

Abstract
Although targeted therapies are often effective systemically, they fail to adequately control brain metastases. In preclinical models of breast cancer that faithfully recapitulate the disparate clinical responses in these micro-environments, we observed that brain metastases evade phosphatidylinositide 3-kinase (PI3K) inhibition despite drug accumulation in the brain lesions. In comparison to extracranial disease, we observed increased HER3 expression and phosphorylation in brain lesions. HER3 blockade overcame the resistance of HER2-amplified and/or PIK3CA-mutant breast cancer brain metastases to PI3K inhibitors, resulting in marked tumor growth delay and improvement in mouse survival. These data provide a mechanistic basis for therapeutic resistance in the brain microenvironment and identify translatable treatment strategies for HER2-amplified and/or PIK3CA-mutant breast cancer brain metastases.
INTRODUCTION
Brain metastases (BMs) from human epidermal growth factor receptor 2 (HER2)–positive breast cancer (BC) patients respond poorly to treatment. Survival rates vary from 4 to 6 months with whole-brain irradiation to about 18 months with multimodality therapy (1). The incidence of BM among these patients is as high as 50% (1). The anti-HER2 antibody trastuzumab is an integral component of current treatment regimens and improves survival of HER2-positive metastatic BC patients. However, clinical data suggest that adjuvant trastuzumab is associated with central nervous system (CNS) metastases as the site of first recurrence in HER2-positive BC patients (2). This is likely because trastuzumab decreases the cumulative incidence of extracranial recurrence but fails to prevent recurrence in the brain, ultimately resulting in an increase in the ratio of BMs to total relapse events (2). This failure is mostly attributed to the lack of drug penetration through the blood-brain barrier (BBB). However, although the BBB is relevant for large molecules, such as trastuzumab, the blood-tumor barrier (BTB) is leakier, permitting drug delivery to BMs, especially at later stages of the disease (3, 4). Consistent with the BTB being leaky, both preclinical and clinical studies have shown some accumulation of anti-HER2 drugs in BMs (5, 6), but it remains unclear whether adequate drug concentrations are achieved in the clinical setting. In addition, BBB-penetrant small-molecule HER2 inhibitors such as lapatinib fail to control BM (7–9). Although delivery of anti-HER2 drugs in BMs remains a major issue, these findings suggest that additional mechanisms play a role in BM resistance. Despite major efforts to understand the failure of HER2-positive tumors to respond to anti-HER2 therapy in the brain micro-environment (10), the mechanisms that underlie this resistance remain unknown (1, 11).
Homo- and heterodimerization of HER2 triggers activation of downstream pathways that are critical for cell survival and proliferation, most notably the phosphatidylinositide 3-kinase (PI3K)–AKT pathway (12, 13). Among various heterodimers, the HER2-HER3 dimer is a potent inducer of the PI3K-AKT pathway and plays a central role in the progression of HER2-positive BCs (14, 15). BCs with PIK3CA mutations and/or HER2 amplification are highly sensitive to single-agent PI3K pathway inhibitors, and these drugs are actively being developed in the clinic for these malignancies (16–18). Although PI3K targeting has been investigated for controlling systemic metastases of BC, mechanisms for its lack of efficacy in the setting of BM are not well understood.
We previously reported a model that mimics the clinical situation; a HER2-amplified BC xenograft treated with the anti-HER2 therapies trastuzumab or lapatinib is sensitive when growing in the mammary fat pad (MFP) but resistant when situated in the brain parenchyma (10). Our findings suggested that this resistance is not mediated by impaired drug delivery in the brain setting. Here, we investigate the effect of a BBB-penetrant PI3K inhibitor on HER2-amplified and/or PIK3CA-mutant breast tumor growth within different microenvironments, identify a mechanism of resistance to either PI3K or HER2 inhibition in the brain setting, and reveal strategies to overcome resistance through combinatorial approaches.
RESULTS
HER2-amplified and/or PIK3CA-mutant BC display differential response to PI3K inhibition in the MFP versus brain parenchyma
We compared the efficacy of the specific pan-Class I PI3K inhibitor buparlisib (BKM120) on HER2-amplified and/or PIK3CA-mutant human BC cells growing as xenografts either in the MFP or in the brain. Buparlisib has activity against BC cells harboring HER2 amplification as well as oncogenic PI3K catalytic subunit alpha (PIK3CA) mutations (19). Furthermore, buparlisib effectively crosses the BBB and penetrates the brain parenchyma (20, 21). The three BC cell line xenografts examined were HER2-amplified BT474, HER2-amplified and PIK3CA-mutant (E545K) MDA-MB-361, and PIK3CA-mutant (H1047R) T47D lines. Each of these cell lines was genetically engineered to express Gaussia luciferase (Gluc), which was used as a surrogate for BM volume (6, 10). Consistent with previous studies, the blood Gluc activity for each of the three tumor models directly correlated with tumor volume (fig. S1A). Buparlisib led to regression of all tumors growing in the MFP (P < 0.05 at day 35); however, the corresponding BMs were resistant to treatment (Fig. 1, A to C). Statistical analysis of time to progression, defined as time to doubling in tumor size measured either directly or via Gluc activity, revealed a significant difference between isogenic tumors growing in the MFP versus the brain for BT474-Gluc and T47D-Gluc tumors (P < 0.05), although there was no significant difference for MDA-MB-361-Gluc tumors (P = 0.1). In BT474-Gluc and T47D-Gluc models, size measurement at 14 days (Fig. 1, A to C) after treatment initiation with buparlisib revealed a significantly higher number of mice with tumor regression for tumors growing in the MFP compared to tumors growing in the brain parenchyma (BT474-Gluc MFP versus BM: 5 of 5 versus 0 of 7; Fisher’s exact test, P = 0.001; T-47D-Gluc MFP versus BM: 7 of 7 versus 0 of 9; Fisher’s exact test, P < 0.0001). MDA-MB-361 tumors growing in the brain exhibited a similar lack of PI3K inhibitor control, however at a later time point than BT474 and T47D BMs. Analysis of response over a period of 28 days revealed that five of six MDA-MB-361 tumors in the MFP regressed after treatment with buparlisib, whereas this was the case for only one of six tumors in the brain (Fisher’s exact test, P = 0.08).
Fig. 1. HER2-amplified and/or PIK3CA-mutant breast tumors display differential response to PI3K inhibition when grown in the MFP versus brain despite similar drug delivery.
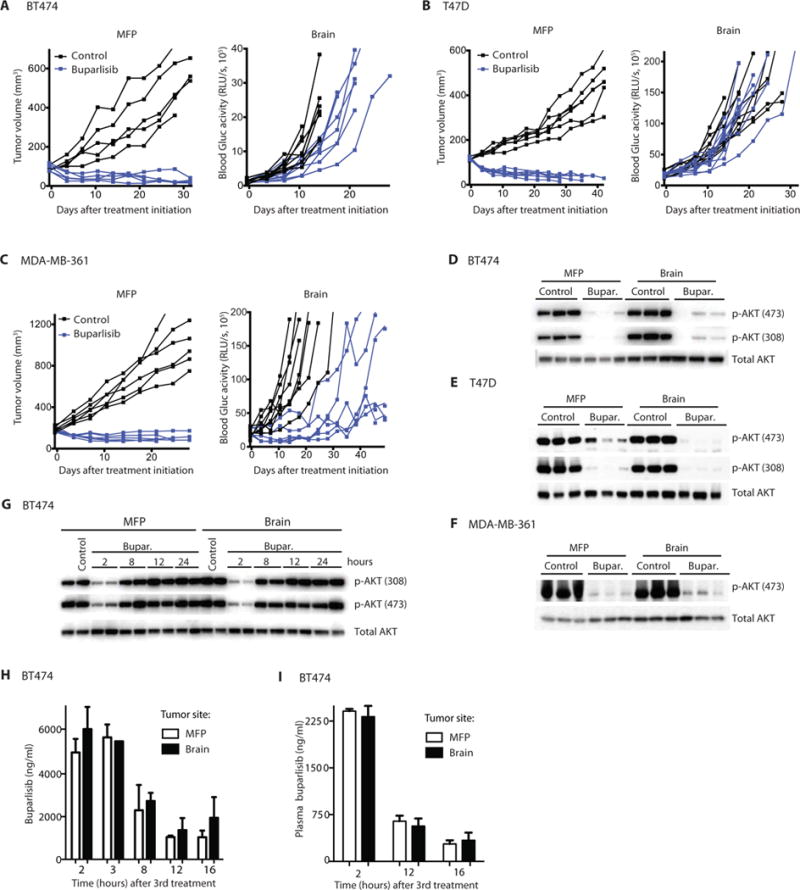
Established HER2-amplified BT474-Gluc (A), PIK3CA-mutant T47D-Gluc (B), and HER2-amplified and PIK3CA-mutant MDA-MB-361-Gluc tumors (C) growing in the MFP (left) or brain (right) were treated with buparlisib (BKM120) or vehicle (PEG 300), and tumor volume (MFP) or blood Gluc activity (brain) was measured at the indicated times (BT474: MFP n = 5, brain n = 7 to 9; MDA-MB-361: MFP n = 6, brain n = 6 to 7; T47D: MFP n = 6 to 7, brain n = 9 to 10). RLU/s, relative light units per second. BT474-Gluc (D), T47D-Gluc (E), or MDA-MB-361-Gluc (F) tumor tissue, collected 2 hours after the final treatment with buparlisib (Bupar.), was analyzed for AKT phosphorylation as a readout of PI3K inhibition. (G) BT474-Gluc tumor tissue, collected at the indicated time points after the third dose of buparlisib (48 hours after the first dose), was analyzed for AKT phosphorylation. (H) The concentration of buparlisib in BT474-Gluc tumor tissue collected after the indicated time points was determined [unpaired t test, 2 hours (n ≥ 4), P = 0.41; 8 hours (n = 2), P = 0.78; 12 hours (n = 2), P = 0.67; 16 hours (n ≥ 3), P = 0.45]. (I) Plasma concentration of buparlisib in mice whose tumor buparlisib concentration was analyzed in (H) [unpaired t test, 2 hours (n ≥ 4), P = 0.64; 12 hours (n = 2), P = 0.66; 16 hours (n = 4), P = 0.68]. Data are means ± SD.
Tumor tissue collected 2 hours after the final dose of buparlisib displayed marked suppression of AKT phosphorylation in both BM and MFP tumors, compared with untreated tumors (Fig. 1, D to F). We next asked if the duration of PI3K inhibition was similar in BM and MFP tumors. A time course of buparlisib treatment in BT474 MFP tumors showed a rebound in phosphorylated AKT (p-AKT) to control levels by 12 hours (Fig. 1G). This rebound after inhibition is consistent with published literature (22, 23). A similar recovery of p-AKT was observed in MDA-MB-361 BM 12 hours after buparlisib treatment (fig. S1B).
Direct measurement of the buparlisib concentrations in the plasma and in breast tumors growing at both sites showed no substantial differences between the MFP tumors and BM (Fig. 1, H and I, and fig. S1C), consistent with previous findings that buparlisib freely crosses the BBB. Therefore, the difference in sensitivity of MFP tumors and BM cannot be explained by impaired drug delivery.
To determine whether the discordance in treatment response was specific for the brain microenvironment, we investigated the efficacy of buparlisib in an additional extracranial microenvironment—the liver parenchyma. We implanted isogenic BT474-Gluc cells in the liver of female nude mice, allowed tumors to reach a similar volume as in the brain, and treated with the same dose of buparlisib (50 mg/kg daily per os). Our studies revealed a marked growth delay of HER2-amplified tumors growing in the liver (fig. S2, A to C), supporting the hypothesis that tumor resistance to PI3K inhibition is specific to the brain microenvironment.
We also examined whether the brain microenvironment might enhance or suppress the proliferation of BC cells, thus reducing the ability of PI3K inhibitors to slow their growth. We found that the growth rates of BC cells in the brain and the MFP were comparable (fig. S3A). Together, these data show that the marked difference in sensitivity to PI3K inhibition is not simply due to a compromised pharmacodynamic or pharmacokinetic profile of buparlisib in BM or to differential growth rates of the isogenic tumors in distinct microenvironments.
HER2-amplified and/or PIK3CA-mutant BC cells require continued exposure to the brain microenvironment to maintain resistance to PI3K inhibition
To determine whether BC cells require constant influence from the brain microenvironment to be resistant to PI3K inhibitors, we isolated cells from BT474-Gluc, MDA-MB-361-Gluc, and T47D-Gluc BMs after dissociating them from the brain parenchyma. After 1 week of ex vivo culture, the vast majority of viable cells were cancer cells, as identified by green fluorescent protein (GFP) expression. These “brain microenvironment–exposed” BC cells were similarly sensitive to buparlisib in vitro as parental cells (fig. S3, B to D). These data suggest that the brain microenvironment does not induce a permanent change in BC cells nor is there preferential growth of buparlisib-resistant clones in the brain. Instead, the BC cells must be situated in the brain microenvironment to maintain resistance to PI3K inhibition.
HER3 is overexpressed and phosphorylated in HER2-amplified and/or PIK3CA-mutant BC BMs in mouse models and human samples
To determine whether there is differential phosphorylation of growth factor receptor tyrosine kinases (RTKs) in xenograft tumors growing in either the brain or the MFP, we performed a phosphoreceptor tyrosine kinase protein array. The array demonstrated a clear increase in the phosphorylation of HER3 and epidermal growth factor receptor (EGFR), but not HER2 and HER4, in BT474 xenografts grown in the brain compared to those in the MFP (Fig. 2A). In addition, we observed an increase of RON RTK. We focused on HER3 as a potential mediator of resistance because of literature demonstrating a role for HER3 in reactivation of downstream pathways in the presence of PI3K inhibitors (22–24). Western blotting confirmed increased expression and phosphorylation of HER3 in BT474-Gluc, T47D-Gluc, and MDA-MB-361-Gluc BM compared with their MFP counterparts (Fig. 2B). HER3 overexpression in BT474-Gluc and T47D-Gluc BM was also confirmed by quantitative polymerase chain reaction (qPCR) (Fig. 2, C and D).
Fig. 2. HER3 is overexpressed and hyperactivated in BMs.
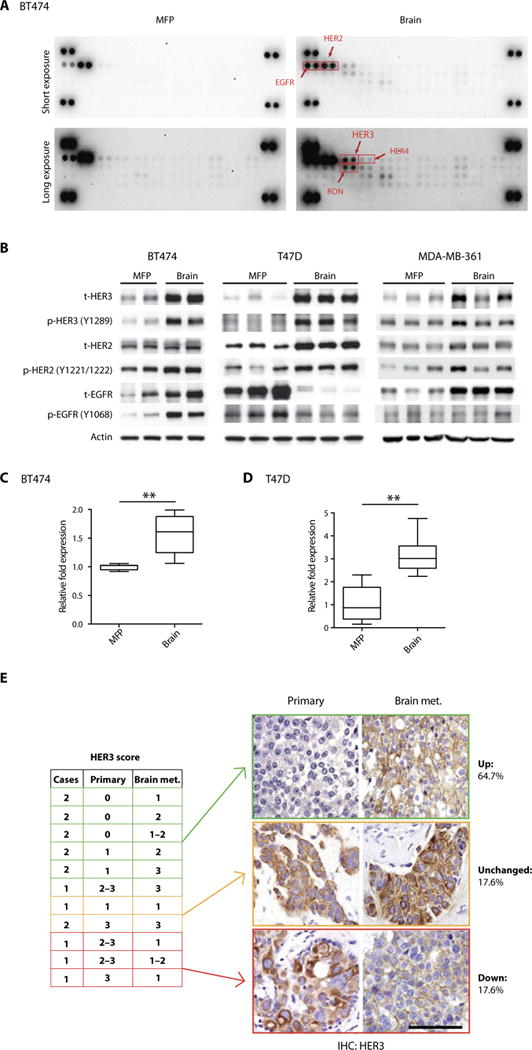
(A) A phosphoreceptor tyrosine kinase array depicts ErbB family members as hyperphosphorylated in HER2-amplified BT474 breast tumors growing in the brain (right) compared with the MFP (left). (B) Breast tumor tissue from BT474, T47D, or MDA-MB-361 MFP or brain tumors was analyzed for phosphorylated and total (t) EGFR, HER2, and HER3. (C and D) Quantification of HER3 mRNA (human) in BT474 (n = 4) and T47D (brain; n = 7 and 9) (unpaired t test, **P < 0.001). Data are means ± SD. (E) Whole-tissue section immunohistochemistry (IHC) analysis of HER3 protein in matched human primary and brain metastatic (met.) HER2-positive breast tumor tissue (left). Representative IHC images of matched human primary and brain metastatic HER2-positive breast tumor tissue stained for HER3 (right). Percentage of HER3 signal in BM and matched primary tumor is depicted (scale bar, 200 μm).
To determine the mechanism of differential HER3 regulation in the brain and MFP, we compared the transcriptomes of BT474 tumors grown in both microenvironments. Gene set enrichment analysis revealed that targets of the transcription factor Forkhead box O3a (FOXO3a)—a known inducer of HER3 transcription (25)—were enriched in the BM compared to MFP (fig. S3, E and F).
Although quantification of p-HER3 revealed an increase in p-HER3 for all BM compared to MFP, the increase was only modest for MDA-MB-361-Gluc tumors (fig. S4, A to C). Notably, MDA-MB-361-Gluc BMs were the least resistant to buparlisib (Fig. 1C). Whereas buparlisib had no impact on the growth of T47D or BT474 BM, the PI3K inhibitor delayed the progression of MDA-MB-361 BM (fig. S4, D to F). Although the brain micro-environment induced resistance to PI3K inhibitors in all models (compared to the MFP), it is notable that the most pronounced resistance was in the models with the highest induction of HER3 phosphorylation.
To assess the clinical relevance of this finding, we determined the expression of HER3 in a cohort of 17 matched primary and BM tissues obtained from patients with HER2-amplified BC. This analysis revealed an increase in HER3 protein in the BM biopsies of 64.7% of patients, compared with their primary tumor (P = 0.047) (Fig. 2E and fig. S5A). A comparison of HER2 measurements in eight of these samples did not reveal a similar differential expression between the two sites (P = 0.65) (fig. S5B). The difference in HER3 protein amounts was also observed in a series of unmatched samples of primary tumors (n = 10), extracranial metastases (n = 9), and BM (n = 13) of HER2-positive BC patients (table S1). Notably, we found statistically significant overexpression of HER3 in BMs compared to primary tumors (P = 0.046), whereas no difference between extracranial metastases and primary tumors was detected (P = 0.16). Collectively, our HER3 evaluation indicates that HER3 can be expressed in primary and extra-cranial sites but is increased in brain metastatic lesions. Our findings are consistent with a previous analysis that demonstrated an increase in HER3 mRNA in human BC BM compared to the corresponding matched primary tumor (26). However, this previous study did not use a genetically defined subset of BCs.
Neuregulins induce in vitro resistance to PI3K inhibitors in HER2-amplified and/or PIK3CA-mutant BC cells, and this is abrogated by antibodies targeting HER3
To identify potential mediators of resistance to PI3K inhibition, we examined the effect of 220 different growth factors on the sensitivity of BT474, MDA-MB-361, and T47D BC cells to buparlisib (table S2). This unbiased screen revealed that the HER3 ligands neuregulin-1 (NRG-1) and NRG-2 were the most potent inducers of resistance to buparlisib in all three cell lines (Fig. 3, A to C, and fig. S6, A and B). Several NRG-1 and NRG-2 isoforms were capable of inducing resistance (fig. S6A).
Fig. 3. NRGs induce in vitro resistance to PI3K inhibition in HER2-amplified and/or PIK3CA-mutant BC cells, and this is dependent on HER3 activation.
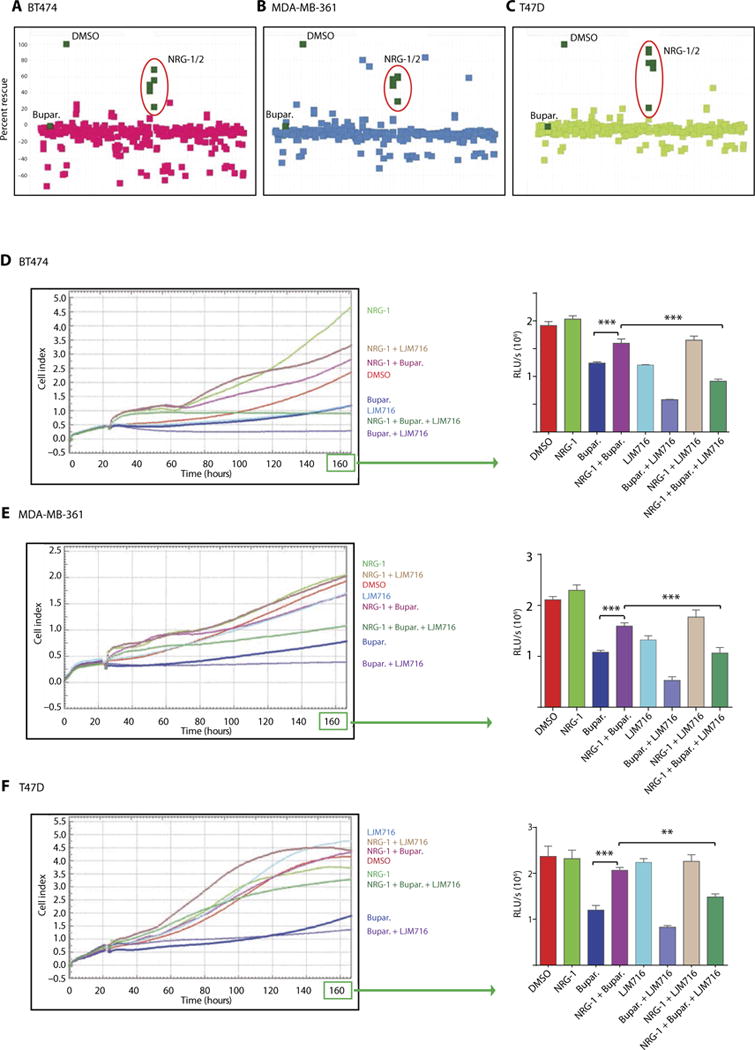
(A to C) A library of secreted proteins was screened for their ability to rescue the inhibition of BT474 (A), MDA-MB-361 (B), and T47D (C) BC cell growth in vitro by buparlisib. Each dot represents the average of three replicates of cells conditioned with the same supernatant. “DMSO” represents cells treated with dimethyl sulfoxide control in the absence of secreted proteins (rescue, buparlisib-treated cells in the presence of growth factor relative to control). (D to F) In vitro xCELLigence growth assay of BT474 (D), MDA-MB-361 (E), or T47D (F) BC cells exposed under the indicated conditions for the indicated time. The y axis is cell index defined as the relative change in measured electrical impedance. The graphs on the right show CellTiter-Glo luminescence viability measurements at the end of the experiments [one-way analysis of variance (ANOVA)/Tukey test, **P = 0.0009, ***P < 0.0001]. Data are means ± SD.
Because HER3 is a receptor for NRG-1, we examined the ability of HER3 inhibition to overcome NRG-1–induced resistance to PI3K inhibition in vitro. To target HER3, we used the anti-HER3 monoclonal antibody LJM716 (Novartis) that locks HER3 in an inactive state (27). NRG-1–induced resistance to buparlisib in all BC cell lines (BT474, MDA-MB-361, and T47D) was overcome by LJM716 in vitro (Fig. 3, D to F). In vitro investigation of HER3 and AKT phosphorylation revealed that NRG-1 blunts buparlisib-mediated inhibition of p-AKT, and the addition of LJM716 to buparlisib in the presence of NRG-1 results in the resuppression of p-AKT (fig. S6C).
On the basis of our findings that NRG-1 induces in vitro resistance to PI3K inhibition and that its receptor (HER3) is overexpressed and activated in BM, we investigated the expression of NRG-1 in BM from HER2-amplified and/or PIK3CA-mutant BC. We used qPCR to determine the expression pattern of two splice isoforms, α and β, representing variations in the EGF signaling domain of NRG-1. Both isoforms can bind and activate HER3 (28). Although NRG-1β is enriched in normal brain tissue and NRG-1α in the normal MFP, there were no differences in NRG-1 mRNA isoforms between BT474 tumors in either organ (fig. S7A). To test whether NRG-1 in BM is produced by cancer cells or stromal cells, we separated HER2-positive cells from HER2-negative cells of BT474-Gluc BM using fluorescence-activated cell sorting (FACS) and performed qPCR for NRG-1α and NRG-1β. Our analysis revealed that NRG-1 is expressed in both the HER2-positive (cancer) and HER2-negative (stromal) cells (fig. S7B) without any significant difference. Together, these data indicate that both stromal and cancer cells within BM express NRG-1 isoforms, and therefore, activation of HER3 can be mediated via autocrine and/or paracrine mechanisms.
HER3 targeting sensitizes HER2-amplified and/or PIK3CA-mutant BC BMs to PI3K inhibition
We next determined whether HER3 targeting would sensitize BM to PI3K inhibition in vivo. We examined the effect of the HER3 antibody LJM716 or the HER2-HER3 dimerization inhibitor pertuzumab (29), alone or in combination with buparlisib, on the growth of BT474, MDA-MB-361, or T47D BC BM. Neither pertuzumab nor LJM716 monotherapy slowed the growth of BM. However, the combination of HER3 inhibition with buparlisib greatly slowed the growth of HER2-driven and/or PI3K-driven BC BM (Fig. 4). The tumor growth delay with the combination therapy improved survival compared to buparlisib monotherapy. In mice bearing BT474-Gluc BM, the median survival was 30 days with buparlisib monotherapy versus 76 days with combination therapies (P<0.001) (Fig. 4A). In mice bearing MDA-MB-361-Gluc BM, median survival was 42 days in the buparlisib monotherapy group versus 84 and >125 days (not reached) in the buparlisib/pertuzumab and buparlisib/LJM716 groups, respectively (P < 0.001) (Fig. 4B). In mice bearing T47D BM, median survival was 54 days with buparlisib monotherapy and >84 days (not reached) with the buparlisib/LJM716 combination therapy (P < 0.01) (Fig. 4C).
Fig. 4. Therapeutic targeting of HER3 sensitizes HER2-amplified and PI3KCA-mutant BC BMs to PI3K inhibition.
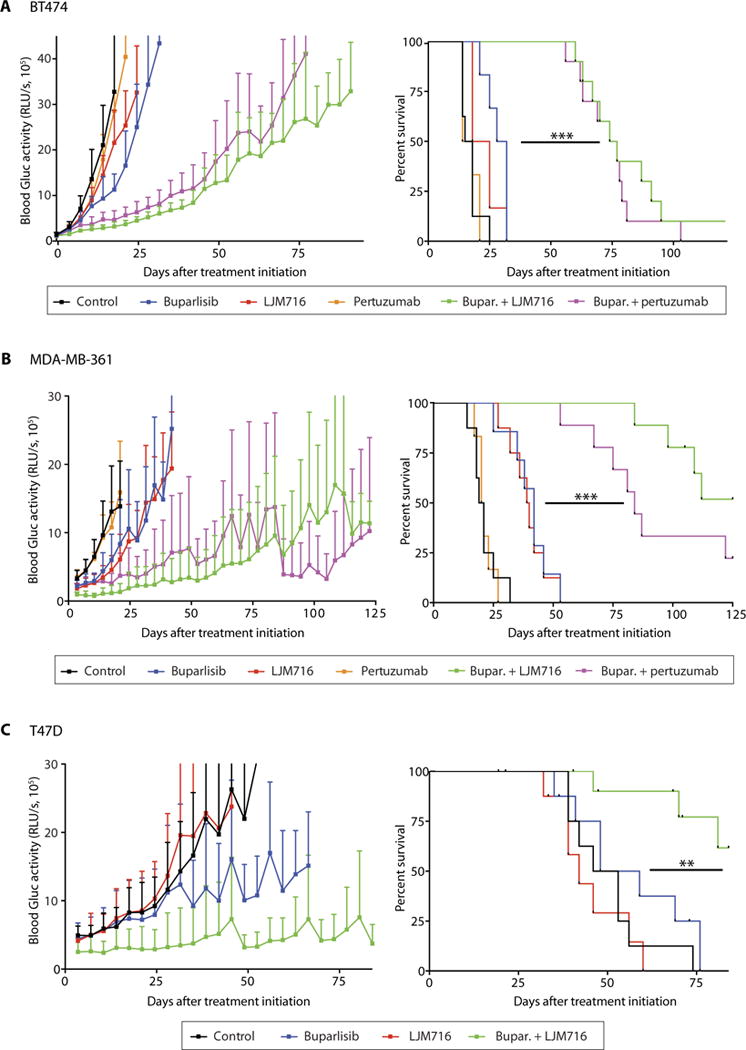
Tumor growth curve (left) and survival analysis (right) of established BT474-Gluc (A), MDA-MB-361 (B), or T47D (C) brain tumors untreated or treated with buparlisib, LJM716, pertuzumab, buparlisib and LJM716, or buparlisib and pertuzumab [log-rank test; (A) BT474-Gluc—n = 6 to 10 per group; buparlisib + LJM716 versus buparlisib: hazard ratio (HR), 0.19; 95% confidence interval (CI), 0.04 to 0.94; P < 0.0001; buparlisib + pertuzumab versus buparlisib: HR, 0.20; 95% CI, 0.04 to 0.97; P < 0.0001; (B) MDA-MB-361—n = 6 to 9 per group; buparlisib + LJM716 versus buparlisib: HR, 0.13; 95% CI, 0.03 to 0.60; P < 0.0001; buparlisib + pertuzumab versus buparlisib: HR, 0.19; 95% CI, 0.04 to 0.78; P < 0.0001; (C) T47D—n = 8 to 11 per group; buparlisib + LJM716 versus buparlisib: HR, 0.19; 95% CI, 0.05 to 0.73; P = 0.005] (**P = 0.005 and ***P < 0.0001). Tumor size data are means ± SD.
HER3 targeting enhances the pharmacodynamic effects of PI3K inhibition in BMs
We next aimed to understand the intracellular signaling changes that underlie the beneficial effect of dual PI3K and HER3 inhibition in BM. We examined PI3K activity directly by using a validated method to measure phosphatidylinositol (3,4,5)-triphosphate (PIP3), the direct product of PI3K, and phosphatidylinositol (4,5)-bisphosphate (PIP2) concentration (16). PIP2 concentration was used as control because PIP2 is the most abundant phosphatidylinositide (30), and its concentration is not influenced by PI3K activity (31). Our studies revealed that 10-day treatment of BT474-Gluc BM with the combination of buparlisib and LJM716 significantly decreased the PIP3/PIP2 ratio compared to buparlisib monotherapy (P = 0.0178; Fig. 5A). Decreases in p-AKT and p-S6 in the combination group were also observed after 3-day treatment (Fig. 5B). These correlated with a decrease in the phosphorylation of downstream PI3K pathway effectors, including AKT, S6, and PRAS40 in the combination group after 10-day treatment (Fig. 5C). Together, our data suggest that HER3 inhibition can overcome brain microenvironment–mediated resistance to PI3K inhibition by suppressing HER3-induced activation of PI3K signaling.
Fig. 5. HER3 blockade inhibits PI3K signaling in BM.
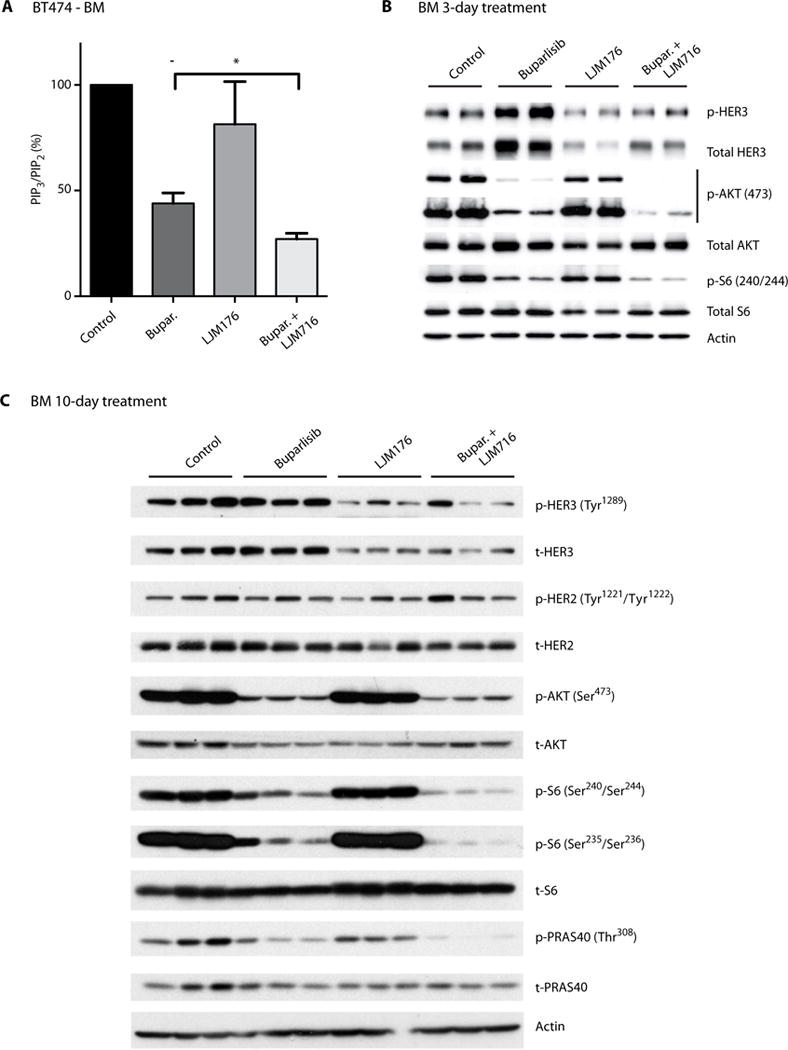
(A) Analysis of PIP3/PIP2 ratio in HER2-amplified BT474-Gluc BM untreated or treated with buparlisib, LJM716, or buparlisib and LJM716. Tumor tissue was collected 3 hours after the 10th treatment with buparlisib (24-hour intervals) and/or fifth dose of LJM716 (48-hour intervals) (unpaired t test, n = 5, *P = 0.0178). Data are means ± SEM. (B) Analysis of HER3, AKT, and S6 phosphorylation in HER2-amplified BT474-Gluc BM untreated or treated for 3 days with buparlisib, LJM716, or their combination. (C) Analysis of HER3, HER2, AKT, S6, and PRAS phosphorylation in HER2-amplified BT474-Gluc brain tumors untreated or treated for 10 days with buparlisib, LJM716, or their combination.
HER3 targeting sensitizes HER2-amplified BC BMs to HER2-targeted therapies
Finally, we tested the effects of targeting HER3 or its dimerization with HER2 on the efficacy of HER2-targeted therapies in the brain microenvironment. The rationale for these studies was threefold: (i) Resistance of BMs to HER2-targeted therapies is a major clinical problem, (ii) BMs show increased expression and activation of HER3, and (iii) blocking HER2-HER3 heterodimerization has been shown to improve the outcomes of patients with HER2-positive metastatic BC treated with trastuzumab (32). Our in vivo studies using the anti-HER2 antibody trastuzumab or the HER2/EGFR small-molecule inhibitor neratinib for the treatment of HER2-amplified BT474-Gluc BM revealed that these treatments alone had minimal efficacy. However, we observed a marked delay in the growth of brain lesions treated with the combination of trastuzumab or neratinib with LJM716 or pertuzumab (Fig. 6, A and B). This delay in tumor growth translated into a marked survival benefit (P < 0.01) (Fig. 6, C and D). In vitro studies confirmed that NRG-1 causes resistance to trastuzumab that can be overcome by LJM716 (fig. S7C).
Fig. 6. Therapeutic targeting of HER3 sensitizes HER2-amplified BC BMs to HER2 inhibition.
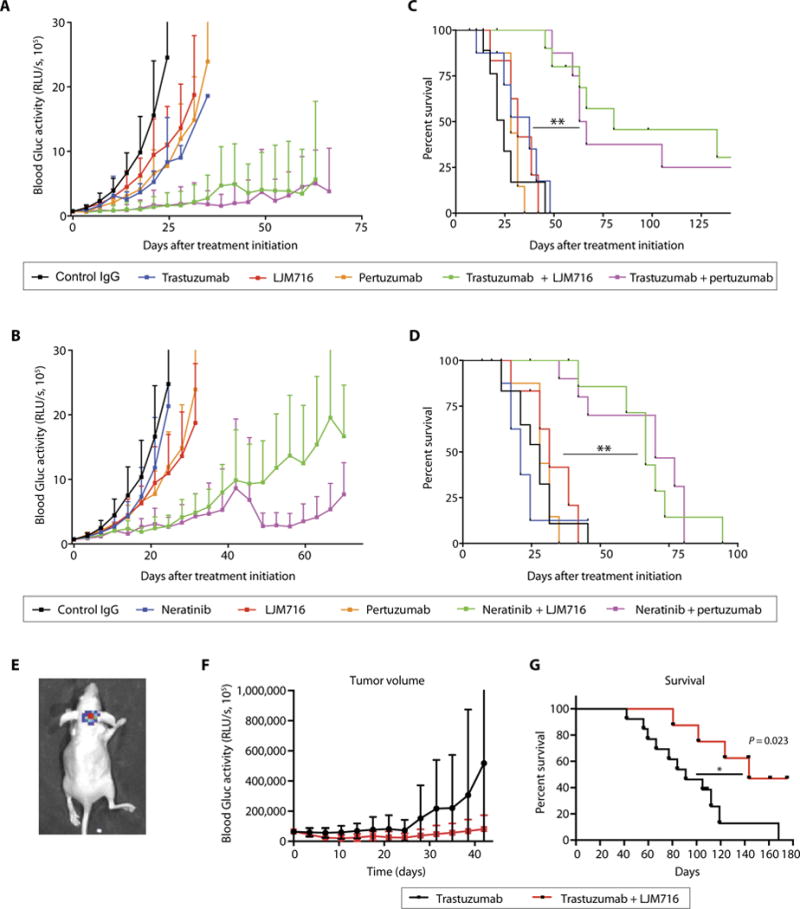
Tumor growth curve (A and B) and survival analysis (C and D) of established BT474-Gluc brain tumors untreated or treated with trastuzumab, neratinib, pertuzumab, LJM716, and indicated combinations (n = 8 to 10 per group; log-rank test; trastuzumab + LJM716 versus trastuzumab: HR, 0.16, 95% CI, 0.03 to 0.83; **P < 0.001; trastuzumab + pertuzumab versus trastuzumab: HR, 0.17; 95% CI, 0.04 to 0.85; **P < 0.001; neratinib + LJM716 versus neratinib: HR, 0.24; 95% CI, 0.06 to 0.90; P < 0.001; neratinib + pertuzumab versus neratinib: HR, 0.24; 95% CI, 0.06 to 0.91; P = 0.001). Tumor size data are means ± SD. IgG, immunoglobulin G. (E) Representative bioluminescence whole-body imaging of brain BT474-Gluc tumors from an intracarotid model. Tumor growth curve (F) and survival analysis (G) of BT474-Gluc brain tumors treated with trastuzumab (n = 13) or trastuzumab + LJM716 (n = 8) in the intracarotid model (log-rank test: HR, 0.30; 95% CI, 0.11 to 0.83; *P = 0.02).
Although the model of direct stereotactic implantation faithfully recapitulates clinical findings (10), it skips early steps of the metastatic process (33). To test whether the model of direct intracranial injection of cancer cells affects the delivery and efficacy of antibodies in brain lesions, we compared the efficacy of trastuzumab alone and in combination with LJM716 in a model of BM that develops after intracarotid injection of BT474-Gluc cells (Fig. 6E) (6). The addition of LJM716 to trastuzumab delayed the growth of BM and resulted in a significant improvement in survival in this model as well (P = 0.02) (Fig. 6, F and G). Our results indicate that, although drug delivery into the brain parenchyma may be a problem, it is not the only explanation for the therapeutic resistance of BMs.
DISCUSSION
Here, we identify a mechanism by which the brain microenvironment specifically induces resistance to PI3K-targeted therapy. Our results agree with previous findings demonstrating resistance to PI3K inhibition in orthotopic PDX models of HER2-positive BC BM (34). We believe that these findings will have a major impact on the way targeted cancer therapies are understood and applied. To date, targeted therapies mostly focus on genetically encoded vulnerabilities of cancer cells. This work demonstrates that the environment in which the cancer cells reside can also affect the efficacy of targeted therapies and that it should be considered during the development of treatment strategies.
Our findings demonstrate that BC resistance to PI3K inhibition when growing in the brain is mediated by enhanced activity of the HER3 signaling pathway. The data indicate that HER3 up-regulation and activation in BM impair the ability of the PI3K inhibitor buparlisib (BKM120) to sufficiently suppress PI3K activity. Blocking HER3 or its dimerization with HER2 reduces the activity of PI3K and its downstream signaling and restores the efficacy of the PI3K inhibitor. Considering the critical role of HER3 in resistance to HER2-targeted therapies, we additionally show that HER3 targeting can overcome resistance of HER2-amplified BM to HER2-targeted therapies. Thus, the observations on the mechanistic role of HER3 activation in causing therapeutic resistance in BMs from HER2-positive BC may be applied to both PI3K-and HER2-targeted therapies.
Although HER3 activation and signaling are similar between our in vitro and in vivo results, our findings do not exclude a ligand-independent role for HER3 activation in brain lesions. NRG-1 has numerous isoforms that are generated by differential promoter activity and alternative splicing, making the characterization of protein expression challenging. Splice variants with exon 7 produce α and β EGF isoforms that show both overlapping and specific biological functions. We observed expression of NRG-1 mRNA (α and β isoforms) in both the brain stroma and tumor cells. This is consistent with studies demonstrating expression of NRG-1 in BC cells (35). Furthermore, NRG-1 is important in the regulation of brain plasticity, and it is expressed in astrocytes and neurons (36). A recent genomic and transcriptomic investigation of BC cells from BMs revealed that tumor cells in brain lesions can lose expression of NRG-1 mRNA, suggesting that HER3-positive tumors can depend on the NRG-1–rich brain microenvironment (37).
Instead of targeting the NRG-1 ligand, we focused on targeting its receptor (HER3) because of presumed toxicities associated with depleting mice of NRG-1 (38). This rationale is supported by preclinical findings indicating that phosphorylated HER3 is associated with resistance to inhibitors of PI3K signaling, including HER2-targeted therapies (22, 23, 39). Moreover, clinical trials showed that targeting the HER2-HER3 interaction in patients with metastatic BC results in improved therapy response and survival(32,40). The treatments used in the present study may also be beneficial for meta-static BC as well as other tumor types that reside in organs that use the NRG/HER3 pathway for growth and/or survival.
Previous descriptive studies reported an overexpression of HER3 in BMs, leading the authors to the conclusion that HER3 has a role in the ability of cancer cells to colonize the brain (26). Analysis of 36 BMs from various cancers revealed that HER3 was the most highly and consistently expressed member of the ERBB/HER family across different primary cancer types and that the HER3 transcript abundance correlated with HER2 (37). Further studies indicate that the NRG-1/HER3/HER2 signaling activates matrix metalloproteinases to promote transendothelial migration of BC cells, suggesting that HER3 is involved in cancer cell migration through the BBB and the development of BMs (41). Although our results do not contradict this hypothesis, the data in this study provide evidence that the expression and activation of HER3 are mediated by the CNS micro-environment. Our results obtained from multiple isogenic HER2-amplified and/or PI3KCA-mutant BC cell lines implanted in different environments reveal that HER3 is critically involved in BC BM resistance to PI3K and HER2 inhibitors, emphasizing the functional importance of the brain microenvironment in driving resistance to targeted therapies.
In addition to HER3, the phosphoreceptor protein array indicated a hyperactivation of EGFR in BT474-Gluc BM. Although increased phosphorylated and total EGFR were confirmed by Western blot in BT474-Gluc BM, T47D-Gluc BM showed less phosphorylated and total EGFR compared to MFP tumors. MDA-MB-361-Gluc BM presented increased total EGFR but undetectable amounts of phosphorylated protein. Although we cannot exclude that EGFR might be involved in the resistance of HER2-amplified and/or PIK3CA-mutant BC to PI3K inhibitionin the brain, the inconsistent pattern of EGFR expression and phosphorylation among the different tumors that are all resistant to buparlisib when in the brain suggests that the role of EGFR in resistance to PI3K inhibition in this setting may not be critical. Furthermore, we found that the HER2-HER3 dimerization inhibitor pertuzumab overcomes the brain microenvironment–mediated resistance to buparlisib. Pertuzumab binds the ECD II (extracellular domain II) dimerization domain of HER2 and blocks the HER2-HER3 interaction, suggesting that EGFR does not play a major role in this setting. To provide more insight, we performed in vivo studies in mice bearing HER2-amplified BT474 tumors in the brain using the dual HER2 and EGFR inhibitor neratinib. Similarly to HER2 targeting (trastuzumab) or PI3K targeting (buparlisib), neratinib monotherapy failed to delay tumor growth in the brain and improve survival. However, the addition of pertuzumab led to a marked survival improvement. Overall, these data support the hypothesis that resistance to HER2/PI3K inhibition is most likely mediated by HER2/HER3 and not by EGFR activation. Finally, the RTK screen revealed increased phosphorylation of RON RTK. RON, also known as macrophage stimulating 1 receptor, is overexpressed in BC and regulates β-catenin, NF-κB (nuclear factor κB), SMAD, and JAK/STAT3 (Janus kinase/signal transducer and activator of transcription 3) signaling pathways (42). Its role in HER2 signaling remains to be determined.
In conclusion, this study shows that the brain microenvironment promotes a specific, mechanism-based resistance to PI3K inhibitors in HER2-amplified and/or PI3KCA-mutant BC cells. The data presented in this work demonstrate that a pathway known to mediate resistance in extracranial disease is hyperactivated a priori in the brain micro-environment, resulting in de novo resistance. Although genetic alterations in cancer cells remain important therapeutic targets, and impaired drug delivery in BMs may affect efficacy of targeted therapies, our data indicate that the tumor microenvironment has a key role in resistance and should be critically considered for the optimization of targeted treatment. Because the lack of efficacy of targeted therapies against BMs is an emerging clinical problem, our findings are of high translational relevance and are expected to have a substantial impact on the development of therapeutic strategies against BMs of HER2-amplified or PIK3CA-mutant BC.
MATERIALS AND METHODS
Study design
This study was designed to test the efficacy of PI3K inhibitors against HER2-positive tumors in different microenvironments, determine mechanisms of resistance to these therapies in the brain, and develop new therapeutic strategies to overcome this resistance. To investigate microenvironment-related differences in therapy response, the growth of isogenic tumors implanted in the brain or the MFP during treatment with the PI3K inhibitor buparlisib was studied and compared. Tumor growth was determined either by direct measurement for tumors in the MFP or by measuring the Gluc activity for tumors growing in the brain. For survival analysis, animals were randomly assigned to treatment groups (control, monotherapies, or combination treatment, as described in this study) when tumors reached a predefined volume. We used a log-rank test to provide at least 80% power to detect a hazard ratio of 0.25 with an α level of 0.05. All calculations were based on findings from our preliminary experiments. Individuals administering the drugs were not blinded to the treatments. Treatments were performed by multiple individuals to ensure reproducibility. The survival endpoint was reached when mice lost at least 20% of their body weight or showed symptoms of neurological impairment. All animals were included in the statistical analysis. Experimental replicates are indicated in the figure legends. Statistical methods are described in “Statistical analysis.”
Cell lines, infections, and culture
BT474, MDA-MB-361, and T47D cells were transduced with an expression cassette encoding Gluc and GFP separated by an internal ribosomal entry site, using a lentiviral vector as previously described (43). All cell lines, purchased from the American Type Culture Collection, were authenticated (Cell Check). BT474-Gluc and T47D-Gluc cells were maintained in RPMI 1640 supplemented with 10% (v/v) fetal bovine serum (FBS) (Atlanta Biologicals). MDA-MB-361-Gluc cells were maintained in Dulbecco’s modified Eagle’s medium/F12 supplemented with 10% (v/v) FBS.
MFP, liver, and brain metastatic xenografts
Female nude mice (8 to 9 weeks of age) were implanted with a 0.36- or 0.72-mg 17β-estradiol pellet (Innovative Research of America) the day before implantation of tumor cells. Pellets were replaced at expiration date. MFP and brain implantations were performed as previously described (10). For liver tumors, 106 BT474-Gluc cells were mixed with Matrigel (1:1 volume ratio) and injected in the subcapsular region of the liver parenchyma in the median lobe. For the intracarotid model, 100,000 BT474 cells were diluted in 50 to 100 μl of phosphate-buffered saline (PBS) and slowly injected into the right carotid artery of mice. All animal procedures were performed according to the guidelines of the Public Health Service Policy on Human Care of Laboratory Animals and in accordance with a protocol approved by the Institutional Animal Care and Use Committee of Massachusetts General Hospital (MGH).
Tumor size monitoring and survival analysis
Cranial windows were implanted into nude mice, as previously described (44). To assess tumor volume, in vivo imaging was performed through a cranial window using a small-animal ultrasonography device (Vevo 2100, FujiFilm VisualSonics Inc.). BT474-Gluc experimental liver metastatic growth was determined by small-animal, high-frequency ultrasonography imaging (Vevo 2100, FujiFilm VisualSonics Inc.). MFP and brain tumor size were measured as previously described (6, 10). Tumor size was measured twice a week by measuring the activity of secreted Gluc in the blood. Measurement of blood Gluc was performed as previously described (43).
Reagents and treatments
Buparlisib (BKM120) was administered at either 30 or 50 mg/kg once a day via oral gavage (per os). LJM716 was administered at 25 mg/kg every 2 days via intraperitoneal injection. Both buparlisib and LJM716 were obtained from Novartis. Trastuzumab and pertuzumab were administered at 15 mg/kg intraperitoneally weekly. Trastuzumab and pertuzumab were obtained from the MGH pharmacy. Neratinib was purchased from LC laboratories and administered at 20 mg/kg once daily per os. Recombinant NRG-1β was obtained from R&D Systems.
Buparlisib concentration measurement
Tumor samples were collected, weighed, and lysed in Lysing Matrix D tube (MP Biomedicals) in RIPA buffer (Cell Signaling Technologies) at fixed weight/volume ratio. The samples were centrifuged at 13,000 rpm and 4°C in a microcentrifuge for 10 min. Supernatant was collected from the lysate and stored at −80°C. The buparlisib concentration in tumor lysate was analyzed by liquid chromatography–tandem mass spectrometry at inVentiv Health Clinical Lab Inc. Normalization was performed by tumor weight–lysis buffer ratio to yield buparlisib exposure (in nanogram per milliliter) in tumor tissue.
Western blotting and human phosphoreceptor tyrosine kinase array
Tumor tissues were homogenized in RIPA buffer (Boston BioProducts) with protease (complete ULTRA tablets, Roche) and phosphatase inhibitors (phosSTOP, Roche) using a LabGEN (Cole-Parmer) tissue homogenizer. Lysates were centrifuged at 14,000 rpm (~18,000g) for 30 min at 4°C. Protein concentration of the supernatant was measured before boiling in Laemmli buffer. The human phosphoreceptor tyrosine kinase array (R&D Systems) was used according to the manufacturers’ recommendations. All antibodies were obtained from Cell Signaling Technologies, unless indicated otherwise.
Gene array
RNA was extracted from BT474-Gluc tumors grown in the BM and MFP. RNA was extracted, processed according to the manufacturer’s instructions, and hybridized to Affymetrix GeneChip Human Genome U133 Plus 2.0 arrays.
Isolation of cancer cells from brain metastatic tumors
Tumor tissue was minced in RPMI and incubated in RPMI + 10% FBS + 1% penicillin/streptomycin (P/S) with collagenase/dispase enzyme mix (1 mg/ml; Roche) at 37°C, shaking for 1 hour. Tissue was then centrifuged at 1500 rpm for 5 min, and supernatant was removed. Pellet was resuspended in RPMI + 10% FBS + 1% P/S and pipetted well to dissociate clumps. Medium was refreshed the following day. FACS was performed using FACSAria III Fusion SORP. HER2-positive cells were sorted with a phycoerythrin-conjugated HER2 antibody (eBiosciences).
Quantitative real-time PCR
RNA was extracted using the RNeasy Mini kit (Qiagen) using the manufacturer’s protocol with optional on-column DNA digestion. Complementary DNA (cDNA) was synthesized by using the iScript Supermix Reverse Transcription system (Bio-Rad). Real-time PCRs were performed with the following primers: HER3, GCCAAGGGCCCAATCTACAA (forward) and TGTCAGATGGGTTTTGCCGA (reverse); NRG-1α and NRG-1β sequences were obtained from Brown et al. (45).
Immunohistochemistry
Paraffin-embedded primary BC and matched BMs were cut into 5-μm-thick serial sections, and staining procedure was carried out following the manufacturer’s IHC protocol (Cell Signaling). Primary antibody: anti-HER3 (1:10 dilution; Cell Signaling Technology) and anti-HER2 (1:200 dilution; Cell Signaling Technology). Sections were counter-stained with hematoxylin and blued in Scott’s tap water substitute (Electron Microscopy Sciences). Matched human primary breast and brain metastatic tumors were scored blindly by an MGH breast pathologist (E.B.). Staining intensity, location, and coverage were all taken into account during the scoring.
Growth factor screening
A collection of cDNA constructs representing secreted and single-pass transmembrane proteins was identified [as previously described (46)] and purchased from Invitrogen Ultimate ORF collection (this library is maintained by DMP BioArchive). pCDNA-DEST40 (Invitrogen) was the plasmid vector for all clones, and all clone inserts were confirmed by full sequencing. For this study, a collection of 338 cDNA constructs, representing 220 distinct secreted and single-pass transmembrane proteins, was used to identify rescued ligands in the present of each compound treatment; each cDNA construct was reverse-transfected using HEK293T/17. After 3 days of incubation, secreted proteins were then transferred to studied cell lines, followed by the addition of compounds for a 96-hour incubation. CellTiter-Glo (CTG) assay was then used for conducting endpoint readout. After plate-to-plate normalization of the CTG reading for triplicates, the rescue percent value for each secreted protein was calculated using the following formula: Rescue % = [median (drug + supe) − median (drug)]/[median (DMSO) − median (drug)]. The statistical probability score of the Rescue % value was also calculated: P = Chidist[z(drug + supe)2, 1], where z(drug + supe) = [median (drug + supe) − median (drug)]/SD(drug + supe), where “drug” stands for 1 μM BKM120, and “supe” stands for the individual supernatant collected from transfected HEK293T/17 expressing one of the 220 secreted proteins. Data points were then plotted using Spot-fire software. Secreted proteins that had a rescue of ≥20% with P ≤ 0.05 were labeled in each scatter plot.
In vitro cell growth assays
The xCELLigence system (ACEA Biosciences Inc.) was used to assess the cell index of T47D, BT474, and MDA-MB-361 cells. One day before drug addition, cells were seeded in 96-well E-plates at a density of 6,000 to 10,000 cells per well in 90 μl of growth medium. Cells were monitored every 15 min for a period of up to 24 hours via the incorporated sensor electrode arrays of the E-Plate 96. After 24 hours of incubation, E-plates were removed, and LJM716 (100 nM) was added and incubated for 1 hour, followed by NRG-1 (10 ng) and/or buparlisib (1 μM). Each treatment was tested in triplicate. Electric impedance was measured in 1-hour intervals after drug addition until the end of the experiment. Cell index value, which is a dimensionless parameter derived as a relative change in measured electrical impedance to represent numbers of attached cells, was calculated from the electric impedance and plotted using the RTCA software provided by the vendor. At the end of the experiment, cell growth and/or viability was determined by measuring cellular adenosine 5′-triphosphate content using the CellTiter-Glo Luminescent Cell Viability assay (Promega) according to the manufacturer’s protocol.
Measurement and analysis of PIP3/PIP2 concentration
Quantification of PIP3/PIP2 was performed as previously described (16). Briefly, frozen tissue of BT474 tumors in the brain was resuspended in 3 ml of 5% tricarboxylic acid/1 mM EDTA, vortexed, and centrifuged at 3000 rpm for 5 min. The supernatant was discarded, and this washing step was repeated one more time. Afterward, neutral lipids were extracted by adding 3 ml of MeOH:CHCl3 (2:1) and continuously vortexed over 10 min at room temperature. Extracts were centrifuged at 3000 rpm for 5 min. The supernatant was discarded, and this extraction step was repeated one more time. The acidic lipids were extracted adding 2.25 ml of MeOH:CHCl3:12 M HCl (80:40:1) with continuous vortexing over 25 min at room temperature. Extracts were centrifuged at 3000 rpm for 5 min, and the supernatant was transferred to a new 15-ml tube; 0.75 ml of CHCl3 and 1.35 ml of 0.1 M HCl were added to the supernatant, vortexed, and centrifuged at 3000 rpm for 5 min. The organic (lower) phase was collected; 1.45 ml was transferred into a vial for PIP3 measurement, and 0.05 ml was transferred into a new vial for PIP2 measurement. All samples were dried in a vacuum dryer for 1 hour. PIP3 samples were resuspended in 120 ml of PBS-Tween plus 3% protein stabilizer (provided by the Echelon kit). PIP2 samples were resuspended in 120 ml of PBS plus 0.25% protein stabilizer. Samples were sonicated in an ice-water bath for 10 min, vortexed for 30 s, and centrifuged for 30 s at 13,000 rpm before being added to the enzyme-linked immunosorbent assay (ELISA). Once phospholipids were isolated from cells, PIP3 and PIP2 concentrations were measured using ELISA kits (Echelon, K-2500s, and K4500) according to the manufacturer’s instructions.
Statistical analysis
Data are expressed as means ± SD unless otherwise noted. A t test (two-tailed with unequal variance) was used when only two variables were present in the analysis. One-way ANOVA and Tukey’s posttest were used to compare pairs of columns. Wilcoxon signed-rank sum test for ordered categorical data was used to analyze the matched human tissue. Survival curves were estimated using the Kaplan-Meier method. P < 0.05 was considered statistically significant. GraphPad Prism or Stata 13.0 was used for statistical analysis.
Supplementary Material
Fig. S1. The blood Gluc activity is directly correlated with tumor volume for each tumor model.
Fig. S2. Liver BT474 tumors respond to buparlisib.
Fig. S3. Microenvironment-dependent regulation of BC cells alters FOXO3 activity.
Fig. S4. MDA-MB-361 brain lesions transiently respond to buparlisib.
Fig. S5. HER3 expression is increased in BMs from HER2-positive BC.
Fig. S6. Several NRG isoforms induce resistance to buparlisib.
Fig. S7. NRG-1 is expressed in MFP and brain lesions.
Table S1. Differential expression of HER3 in intracranial and extracranial lesions from unmatched patient clinical samples.
Table S2. List of recombinant human growth factors screened for their ability to induce resistance to buparlisib.
Acknowledgments
We thank S. Roberge, J. Kahn, C. Smith, and P. Huang for help with the experiments and interpretation of data, and I. Garkatsev and B. Seed for the helpful suggestions. We thank A. Wheless for the administrative support with the retrieval of the human matched samples of primary tumors and BM.
Funding: Supported in part by the U.S. Department of Defense Breast Cancer Research Innovator Award W81XWH-10-1-0016 (to R.K.J.); the U.S. National Cancer Institute grants R01-CA126642 and R35-CA197743 (to R.K.J.), P01-CA080124 (to R.K.J. and D.F.), R01-CA096915 (to D.F.), and Breast SPORE grant P50 CA098131 (to C.L.A.); the Ludwig Center at Harvard (to R.K.J.); National Foundation for Cancer Research (NFCR) (R.K.J.); the MIT-HCC (Massachusetts Institute of Technology–Harvard Cancer Center) Bridge grant (to R.K.J.); the Federal Share Proton Beam Program Income (to R.K.J.); grant T32-CA073479 (to D.P.K.); the NIH/National Cancer Institute grant R01CA137008 (to J.A.E.); the German Research Foundation grant (Deutsche Forschungsgemeinschaft) AS 422-2/1 (to V.A.); and the Susan G. Komen for the Cure (GBF) and Dutch Cancer Society Grant Trustees (KWF Stagebeurs Buitenland) (to R.R.R). M.B.S. is supported by the National Cancer Institute F30-CA200345.
Footnotes
Author contributions: D.P.K., V.A., G.B.F., J.A.E., and R.K.J. designed the research and wrote the manuscript. Q.S., M.B., S.G., Z.A.C., D.G.D., R.S., A.H., and D.F. assisted with the research design. D.P.K., V.A., G.B.F., Q.S., M.B., S.G., X.Q., R.S., Z.A.C., R.R.R., D.B., B.P., Y.S., C.C., K.N., C.S.F.W., J.K., R.D., J.T., and A.T. performed the experiments. C.R.M., M.B.S., C.K.A., M.S., E.B., M.V.E., and C.L.A. collected the human specimens.
Competing interests: R.K.J. received consultant fees from Enlight, Ophthotech, SPARC, and SynDevRx. R.K.J. owns equity in Enlight, Ophthotech, SynDevRx, and XTuit and serves on the Board of Directors of XTuit and the Boards of Trustees of Tekla Healthcare Investors, Tekla Life Sciences Investors, the Tekla Healthcare Opportunities Fund, and the Tekla World Healthcare Fund. J.A.E. is a paid consultant for Novartis, Sanofi-Aventis, AstraZeneca, Chugai, Amgen, Genentech, GSK, Merck, and Pfizer and holds equity in Gatekeeper Pharmaceuticals. J.A.E. also received research support from Novartis and is now an employee of Novartis. D.P.K., V.A., G.B.F., Q.S., D.F., and R.K.J. are inventors on patent application (PAT056561) held/submitted by D.P.K., V.A., G.B.F., Q.S., D.F., and R.K.J. (MGH/Harvard Medical School and Novartis) that covers the finding that a combination of a PI3K inhibitor and a Her3 antagonist can be used to treat BC BMs.
Data and materials availability: Gene array raw data are available at Gene Expression Omnibus (accession number GSE86849). Data were normalized using the robust multiarray average (PMID: 12925520). The gene set of FOXO3a targets used for gene set enrichment analysis (PMID: 16199517) (with default parameters) can be obtained here: http://software.broadinstitute.org/gsea/msigdb/cards/V$FOXO3_01.html. BKM120 and LJM716 are available from Novartis under a material transfer agreement with J. Engelman (MGH).
SUPPLEMENTARY MATERIALS
www.sciencetranslationalmedicine.org/cgi/content/full/9/391/eaal4682/DC1
REFERENCES AND NOTES
- 1.Kodack DP, Askoxylakis V, Ferraro GB, Fukumura D, Jain RK. Emerging strategies for treating brain metastases from breast cancer. Cancer Cell. 2015;27:163–175. doi: 10.1016/j.ccell.2015.01.001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Olson EM, Abdel-Rasoul M, Maly J, Wu CS, Lin NU, Shapiro CL. Incidence and risk of central nervous system metastases as site of first recurrence in patients with HER2-positive breast cancer treated with adjuvant trastuzumab. Ann Oncol. 2013;24:1526–1533. doi: 10.1093/annonc/mdt036. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Murrell DH, Foster PJ, Chambers AF. Brain metastases from breast cancer: Lessons from experimental magnetic resonance imaging studies and clinical implications. J Mol Med. 2014;92:5–12. doi: 10.1007/s00109-013-1108-z. [DOI] [PubMed] [Google Scholar]
- 4.Dasararaju R, Mehta A. Current advances in understanding and managing secondary brain metastasis. CNS Oncol. 2013;2:75–85. doi: 10.2217/cns.12.33. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Tamura K, Kurihara H, Yonemori K, Tsuda H, Suzuki J, Kono Y, Honda N, Kodaira M, Yamamoto H, Yunokawa M, Shimizu C, Hasegawa K, Kanayama Y, Nozaki S, Kinoshita T, Wada Y, Tazawa S, Takahashi K, Watanabe Y, Fujiwara Y. 64Cu-DOTA-trastuzumab PET imaging in patients with HER2-positive breast cancer. J Nucl Med. 2013;54:1869–1875. doi: 10.2967/jnumed.112.118612. [DOI] [PubMed] [Google Scholar]
- 6.Askoxylakis V, Ferraro GB, Kodack DP, Badeaux M, Shankaraiah RC, Seano G, Kloepper J, Vardam T, Martin JD, Naxerova K, Bezwada D, Qi X, Selig MK, Brachtel E, Duda DG, Huang P, Fukumura D, Engelman JA, Jain RK. Preclinical efficacy of Ado-trastuzumab emtansine in the brain microenvironment. J Natl Cancer Inst. 2016;108:djv313. doi: 10.1093/jnci/djv313. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Bachelot T, Romieu G, Campone M, Diéras V, Cropet C, Dalenc F, Jimenez M, Le Rhun E, Pierga JY, Gonçalves A, Leheurteur M, Domont J, Gutierrez M, Curé H, Ferrero JM, Labbe-Devilliers C. Lapatinib plus capecitabine in patients with previously untreated brain metastases from HER2-positive metastatic breast cancer (LANDSCAPE): A single-group phase 2 study. Lancet Oncol. 2013;14:64–71. doi: 10.1016/S1470-2045(12)70432-1. [DOI] [PubMed] [Google Scholar]
- 8.Lin NU, Carey LA, Liu MC, Younger J, Come SE, Ewend M, Harris GJ, Bullitt E, Van den Abbeele AD, Henson JW, Li X, Gelman R, Burstein HJ, Kasparian E, Kirsch DG, Crawford A, Hochberg F, Winer EP. Phase II trial of lapatinib for brain metastases in patients with human epidermal growth factor receptor 2–positive breast cancer. J Clin Oncol. 2008;26:1993–1999. doi: 10.1200/JCO.2007.12.3588. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Lin NU, Diéras V, Paul D, Lossignol D, Christodoulou C, Stemmler HJ, Roché H, Liu MC, Greil R, Ciruelos E, Loibl S, Gori S, Wardley A, Yardley D, Brufsky A, Blum JL, Rubin SD, Dharan B, Steplewski K, Zembryki D, Oliva C, Roychowdhury D, Paoletti P, Winer EP. Multicenter phase II study of lapatinib in patients with brain metastases from HER2-positive breast cancer. Clin Cancer Res. 2009;15:1452–1459. doi: 10.1158/1078-0432.CCR-08-1080. [DOI] [PubMed] [Google Scholar]
- 10.Kodack DP, Chung E, Yamashita H, Incio J, Duyverman AMMJ, Song Y, Farrar CT, Huang Y, Ager E, Kamoun W, Goel S, Snuderl M, Lussiez A, Hiddingh L, Mahmood S, Tannous BA, Eichler AF, Fukumura D, Engelman JA, Jain RK. Combined targeting of HER2 and VEGFR2 for effective treatment of HER2-amplified breast cancer brain metastases. Proc Natl Acad Sci USA. 2012;109:E3119–E3127. doi: 10.1073/pnas.1216078109. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Eichler AF, Chung E, Kodack DP, Loeffler JS, Fukumura D, Jain RK. The biology of brain metastases—Translation to new therapies. Nat Rev Clin Oncol. 2011;8:344–356. doi: 10.1038/nrclinonc.2011.58. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Zhou BP, Liao Y, Xia W, Spohn B, Lee MH, Hung MC. Cytoplasmic localization of p21Cip1/WAF1 by Akt-induced phosphorylation in HER-2/neu-overexpressing cells. Nat Cell Biol. 2001;3:245–252. doi: 10.1038/35060032. [DOI] [PubMed] [Google Scholar]
- 13.Zhou BP, Liao Y, Xia W, Zou Y, Spohn B, Hung MC. HER-2/neu induces p53 ubiquitination via Akt-mediated MDM2 phosphorylation. Nat Cell Biol. 2001;3:973–982. doi: 10.1038/ncb1101-973. [DOI] [PubMed] [Google Scholar]
- 14.Alimandi M, Romano A, Curia MC, Muraro R, Fedi P, Aaronson SA, Di Fiore PP, Kraus MH. Cooperative signaling of ErbB3 and ErbB2 in neoplastic transformation and human mammary carcinomas. Oncogene. 1995;10:1813–1821. [PubMed] [Google Scholar]
- 15.Lee-Hoeflich ST, Crocker L, Yao E, Pham T, Munroe X, Hoeflich KP, Sliwkowski MX, Stern HM. A central role for HER3 in HER2-amplified breast cancer: Implications for targeted therapy. Cancer Res. 2008;68:5878–5887. doi: 10.1158/0008-5472.CAN-08-0380. [DOI] [PubMed] [Google Scholar]
- 16.Costa C, Ebi H, Martini M, Beausoleil SA, Faber AC, Jakubik CT, Huang A, Wang Y, Nishtala M, Hall B, Rikova K, Zhao J, Hirsch E, Benes CH, Engelman JA. Measurement of PIP3 levels reveals an unexpected role for p110β in early adaptive responses to p110α-specific inhibitors in luminal breast cancer. Cancer Cell. 2015;27:97–108. doi: 10.1016/j.ccell.2014.11.007. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Fritsch C, Huang A, Chatenay-Rivauday C, Schnell C, Reddy A, Liu M, Kauffmann A, Guthy D, Erdmann D, De Pover A, Furet P, Gao H, Ferretti S, Wang Y, Trappe J, Brachmann SM, Maira SM, Wilson C, Boehm M, Garcia-Echeverria C, Chene P, Wiesmann M, Cozens R, Lehar J, Schlegel R, Caravatti G, Hofmann F, Sellers WR. Characterization of the novel and specific PI3Kα inhibitor NVP-BYL719 and development of the patient stratification strategy for clinical trials. Mol Cancer Ther. 2014;13:1117–1129. doi: 10.1158/1535-7163.MCT-13-0865. [DOI] [PubMed] [Google Scholar]
- 18.Huang A, Fritsch C, Wilson C, Reddy A, Liu M, Lehar J, Quadt C, Hofmann F, Schlegel R. Single agent activity of PIK3CA inhibitor BYL719 in a broad cancer cell line panel. Cancer Res. 2012;72:3749. [Google Scholar]
- 19.Maira SM, Pecchi S, Huang A, Burger M, Knapp M, Sterker D, Schnell C, Guthy D, Nagel T, Wiesmann M, Brachmann S, Fritsch C, Dorsch M, Chène P, Shoemaker K, De Pover A, Menezes D, Martiny-Baron G, Fabbro D, Wilson CJ, Schlegel R, Hofmann F, García-Echeverría C, Sellers WR, Voliva CF. Identification and characterization of NVP-BKM120, an orally available pan-class I PI3-kinase inhibitor. Mol Cancer Ther. 2012;11:317–328. doi: 10.1158/1535-7163.MCT-11-0474. [DOI] [PubMed] [Google Scholar]
- 20.Bendell JC, Rodon J, Burris HA, de Jonge M, Verweij J, Birle D, Demanse D, De Buck SS, Ru QC, Peters M, Goldbrunner M, Baselga J. Phase I, dose-escalation study of BKM120, an oral pan-Class I PI3K inhibitor, in patients with advanced solid tumors. J Clin Oncol. 2012;30:282–290. doi: 10.1200/JCO.2011.36.1360. [DOI] [PubMed] [Google Scholar]
- 21.Koul D, Fu J, Shen R, LaFortune TA, Wang S, Tiao N, Kim YW, Liu JL, Ramnarian D, Yuan Y, Garcia-Echevrria C, Maira SM, Yung WKA. Antitumor activity of NVP-BKM120— A selective pan class I PI3 kinase inhibitor showed differential forms of cell death based on p53 status of glioma cells. Clin Cancer Res. 2012;18:184–195. doi: 10.1158/1078-0432.CCR-11-1558. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Chandarlapaty S, Sawai A, Scaltriti M, Rodrik-Outmezguine V, Grbovic-Huezo O, Serra V, Majumder PK, Baselga J, Rosen N. AKT inhibition relieves feedback suppression of receptor tyrosine kinase expression and activity. Cancer Cell. 2011;19:58–71. doi: 10.1016/j.ccr.2010.10.031. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Chakrabarty A, Sánchez V, Kuba MG, Rinehart C, Arteaga CL. Feedback upregulation of HER3 (ErbB3) expression and activity attenuates antitumor effect of PI3K inhibitors. Proc Natl Acad Sci USA. 2012;109:2718–2723. doi: 10.1073/pnas.1018001108. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Garrett JT, Olivares MG, Rinehart C, Granja-Ingram ND, Sánchez V, Chakrabarty A, Dave B, Cook RS, Pao W, McKinely E, Manning HC, Chang J, Arteaga CL. Transcriptional and posttranslational up-regulation of HER3 (ErbB3) compensates for inhibition of the HER2 tyrosine kinase. Proc Natl Acad Sci USA. 2011;108:5021–5026. doi: 10.1073/pnas.1016140108. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Amin DN, Sergina N, Lim L, Goga A, Moasser MM. HER3 signalling is regulated through a multitude of redundant mechanisms in HER2-driven tumour cells. Biochem J. 2012;447:417–425. doi: 10.1042/BJ20120724. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Da Silva L, Simpson PT, Smart CE, Cocciardi S, Waddell N, Lane A, Morrison BJ, Cristina Vargas A, Healey S, Beesley J, Pakkiri P, Parry S, Kurniawan N, Reid L, Keith P, Faria P, Pereira E, Skalova A, Bilous M, Balleine RL, Do H, Dobrovic A, Fox S, Franco M, Reynolds B, Kum Khanna K, Cummings M, Chenevix-Trench G, Lakhani SR. HER3 and downstream pathways are involved in colonization of brain metastases from breast cancer. Breast Cancer Res. 2010;12:R46. doi: 10.1186/bcr2603. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Garner AP, Bialucha CU, Sprague ER, Garrett JT, Sheng Q, Li S, Sineshchekova O, Saxena P, Sutton CR, Chen D, Chen Y, Wang H, Liang J, Das R, Mosher R, Gu J, Huang A, Haubst N, Zehetmeier C, Haberl M, Elis W, Kunz C, Heidt AB, Herlihy K, Murtie J, Schuller A, Arteaga CL, Sellers WR, Ettenberg SA. An antibody that locks HER3 in the inactive conformation inhibits tumor growth driven by HER2 or neuregulin. Cancer Res. 2013;73:6024–6035. doi: 10.1158/0008-5472.CAN-13-1198. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Hobbs SS, Coffing SL, Le ATD, Cameron EM, Williams EE, Andrew M, Blommel EN, Hammer RP, Chang H, Riese DJ., II Neuregulin isoforms exhibit distinct patterns of ErbB family receptor activation. Oncogene. 2002;21:8442–8452. doi: 10.1038/sj.onc.1205960. [DOI] [PubMed] [Google Scholar]
- 29.Agus DB, Akita RW, Fox WD, Lewis GD, Higgins B, Pisacane PI, Lofgren JA, Tindell C, Evans DP, Maiese K, Scher HI, Sliwkowski MX. Targeting ligand-activated ErbB2 signaling inhibits breast and prostate tumor growth. Cancer Cell. 2002;2:127–137. doi: 10.1016/s1535-6108(02)00097-1. [DOI] [PubMed] [Google Scholar]
- 30.Guillou H, Stephens LR, Hawkins PT. Quantitative measurement of phosphatidylinositol 3,4,5-trisphosphate. Methods Enzymol. 2007;434:117–130. doi: 10.1016/S0076-6879(07)34007-X. [DOI] [PubMed] [Google Scholar]
- 31.Condliffe AM, Davidson K, Anderson KE, Ellson CD, Crabbe T, Okkenhaug K, Vanhaesebroeck B, Turner M, Webb L, Wymann MP, Hirsch E, Ruckle T, Camps M, Rommel C, Jackson SP, Chilvers ER, Stephens LR, Hawkins PT. Sequential activation of class IB and class IA PI3K is important for the primed respiratory burst of human but not murine neutrophils. Blood. 2005;106:1432–1440. doi: 10.1182/blood-2005-03-0944. [DOI] [PubMed] [Google Scholar]
- 32.Baselga J, Cortés J, Kim S-B, Im S-A, Hegg R, Im Y-H, Roman L, Luiz Pedrini J, Pienkowski T, Knott A, Clark E, Benyunes MC, Ross G, Swain SM, CLEOPATRA Study Group Pertuzumab plus trastuzumab plus docetaxel for metastatic breast cancer. N Engl J Med. 2012;366:109–119. doi: 10.1056/NEJMoa1113216. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Kienast Y, von Baumgarten L, Fuhrmann M, Klinkert WEF, Goldbrunner R, Herms J, Winkler F. Real-time imaging reveals the single steps of brain metastasis formation. Nat Med. 2010;16:116–122. doi: 10.1038/nm.2072. [DOI] [PubMed] [Google Scholar]
- 34.Ni J, Ramkissoon SH, Xie S, Goel S, Stover DG, Guo H, Luu V, Marco E, Ramkissoon LA, Jee Kang Y, Hayashi M, Nguyen Q-D, Ligon AH, Du R, Claus EB, Alexander BM, Yuan G-C, Wang ZC, Iglehart JD, Krop IE, Roberts TM, Winer EP, Lin NU, Ligon KL, Zhao JJ. Combination inhibition of PI3K and mTORC1 yields durable remissions in mice bearing orthotopic patient-derived xenografts of HER2-positive breast cancer brain metastases. Nat Med. 2016;22:723–726. doi: 10.1038/nm.4120. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Dunn M, Sinha P, Campbell R, Blackburn E, Levinson N, Rampaul R, Bates T, Humphreys S, Gullick WJ. Co-expression of neuregulins 1, 2, 3 and 4 in human breast cancer. J Pathol. 2004;203:672–680. doi: 10.1002/path.1561. [DOI] [PubMed] [Google Scholar]
- 36.Mei L, Nave KA. Neuregulin-ERBB signaling in the nervous system and neuropsychiatric diseases. Neuron. 2014;83:27–49. doi: 10.1016/j.neuron.2014.06.007. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Saunus JM, Quinn MCJ, Patch A-M, Pearson JV, Bailey PJ, Nones K, McCart Reed AE, Miller D, Wilson PJ, Al-Ejeh F, Mariasegaram M, Lau Q, Withers T, Jeffree RL, Reid LE, Da Silva L, Matsika A, Niland CM, Cummings MC, Bruxner TJC, Christ AN, Harliwong I, Idrisoglu S, Manning S, Nourse C, Nourbakhsh E, Wani S, Anderson MJ, Fink JL, Holmes O, Kazakoff S, Leonard C, Newell F, Taylor D, Waddell N, Wood S, Xu Q, Kassahn KS, Narayanan V, Taib NA, Teo S-H, Chow YP, kConFab. Jat PS, Brandner S, Flanagan AM, Khanna KK, Chenevix-Trench G, Grimmond SM, Simpson PT, Waddell N, Lakhani SR. Integrated genomic and transcriptomic analysis of human brain metastases identifies alterations of potential clinical significance. J Pathol. 2015;237:363–378. doi: 10.1002/path.4583. [DOI] [PubMed] [Google Scholar]
- 38.Meyer D, Birchmeier C. Multiple essential functions of neuregulin in development. Nature. 1995;378:386–390. doi: 10.1038/378386a0. [DOI] [PubMed] [Google Scholar]
- 39.Serra V, Scaltriti M, Prudkin L, Eichhorn PJA, Ibrahim YH, Chandarlapaty S, Markman B, Rodriguez O, Guzman M, Rodriguez S, Gili M, Russillo M, Parra JL, Singh S, Arribas J, Rosen N, Baselga J. PI3K inhibition results in enhanced HER signaling and acquired ERK dependency in HER2-overexpressing breast cancer. Oncogene. 2011;30:2547–2557. doi: 10.1038/onc.2010.626. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Verma S, Joy AA, Rayson D, McLeod D, Brezden-Masley C, Boileau JF, Gelmon KA. HER story: The next chapter in HER-2-directed therapy for advanced breast cancer. Oncologist. 2013;18:1153–1166. doi: 10.1634/theoncologist.2013-0217. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.Momeny M, Saunus JM, Marturana F, McCart Reed AE, Black D, Sala G, Iacobelli S, Holland JD, Yu D, Da Silva L, Simpson PT, Khanna KK, Chenevix-Trench G, Lakhani SR. Heregulin-HER3-HER2 signaling promotes matrix metalloproteinase-dependent blood-brain-barrier transendothelial migration of human breast cancer cell lines. Oncotarget. 2015;6:3932–3946. doi: 10.18632/oncotarget.2846. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42.Yao HP, Zhou YQ, Zhang R, Wang MH. MSP-RON signalling in cancer: Pathogenesis and therapeutic potential. Nat Rev Cancer. 2013;13:466–481. doi: 10.1038/nrc3545. [DOI] [PubMed] [Google Scholar]
- 43.Yao HP, Zhou YQ, Zhang R, Wang MH. Secreted Gaussia luciferase as a biomarker for monitoring tumor progression and treatment response of systemic metastases. PLOS ONE. 2009;4:e8316. doi: 10.1371/journal.pone.0008316. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44.Yuan F, Salehi HA, Boucher Y, Vasthare US, Tuma RF, Jain RK. Vascular permeability and microcirculation of gliomas and mammary carcinomas transplanted in rat and mouse cranial windows. Cancer Res. 1994;54:4564–4568. [PubMed] [Google Scholar]
- 45.Brown DJ, Lin B, Holguin B. Expression of neuregulin 1, a member of the epidermal growth factor family, is expressed as multiple splice variants in the adult human cornea. Invest Ophthalmol Vis Sci. 2004;45:3021–3029. doi: 10.1167/iovs.04-0229. [DOI] [PubMed] [Google Scholar]
- 46.Harbinski F, Craig VJ, Sanghavi S, Jeffery D, Liu L, Ann Sheppard K, Wagner S, Stamm C, Buness A, Chatenay-Rivauday C, Yao Y, He F, Lu CX, Guagnano V, Metz T, Finan PM, Hofmann F, Sellers WR, Porter JA, Myer VE, Graus-Porta D, Wilson CJ, Buckler A, Tiedt R. Rescue screens with secreted proteins reveal compensatory potential of receptor tyrosine kinases in driving cancer growth. Cancer Discov. 2012;2:948–959. doi: 10.1158/2159-8290.CD-12-0237. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Fig. S1. The blood Gluc activity is directly correlated with tumor volume for each tumor model.
Fig. S2. Liver BT474 tumors respond to buparlisib.
Fig. S3. Microenvironment-dependent regulation of BC cells alters FOXO3 activity.
Fig. S4. MDA-MB-361 brain lesions transiently respond to buparlisib.
Fig. S5. HER3 expression is increased in BMs from HER2-positive BC.
Fig. S6. Several NRG isoforms induce resistance to buparlisib.
Fig. S7. NRG-1 is expressed in MFP and brain lesions.
Table S1. Differential expression of HER3 in intracranial and extracranial lesions from unmatched patient clinical samples.
Table S2. List of recombinant human growth factors screened for their ability to induce resistance to buparlisib.