the opportunity to recover a wider range of mutant alleles than can be specified on a repair template. Whereas RAD gene mutants bypass the repair template requirements in S. cerevisiae, they do not do so in C. albicans, suggesting that S. cerevisiae and C. albicans have distinct repair preferences.
In C. albicans transformations lacking a repair template, there is almost always a second copy of the gene to provide homology for repair of DSBs. Without a repair template, CRISPR mutagenesis of a strain heterozygous for ade2Δ/ADE2 resulted in homozygosis of the ade2Δ allele, either by use of the ade2Δ allele as a repair template (gene conversion) or through homozygosis of part or all of the ade2Δ chromosome via mitotic recombination or a 2n-1 intermediate. As C. albicans is polymorphic at many loci (designated A and B alleles) (26), this CRISPR mutagenesis would render the strain homozygous for that allele (i.e., A/A or B/B). This feature could be utilized for large-scale LOH studies (to uncover allelic differences) and/or genetic mapping studies, similarly to the concept presented by Sadhu et al. (27). This is particularly useful for C. albicans genetics, as it may circumvent its apparent lack of meiosis that such studies have classically relied upon.
During the preparation of the manuscript, a description of an alternative C. albicans CRISPR gene drive system was published (28). This system was presented as a means to study synthetic genetic interactions, though it could also be used for creating single homozygous knockouts or for creating double homozygous knockouts via mating. In contrast, the architecture of our system allows for multiple gene homozygous knockouts by serial vector recycling and disrupts only the target locus in the genome. Additionally, our inducible vectors represent a novel tool for the creation of conditional mutants.
The Unified Solo vectors provide a straightforward avenue to making deletion libraries in fungal species, especially in those such as C. albicans where manipulation by traditional gene replacement methods has been challenging. Making deletions using the first-generation Candida CRISPR vectors required cotransformation of a repair template. Consequently, to make a CRISPR replacement library, repair templates would have to be synthesized for each transformation and cotransformed as a separated arrayed set for any given isolate, as cotransformation of a matched CRISPR/guide module and repair template would be unlikely in a complex pooled library. Our Unified Solo vectors solve this problem by combining the repair template, Cas9, and a guide RNA on a single plasmid that either integrates into the genome at the target mutagenesis site (in the case of C. albicans) or maintains itself as an episome (in the case of S. cerevisiae or N. castellii). Thus, every cell that gets a particular guide RNA also receives its corresponding repair template. Furthermore, libraries of plasmids targeting every locus could be pooled in a single transformation to screen for phenotypes of interest, with guide sequences serving as barcodes to identify a mutant’s abundance (in the case of a pool) or identity (in the case of a clone). The inducible gene drive system permits the creation of on-demand conditional knockouts that can be used for identification of essential genes, pathogenicity factors in hosts, or other developmentally regulated processes. Moreover, each of these systems can recycle the dominant marker, simplifying subsequent mutagenesis. This model of library generation and phenotypic screening has the potential to be broadly applicable.
MATERIALS AND METHODS
Transformation.
A hybrid lithium acetate/electroporation protocol was used for C. albicans, S. cerevisiae, and C. glabrata. Briefly, a 5-ml yeast extract-peptone-dextrose (YPD) culture of each strain was grown overnight at 30°C on a roller drum to saturation. Cells were pelleted by centrifugation and resuspended in 2 ml of 100 mM lithium acetate–10 mM Tris (pH 8.0)–1 mM EDTA (pH 8.0) to which 50 µl 1 M dithiothreitol (DTT) was added. These cells were incubated on a 30°C roller drum for 1.5 to 15 h. Next, cells were washed twice in 5 ml of ice-cold water, washed once in 5 ml of ice-cold 1 M sorbitol, and resuspended in 500 µl of ice-cold 1 M sorbitol. Cells were kept on ice while transformation mixes were prepared. Cell suspension (40 µl) was added to aliquoted DNA, mixed by pipetting, placed in chilled Bio-Rad gene pulser cuvettes (Bio-Rad catalog no. 1652086), and electroporated on a Bio-Rad Gene pulser with a capacitance of 25 µF and resistance of 200 Ω at 1.8 kV. YPD medium (100 µl) was immediately added to the cuvette, and the cell mixture is incubated for between 1 and 12 h at room temperature before plating onto appropriate selective media.
N. castellii was transformed using a modified version of the lithium acetate method previously described by Gietz and Schiestl (29). A 5-ml YPD culture of N. castellii was grown overnight at 30°C on a roller drum. This preculture was then used to inoculate 50 ml of YPD medium to reach an optical density at 600 nm (OD600) of 0.2. The culture was then grown at 30°C with vigorous shaking in a baffled flask until it reached an OD600 of 0.8. Cells were collected by centrifugation, washed once in 25 ml of water, and resuspended in 1 ml of 100 mM LiAc–100 mM Tris-Cl (pH 7.5). This suspension was transferred to a microcentrifuge tube and centrifuged at top speed for 15 s. The pellet was resuspended in 400 µl of 100 mM LiAc–100 mM Tris-Cl (pH 7.5) and incubated at room temperature for 20 min. For each transformation, 50 µl of cell suspension was added to a microcentrifuge tube. To each transformation tube, the following reagents were added in the following order, with vortex mixing performed after each addition: 240 µl 50% polyethylene glycol (PEG) 3350, 36 µl 1 M LiAc, 12.5 µl boiled single-stranded DNA (ssDNA) (10 mg/ml), and 1.5 µg each plasmid DNA and repair template. Transformation mixes were incubated at 23°C for 30 min with rotation and then heat shocked at 37°C for 20 min. Heat-shocked cells were centrifuged at 8,000 rpm for 15 s, and transformation liquid was removed. Cells were washed with 500 µl of water and resuspended in 200 µl of water. At least 25 µl of this suspension was plated onto selective media.
Strains and growth conditions.
Strains used in this study are listed in Table S2 in the supplemental material. C. albicans strain SC5314, C. glabrata strains BG2 and CLIB138, S. cerevisiae strains BY4741 and FermPro, and N. castellii strains DBP069 and DBP596 were used for experiments. Yeast cells were grown in YPD medium (1% Difco Bacto yeast extract, 2% Difco Bacto peptone, 2% glucose) and selected with nourseothricin (Nat) at between 100 and 200 µg/ml. Flipout of the Natr gene from C. albicans vectors was induced by overnight growth in Difco yeast carbon base supplemented with bovine serum albumin (4%) overnight or by overnight growth in YP maltose medium (1% Difco Bacto yeast extract, 2% Difco Bacto peptone, 2% maltose). Episomal plasmid loss experiments were performed by overnight growth in nonselective liquid YPD medium. Drug-sensitive isolates, which had either flipped out the cassette or lost the plasmid, were identified by plating for single colonies on nonselective media and subsequent identification by replica plating to selective media.
Filamentation experiments were performed with yeast grown overnight in liquid YPD medium, washed twice with RPMI 1640 medium (Thermo Fisher catalog no. 22400-105) supplemented with 10% fetal bovine serum (FBS), and incubated in RPMI medium–10% FBS for the indicated time starting at an optical density of 0.1. CRISPR-mutagenized loci were verified by agarose gel electrophoresis and sequencing of PCR products amplified from target loci.
Plasmids/DNA.
Plasmids for CaCas9 system vectors are listed in Table S1. All key components were verified by sequencing and restriction analysis. Vectors and sequences will be available on Addgene. Transformations were performed with between 0.3 and 5 µg of plasmid DNA (smaller amounts were used for episomal plasmids and larger amounts for linear vectors for integration) and with between 1 and 5 µg of a repair template (where applicable). Repair templates were generated with 60-bp oligonucleotide primers containing 20-bp overlaps at their 3′ ends centered at the mutation point, which consisted of either stop codons or an open reading frame (ORF) deletion. N. castellii repair templates were 500 bp in length. Oligonucleotide sequences are listed in Table S3. Primers were extended by thermocycling performed with Ex Taq (TaKaRa catalog no. RR001A), and double-stranded products were purified directly from the PCR using a QIAquick gel extraction kit with isopropanol. Phosphorylated and annealed guide sequence-containing primers were ligated into calf intestinal phosphatase (CIP)-treated, BsmBI-digested parent vectors as described previously (9). The correct clones were identified by sequencing.
Plasmids used in this study. Download TABLE S1, PDF file, 0.1 MB (81.7KB, pdf) .
Copyright © 2018 Vyas et al.
This content is distributed under the terms of the Creative Commons Attribution 4.0 International license.
Strains used in this study. Download TABLE S2, PDF file, 0.1 MB (79.8KB, pdf) .
Copyright © 2018 Vyas et al.
This content is distributed under the terms of the Creative Commons Attribution 4.0 International license.
Oligonucleotides used for repair template generation. Download TABLE S3, PDF file, 0.05 MB (52KB, pdf) .
Copyright © 2018 Vyas et al.
This content is distributed under the terms of the Creative Commons Attribution 4.0 International license.
Computational analysis for guide design.
We designed guide sequences corresponding to each annotated gene in the genomes of C. albicans, C. glabrata, S. cerevisiae, and N. castellii (see the supplemental guide tables [DOI https://doi.org/10.17605/OSF.IO/ARDTX] available at http://osf.io/ARDTX/), using updated guide selection and scoring methods (21) (summarized in Table 1).
For C. glabrata, S. cerevisiae, and N. castellii, we listed all 20 nucleotides (nt) followed by NGG (or CCN plus 20 nt) in the genome. We removed any targets that had 6 instances of "T" in the 20 nt before the NGG because their presence would result in premature termination from polymerase (Pol) III promoters. We kept only the guides that were hitting the coding regions of genes. To find putative off-targets, we took the 14 nt closest to the protospacer-adjacent-motif (PAM) sequence, constructed the four possible sequences of the PAM, and mapped these with Bowtie, allowing up to 3 mismatches. For each original guide, we calculated the on-target score using rule set 2 for sgRNA on-target activity script (described in reference 21) as well as all the possible off-target scores for all the places in the genome that the 14-nt NGG hit with ≤3 mismatches (cutting frequency determination [CFD] scoring) (21). We defined guides as having no off-targets if the off-target scores at other locations were lower than 0.2. To find guides that targeted more than one gene, we followed the requirements that the off-target score for the other genes hit was 0.5 or higher and that there were no other hits with off-target scores higher than 0.2 and lower than 0.5. We also removed guides with off-targets in intergenic regions.
For C. albicans, using rule set 2 for the sgRNA on-target activity script (21), we rescored the guides uniquely targeting the two alleles that we had previously designed (9). Additionally, to assess whether those guides could be used on the Unified Solo vector, we checked that the guide was not targeting the vector and the absence of BsmBI sites in the guide.
Sequences (FASTA files) and annotations (GFF files) for C. albicans and C. glabrata were downloaded from the Candida Genome Database (30). C. albicans sequences from strain SC5314 (assembly 22, version A22-s05-m01-r03) and C. glabrata sequences from strain CBS138 (version s02-m07-r06) were used for guide selection. N. castellii guide design was based on sequences (FASTA files) and annotations (GFF files) for strain CBS4309 (assembly ASM23724v1.31) obtained from Ensembl (31). S. cerevisiae guide design was based on sequences (FASTA files) and annotations (GFF files) downloaded from the Saccharomyces Genome Database (32). The sequences were from strain S288C (version sacCer3/SGD_2010).
Data availability.
Species-specific guide sequence tables (for single-gene, multigene, and/or allele-specific guides) are available at the Open Science Frameworks repository (DOI https://doi.org/10.17605/OSF.IO/ARDTX; https://OSF.IO/ARDTX).
ACKNOWLEDGMENTS
We thank Felix Lam, Juliana Leung, and George Bell for comments on the manuscript.
This work was supported by NIH GM035010 (G.R.F.), GM118135 (D.P.B.), and R15AI130950 (D.A.B.). G.G.B. was supported by NIH F32 GM108201. M.A.G. was partially supported by a National Science Foundation Graduate Research Fellowship under grant 1122374.
REFERENCES
- 1.Wright AV, Nuñez JK, Doudna JA. 2016. Biology and applications of CRISPR systems: harnessing nature’s toolbox for genome engineering. Cell 164:29–44. doi: 10.1016/j.cell.2015.12.035. [DOI] [PubMed] [Google Scholar]
- 2.Shalem O, Sanjana NE, Zhang F. 2015. High-throughput functional genomics using CRISPR-Cas9. Nat Rev Genet 16:299–311. doi: 10.1038/nrg3899. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Mali P, Esvelt KM, Church GM. 2013. Cas9 as a versatile tool for engineering biology. Nat Methods 10:957–963. doi: 10.1038/nmeth.2649. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Jinek M, Chylinski K, Fonfara I, Hauer M, Doudna JA, Charpentier E. 2012. A programmable dual-RNA-guided DNA endonuclease in adaptive bacterial immunity. Science 337:816–821. doi: 10.1126/science.1225829. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Mali P, Yang L, Esvelt KM, Aach J, Guell M, DiCarlo JE, Norville JE, Church GM. 2013. RNA-guided human genome engineering via Cas9. Science 339:823–826. doi: 10.1126/science.1232033. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Cong L, Ran FA, Cox D, Lin S, Barretto R, Habib N, Hsu PD, Wu X, Jiang W, Marraffini LA, Zhang F. 2013. Multiplex genome engineering using CRISPR/Cas systems. Science 339:819–823. doi: 10.1126/science.1231143. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.DiCarlo JE, Norville JE, Mali P, Rios X, Aach J, Church GM. 2013. Genome engineering in Saccharomyces cerevisiae using CRISPR-Cas systems. Nucleic Acids Res 41:4336–4343. doi: 10.1093/nar/gkt135. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Ramakrishna S, Kwaku Dad AB, Beloor J, Gopalappa R, Lee SK, Kim H. 2014. Gene disruption by cell-penetrating peptide-mediated delivery of Cas9 protein and guide RNA. Genome Res 24:1020–1027. doi: 10.1101/gr.171264.113. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Vyas VK, Barrasa MI, Fink GR. 2015. A Candida albicans CRISPR system permits genetic engineering of essential genes and gene families. Sci Adv 1:e1500248. doi: 10.1126/sciadv.1500248. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Ng H, Dean N. 2017. Dramatic improvement of CRISPR/Cas9 editing in Candida albicans by increased single guide RNA expression. mSphere 2:e00385-16. doi: 10.1128/mSphere.00385-16. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Nguyen N, Quail MMF, Hernday AD. 2017. An efficient, rapid, and recyclable system for CRISPR-mediated genome editing in Candida albicans. mSphere 2:e00149-17. doi: 10.1128/mSphereDirect.00149-17. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Huang MY, Mitchell AP. 2017. Marker recycling in Candida albicans through CRISPR-Cas9-induced marker excision. mSphere 2:e00050-17. doi: 10.1128/mSphere.00050-17. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Min K, Ichikawa Y, Woolford CA, Mitchell AP. 2016. Candida albicans Gene deletion with a transient CRISPR-Cas9 system. mSphere 1:e00130-16. doi: 10.1128/mSphere.00130-16. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Ko HC, Hsiao TY, Chen CT, Yang YL. 2013. Candida albicans ENO1 null mutants exhibit altered drug susceptibility, hyphal formation, and virulence. J Microbiol 51:345–351. doi: 10.1007/s12275-013-2577-z. [DOI] [PubMed] [Google Scholar]
- 15.Gerami-Nejad M, Zacchi LF, McClellan M, Matter K, Berman J. 2013. Shuttle vectors for facile gap repair cloning and integration into a neutral locus in Candida albicans. Microbiology 159:565–579. doi: 10.1099/mic.0.064097-0. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.DiCarlo JE, Chavez A, Dietz SL, Esvelt KM, Church GM. 2015. Safeguarding CRISPR-Cas9 gene drives in yeast. Nat Biotechnol 33:1250–1255. doi: 10.1038/nbt.3412. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Enkler L, Richer D, Marchand AL, Ferrandon D, Jossinet F. 2016. Genome engineering in the yeast pathogen Candida glabrata using the CRISPR-Cas9 system. Sci Rep 6:35766. doi: 10.1038/srep35766. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Boulton SJ, Jackson SP. 1996. Identification of a Saccharomyces cerevisiae Ku80 homologue: roles in DNA double strand break rejoining and in telomeric maintenance. Nucleic Acids Res 24:4639–4648. doi: 10.1093/nar/24.23.4639. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Milne GT, Jin S, Shannon KB, Weaver DT. 1996. Mutations in two Ku homologs define a DNA end-joining repair pathway in Saccharomyces cerevisiae. Mol Cell Biol 16:4189–4198. doi: 10.1128/MCB.16.8.4189. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Feri A, Loll-Krippleber R, Commere PH, Maufrais C, Sertour N, Schwartz K, Sherlock G, Bougnoux ME, d’Enfert C, Legrand M. 2016. Analysis of repair mechanisms following an induced double-strand break uncovers recessive deleterious Alleles in the Candida albicans diploid genome. MBio 7:e01109-16. doi: 10.1128/mBio.01109-16. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Doench JG, Fusi N, Sullender M, Hegde M, Vaimberg EW, Donovan KF, Smith I, Tothova Z, Wilen C, Orchard R, Virgin HW, Listgarten J, Root DE. 2016. Optimized sgRNA design to maximize activity and minimize off-target effects of CRISPR-Cas9. Nat Biotechnol 34:184–191. doi: 10.1038/nbt.3437. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Norton EL, Sherwood RK, Bennett RJ. 2017. Development of a CRISPR-Cas9 system for efficient genome editing of Candida lusitaniae. mSphere 2:e00217-17. doi: 10.1128/mSphere.00217-17. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Golic KG. 1991. Site-specific recombination between homologous chromosomes in Drosophila. Science 252:958–961. doi: 10.1126/science.2035025. [DOI] [PubMed] [Google Scholar]
- 24.Van Deursen J, Fornerod M, Van Rees B, Grosveld G. 1995. Cre-mediated site-specific translocation between nonhomologous mouse chromosomes. Proc Natl Acad Sci U S A 92:7376–7380. doi: 10.1073/pnas.92.16.7376. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Golic KG, Golic MM. 1996. Engineering the Drosophila genome: chromosome rearrangements by design. Genetics 144:1693–1711. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Muzzey D, Schwartz K, Weissman JS, Sherlock G. 2013. Assembly of a phased diploid Candida albicans genome facilitates allele-specific measurements and provides a simple model for repeat and indel structure. Genome Biol 14:R97. doi: 10.1186/gb-2013-14-9-r97. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Sadhu MJ, Bloom JS, Day L, Kruglyak L. 2016. CRISPR-directed mitotic recombination enables genetic mapping without crosses. Science 352:1113–1116. doi: 10.1126/science.aaf5124. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Shapiro RS, Chavez A, Porter CBM, Hamblin M, Kaas CS, DiCarlo JE, Zeng G, Xu X, Revtovich AV, Kirienko NV, Wang Y, Church GM, Collins JJ. 2018. A CRISPR-Cas9-based gene drive platform for genetic interaction analysis in Candida albicans. Nat Microbiol 3:73–82. doi: 10.1038/s41564-017-0043-0. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Gietz RD, Schiestl RH. 2007. High-efficiency yeast transformation using the LiAc/SS carrier DNA/PEG method. Nat Protoc 2:31–34. doi: 10.1038/nprot.2007.13. [DOI] [PubMed] [Google Scholar]
- 30.Skrzypek MS, Binkley J, Binkley G, Miyasato SR, Simison M, Sherlock G. 2017. The Candida Genome Database (CGD): incorporation of assembly 22, systematic identifiers and visualization of high throughput sequencing data. Nucleic Acids Res 45:D592–D596. doi: 10.1093/nar/gkw924. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Zerbino DR, Achuthan P, Akanni W, Amode MR, Barrell D, Bhai J, Billis K, Cummins C, Gall A, Girón CG, Gil L, Gordon L, Haggerty L, Haskell E, Hourlier T, Izuogu OG, Janacek SH, Juettemann T, To JK, Laird MR, Lavidas I, Liu Z, Loveland JE, Maurel T, McLaren W, Moore B, Mudge J, Murphy DN, Newman V, Nuhn M, Ogeh D, Ong CK, Parker A, Patricio M, Riat HS, Schuilenburg H, Sheppard D, Sparrow H, Taylor K, Thormann A, Vullo A, Walts B, Zadissa A, Frankish A, Hunt SE, Kostadima M, Langridge N, Martin FJ, Muffato M, Perry E, et al. . 2018. Ensembl 2018. Nucleic Acids Res 46:D754–D761. doi: 10.1093/nar/gkx1098. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Engel SR, Dietrich FS, Fisk DG, Binkley G, Balakrishnan R, Costanzo MC, Dwight SS, Hitz BC, Karra K, Nash RS, Weng S, Wong ED, Lloyd P, Skrzypek MS, Miyasato SR, Simison M, Cherry JM. 2014. The reference genome sequence of Saccharomyces cerevisiae: then and now. G3 (Bethesda) 4:389–398. doi: 10.1534/g3.113.008995. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
CRISPR mutagenesis in S. cerevisiae with multiple guides and repair templates. (A) S. cerevisiae strain L6437 was cotransformed with plasmids pV1386 and pGB9 alongside appropriate stop repair templates, as indicated. Transformants displayed either the ade2 genotype or leu2 genotype but not the ade2 leu2 genotype. (B) Strain L6437 was cotransformed with plasmids as described above except that a consolidated repair template was used that linked the stop repair templates described in the panel A legend into a single molecule. While most transformants were either ade2 or leu2, ade2 leu2 double mutants were identified. (C) PCR genotyping identified sequences consistent with translocation between the ADE2 and LEU2 chromosomes. Download FIG S1, PDF file, 0.1 MB (67.4KB, pdf) .
Copyright © 2018 Vyas et al.
This content is distributed under the terms of the Creative Commons Attribution 4.0 International license.
Targeting multiple genes in one transformation using CRISPR. Download TEXT S1, PDF file, 0.1 MB (75.8KB, pdf) .
Copyright © 2018 Vyas et al.
This content is distributed under the terms of the Creative Commons Attribution 4.0 International license.
Plasmids used in this study. Download TABLE S1, PDF file, 0.1 MB (81.7KB, pdf) .
Copyright © 2018 Vyas et al.
This content is distributed under the terms of the Creative Commons Attribution 4.0 International license.
Strains used in this study. Download TABLE S2, PDF file, 0.1 MB (79.8KB, pdf) .
Copyright © 2018 Vyas et al.
This content is distributed under the terms of the Creative Commons Attribution 4.0 International license.
Oligonucleotides used for repair template generation. Download TABLE S3, PDF file, 0.05 MB (52KB, pdf) .
Copyright © 2018 Vyas et al.
This content is distributed under the terms of the Creative Commons Attribution 4.0 International license.
Data Availability Statement
Species-specific guide sequence tables (for single-gene, multigene, and/or allele-specific guides) are available at the Open Science Frameworks repository (DOI https://doi.org/10.17605/OSF.IO/ARDTX; https://OSF.IO/ARDTX).