

De novo heterozygous mutations in STXBP1 cause early infantile epileptic encephalopathy. Kovačević et al. analyse the underlying disease mechanisms in silico, in vitro and in vivo, and show that protein instability, haploinsufficiency and cortical hyperexcitability explain STXBP1-encephalopathy. In addition, they demonstrate the construct, face and predictive validity of Stxbp1+/- mice.
Keywords: STXBP1-encephalopathy, haploinsufficiency, epilepsy, video-EEG, behavioural inflexibility
Abstract
De novo heterozygous mutations in STXBP1/Munc18-1 cause early infantile epileptic encephalopathies (EIEE4, OMIM #612164) characterized by infantile epilepsy, developmental delay, intellectual disability, and can include autistic features. We characterized the cellular deficits for an allelic series of seven STXBP1 mutations and developed four mouse models that recapitulate the abnormal EEG activity and cognitive aspects of human STXBP1-encephalopathy. Disease-causing STXBP1 variants supported synaptic transmission to a variable extent on a null background, but had no effect when overexpressed on a heterozygous background. All disease variants had severely decreased protein levels. Together, these cellular studies suggest that impaired protein stability and STXBP1 haploinsufficiency explain STXBP1-encephalopathy and that, therefore, Stxbp1+/− mice provide a valid mouse model. Simultaneous video and EEG recordings revealed that Stxbp1+/− mice with different genomic backgrounds recapitulate the seizure/spasm phenotype observed in humans, characterized by myoclonic jerks and spike-wave discharges that were suppressed by the antiepileptic drug levetiracetam. Mice heterozygous for Stxbp1 in GABAergic neurons only, showed impaired viability, 50% died within 2–3 weeks, and the rest showed stronger epileptic activity. c-Fos staining implicated neocortical areas, but not other brain regions, as the seizure foci. Stxbp1+/− mice showed impaired cognitive performance, hyperactivity and anxiety-like behaviour, without altered social behaviour. Taken together, these data demonstrate the construct, face and predictive validity of Stxbp1+/− mice and point to protein instability, haploinsufficiency and imbalanced excitation in neocortex, as the underlying mechanism of STXBP1-encephalopathy. The mouse models reported here are valid models for development of therapeutic interventions targeting STXBP1-encephalopathy.
Introduction
Disturbances in synaptic function are a key feature of many neurological, neurodevelopmental and psychiatric disorders [‘synaptopathies’ (Brose et al., 2010)]. Genetic studies have implicated mutations in genes encoding synaptic proteins in autism spectrum disorders, developmental delay, intellectual disability and epilepsy. Mechanistic studies have identified synaptic deficits in model systems and relevant behavioural phenotypes in mouse models (Brose et al., 2010).
One poorly understood synaptopathy is a spectrum of disorders caused by mutations in the STXBP1 (syntaxin-binding protein 1, encoding the presynaptic protein Munc18-1), together referred to as STXBP1-encephalopathies, one of the early infantile epileptic encephalopathies (EIEE4, OMIM #612164). STXBP1-encephalopathies are caused by de novo heterozygous missense or truncating mutations or micro-deletions, first reported by Saitsu et al. (2008), and include intellectual disability, epilepsy, autism spectrum disorders, and involuntary movements (spasms and jerks) (Hamdan et al., 2009; de Rubeis et al., 2014). A meta-analysis of 147 patients with STXBP1-encephalopathy reported in literature up to 2016 found that all patients had intellectual disability, 95% had epilepsy and around 20% were diagnosed with autism (Stamberger et al., 2016). In patients with STXBP1-encephalopathy, epilepsy is characterized by early onset of tonic spasms, often immediately after birth, and intractable seizures with multifocal epileptic activity on EEG (Saitsu et al., 2008).
To help understand how STXBP1/Munc18-1 mutations lead to such complex and diverse pathology, the availability of cellular and in vivo models is critical. Null mutant nematodes (Brenner, 1974), flies (Harrison et al., 1994), mice (Verhage et al., 2000) and zebrafish (Grone et al., 2016) have been studied, but more precise in vivo models, that mimic haploinsufficiency, have not been described so far. Inactivation of two copies of one stxbp1 gene in fish, a species that underwent an additional genome duplication, might be considered such a model (Grone et al., 2016), but functional diversification of duplicated genes since the duplication event, still make this situation different from inactivation of one copy of the only STXBP1 gene in mammals. Several studies have tested single STXBP1 human disease associated mutations in cell-free experiments or in cultured neurons (i.e. C180Y, V84D, M443R, G544D and R367X heterozygous mutations). These studies proposed different pathogenic mechanisms: altered affinity for its binding partner, syntaxin-1 (Saitsu et al., 2008; Yamashita et al., 2016), impaired protein stability (Saitsu et al., 2008), or a propensity of protein to aggregate (Chai et al., 2016). Finally, dominant negative effects cannot be excluded. Hence, different working mechanisms have been proposed to explain STXBP1-encephalopathy, even for the same mutations, but no in vivo models have been described.
The aim of this study was to systematically compare STXBP1 patient mutations at the cellular level, to characterize impairments in their cellular function and to generate mouse models for STXBP1-encephalopathy. Seven human disease-causing mutations were introduced into the STXBP1 sequence and expressed on a Stxbp1 null background to assess basic protein function and on a heterozygous background to mimic the human patient situation. Protein stability of these variants was assessed in HEK293 cells and primary neurons. Four Stxbp1+/− mouse models were generated to model Stxbp1 haploinsufficiency on different genomic backgrounds or to test selective haploinsufficiency in GABAergic cells. The translational value of these models was tested using EEG, a battery of cognitive and other behavioural tests. Our data suggest that cellular Munc18-1 instability is the probable explanation for Stxbp1 haploinsufficiency. Seizures, tonic spasms and EEG-abnormalities observed in humans are all recapitulated in Stxbp1+/− mice and were suppressed by the antiepileptic drug levetiracetam. Cognitive deficits and hyperactivity were also observed in these mouse models, but not autism related social deficits. Our data suggest protein instability, haploinsufficiency and cortical imbalanced excitability as the underlying mechanism of the observed phenotype implicating Stxbp1+/− mice as a valid model.
Materials and methods
Study design
This study was designed to investigate the effects of different STXBP1 mutations found in patients on neuronal function and to generate and characterize mouse models for STXBP1-encephalopathy with respect to construct-, face-, and predictive validity. The study is a three-layered analysis: in silico, in vitro and in vivo. The study followed the ARRIVE guidelines (https://www.nc3rs.org.uk/arrive-guidelines). We systematically compare public resources of human exome sequencing data and analyse the incidence of STXBP1 mutations. In vitro experiments were performed in the cultures obtained from Stxbp1+/− and Stxbp1−/− mice bred on a C57BL/6J background. Number of animals used for in vitro experiments was <5 mice per group. Number of replicates for in vitro experiments is indicated in the figures. Sample size for the behavioural experiment was between 7 and 13 male mice per group. When the several batches of the same line and same genotype were tested, data were matched. Total number of mice used in the behavioural study is indicated in the ‘Animals’ section and the exact number of mice per group is indicated in the figures. During the behavioural experiments, the mouse identification number, but not the genotype of the mouse, was available to the researcher. The mouse identification number was connected with the right genotype in the database. Analysis of behavioural data was performed blinded to genotype and two persons independently analysed EEG/electrocorticography (ECoG) and video-monitoring data. Before any analysis was performed, behavioural data were examined for outliers [>3 times the standard deviation (SD) from the strain mean] and outlier data were removed. The number of outliers per group is indicated in Supplementary Table 1.
Generation of lentiviral particles
Generation of lentiviral constructs driven by the human synapsin promoter, generation of virus stocks, and neuron infections were performed as published (Wierda et al., 2007). STXBP1 human disease variants were generated using Quickchange (Stratagene) and sequence verified. For expression in Stxbp1 heterozygote neurons Stxbp1 wild-type and mutants were cloned into lenti vector pSyn-Munc18-T2A-CreEGFP in which synapsin drives expression in neurons of Munc18-1 and of CreEGFP via the cleavage-peptide sequence T2A. Viral particles were titrated in HEK293 (ATCC) cells and equal titres were used to infect Stxbp1 null mutant or heterozygote neurons at in vitro Day 0.
Cell culture and electrophysiology
For quantification of Munc18-1 levels, embryonic cortical neurons from Stxbp1 null mutant mice (embryonic Day 18) were grown on glass coverslips (150 000 cells/12 wells) coated with poly-l-ornithine (Sigma, P4957; 5 μg/ml) and laminin (Sigma, L2020; 2.5 μg/ml). Autaptic neuronal culture and electrophysiological recordings were performed as described (Toonen et al., 2006; Wierda et al., 2007). Briefly, cortices were dissected in Hank’s balanced salt solution (HBSS; Sigma, H9394) supplemented with 10 mM HEPES and digested with 0.25% trypsin (Life Technologies, 15090-046) at 37°C for 20 min. After trituration, cells were plated in a 12-well plate at a density of 1000 cells/well for Stxbp1+/− neurons and 6000 cells/well for Stxbp1 null neurons on top of pre-grown rat glia islands on 18 mm coverslips. The 6-fold higher density for Stxbp1 null neurons was necessary to ensure similar neuronal density as it was for Stxbp1+/− neurons in mature cultures 2 weeks later, due to cell death of Stxbp1 null neurons before viral expression of Munc18 (variants) rescues cell viability. Cultures were grown in Neurobasal® supplemented with 2% B27 (Life Technologies, 17504-044), 18 mM HEPES (Life Technologies 15630-056), 0.5 mM GlutaMAX™ (Life Technologies 35050-038) and 0.1% penicillin/streptomycin (Life Technologies, 15140-122). Cells were whole-cell voltage clamped at −70 mV using Axopatch 200A. Recordings were performed on in vitro Days 14–17 with borosilicate glass pipettes (2.5–4 mΩ) containing (in mM): 125 K+-gluconate, 10 NaCl, 4.6 MgCl2, 4 K2-ATP, 1 EGTA, 15 creatine phosphate, 20 U/ml phosphocreatine kinase (pH 7.30, 300 mOsm). Action potentials were induced by 0.5 ms depolarizing steps to +30 mV. Digidata 1322A and Clampex 9.0 were used for signal acquisition. All recordings were performed at room temperature. Custom-made MatLab routines, Clampfit 9.0 and Minianalysis software were used for offline analysis. Readily releasable pool size was estimated by back extrapolation of cumulative excitatory postsynaptic current (EPSC) charge during a 40 Hz 100 action potential train as in Meijer et al. (2012).
Immunocytochemistry and confocal microscopy
Cultures were fixed at in vitro Day 14 with 4% formaldehyde, permeabilized with 0.5% Triton™ X-100 and stained with primary antibodies for 1 h at room temperature, washed with phosphate-buffered saline (PBS) and stained for 2 h at room temperature with Alexa conjugated secondary antibodies (1:1000, Invitrogen). Primary antibodies were used at the following concentrations: Munc18-1 (SySy 116.003; 1:1000), MAP2 (Abcam ab5392, 1:1000), VAMP2 (SySy 104 211, 1:2000), Syntaxin-1 (not commercial polyclonal I379), Synapsin (not commercial polyclonal E028). Coverslips were mounted with DABCO-Mowiol® (Sigma, 81381) and examined on a Zeiss LSM510 confocal microscope. Images were acquired with a 40× oil objective (N.A. 1.3) and 0.7× mechanical zoom and analysed in MATLAB with SynD (Schmitz et al., 2011). Somatic protein levels were quantified by placing six 10 × 10-pixel regions of interest in the cell soma. Synaptic protein levels were quantified by placing single regions of interest on all VAMP2-positive synapses as described in Schmitz et al. (2011).
Pharmacological treatments in heterologous cells and neurons
For protein stability assays, HEK293 cells were infected with lentiviral particles expressing piCreEGFP2aMunc18 wild-type or EIEE mutants in Opti-MEM® (Life Technologies; 31985-070). After 2 days, cells were replated in Dulbecco’s modified Eagle medium with 10% foetal bovine serum. After 8 h, cycloheximide treatments were performed by adding cycloheximide (100 µg/ml; Sigma) or dimethyl sulphoxide (DMSO) as control after which cells were lysed in SDS sample buffer after 0, 6, 12, 24 and 30 h. Munc18-1 levels were assessed after SDS-PAGE and transfer to nitrocellulose membranes using Munc18-1 rabbit polyclonal (not commercial polyclonal 2701, 1:1000), mouse monoclonal GFP (eBioscience; 1:1000) and mouse monoclonal actin (Chemicon; 1:10000) primary antibodies and IRDye® 680LT-anti mouse and/or IRDye® 800CW-anti rabbit as secondary antibody (both 1:5000). Blots were scanned on an Odyssey CLx Imaging system (LI-COR) and quantified with Image Studio Lite software (LI-COR Biotechnology, Germany). For protease inhibition experiments, cells were incubated for 15 h with 100 µg/ml cycloheximide and 10 µM MG132 (BioConnect; sc-201270), 25 µM Leupeptin (Sigma; L2884) or 1 µM Pepstatin A (Sigma; P5318) or DMSO (1:500) after which cells were processed as described above. To test protein stability of Munc18-1 wild-type and EIEE mutants in neurons, in vitro Day 0 Stxbp1 null cortical neurons were first infected in suspension with pLentiM18wtEGFP to rescue cell lethality of Stxbp1 null neurons and incubated for 1.5 h at 37°C; 5% CO2 after which neurons were plated at a density of 300 000 cells per well on poly-l-ornithine/laminin coated 6-well plates. At in vitro Day 4, cells were infected with piCreEGFP2aM18 (wild-type or EIEE mutants) and after 10 days (in vitro Day 14) 100 µg/ml cycloheximide was added and cells were lysed after 0, 12, 24 and 36 h, after which cells were processed as described above.
Animals
All animals were kept in standard husbandry conditions. Animals aged 8–10 weeks were separately housed on sawdust in the standard Makrolon type II cages on a 12-h light-dark cycle (light was on at 7:00 h). Food and water were available ad libitum. Behavioural experiments were performed on male mice aged between 8 and 12 weeks. All experiments were approved by the local animal research committee and complied with the European Council Directive (86/609/EEC).
Nine batches of congenic BL6 male mice, four batches of conditional null mice, where a single copy of a floxed allele was excised using Cre expression (Stxbp1cre/+), and one batch of reverse 129Sv mice were used. The total number of animals used was 181 for congenic BL6 line, 62 for the Stxbp1cre/+ line and 24 for reverse 129Sv balanced number of mice between genotypes (Stxbp1+/+ and Stxbp1+/− mice). One batch of 28 Gad2-Stxbp1cre/+ mice was generated in which a high mortality was observed, and for ethical reasons breeding of these mice was discontinued. Distribution of mice per batch and per experiment is shown in Supplementary Table 1.
High-density mouse genotyping
To determine the extent of residual 129S1Sv/J genome sequence flanking gene-targeted regions on a BL6 background, we ran a high-density mouse genotyping array. The ‘MegaMUGA’ array (https://www.ncbi.nlm.nih.gov/pmc/articles/PMC4751547/) contains 77 808 strain-informative single nucleotide polymorphism (SNP) markers, allowing the direct mapping of coordinates of residual 129S1Sv/J sequence. These coordinates were entered into the Genes and Marker Query on the Mouse Genome Informatics database (http://www.informatics.jax.org/marker/) to identify protein coding genes within the flanking 129S1Sv/J sequence.
Next, we used a recently developed web tool: ‘Me-PaMuFind-It’ (http://me-pamufind-it.org/) to generate a list of potential genes from the 129S1Sv/J genome that contained mutations, and remained after 20 generations of backcrossing, called passenger mutations (Vanden Berghe et al., 2015). It should be noted, however, that our congenic BL6 line was backcrossed for 40 generations and the flanking gene region was smaller than one predicted for the 20th generation by the web tool. Finally, comparing the list of protein coding genes within flanking gene region revealed by high-density genomic analysis with the list of potential passenger mutations obtained from web tool, we predicted passenger mutations in the samples that potentially influenced the phenotypic outcome in mutant mouse.
Immunohistochemistry
Stxbp1cre / +, congenic BL6 Stxbp1+/− mice and controls (n = 4, 5 and 5, respectively) were sacrificed by cervical dislocation. Brains were fixed in 4% PFA (paraformaldehyde) and cryoprotected in a 30% sucrose solution overnight. Brains were blocked in the coronal plane, frozen on dry ice, and sectioned at 50 µm on a cryostat. Coronal brain sections were stained with c-Fos primary antibody (Santa Cruz, sc-52; 1:800/1:500). After overnight incubation, sections were incubated with biotinylated goat anti-rabbit secondary antibody (#65-6140, Invitrogen; 1:400). The sections were then incubated in avidin-biotin peroxidase complex (Vectastain ABC, Vector Laboratories; 1:800) and peroxidase labelling was visualized by DAB/nickel substrate working solution (DAB Peroxidase Substrate, SK-4100; Vector Laboratories). Images were obtained using a Leica light microscope (×10 magnification) and number of c-Fos positive cells was counted using ImageJ software. A detailed description of the protocol and analysis can be found in the Supplementary material.
Video monitoring
Heterozygous mice and their controls from all three lines: conditional Stxbp1 line, congenic BL6 Stxbp1 line and reverse 129Sv Stxbp1 line were individually housed and their behaviour was recorded during 4–9 h. Detection of spontaneous abnormal behavioural events was done off-line by two researchers unaware of the mice genotype. Preliminary analysis revealed two types of behaviours observed in all Stxbp1+/− mice, but not in their controls. These two behavioural events were described as twitch and jump. A twitch was defined as a sudden, myoclonic jerk of the animal’s body from resting state (apparent sleep) that did not cause the mouse to wake (Fig. 5A and Supplementary Video 1). Jumps were differentiated from twitches by a change in the position of the mouse in the cage. In contrast to twitches, jumps were followed by activity (Fig. 5B and Supplementary Video 2) and sometimes accompanied by a Straub tail reaction. In total 42 mice were analysed (more details of batches in Supplementary Table 1).
Figure 5.

Video and EEG recordings revealed epileptic-like events in Stxbp1+/− mice. (A) Video monitoring revealed sudden jerks referred as twitches in Stxbp1+/− mice. (B) Video monitoring revealed sudden jumps in Stxbp1+/− mice, sometimes accompanied by Straub tail responses previously reported as a common phenomenon observed after seizure onset (Wagnon et al., 2015). (C) Distribution of motor effects of epileptic-like neural activity (twitches and jumps) during 3-h video-monitoring in Stxbp1+/− mice from three lines: floxed, congenic BL6 and reverse 129Sv line. These motor events were never found in control mice (Stxbp1+/+). (D) Monitoring for 24 h showed that most of the twitches and jumps in congenic BL6 Stxbp1+/− mouse occurred during the light phase (twitch: 22/34 and jump: 2/3) of the day/night cycle. (E) Average number of behavioural epileptic-like events per hour per line of Stxbp1+/− mice. (F) Positions of the recording electrodes and ground electrode relative to Bregma. (G) Representative example of ECoG traces in a congenic BL6 Stxbp1+/− mouse during the slow-wave sleep, awake state and spike-wave discharges. The red trace is an expanded ECoG trace of spike-wave discharge. (H) Power spectrum of slow wave sleep (SWS), spike wave discharge (SWD) and wake state. (I) Occurrence of behavioural epileptic events (twitches and jumps) and spike-wave discharges detected in cortical and hippocampal EEG traces during 3-h recording in Stxbp1cre/+ and congenic BL6 Stxbp1+/− mice. (J) Predicted probability of coincidence of 3/13 twitches and 1/7 jumps with spike-wave discharge detected in a representative congenic BL6 Stxbp1+/− mouse. Probability lower than 5% (grey line) was considered as concurrence. (K) Probability of concurrence of behavioural epileptic events and spike-wave discharges within 10 s presented as a negative logarithm for Stxbp1cre/+ and congenic BL6 Stxbp1+/− mice. (L) Total number of detected spike-wave discharges and epileptic events in the 12-h light phase. The number of mice: three Stxbp1cre/+ mice, four congenic BL6 Stxbp1+/− mice for cortical recording and two congenic BL6 Stxbp1+/− mice for hippocampal recording. (M) Average frequency of detected spike-wave discharges during 6 h of recording after administration of saline, first levetiracetam dose (LEV I: 50 mg/kg, i.p.) and fifth levetiracetam dose (LEV V: 5 days, 50 mg/kg per day, i.p.). Different greyscale circles represent individual mice. *P < 0.05, **P < 0.01 compared to saline administration.
Simultaneous radiotelemetric EEG-video monitoring and levetiracetam treatment
ECoG transmitters (ETA-F10; specification: https://www.datasci.com/products/implantable-telemetry/mouse-(miniature)/eta-f10) were implanted under deep anaesthesia induced and maintained by isoflurane. The recording electrode was positioned above the motor cortex (2.2 mm anterior, 1 mm lateral) and the ground electrode was positioned above the cerebellum (6 mm posterior, 1 mm lateral) using the stainless screws. The transmitter was placed subcutaneously (s.c.) in the abdominal pocket. All animals received pre- and postoperative analgesic treatment (buprenorphine, 0.005 mg/kg, s.c.). Simultaneous ECoG recording and video-monitoring of the animals were performed during at least 24 h started after 7 days recovery period.
For intrahippocampal EEG recordings mice were analysed with a telemetric EEG/video-monitoring system (TL11M2-EET, DSI) as described before (Pitsch et al., 2017). Briefly, intraperitoneally (i.p.) narcotized male mice [16 mg/kg xylazine (xylariem, Ecuphar) and 100 mg/kg ketamine i.p. (ketamine 10%, WDT)] were implanted with depth EEG electrodes at an age of 70 days with subcutaneous transmitter placement on the right abdominal side. Depth electrodes were positioned in left and right hippocampal CA1 (−2 AP, ±1.4 ML, 1.3 DV) (Paxinos and Franklin, 2012). Reference electrodes were placed with a stainless screw in contact with the cerebellar cortex of the simple lobe at the midline (−6 AP, 0 ML, 0 DV). All implanted mice received analgesic treatment before and once per day for 3 days post-operation (5 mg/kg ketoprofen, subcutaneously; Gabrilen®, MIBE). Simultaneous EEG and video monitoring was carried out for 16 days. Visual inspection of recordings revealed no significant differences between days. Thus, EEG and video monitoring over a period of 24 h was analysed in detail.
A group of congenic BL6 Sxbp1+/− mice with cortically implanted ECoG transmitters was subjected to treatment with antiepileptic drug, levetiracetam (Sigma Aldrich). Treatment started after a recovery period with intraperitoneal injection of saline (0.9% sodium chloride) at the beginning of the light phase. One day after saline injection, mice were injected with levetiracetam (50 mg/kg, i.p.) and treatment continued once daily for 5 days (levetiracetam 50 mg/kg, i.p.). ECoG/video monitoring was performed for at least 6 h, beginning 60 min after saline injection, first and fifth levetiracetam injections. Levetiracetam dose, pretreatment time and duration of assessment of drug effects were based on previous studies (Löscher and Hönack, 1993; Doheny et al., 1999).
Two observers, unaware of the mouse genotype, independently analysed EEG and ECoG signal and video records. Visual inspection of electrographic signal revealed spike-wave discharges with narrow peak frequency ∼7 Hz as the potential pathological electrographic activity. Probability of concurrence of spike-wave discharges longer than 1.5 s and behavioural epileptic-like events (previously described twitches and jumps) within ±10 s was analysed by calculating probability of coincidence, a concurrence of the events without apparent causal connection. Probability of 0.05 was taken as the borderline for the connection. In total 15 mice were analysed. More details about surgery and analysis can be found in the Supplementary material.
Visual analysis of the ECoG signal was complemented with an automated detection of potentially pathological events using the Event Classifier application in Neuroarchiver software (Open Source Instruments) during 24 h of recording. Video and ECoG recordings were used to select abnormal ECoG events (spike-wave discharges) that were accompanied with behavioural manifestation. The selected events were classified based on several metrics using Event Classifier and included into the library of potentially pathological events (Supplementary Fig. 5A). The generated library allowed fast, automatic identification of similar abnormal electrographic events. Detailed description of event sorting is available at: http://www.opensourceinstruments.com/Electronics/A3018/Seizure_Detection.html#Similarity%20of%20Events.
Behavioural phenotyping
Behavioural phenotyping of all Stxbp1 mice started with the assessment of the spontaneous behaviour in an automated home-cage environment (PhenoTyper model 3000, Noldus Information Technology). This system allows the assessment of several aspects of animal behaviour over a period of a few days, without human interference. Spontaneous behaviour was automatically monitored for 3 days in the PhenoTyper (Maroteaux et al., 2012; Loos et al., 2014). The observation of spontaneous behaviour was followed by the assessment of avoidance learning using Shelter task (Maroteaux et al., 2012) or discrimination and reversal learning using the CognitionWall task in the same automated home-cage (Remmelink et al., 2016).
After testing in the PhenoTyper, the test battery was performed on 26 congenic BL6 Stxbp1 mice (Batch 1) and 24 Stxbp1cre/+ mice (Batch 1) aged between 8 and 10 weeks old in order to test muscle strength, anxiety, learning and memory. Mice were weighed and acclimatized to their new housing for 1 week prior to testing. All tests were performed during the light phase, starting from the least stressful: novelty induced hypophagia, grip strength meter, elevated plus maze, open field, dark-light box, rotarod, and Barnes maze. Briefly, novelty induced hypophagia test was performed to assess anxiety-related behaviour, by measuring time to eat the cracker in the new cage. At least 1 day later, grip strength meter was performed to assess the muscle strength by measuring the force applied in grasping a pull bar. Additionally, three anxiety-related paradigms were performed: elevated plus maze, open field, and dark-light box; these tests are based on the natural aversion of mice for open and/or bright spaces. The anxiety-related parameters in these tests were: latency, time spent, and number of visits to the anxiety-related compartments (open arms, centre zone, bright compartment). Accelerating rotarod was performed to assess motor coordination and motor learning in mice; the parameter monitored was maximum rotation per minute (rpm) reached in each trial. The assessment of spatial learning and memory and reversal learning was done using the Barnes maze test. Mice were trained to choose one of the 24 holes in the Barnes maze as the escape hole. Latency to find the escape hole and distance travelled to the escape were measured as the parameters of learning. Memory was assessed by the probe trial when the escape hole was removed and hole visits per octant was measured. Reversal learning was checked by relocating the escape hole to the diametrically opposite position.
New batches of congenic BL6 and Stxbp1cre/+ mice were used to perform additional tests that needed to be done separately because of possible interference with other tests: congenic Stxbp1 BL6 mice, from Batches 2 to 6, were used for modified Barnes maze, 5-choice serial reaction time test, three-chamber test, Shelter task, CognitionWall; and Batch 2 from the Stxbp1cre/+ line was used to perform three-chamber test and CognitionWall test. The modified Barnes maze test is designed to improve assessment of spatial learning by avoiding development of serial strategy. The maze contains 44 holes divided into three rings, instead of 24 holes in one perimeter. The training procedure and parameters analysed were similar to the Barnes maze test. The various aspects of attention and impulsivity were assessed using the 5-choice serial reaction time test by calculating response accuracy, percentage of premature responses and correct response latency. The three-chamber test was applied for assaying sociability and preference for social novelty in mice. The parameters analysed were time spent in the mouse/novel mouse zone.
The reverse Stxbp1 129Sv mice (24 mice in total) were tested in the similar test battery as Stxbp1cre/+ and congenic BL6 mice. However, before the beginning of the behavioural experiments, reverse 129Sv animals were recorded in their home-cage for assessment of epileptic-like activity. In addition, considering very low activity of the 129Sv strain, the rotarod and three-chamber test were not performed and instead of the Barnes maze test, the Morris water maze was done. A detailed description of the behavioural experiment is provided in the Supplementary material.
Statistical analysis
All statistical analyses were performed using IBM SPSS statistic 20 (IBM corporation, Armonk, NY, USA). The effect of genetic background and genotype were assessed using two-way ANOVA, or repeated measure ANOVA. When the assessment of genetic background effect was not applicable, the genotype differences were compared using parametric tests (t-test, ANOVA, repeated-measures ANOVA) whenever normality and homoscedasticity criteria were met. Otherwise, non-parametric tests were performed (Kruskal-Wallis, Mann-Whitney U-test). To assess genotype effect in CognitionWall DL/RL task, the log rank test was performed for two Kaplan-Meier survival curves. An error probability level of P < 0.05 was accepted as statistically significant throughout the study of the classical behavioural tests. For all given level of analysis of PhenoTyper spontaneous behaviour data, statistical analysis was based on estimated false discovery rate (FDR), P-values were corrected by minimum positive FDR with a threshold set at 5% (Verhoeven et al., 2005).
Results
Human genomics data imply a crucial role of STXBP1 in normal development
We performed an in silico analysis of publicly available genomics data on STXBP1, especially from the Exome Aggregation Consortium (ExAC, Lek et al., 2016) and at http://gnomad.broadinstitute.org. No loss-of-function mutations were observed in over 60 000 humans (pLI = 1, Lek et al., 2016), while, based on its size, 25.5 mutations are predicted. The clinically-observed, heterozygous, and pathogenic mutations in STXBP1: C180Y, V84D, M443R, G544D, R367X, T574P, and C522R, (Fig. 1A) are all ultra-rare (Lek et al., 2016), and were not observed in over 140 000 human whole exomes and genomes at http://gnomad.broadinstitute.org. Our recent analysis of all available whole exome sequence data implicated 288 genes, and STXBP1 was the only gene implicated in all neurodevelopmental disorders studied: developmental delay (q = 4 × 10−17), epilepsy (q = 3 × 10−8), intellectual disability (q = 0.008), and autism (q = 0.10, Nguyen et al., 2017). These data indicate that STXBP1 is exceptionally intolerant to loss-of-function mutations and therefore predicted to be exceptionally important in normal human development and for brain function. Heterozygous mutations certainly occur, but the phenotypic consequences are highly adverse with strong negative selection pressure.
Figure 1.

Morphological and electrophysiological characteristics of dissociated hippocampal neurons expressing the human disease variants in Stxbp1 null mouse neurons. (A) Schematic overview of some of the previously discovered STXBP1 truncations, deletions and missense mutations in human patients showing the seven mutations tested in colour-coded boxes. (B) Dissociated cortical neurons were stained for Munc18-1, dendritic marker MAP2 and synaptic marker synaptobrevin (VAMP). Examples represent Stxbp1 null neurons expressing wild-type Munc18-1 (WT), C180Y or M433R. (C–G) Morphological and synaptic characteristics of Munc18-1 wild-type neurons, Stxbp1+/− neurons and null mutant neurons expressing one of the human disease variants: (C) mean synapse density calculated as the ratio of synapse number and total dendritic length. (D) Mean soma Munc18-1 level. (E) Mean synaptic Munc18-1 level. (F and G) Mean somatic and synaptic syntaxin-1 level. (H) Example traces of evoked release from wild-type neurons, Stxbp1 null neurons expressing C180Y and M433R upon a single action potential stimulation. (I) Mean EPSC amplitude expressed as the ratio of the wild-type value. (J) Paired-pulse ratio (calculated as the ratio of second EPSC and first EPSC) depending on the pulse interval. (K) Frequency of spontaneous release normalized to the wild-type values. (L). Amplitude of spontaneous release normalized to the wild-type values. (M) Example traces of spontaneous release in Stxbp1 null neurons expressing Munc18-1 wild-type, C180Y or M433R human variants. (N) The size of the readily releasable pool derived from back extrapolation of cumulative total charge released during 40 Hz train, 100 APs. The number of analysed cells is indicated in the bars. *P < 0.05, **P < 0.01, ***P < 0.001 versus infected wild-type control. Explanation of statistical analysis is provided in the Supplementary material. mEPSC = mini excitatory postsynaptic current; RRP = readily releasable pool.
Expression of human disease variants produces diverse phenotypes consistent with haploinsufficiency
To assess the functional impact of STXBP1 mutations found in human patients, we generated seven patient mutations in the Munc18-1 protein (Fig. 1A) and expressed each of these in null mutant mouse neurons in culture with wild-type Munc18-1 protein as control (Fig. 1B and Supplementary Fig. 1). In this way, neurons rely exclusively on one of these variants and the loss of basic protein function can be compared quantitatively to wild-type Munc18-1 protein for each of the seven selected mutations. In these initial experiments, we used microdot cultures, where single neurons grow on islands of rat astrocytes. This reduced and standardized preparation allows quantitative comparisons between neurons expressing different variants for many morphological and functional parameters (Wierda et al., 2007; Schmitz et al., 2011; Santos et al., 2017). Subsequently, we tested a selection of these variants in neurons carrying a single copy of the Stxbp1 gene to model the situation in patients (see below).
The most firmly established molecular function of Munc18-1 is to prepare synaptic vesicles for release in the presynaptic nerve terminal (reviewed in Toonen and Verhage, 2007). Consequently, in the absence of Munc18-1, neurons have no synaptic activity, not even spontaneous activity (Verhage et al., 2000) and die within a few days in vitro and in vivo (Verhage et al., 2000; Heeroma et al., 2004; Santos et al., 2017). Expression of five human variants in Stxbp1 null mutant neurons rescued neuronal viability: C180Y, M433R, G544D, T574P and C522R (Fig. 1C). However, few null mutant neurons expressing R388X survived and no neurons expressing V84D survived. Neurons expressing one of the five variants that rescued viability, also formed synapses and had a normal dendritic length (Supplementary Fig. 2); however, neurons expressing M443R and G544D variants had lower synaptic density compared to the wild-type viral expression (P < 0.001, P = 0.005, respectively; Fig. 1C). These data show that in a reduced neuronal model system, three of the selected Munc18-1 variants behave like wild-type Munc18-1 protein when analysed for survival and morphological parameters, but two variants, R388X and V84D, did not rescue viability, and two show mild impairments in synapse formation or maintenance. Hence, Munc18-1 variants that produce similar clinical features, behave rather differently under conditions where neurons rely solely on a given variant.
One of the proposed effects of point mutations in Munc18-1 is a reduced stability of the mutant protein (Saitsu et al., 2008; Stamberger et al., 2016). Therefore we analysed cellular protein levels for all five variants that rescued viability. Expression of wild-type Munc18-1 in null mutant neurons using lentiviral vectors produced ∼2-fold higher expression levels in the soma as compared to endogenous Munc18-1 in wild-type neurons (Fig. 1D). However, expression of C180Y, T574P and C522R using the same viral infection produced significantly lower protein levels in the soma relative to wild-type viral expression (P = 0.003, P = 0.017 and P = 0.010, respectively; Fig. 1D). Somatic expression levels of the two mutants that had lower synaptic density, M443R and G544D (Fig. 1C), were similar to over-expression of wild-type Munc18-1 (M443R) or at least as high as uninfected wild-type neurons (G544D; Fig. 1D).
Targeting of Munc18-1 variants to the nerve terminal was assessed by quantifying immunoreactivity in synapses using staining for the synapse marker synapsin as a mask. The synaptic Munc18-1 levels of all five variants were significantly lower than wild-type viral expression [F(5,125) = 29.77, P < 0.001; Fig. 1E]. Loss of Munc18-1 expression is known to reduce the cellular level of the SNARE protein syntaxin-1 (Voets et al., 2001; Toonen et al., 2005). The syntaxin-1 expression level was slightly reduced in the soma for C180Y and G544D variants (Fig. 1F) and more profoundly in the synapse of neurons expressing M443R and G544D variants (Fig. 1G), while the synaptic level of another presynaptic protein, VAMP/synaptobrevin 2 was unaltered for all variants (Supplementary Fig. 2). These data suggest that the Munc18-1 variants C180Y, T574P and C522R have reduced stability and that the remaining M443R and G544D are impaired in synaptic targeting of both Munc18-1 and syntaxin-1. This may explain the reduced synapse density for these two variants (Fig. 1C). However, the synaptic Munc18-1 level is comparable to heterozygous synapses and might therefore be sufficient to support basal synaptic transmission (Toonen et al. 2006).
We assessed synaptic transmission using patch clamp recordings of Stxbp1 null mutant neurons expressing one of the four Munc18-1 variants tested above (C180Y, M443R, T574P, C522R) and compared these to neurons expressing wild-type Munc18-1. Null mutant neurons expressing C180Y, T574P or C522R showed normal evoked responses, but neurons expressing M443R showed a 50% reduced response (Fig. 1H and I). C180Y and C522R showed similar paired pulse plasticity, but M443R and T574P showed increased paired pulse plasticity, suggesting reduced release probability (Fig. 1J). Null mutant neurons expressing the M443R variant also exhibited a severely reduced frequency of spontaneous synaptic events (Fig. 1K and M). While evoked synaptic transmission was normal in C180Y expressing null mutant neurons, the spontaneous event frequency was reduced (Fig. 1K and M). Neurons expressing T574P and C522R variants did not show significantly reduced spontaneous frequency (Fig. 1K and M). In all cases the amplitude of spontaneous events was normal (Fig. 1L), confirming the presynaptic origin of these phenotypes. Finally, to assess the size of the readily releasable pool and vesicle recruitment rates, we plotted the synaptic responses to trains of action potentials cumulatively (Supplementary Fig. 2). We observed different recruitment rates and pool sizes among the four variants tested, but no significant differences (Fig. 1N). Taken together, these morphological and functional data on the null mutant background show that different variants of the Munc18-1 protein found in patients have retained basic protein function to different extents, with most affected V84D > R388X > M443R and G544D > C180Y > T574P and C522R. Given the fact that these mutations all produce similar clinical features, haploinsufficiency becomes the most plausible explanation for STXBP1-encephalopathy pathogenesis.
Expression of human disease variants in heterozygous neurons supports normal synaptic transmission
To model the situation in patients as precisely as possible, we expressed the same seven variants tested above on the null background, now in Stxbp1 heterozygous neurons. Hence, in this case, neurons express both wild-type Munc18-1 (from a single endogenous allele) and one of the variants, as is the case in patients. Under these conditions, all variants supported normal synaptic development (Fig. 2A and B), also neurons expressing V84D or R388X, which were shown not to rescue viability when expressed on the null background (Fig. 1). Analysis of Munc18-1 levels confirmed that heterozygous neurons have ∼50% lower Munc18-1 expression levels in soma and synapses as compared to wild-type neurons (Fig. 1D and E), in line with previous analyses (Toonen et al., 2006). Expression of none of the Munc18-1 variants produced a significant increase in somatic or synaptic Munc18-1 levels. Both somatic and synaptic Munc18-1 levels remained below that of wild-type neurons (P < 0.001 and P < 0.001 for all variants compared to wild-type viral expression, Fig. 2C and D), while the synaptic VAMP/synaptobrevin 2 level was normal in all cases [F(6,173) = 2.03, P = 0.064; Supplementary Fig. 2]. Together these data provide further support for the conclusion that all seven Munc18-1 variants tested have a reduced cellular stability. The fact that synaptogenesis is normal in heterozygous neurons expressing the most severely affected mutants V84D and R388X, argues against dominant negative effects.
Figure 2.

Morphological and electrophysiological characteristics of dissociated hippocampal neurons expressing the human disease variants in Stxbp1+/− mouse neurons. (A) Dissociated cortical neurons were stained for Munc18-1 and synaptic marker synaptobrevin (VAMP). Examples represent Stxbp1+/− neurons expressing wild-type Munc18-1 (WT), C180Y or M433R human variants. (B–D) Morphological and synaptic characteristics of Stxbp1+/− neurons expressing wild-type Munc18-1 or one of the human disease variants: (B) mean synapse density: calculated as the ratio of synapse number and total dendritic length. (C and D) Mean somatic and synaptic Munc18-1 level. (E) Example traces of evoked release upon a single action potential stimulation from Stxbp1+/− neurons expressing wild-type Munc18-1, C180Y, M433R, V84D, G544D or R388X human variants. (F) Mean EPSC amplitude expressed as the ratio of the infected wild-type values. (G) Paired-pulse ratio (second EPSC/first EPSC) depending on the pulse interval. (H) Readily releasable pool estimate derived from back-extrapolation of cumulative total charge released during 40 Hz train, 100 APs (I) Frequency of spontaneous release expressed as the ratio of infected wild-type values. (J) Amplitude of spontaneous release expressed as the ratio of infected wild-type values. (K) Example traces of spontaneous release in Stxbp1+/− neurons expressing wild-type Munc18-1, C180Y, M433R, G544D, R388X or V84D human variants. The number of analysed cells is indicated in the bars. ***P < 0.001 versus infected wild-type control. mEPSC = mini excitatory postsynaptic current; RRP = readily releasable pool.
Heterozygous neurons expressing each of the seven Munc18-1 variants showed normal synaptic transmission. Evoked responses [F(5,262) = 1.58, P = 0.166; Fig. 2E and F], paired pulse plasticity (Fig. 2G) and frequency and amplitude of spontaneous events [F(5,240) = 1.64, P = 0.149 and F(5,240) = 0.83, P = 0.526, respectively; Fig. 2I–K] were all similar between heterozygous neurons expressing wild-type Munc18-1 or one of the variants found in patients. Cumulative plots of the total charge during action potential trains revealed no differences in vesicle recruitment rate or the size of the readily releasable pool [F(5,237) = 1.06, P = 0.383; Fig. 2H and Supplementary Fig. 2). Hence, while the different Munc18-1 variants have retained basic protein function to a different extent, all of them support normal synaptic transmission when combined with a single wild-type allele, as is the case in patients. Taken together, these data show that expression of different Munc18-1 variants with different basic protein function and cellular stability, do not lead to differences in synaptic transmission on the time scale of the in vitro experiments performed here in the presence of one wild-type Munc18-1 allele.
Reduced protein stability for all Munc18-1 variants found in patients
Analyses of the expression of Munc18-1 variants in Stxbp1 null and heterozygous neurons produced evidence for reduced protein stability for most variants. We tested this more systematically by expression of C180Y, M443R, C522R and T574P variants in heterologous cells (HEK293) as a part of a bicistronic expression cassette where each of the variants is expressed as a single mRNA of a variant fused to GFP, separated by a 2A sequence (Ryan et al., 1991). Upon translation, the single mRNA generates two separate proteins in a 1:1 ratio by ribosome skipping. In this way, GFP intensity can be used as an in-cell control across all variants and was used to normalize expression levels of the variants. All mutants showed a distribution similar to wild-type protein, albeit much weaker, with no signs of accumulation in specific compartments (Fig. 3A and Supplementary Fig. 3). All four Munc18-1 variants and the wild-type protein showed complete cleavage of the 2A sequence and no signs of read-through (Fig. 3B). An ∼80% lower level was detected for all mutants, as compared to the wild-type protein (Fig. 3A and B). Hence, all Munc18-1 variants found in patients show a severe reduction in protein stability in heterologous cells.
Figure 3.

Cellular stability of wild-type and human disease variants of Munc18-1 in HEK293 cells and neurons. (A) Immunochemistry of HEK293 cells infected with wild-type Munc18-1 (WT), C180Y, M443R, C522R and T574P constructs stained for Munc18-1, EGFP and Golgi marker (GM130). (B) Normalized Munc18-1 levels in HEK293 cells after viral infection with wild-type, C180Y, M443R, C522R and T574P constructs. The inset shows representative western blot of HEK293 cells after viral infection; n = 5, 5, 5, 2 and 2, respectively. (C) Western blot analysis of Munc18-1 protein levels 0, 6, 12, 24 and 30 h after block of protein synthesis with cycloheximide for HEK293 cells infected with wild-type, C180Y, M433R, C522R or T574P constructs. The infection with wild-type construct was used as a control for all performed western blot analysis. (D) Quantitative analysis of the Munc18-1 protein expression from western blots in HEK cells represented in C. (E) Western blot analysis of Munc18-1 protein levels 0, 12, 24 and 36 h after block of protein synthesis with cycloheximide for wild-type, C180Y, M433R, C522R or T574P constructs in Stxbp1 null neurons. The infection with wild-type construct was used as a control for all performed western blot analysis. (F) Quantitative analysis of the Munc18-1 protein expression from western blots in neurons represented in E. (G–I) Normalized Munc18-1 protein levels from three constructs expressed in HEK cells treated with MG132, Leupeptin or Pepstatin; n = 3, 2 and 2, respectively.
To further document this reduced stability, we performed pulse-chase experiments in HEK cells and primary mouse neurons by inhibiting protein synthesis with 100 µg/ml cycloheximide at t = 0 and quantifying the cellular Munc18-1 levels at different time points afterwards using immunoprecipitation of the denatured protein, which is almost 100% efficient for this protein/antibody combination (de Vries et al., 2000). Both in HEK293 cells (Fig. 3C and D) and in primary mouse neurons (Fig. 3E and F), the major difference in initial protein level between wild-type Munc18-1 and all the variants found in patients was confirmed. The half-life in heterologous cells was much shorter (10-fold decrease in 30 h) than in primary mouse neurons (2-fold over the same period; cf.Fig. 3D and F). To identify the cellular pathways responsible, we tested inhibition of the proteasome system using 10 µM MG132 or the lysosomal system using 25 µM Leupeptin/1 µM Pepstatin A for two selected Munc18-1 variants. No evidence for the involvement of these two cellular pathways was observed (Fig. 3G–I). These data show that all Munc18-1 variants found in patients have a severely reduced cellular stability, especially in heterologous cells, but also in primary neurons. We found no evidence for dominant negative phenotypes and we did not identify specific locations or pathways in the cell that could be linked to the impaired stability of these mutants. Taken together, these data provide strong support for haploinsufficiency as the best explanation for the observed clinical phenotype.
Generation and genomic analysis of Stxbp1+/− mouse lines
The in vitro data support a haploinsufficiency model to explain STXBP1 pathogenesis and not other explanations (e.g. dominant negative effects). Therefore, we studied Stxbp1 heterozygous mice (Stxbp1+/−) as a model for haploinsufficiency in STXBP1-encephalopathy.
Four Stxbp1+/− mouse lines were generated (Fig. 4A–D): (i) a Stxbp1cre/+ line where one copy of the gene was flanked by loxP sites and subsequently deleted using Cre-expression in the germline; (ii) a congenic BL6 Stxbp1+/− line; (iii) a reverse 129Sv Stxbp1+/− line; and (iv) a Gad2-Stxbp1cre/+ line. The Stxbp1cre/+ line was created using Cre-loxP-mediated recombination in the germ line. Cre-deleter mice were crossed with Stxbp1+/lox mutants, which were generated on a C57BL/6J background (Heeroma et al., 2004) and bred further on C57BL/6J background, generating a Stxbp1+/− mouse line without flanking gene variations. The congenic BL6 Stxbp1+/− line was originally generated by deleting exons 2–6, preventing translation from amino acid 14 onwards, in the embryonic stem cells derived from a 129S1/SvImJ strain (Fig. 4B; Verhage et al., 2000) 20 years ago and back-crossed to C57BL/6J mice for >40 generations. The reverse 129Sv Stxbp1+/− line was created by mating male congenic BL6 Stxbp1+/− mice with females of the inbred 129S1/SvImJ strain for three generations (Fig. 4C). For the fourth mouse line, Gad2-Stxbp1cre/+ mice, a selective heterozygous inactivation of Stxbp1 expression in GABAergic cells was achieved by crossing Gad2TM2-Cre mice with Stxbp1+/lox mutants (Fig. 4D). All these mice were viable and fertile, except the Gad2-Stxbp1cre/+ mice where around 50% of the mice did not survive beyond postnatal Day 14 (Supplementary Fig. 4, see below).
Figure 4.

Generation, genomic analysis, routine behavioural observation and spontaneous activity of Stxbp1+/− mice. (A–D) Generations of four lines of Stxbp1+/− mice: conditional, congenic BL6, reverse 129Sv and Gad2- Stxbp1 line. Chromosomes bearing Stxbp1 mutation originating from the C57BL/6J genetic background are shown in grey, and those from the 129/SvJ genetic background are shown in orange. Stxbp1 mutation is represented with grey triangle; LoxP sites are represented with black rectangles and Cre-deleter lines are represented with scissors (red scissors: Cre expressed in all neuron, blue scissors: Gad2tm2-Cre mice with Cre-recombinase expressed in only GABAergic neurons). The flanking gene region is represented as orange region in the grey chromosome in the congenic BL6 line and vice versa in the reverse 129Sv line (adapted from Wolfer et al., 2002). (E and F) High-density genomic analysis showed the size and position of flanking gene region in Stxbp1+/− samples from congenic BL6 and reverse 129Sv lines. ‘Me-PaMuFind-It’ web tool (http://me-pamufind-it.org/) revealed three genes with passenger mutations from 129Sv genetic background within flanking genes region in congenic BL6 Stxbp1+/− samples. (G) Stxbp1+/− mice showed lower body weight compared to their controls. (H) Grip strength was normal in Stxbp1+/− mice. (I) Motor coordination and motor learning in Stxbp1+/− mice was normal, as assessed on the rotarod. (J and K) Circadian rhythm assessed by changes in the activity in anticipation of (filled bars), and response to (open bars), day/night transitions and proportion of time spent outside the shelter was normal in Stxbp1+/− mice. (L) Proportion of activity duration during the first 3 h of the dark phase days in the home-cage environment (PhenoTyper) showed increased activity of Stxbp1+/− mice from all three lines during the first dark phase. (M) Proportion of activity duration in the PhenoTyper during the light phases was overall lower for Stxbp1+/− mice (conditional mice, congenic BL6 and reverse 129Sv). The number of animals assigned is shown in the graphs. *P < 0.05; ***P < 0.001.
High-density genomic analysis revealed that after 40 generations, the 129/SvJ region flanking the Stxbp1 locus extended <10 Mbp upstream and downstream of the Stxbp1 gene and contained 240 protein coding genes (Fig. 4E). The ‘Me-PaMuFind-It’ web tool (Vanden Berghe et al., 2015) identified 10 potential 129-derived passenger mutations in protein coding genes in the 129Sv/J-derived genomic region surrounding the Stxbp1 gene. However, comparing the list of potential passenger mutations predicted by the ‘Me-PaMuFind-It’ web tool (see ‘Material and methods’ section) with the list of protein coding genes within the flanking gene region identified in our high-density genomic analysis we predict that three 129-derived passenger mutations are still present in our congenic BL6 mouse model: Card9, Rapgef1 and Dolpp1. To our knowledge there is no evidence that these mutations are functional.
The high-density genomic analysis performed on the samples from the third generation of reverse 129/SvJ Stxbp1+/− mice showed the expected percentage of residual genetic material from C57BL/6J background (for the third generation, the expected percentage was 12.5%, and 13.0 ± 2.7% was observed, n = 5); The C57BL/6J background genes were aggregated around previously detected flanking genes region of Stxbp1 gene on the chromosome 2 (Fig. 4F). Taken together, we derive the following conclusions regarding these mouse lines: (i) the Stxbp1cre/+ line can be considered the most appropriate haploinsufficiency model on a C57BL/6J background, with no flanking gene issues. Its only imperfection is the presence of one remaining LoxP site; (ii) the congenic BL6 Stxbp1+/− line can be exploited to test phenotypic effects of flanking genes (∼1% of the genome), a common problem for many behavioural studies using traditional knock-out mice (Gerlai, 2001; Wolfer et al., 2002; Vanden Berghe et al., 2015); (iii) the reverse 129Sv Stxbp1+/− line can be used to test differences in phenotypic expression of the same genotypes (Stxbp1 heterozygosity), in different genomic backgrounds (C57BL/6J versus 129/SvJ); and (iv) the Gad2-Stxbp1cre/+ can be used to specifically test phenotypic effects of Stxbp1 heterozygosity in inhibitory neurons.
Stxbp1 +/− mice show normal general behaviour
Eight-week-old Stxbp1cre/+, congenic BL6 Stxbp1+/− and reverse 129Sv Stxbp1+/− mice were viable and did not show overt phenotypic abnormalities. However, Stxbp1+/− mice of all three lines showed a small but significant reduction in body weight at adulthood [genotype: F(1,92) = 26.20, P < 0.001] (Fig. 4G). All three Stxbp1+/− lines had similar muscle strength as assessed by the grip strength test at around 12 weeks of age and motor coordination tested on the rotarod at around 12 weeks of age (Fig. 4H and I). The diurnal rhythm of Stxbp1+/− mice was assessed by monitoring undisturbed behaviour in an automated home-cage system (PhenoTyper) enriched with a shelter for three consecutive days. It has been described before that mice tend to sleep/rest inside the shelter and they spend time outside the shelter during the active period (Maroteaux et al., 2012; Loos et al., 2014). The change of activity in anticipation of and response to light/dark transitions and the changes in the time spent outside the shelter in the PhenoTyper were similar between genotypes (Fig. 4J and K), except for increased activity in response to dark phase observed for reverse 129Sv Stxbp1+/− mouse lines (P < 0.001). Taken together, our data suggest that besides the slightly lower body weight, there were no general abnormalities in motor performance, spontaneous activity and diurnal behaviour in Stxbp1+/− mouse lines.
Stxbp1 haploinsufficiency influences spontaneous activity
To assess spontaneous behaviour, we analysed a set of 115 behavioural parameters obtained after 3 days continuous video-monitoring in the PhenoTyper without human intervention. These parameters are divided in six main categories: activity, dark-light ratio, habituation, kinematics, sheltering, and phase transition (Loos et al., 2014). The analysis showed significant genotype effects mostly for activity and kinematics parameters in all Stxbp1+/− mouse models (Supplementary Tables 2–4). Activity of all mice showed striking differences in relation to the time of the day, in particular during the first 3 h of the dark phase, the following 9 h of the dark phase and the light phase (Fig. 4L, M and Supplementary Fig. 6). The activity of all Stxbp1+/− mouse models was higher relative to control littermates during the first 3 h of the first dark phase in the PhenoTyper for all three lines [genotype: F(1,176) = 35.25, P < 0.001; Fig. 4L and Supplementary Fig. 6], but not for the remaining 9 h of the dark phase (Supplementary Fig. 6). The reverse 129Sv line, but not the other lines, also showed increased activity during the first 3 h of the second and third dark phase (P < 0.001 and P = 0.044, respectively; Fig. 4L). Activity during the light phase was slightly lower when comparing Stxbp1+/− mouse lines with their wild-type controls [F(1,171) = 6.46, P = 0.012; Fig. 4M]. In general, irrespective of Stxbp1 genotype, the reverse 129Sv line mice had lower activity compared to congenic BL6 and Stxbp1cre/+ mice (Supplementary Tables 2–4) as has been observed before Loos et al. (2014). Taken together, these data reveal an increased initial activity, probably in response to novelty, and a corresponding decreased activity during the subsequent light phase as a common feature of all Stxbp1 mouse lines. For the 129Sv background, the increased activity during the initial hours of the dark phase was also observed on the subsequent 2 days.
Stxbp1 +/− mice show epilepsy-like behaviour and EEG abnormalities that could be suppressed with levetiracetam
STXBP1 haploinsufficiency in humans leads to epileptic spasms and tonic seizures, occurred mainly during sleep (Saitsu et al., 2010; Guacci et al., 2016). To investigate similar phenotypes in Stxbp1+/− mice, we performed video-monitoring of Stxbp1+/− mice. These mice showed two types of the spontaneous abnormal behaviours: twitches (Fig. 5A and Supplementary Video 1); and jumps (Fig. 5B and Supplementary Video 2). These two types of events were observed when mice were in an inactive state (apparent sleep) and appeared to occur in a clustered pattern, but not strictly correlated (Fig. 5C). Twitches and jumps were never observed in control littermates by two independent observers unaware of the genotype (n = 18, t = 4 h/animal). Twitches and jumps occurred predominantly, but not exclusively, during the light phase, the inactive phase of mice (Fig. 5D). The average incidence of twitches and jumps was ∼1 twitch/h and ∼1 jump/1.5 h and was similar in the three genomic backgrounds tested, although twitches tended to be more frequent in the Stxbp1cre/+ line (Fig. 5E). Hence, abnormal motor activity in three Stxbp1+/− mouse lines resemble tonic spasms and myoclonic jerks previously reported in human patients, also the fact that they occur predominantly during rest/sleep.
To characterize the epileptic phenotype in Stxbp1+/− mice further, we performed simultaneous video- and wireless EEG-monitoring in Stxbp1+/− mouse models and controls. EEG electrodes were implanted in the hippocampal CA1 region (n = 2 BL6 Stxbp1+/− and n = 2 control mice) or neocortex (n = 4 BL6 Stxbp1+/−, n = 3 Stxbp1cre/+, n = 4 control mice, Fig. 5F). Both hippocampal and cortical recordings revealed frequent, high amplitude spike-wave discharges in all Stxbp1+/− mice that were visually not detected in control mice (Fig. 5G). The spike-wave discharges were characterized by high amplitude, low-voltage ECoG oscillations with a relatively narrow frequency band at ∼7 Hz, and duration usually between 1.5 and 2 s (Fig. 5H). Spike-wave discharges were distinguished from slow-wave sleep by its clear peak at ∼7 Hz and from awake state or paradoxical sleep by larger amplitudes (Fig. 5G and H).
Simultaneous video and wireless EEG-monitoring during the 12 h of the light phase revealed that some of the observed twitches and jumps (Fig. 5A and B) coincided with spike-wave discharges (Fig. 5I). To assess the strength of the correlation between behavioural and EEG events, we performed a simulation study. The probability that two types of events occurred within ±10 s by coincidence was far below 0.05 for twitches and around the limit of 0.05 for jumps (Fig. 5J and K). Finally, the total number of events detected during the 12-h light phase was 49.4 ± 10.0, 17.6 ± 2.4 and 8.7 ± 1.2 for spike-wave discharges longer than 1.5 s, twitches, and jumps, respectively (Fig. 5L). Comparison of spike-wave discharge incidence between Stxbp1+/− mice and control littermates during 24 h recordings revealed that the total number of spike-wave discharge-like events is almost 100 times higher in Stxbp1+/− mice (305 ± 54.5) as compared to control mice (5.6 ± 4.9, Supplementary Fig. 5). Together, these data show that the observed EEG/ECoG abnormalities were strongly correlated with twitches and a correlation with jumps was suggestive, but that not all spike-wave discharges detected in cortex and hippocampus resulted in behavioural manifestation and vice versa.
Levetiracetam is a commonly prescribed antiepileptic drug with a presynaptic mode of action binding to synaptic vesicle protein SV2A (Lynch et al., 2004), and therefore, seems an excellent candidate to inhibit EEG abnormalities in the mouse models for disorders caused by mutations in the presynaptic gene Stxbp1. Acute treatment with levetiracetam significantly reduced the number of spike-wave discharges (P = 0.004 compared to the saline treatment, Fig. 5M). Prolonged levetiracetam treatment for 5 days had a similar effect as acute administration: a significant reduction of spike-wave discharges (P = 0.036 compared to saline treatment, Fig. 5M). Hence, levetiracetam effectively reduced ECoG abnormalities in Stxbp1 mouse models.
The fourth mouse line, Gad2-Stxbp1cre/+, was not healthy enough for systematic analyses. Half the Gad2-Stxbp1cre/+ mice died between the second and the third postnatal week, and the remaining animals showed strong epileptiform activity on ECoG, much more severe than the other three mouse lines (Supplementary Fig. 4). c-Fos expression was also increased much more than the other three mouse lines in several brain regions (Supplementary Fig. 4). For ethical reasons, these mice were sacrificed before further analyses could take place. Therefore, data obtained using this mouse line remain somewhat anecdotal. However, it is evident that Stxbp1 heterozygosity in GABAergic neurons only, produced much stronger phenotypes than Stxbp1 heterozygosity in all neurons.
Increased c-Fos expression in cortical regions of Stxbp1+/− mice
c-Fos is a well-established marker of neural activation. c-Fos levels can be rapidly (within 30–60 min) and transiently increased by diverse stimuli, including seizures (Herrera and Robertson, 1996). To study the pattern of c-Fos expression in Stxbp1+/− mice, we performed c-Fos immunostaining of brain slices of congenic BL6 and Stxbp1cre/+ mice and analysed c-Fos expression in 10 brain regions. Semi-quantitative analysis of c-Fos-positive cells showed increased c-Fos expression in prefrontal cortex, motor cortex and somatosensory cortex in both Stxbp1 mouse models, but not in seven other regions (Fig. 6A–S). Some variation in the localization of increased c-Fos expression was detected between the two mouse models and between individual mice. First, increased expression was detected in the prefrontal cortex of three congenic BL6 Stxbp1+/− mice, but not in Stxbp1cre/+ mice (Fig. 6A–F). Second, increased expression in the motor cortex was detected in two Stxbp1cre/+ and two congenic BL6 Stxbp1+/−, but not in the remaining mice (Fig. 6G–L). Third, increased expression was detected in the somatosensory cortex of one Stxbp1cre/+ and one congenic BL6 Stxbp1+/− mouse, but not in the remaining mice (Fig. 6M–R). These data indicate a robust increase of c-Fos expression in the specific brain regions. This increased expression was probably correlated to the recent seizure history.
Figure 6.
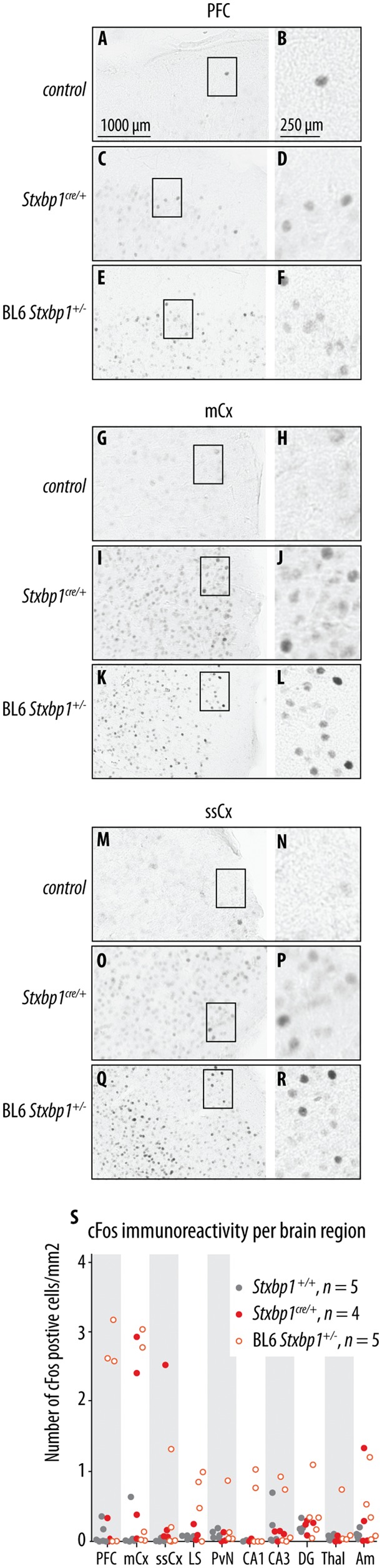
c-Fos expression in Stxbp1+/− mice. (A–R) Representatives of c-Fos expression in prefrontal cortex (PFC), primary motor cortex (mCx) and somatosensory cortex (ssCx) for Stxbp1+/+ mice (control), Stxbp1cre/+ and congenic BL6 Stxbp1+/− mice. (S) Number of c-Fos positive cells per brain region for Stxbp1+/+, Stxbp1cre/+, and congenic BL6 Stxbp1+/− mice. Scale bars and the number of samples are provided in the figure.
Stxbp1 haploinsufficiency affects behavioural flexibility but not spatial learning and attention in mice
In addition to seizures, STXBP1-encephalopathies are characterized by additional symptoms, most notably cognitive deficits, developmental delay and autistic-like traits (Deprez et al., 2010; Saitsu et al., 2010; Hamdan et al., 2011; Stamberger et al., 2016). Therefore, we tested multiple aspects of learning and memory, impulsivity and attention in Stxbp1+/− mice. To evaluate spatial learning, memory and behavioural flexibility, we tested conditional and congenic BL6 mice in the Barnes maze, a dry and less stressful version of a spatial maze (Harrison et al., 2009). Mice from reverse 129Sv line were tested in the Morris water maze instead, given their very low activity in the PhenoTyper and previously reported poor performance during the probe trial in the Barnes maze (Holmes et al., 2002). To evaluate cognitive phenotypes in Stxbp1+/− mice further, two automated cognitive tasks were performed in the PhenoTyper: the CognitionWall task for assessment of discrimination and reversal learning (Remmelink et al., 2016) and the Shelter task for assessment of avoidance learning (Maroteaux et al., 2012). Additionally, attention and impulsivity were tested in the congenic BL6 line using the 5-choice serial reaction time task.
Spatial-learning paradigms showed that Stxbp1+/− mice from all three lines have similar learning curves as their controls: a reduction in the escape latency and distance moved to reach the target with increasing number of training sessions (Fig. 7A, B, E, F, G and H). Congenic BL6 Stxbp1+/− mice showed a mild delay in the learning phase [F(1,21) = 5.70, P = 0.026; Fig. 7A] but tended to visit more holes in the target octant compared to control mice during the first probe trial [t(21) = −1.80, P = 0.086; Fig. 7C and D]. However, this mild delay in learning was not observed in the second cohort of congenic BL6 mice tested in the Barnes maze to analyse long-term memory, nor in the group tested in a novel version of the Barnes maze with increased complexity (Supplementary Fig. 7). Finally, reverse 129Sv Stxbp1+/− mice also showed a normal learning curve in the Morris water maze (Fig. 7G–I). Hence, these data suggest mild behavioural differences during the acquisition of spatial memory in Stxbp1+/− mice, independent on the genetic background and paradigm used.
Figure 7.

Learning and memory in Stxbp1+/− mice in classical spatial paradigms and recently developed automated tasks in the PhenoTyper. (A and B) Latency and distance travelled to find the escape hole in the Barnes maze during the learning phase for congenic BL6 Stxbp1+/− mice. Congenic BL6 Stxbp1+/− (HZ BL6) showed longer latency to find new escape hole during the learning phase (P = 0.026) and during R1 (P < 0.001) and travelled longer distance to the new escape hole (R1-R3: P = 0.024) compared to their controls (WT BL6). (C) HZ BL6 mice showed narrower distribution of holes visit around the target hole during the first probe trial (P1) compared to wild-type BL6. (D) Probability of hole visits in the old target octant during the P1 and P2 tended to be higher for HZ BL6 mice compared to their controls (P = 0.086 and P = 0.060, respectively) and there were no differences in the probability of hole visits in the new target octant during the P2. (E and F) Latency and distance travelled to find the escape hole in the Barnes maze during the learning phase were similar for Stxbp1cre/+ mice (HZ cond) and their controls (wild-type cond). (G and H) Escape latency and distance travelled to the platform during the training in the Morris water maze was similar for reverse 129Sv Stxbp1+/− mice and control mice. (I) Time spent per quadrant during the probe trial was similar for reverse 129Sv Stxbp1+/− mice and control mice. (J) Schematic overview of the CognitionWall DL/RL task. (K and L) Kaplan-Meier survival curves shows the fraction of congenic BL6 and conditional Stxbp1 mice that reached the 80% criterion as a function of hole entries during the DL and RL phases. (M) Average number of entries made per group to reach 80% criterion during the DL and RL phases. HZ BL6 mice reached the 80% criterion during RL with lower number of entries compared to control (P = 0.004). (N) Schematic overview of the Shelter task protocol in the PhenoTyper to assess avoidance learning. (O and P) The preference index during the dark phases of the avoidance learning task was similar for HZ BL6 and HZ cond mice and their controls. Insets represent the learning effect on the preference index (D5/D6/D7-D4). The insets in graph O shows that congenic BL6 Stxbp1+/− mice showed a stronger learning effect compared to their controls (P = 0.012). (Q and R) The aversion index during the dark phases of avoidance learning task showed trend toward lower values for HZ BL6 mice compared to their controls (P = 0.101). The aversion index was similar between HZ cond mice and their controls. *P < 0.05; **P < 0.01; ***P < 0.001 compared to respective control. Statistical analysis is explained in the Supplementary material.
After acquisition training sessions, reversal learning was tested by relocation of the target hole to the diametrically opposite side of the maze. Congenic BL6 Stxbp1+/− mice showed significantly longer escape latency during the first reversal trial [trial × genotype: F(1.583, 33.245) = 5.05, P = 0.018, post hoc R1: P < 0.001; Fig. 7A] and longer distance travelled to the new target hole [F(1,21) = 5.88, P = 0.024; Fig. 7B]. During the second probe trial, congenic BL6 Stxbp1+/− mice tended to visit more holes in the old target octant then control littermates [t(21) = 1.98, P = 0.060; Fig. 7C and D] and they showed the same probability of visiting holes in the new target octant as their controls [t(21) = 1.15, P = 0.264. Fig. 7C and D]. Additionally, during the third probe trial, performed after three additional reversal trials, no differences in the probability of holes visits were found between congenic BL6 Stxbp1+/− mice and their controls [t(21) = 0.93, P = 0.364 Supplementary Fig. 7A and B]. Stxbp1cre/+ mice also showed a tendency to travel a longer distance than control littermates to find a new target hole during the first three reversal trials [F(1,21) = 3.61, P = 0.071 Fig. 7F]. Taken together, these data showed that Stxbp1+/− mice were slower to reverse previously learned strategy, but did finally acquire/maintain the new strategy, suggesting impaired behavioural flexibility.
To explore discrimination and reversal learning further, a newly developed 4-day automated home-cage task was performed (CognitionWall test, Remmelink et al., 2016). This task uses an operant wall with three entry holes placed in the PhenoTyper in front of a reward dispenser (Fig. 7J). During the first 2 days, mice had to learn to earn food rewards by passing through the left hole (discrimination learning) and during Days 3 and 4 the right hole was rewarded (reversal learning). During discrimination learning, Stxbp1cre/+ and congenic BL6 Stxbp1+/− mice showed a similar distribution of entries made to reach the criterion of 80% correct entries as compared to control mice (P = 0.952 and P = 0.147, respectively; Fig. 7K and M). During reversal learning, congenic BL6 Stxbp1+/− mice made significantly fewer entries to reach the 80% criterion (P = 0.004; Fig. 7L and M), while Stxbp1cre/+ mice required a similar number of entries as their controls (P = 0.752; Fig. 7L and M). Thus, congenic BL6 Stxbp1+/− mice showed facilitated reversal learning in this test, while the Stxbp1cre/+ mice showed normal performance.
To assess avoidance learning, the Shelter task (Maroteaux et al., 2012), an automated PhenoTyper assay, was performed. In this test, avoidance learning is studied in an enriched home-cage with a shelter with two entrances, by first assessing the preferred entrance during the first 4 days in the cage of each individual mouse and subsequently applying an aversive stimulus (bright illumination) every time an animal enters the shelter through its preferred entrance (Fig. 7N). The changes in the preference index (fraction of entries via the preferred entrance) were considered as a measure of cognitive response and the changes in the aversion index (a change in the time spent in now illuminated shelter) as the averseness of the bright illumination. Stxbp1cre/+ and congenic BL6 Stxbp1+/− mice showed a decrease of preference index over days comparable with control mice [genotype effect preference index: F(1,351) = 0.62, P = 0.433; F(1,63) = 0.003, P = 0.955, respectively, Fig. 7O and P]. However, the learning effect of congenic BL6 Stxbp1+/− mice was significantly bigger compared to control mice during all three dark phases [F(1,351) = 6.35, P = 0.012; Fig. 7O inset]. Additionally, congenic BL6 Stxbp1+/− mice showed a trend toward lower values of aversion index over days [genotype: F(1,351) = 2.74, P = 0.101; Fig. 7Q]. Reverse 129Sv Stxbp1 mice were also tested in the Shelter task. However, these mice showed inconsistent behaviour (Supplementary Fig. 8A). Together, the Shelter task data indicate an enhanced cognitive response in congenic BL6 Stxbp1+/− mice but a normal cognitive and aversive response in Stxbp1cre/+ mice.
To assess the attentional capacity and impulsivity in congenic BL6 Stxbp1 mice, the 5-choice serial reaction time task was performed. No significant effects of genotype on response accuracy, variability and omission errors (P = 0.82, P = 0.33 and P = 0.68, respectively) and no differences in the number of impulsive responses (P = 0.68; Supplementary Fig. 7 and Supplementary Table 5) were observed. Motivation to execute the task, as assessed by measuring the latency to a correct response and the reward latency were similar between congenic BL6 Stxbp1+/− mice and their controls (P = 0.63, P = 0.12; Supplementary Fig. 7 and Supplementary Table 5). Hence, attention performance and impulsivity was normal in congenic BL6 Stxbp1+/− mice.
These cognitive tests suggest that Stxbp1 haploinsufficiency impairs behavioural flexibility and promotes focused searching strategy in the Barnes maze in Stxbp1cre/+ and congenic BL6 Stxbp1+/− mice, without affecting spatial learning in all three mouse models. The facilitated reversal learning and enhanced aversive cognitive response, accompanied with increased aversion in congenic BL6 Stxbp1+/− mice, suggest a background-specific effect of Stxbp1 haploinsufficiency in these behavioural domains. These background-specific effects were not explained by altered attention or impulsivity.
Stxbp1 haploinsufficiency induces anxiety-like behaviour in mice and does not affect social behaviour
Analysis of spontaneous behaviour during 3 days in the PhenoTyper revealed that Stxbp1cre/+ and congenic BL6 Stxbp1+/− mice spent less time on top of the shelter compared to control mice (P = 0.002 and P < 0.001, respectively; Fig. 8A and B). As this parameter can be interpreted as index of anxiety-related behaviour, we tested anxiety more systematically in Stxbp1+/− mouse models using well-established behavioural paradigms: the elevated plus maze, the dark-light box, the open field and novelty-induced hypophagia.
Figure 8.

Anxiety-related phenotype and social behaviour in Stxbp1+/− mice. (A) Schematic overview of the PhenoTyper home-cage. (B) On shelter duration during the dark phase was shorter for Stxbp1cre/+ and congenic BL6 Stxbp1+/− (HZ cond and HZ BL6) mice compared to their controls. (C–F) Elevated plus maze (EPM) data. (C) Schematic overview of the EPM. (D–F) Latency to enter open arms was longer, time spent on the open arms and number of visits to the open arms were lower for Stxbp1cre/+ and congenic BL6 Stxbp1+/− mice compared to their controls. (G–J) Dark-light box (DLB) data. (G) Schematic overview of the DLB. (H) Latency to visit bright compartment (BC) was longer for congenic BL6 Stxbp1+/− mice compared to their controls. (I–J) Time spent in the bright compartment and number of visits to the bright compartment was lower for Stxbp1cre/+ and congenic BL6 Stxbp1+/− mice compared to their controls. (K–O) Open field data. (K) Schematic overview of the open field. (L) Latency to visit centre zone was longer for Stxbp1cre/+ and congenic BL- Stxbp1+/− mice compared to their controls. (M and N) Percentage of distance moved in the centre zone and number of visits to centre zone was similar for Stxbp1cre/+ and congenic BL6 Stxbp1+/− mice and respective control mice. (O) The total distance moved was longer for reverse 129Sv Stxbp1+/− mice compared to their control, and it was similar for Stxbp1cre/+ and congenic BL6 Stxbp1+/− mice and respective control mice. (P and Q) Novelty induced hypophagia test (NIHP) data. (P) Schematic overview of the NIHP test. (Q) Latency to consume highly palatable food (cracker) was similar for Stxbp1cre/+ and congenic BL6 Stxbp1+/− mice and the respective controls. (R) Schematic overview of three-chamber test protocol. (S) During test for sociability (phase III), Stxbp1cre/+, congenic BL6 Stxbp1+/− mice and their respective controls spent more time in the mouse zone than in the object containing zone. (T) During test for social novelty (phase IV), Stxbp1cre/+, congenic BL6 Stxbp1+/− mice and their respective controls spent more time in the zone with novel mouse than in the zone with familiar mouse. There were no differences between genotypes. *P < 0.05, **P < 0.01, ***P < 0.001 for comparison between zones.
In the elevated plus maze (Fig. 8C), Stxbp1cre/+ and congenic Bl6-Stxbp1+/− mice showed delayed latency to enter the open arms [F(1,44) = 29.67, P < 0.001; Fig. 8D], spent less time on the open arms [F(1,44) = 19.06, P < 0.001; Fig. 8E] and visited the open arms less frequently [F(1,44) = 22.79, P < 0.001; Fig. 8F] compared to control mice. No differences were observed in total distance travelled between genotypes [F(1,44) = 0.220, P = 0.641]. In the dark-light box test (Fig. 8G), congenic BL6 Stxbp1+/− mice showed longer latency to visit the bright compartment (P < 0.001; Fig. 8H). Both Stxbp1cre/+ and congenic BL6 Stxbp1+/− mice spent less time in the bright compartment (P = 0.024 and P < 0.001, respectively; Fig. 8I) and made fewer visits to the bright compartment [F(1,44) = 53.68, P < 0.001; Fig. 8J] compared to control mice. In the open field test (Fig. 8K), Stxbp1cre/+and congenic BL6 Stxbp1+/− mice both showed a longer latency to visit the centre zone of the arena [F(1,44) = 6.98, P = 0.011; Fig. 8L] and showed a trend toward lower percentage of distance moved in the centre zone [F(1,44) = 4.07, P = 0.05; Fig. 8M], while making a similar number of visits to the centre zone [F(1,44) = 2.04, P = 0.160; Fig. 8N] and traveling similar total distance as control mice [F(1,44) = 1.25, P = 0.270; Fig. 8O]. We also tested how soon after being introduced into a novel home-cage, Stxbp1+/− mice started to consume a familiar palatable piece of food (a cream cracker) to assess novelty-induced hypophagia (Fig. 8P). This test showed a similar latency to eat between genotypes [F(1,43) = 0.79, P = 0.380; Fig. 8Q]. Analysis of anxiety-related behaviours using the same apparatuses and protocols as used for congenic BL6 and conditional lines was not possible for the reverse 129Sv line of Stxbp1 mice due to the fact that the majority of mice, both Stxbp1+/− and controls, did not visit anxiety-related compartments at all, as observed before (reviewed in Crawley, 2008), and did not climb on the shelter in the PhenoTyper (Supplementary Table 6 and Supplementary Fig. 8B). Altogether, these data indicate an increased anxiety-like behaviour in Stxbp1cre/+ and congenic BL6 Stxbp1+/− mice that was not confounded by changes in motor activity.
Autism-related traits have been described in some STXBP1-encephalopathy patients (Milh et al., 2011; Campbell et al., 2012). Therefore, we tested social behaviour of Stxbp1 mouse models in the three-chamber test (Fig. 8R). During the sociability phase, Stxbp1cre/+ and congenic BL6 Stxbp1+/− mice, as well as their controls, spent significantly more time in the zone around an inverted mesh wire cup that contained a male conspecific compared to the time spent in the zone around an empty cup (conditional Stxbp1+/+: P = 0.006, Stxbp1cre/+: P < 0.001, congenic BL6 Stxbp1+/+: P < 0.001, congenic BL6 Stxbp1+/−: P = 0.001; Fig. 8S). Subsequently, a novel mouse was introduced in the previously empty cup and preference for social novelty was assessed. All tested animals, independent of the genotype and genetic background, spent significantly more time around the zone with the novel stimulus mouse (conditional Stxbp1+/+: P = 0.020, Stxbp1cre/+: P < 0.001, congenic BL6 Stxbp1+/+: P = 0.026, congenic BL6 Stxbp1+/−: P < 0.001; Fig. 8T). Hence, Stxbp1+/− mice with different genetic backgrounds show normal sociability and preference for social novelty.
Discussion
In this study, in vitro experiments suggest that human disease-associated STXBP1 mutations lead to decreased protein stability and consequently to reduced cellular protein levels and Stxbp1+/− mice are presented as in vivo models for human STXBP1-encephalopathies. Haploinsufficiency of Stxbp1 in mice recapitulated the STXBP1-phenotype observed in humans, with epileptic spasms, often during inactivity (sleep) and accompanied by slow wave discharges in the EEG/ECoG that were partially suppressed by the antiepileptic drug levetiracetam. These mouse models also showed impaired behavioural flexibility, but not impaired spatial learning, and an increased level of anxiety accompanied with hyperactivity. The latter two phenotypes have not yet been reported in patients. Finally, the present study demonstrates that the interplay between genetic background and Stxbp1 dysfunction modulates the behavioural phenotype, leading to differences in cognitive traits depending on the genomic background.
Reduced protein stability and haploinsufficiency explain STXBP1-encephalopathy
When expressed on a null background, different disease-causing STXBP1 variants supported synaptic transmission to different extents. These results are approximately as predicted. R388X and V84D are predicted to be highly disruptive, given the known loss of syntaxin-1 binding upon C-terminal deletions by R388X (Hata and Südhof, 1995), while V84D adds a charge to a internally oriented residue (Misura et al., 2000). On the other hand, amino acid replacement with the same polarity/charge is predicted to have little effect, such as C180Y. Interestingly, this latter mutant was previously shown to be thermolabile in vitro and when expressed in non-neuronal cells (Martin et al., 2014), but supported normal synaptic transmission in primary neurons in this study (cultured at 37°C). Despite the very different support of synaptic transmission, patients have similar clinical symptoms and disease severity does not correlate with (predicted) disruptive capacity (reviewed by Stamberger et al., 2016). This lack of correlation between the extent of molecular functionality and clinical symptoms suggests that haploinsufficiency may explain STXBP1-encephalopathy. The fact that none of the disease-causing variants inhibited synaptic transmission when over-expressed on the heterozygous background excludes dominant-negative scenarios.
While the disruptive capacity of the different mutations is very different, all disease-causing variants are similar in having a markedly reduced stability in cells. This finding further supports haploinsufficiency as a plausible explanation for STXBP1-encephalopathy. The use of the 2A bi-cistronic ribosome-skipping approach (Ryan et al., 1991) excludes reduced mRNA stability/half-life as an explanation for the markedly reduced cellular levels, because protein levels were quantified using the equimolar GFP expression. The reduced protein levels were detected in synapses, but also in somata, suggesting that reduced protein levels are not the consequence of ‘late’ local breakdown at synapses, by physiological mechanisms such a poly-ubiquitination, which has been reported for Munc18-1 (Martin et al., 2014; Schmitz et al., 2016). Instead, cellular levels of disease-causing variants appear to be reduced directly after biogenesis in the soma, except for one, M443R, which might have a synaptic targeting defect (Fig. 1D and E). This might be caused by intrinsic differences in stability, such as thermo-instability (Saitsu et al., 2008; Martin et al., 2014) and/or by physiological quality control mechanisms. Interestingly, experimental mutations, which we have generated and tested in past studies, were often found to be expressed at normal levels, for instance S241A (Schmitz et al., 2016), L130K and F115E (Meijer et al., 2012) and S306A/S313A (Wierda et al., 2007) and physiological mechanisms of protein breakdown were implicated for mutations that showed lower cellular level, for instance S241D (Schmitz et al., 2016). Hence, disease-causing mutations share an unknown feature that experimental mutations do not show, that leads to their reduced cellular level, that cannot be explained by reduced mRNA stability and that seems to operate directly after biosynthesis, due to intrinsic stability differences and/or by cellular quality control.
Despite reduced stability, two disease-associated STXBP1 variants, C180Y and C522R showed normal synaptic transmission on the null background. This is consistent with previous observations that a 50% reduced protein level does not lead to reduced (single) evoked responses (Toonen et al., 2006). Cellular Munc18-1 levels in neurons expressing disease-causing variants are roughly 50% reduced (Fig. 1D and E). Similarly, the normal (single) evoked responses when Munc18-1 levels are 50% reduced (Toonen et al., 2006), also explains that all disease-causing variants showed normal (single) evoked responses when expressed on a heterozygous background (Fig. 2). Thus synapses can support normal synaptic transmission with a single (wild-type) allele, at least for one to two responses (Toonen et al., 2006). However, during more intense, repetitive stimulation, we previously showed that when Munc18-1 levels are 50% reduced, synapses show deeper rundown of synaptic responses (early fatigue) and that this rundown is more pronounced in GABAergic than in glutamatergic neurons (Toonen et al., 2006). Such a differential effect in excitatory versus inhibitory synapses provides a plausible explanation for the epileptic traits observed in mice and humans with reduced neuronal Munc18-1 levels.
Cortical excitability imbalance explains epilepsy in STXBP1-encephalopathy
The epilepsy phenotype in STXBP1-encephalopathy patients is characterized with early onset infantile spasm, often during sleep, with focal and multifocal EEG activity in most patients and suppression-burst patterns in some patients (Saitsu et al., 2008, 2010; Hamdan et al., 2009; Deprez et al., 2010; Otsuka et al., 2010; Mignot et al., 2011; Milh et al., 2011; Vatta et al., 2012; Weckhuysen et al., 2013; Barcia et al., 2014; Keogh et al., 2015; Yamamoto et al., 2016). The epilepsy-like phenotype in Stxbp1+/− mice mimics these clinical features, with sudden myoclonic jerks (twitches), typically during rest (sleep) accompanied in some cases with spike-wave discharges that were suppressed by acute or prolonged levetiracetam treatment. The fact that correlation between behavioural manifestations and abnormal ECoG activity was not complete is compatible with the multifocal epileptic activity described in patients: some (correlated) events may not be detected with the single electrode used in our mouse studies. Interestingly, excessive neuronal activity was limited to different areas of the neocortex (motor-, somatosensory- and prefrontal-cortex) and was not observed in the hippocampus or lower brain areas. Hence, the neocortex might be the major source for seizure generation, at least in mouse models for STXBP1-encephalopathy.
The conclusion from our previous study that excitatory and inhibitory synapses are differentially affected by Stxbp1 heterozygocity (Toonen et al., 2006, see also above), provides a plausible explanation for the epileptic traits observed in mice and humans with reduced neuronal Munc18-1 levels. The general information obtained from Gad2- Stxbp1cre/+ mice suggests that these mice suffered from more severe and frequent seizures compared to pan-neuronal Stxbp1+/− mice and abnormal ECoG activity. Taken together, these data suggest that imbalanced excitation in the neocortex, due to differentially affected excitatory and inhibitory neurotransmission (Toonen et al., 2006) may explain the epilepsy features when one copy of the Stxbp1 gene is mutated or inactivated.
Changes in presynaptic release machinery cause epilepsy and supports the ‘synapthopathy’ concept
Mutations in several presynaptic genes, STX1B, SNAP25, SYN1 and SYN2, cause epilepsy in mice and humans (Li et al., 1995; Garcia et al., 2004; Gerber et al., 2008; Fassio et al., 2011; Rohena et al., 2013; Corradini et al., 2014; Schubert et al., 2014). The clinical features are rather different among these different forms of epilepsy. However, Snap25S187A/S187A mice, showed similar spike-wave discharge patterns (Watanabe et al., 2015) as shown here for Stxbp1 models. In contrast to Stxbp1+/− mice, absence epilepsy and seizures in Snap25S187A/S187A mice did not occur mainly during the inactive period (sleep) and electrographic epileptiform activity was not detected in the hippocampus (Crunelli and Leresche, 2002; Onat et al., 2013). Furthermore, the antiepileptic drug, levetiracetam that binds the presynaptic protein SV2A and modulates synaptic transmission (Lynch et al., 2004) is effective in two STXBP1-encephalopathy patients (Vatta et al., 2012; Dilena et al., 2016). Levetiracetam also suppresses ECoG abnormalities in Stxbp1+/− mice. Hence, several lines of evidence suggest that changes in presynaptic proteins that regulate synaptic transmission produce epilepsy phenotypes, which can be alleviated by drugs that act presynaptically. This provides further support for the ‘synaptopathy’ model for some forms of epilepsy.
Stxbp1 +/− mice show altered cognition and effect of the flanking genes on avoidance and late reversal learning
Most patients with STXBP1-encephalopathy have severe intellectual disabilities and developmental delay (for a review see Stamberger et al., 2016). In Stxbp1cre/+ and congenic BL6 Stxbp1+/− mice we observed an altered behavioural flexibility, but not altered spatial learning. In the Barnes maze, Stxbp1cre/+ and congenic BL6 Stxbp1+/− mice were unable to suppress a previously learned strategy, suggesting impaired extinction component of behavioural flexibility. However, in the CognitionWall RL task, the congenic BL6 Stxbp1+/− line performed better than control after 2 days of continued reversal learning. This suggests that reversal learning after prolonged training in the home-cage environment was in fact enhanced in the congenic BL6 Stxbp1+/− line. Furthermore, the Shelter task suggested stronger avoidance learning and increased aversion in the same line. Higher aversion, improved avoidance learning and reversal learning after prolonged time in the home-cage in the congenic BL6 Stxbp1+/− line, but not in Stxbp1cre/+ mice, could be the consequence of Sv129-derived flanking genes expressed in the brain (especially Rapgef1 and Dolpp1). Alternatively, it has been shown that stress can cause hyper-anxious state and either impair extinction or enhance the late phase of reversal learning, associated with alteration in the ventromedial prefrontal cortex (Cerqueira et al., 2007; Dias-Ferreira et al., 2009; Graybeal et al., 2011, 2014). Considering the increased anxiety observed in Stxbp1cre/+ and congenic BL6 Stxbp1+/− mice and excessive neural activity in prefrontal cortex detected selectively in congenic BL6 Stxbp1+/− mice, we postulate that the neural mechanism similar to the stress-induced effect underlies altered behavioural flexibility in both Stxbp1+/− lines and increased avoidance learning and facilitated reversal learning in congenic BL6 Stxbp1+/−. Taken together, our data on all Stxbp1+/− lines show that these mouse models recapitulate deficit in domain of cognitive flexibility, but not spatial learning, i.e. a partial penetrance of the cognitive dimension of STXBP1-encephalopathy phenotype in mice.
Stxbp1 +/− mouse models show hyperactivity and anxiety but not deficits in sociability
High prevalence of anxiety and autism spectrum disorders in epilepsy patients suggests that these disorders share underlying pathophysiology (Beyenburg et al., 2005; Geschwind and Levitt, 2007). In addition, mutations in several genes encoding synaptic proteins are implicated in both epilepsy and autism (de Rubeis et al., 2014). A recent review (Stamberger et al., 2016) reports autistic features and other behavioural problems, such as hyperactivity in some patients with STXBP1-encephalopathy. Stxbp1+/− mouse models show increased spontaneous motor activity and increased anxiety, but no impairment of sociability. Although Stxbp1+/− mice showed normal circadian rhythm, they displayed increased motor activity in a novel environment during the dark phase and decreased activity during the light phase. On the other hand, motor activity of Stxbp1+/− mice was normal in classical tests. This lack of effect may be due to the fact that all classical tests were performed during the light phase. Interestingly, hyperactivity has been previously reported in several mouse models of autism (Peñagarikano et al., 2011; Rothwell et al., 2014). Stxbp1+/− mice display two phenotypic characteristics also relevant for autism: impaired behavioural flexibility and tendency towards more repetitive response (discussed above). On the other hand, Stxbp1+/− mice did not show impairments in sociability, preference for social novelty, or enhanced motor learning as core elements of autistic behaviour (Rothwell et al., 2014). These data suggest that Stxbp1+/− mice resemble an autistic phenotype in terms of hyperactivity and behavioural inflexibility but not the social-aspects of autistic behaviour.
Although it has been recognized that anxiety is a common comorbidity of epilepsy in patients (Beyenburg et al., 2005), it has not been reported systematically in STXBP1-encephalopathy. However, Stxbp1+/− mice showed robust anxiety-like phenotypes in several classical paradigms and also in the novel, automated tests in the home-cage. These data are in line with increased anxiety previously observed in Stxbp1+/− mice, measured by enhanced heart rate responsiveness (Hager et al., 2014). Our data are consistent with findings from other genetic mouse model of epilepsy, such as Scn1a+/−, Snap25S187A/S187A, Arx(GCG)7/Y, which report hyperactivity and anxiety as related neuropsychiatric comorbidities (Yu et al., 2006; Kitamura et al., 2009; Kataoka et al., 2011; Han et al., 2012; Watanabe et al., 2015). In general, the reverse 129Sv model mice were hypoactive and showed high anxiety-related traits in accordance with previous studies (Homanics et al., 1999; Crawley, 2008). Similar to human behavioural data, the present study suggests that phenotypic spectrum of Stxbp1+/− mice is broad and includes hyperactivity and anxiety.
Conclusion
Together, the present study indicates that Stxbp1+/− mice represent animal models with construct (same genetic cause of disease) and face (analogy with human symptoms) validity for STXBP1-encephalopathy. In addition, the effectivity of levetiracetam demonstrates predictive validity of Stxbp1+/− mouse models for further therapy design. Protein instability due to disease-causing mutations in Munc18-1 and Stxbp1 haploinsufficiency together explain STXBP1-encephalopathy, probably by imbalanced excitability in the neocortex, as suggested by increased c-Fos expression, and the previously observed enhanced synaptic fatigue, especially in GABAergic synapses (Toonen et al., 2006), and strongly enhanced epilepsy traits in mice with single allele deletion of Stxbp1 expression in GABAergic neurons. Finally, in addition to epileptic activity and cognitive phenotypes, Stxbp1+/− mice showed increased anxiety, and hyperactivity, which has not been systematically reported in STXBP1-encephalopathy patients.
Web resources
Data are available at: http://syli.cz/kovacevic.
Supplementary Material
Acknowledgements
We thank Robbert Zalm for expert help with virus production and cloning and Joost Hoetjes, Joke Wortel, Nina Straat, Christiaan van der Meer, Sita van der Wal and Frank den Oudsten for breeding and genotyping mutant mice. We thank Joke Wortel and Nina Straat for animal perfusion and c-Fos staining. We thank Yayan Veldman for help with the analysis of video recordings. We thank Jurjen Broeke for help making representative supplementary videos. We thank Dušan Kovačević for creating a Python program to calculate the probability of coincidence and Eus Van Someren and Vladyslav Vyazovskiy for their help in understanding and interpretation of EEG data.
Funding
This work was supported by Agentschap NL (NeuroBSIK Mouse Phenomics Consortium, BSIK03053 to M.V.), the Netherlands Organization for Scientific Research ZonMw Pionier/VICI 900-01-001 and by the European Union ERC Advanced Grant 322966 and HEALTH-F2-2009-242167 SynSys (to M.V.), the German Research Council (DFG, SFB1089, SCHO 820/4-1, SCHO 820/6-1 to S.S.), EU (Epitarget, GA602102 to S.S.).
Conflict of interest
The authors declare no conflict of interest. At the time of the experiments described in this manuscript, M.L., E.R. and B.K. were full time employees of Sylics (Synaptologics BV), a private, VU University spin-off company that offers mouse phenotyping services. Sylics provided funding for J.K. but had no influence on the scientific decisions made by her and others involved in her work. M.V. participates in a holding that owns Sylics shares and has received consulting fees from Sylics.
Supplementary material
Supplementary material is available at Brain online.
Glossary
Abbreviations
- ECoG
electrocorticography
- EIEE
early infantile epileptic encephalopathy
- EPSC
excitatory postsynaptic current
References
- Barcia G, Chemaly N, Gobin S, Milh M, Van Bogaert P, Barnerias C, et al. Early epileptic encephalopathies associated with STXBP1 mutations: could we better delineate the phenotype? Eur J Med Genet 2014; 57: 15–20. [DOI] [PubMed] [Google Scholar]
- Beyenburg S, Mitchell AJ, Schmidt D, Elger CE, Reuber M. Anxiety in patients with epilepsy: systematic review and suggestions for clinical management [Review]. Epilepsy Behav 2005; 7: 161–71. [DOI] [PubMed] [Google Scholar]
- Brenner S. The genetics of Caenorhabditis elegans. Genetics 1974; 77: 71–94. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Brose N, O'Connor V, Skehel P. Synaptopathy: dysfunction of synaptic function? Biochem Soc Trans 2010; 38: 443–4. [DOI] [PubMed] [Google Scholar]
- Campbell IM, Yatsenko SA, Hixson P, Reimschisel T, Thomas M, Wilson W, et al. Novel 9q34.11 gene deletions encompassing combinations of four Mendelian disease genes: STXBP1, SPTAN1, ENG, and TOR1A. Genet Med 2012; 14: 868–76. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Cerqueira JJ, Mailliet F, Almeida OF, Jay TM, Sousa N. The prefrontal cortex as a key target of the maladaptive response to stress. J Neurosci 2007; 27: 2781–7. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Chai YJ, Sierecki E, Tomatis VM, Gormal RS, Giles N, Morrow IC, et al. Munc18-1 is a molecular chaperone for α-synuclein, controlling its self-replicating aggregation. J Cell Biol 2016; 214: 705–18. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Corradini I, Donzelli A, Antonucci F, Welzl H, Loos M, Martucci R, et al. Epileptiform activity and cognitive deficits in SNAP-25(+/-) mice are normalized by antiepileptic drugs. Cereb Cortex 2014; 24: 364–76. [DOI] [PubMed] [Google Scholar]
- Crawley JN. Behavioral phenotyping strategies for mutant mice. Neuron 2008; 57: 809–18. [DOI] [PubMed] [Google Scholar]
- Crunelli V, Leresche N. Childhood absence epilepsy: genes, channels, neurons and networks [Review]. Nat Rev Neurosci 2002; 3: 371–82. [DOI] [PubMed] [Google Scholar]
- de Rubeis S, He X, Goldberg AP, Poultney CS, Samocha K, Cicek AE, et al. Synaptic, transcriptional and chromatin genes disrupted in autism. Nature 2014; 515: 209–15. [DOI] [PMC free article] [PubMed] [Google Scholar]
- de Vries KJ, Geijtenbeek A, Brian EC, de Graan PN, Ghijsen WE, Verhage M. Dynamics of munc18-1 phosphorylation/dephosphorylation in rat brain nerve terminals. Eur J Neurosci 2000; 12: 385–90. [DOI] [PubMed] [Google Scholar]
- Deprez L, Weckhuysen S, Holmgren P, Suls A, Van Dyck T, Goossens D, et al. Clinical spectrum of early-onset epileptic encephalopathies associated with STXBP1 mutations. Neurology 2010; 75: 1159–65. [DOI] [PubMed] [Google Scholar]
- Dias-Ferreira E, Sousa JC, Melo I, Morgado P, Mesquita AR, Cerqueira JJ, et al. Chronic stress causes frontostriatal reorganization and affects decision-making. Science 2009; 325: 621–5. [DOI] [PubMed] [Google Scholar]
- Dilena R, Striano P, Traverso M, Viri M, Cristofori G, Tadini L, et al. Dramatic effect of levetiracetam in early-onset epileptic encephalopathy due to STXBP1 mutation. Brain Dev 2016; 38: 128–31. [DOI] [PubMed] [Google Scholar]
- Doheny HC, Ratnaraj N, Whittington MA, Jefferys JG, Patsalos PN. Blood and cerebrospinal fluid pharmacokinetics of the novel anticonvulsant levetiracetam (ucb L059) in the rat. Epilepsy Res 1999; 34: 161–8. [DOI] [PubMed] [Google Scholar]
- Fassio A, Patry L, Congia S, Onofri F, Piton A, Gauthier J, et al. SYN1 loss-of-function mutations in autism and partial epilepsy cause impaired synaptic function. Hum Mol Genet 2011; 20: 2297–307. [DOI] [PubMed] [Google Scholar]
- Garcia CC, Blair HJ, Seager M, Coulthard A, Tennant S, Buddles M, et al. Identification of a mutation in synapsin I, a synaptic vesicle protein, in a family with epilepsy. J Med Genet 2004; 41: 183–6. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gerber SH, Rah JC, Min SW, Liu X, de Wit H, Dulubova I, et al. Conformational switch of syntaxin-1 controls synaptic vesicle fusion. Science 2008; 321: 1507–10. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gerlai R. Gene targeting: technical confounds and potential solutions in behavioral brain research [Review]. Behav Brain Res 2001; 125: 13–21. [DOI] [PubMed] [Google Scholar]
- Geschwind DH, Levitt P. Autism spectrum disorders: developmental disconnection syndromes. Curr Opin Neurobiol 2007; 17: 103–11. [DOI] [PubMed] [Google Scholar]
- Graybeal C, Feyder M, Schulman E, Saksida LM, Bussey TJ, Brigman JL, et al. Paradoxical reversal learning enhancement by stress or prefrontal cortical damage: rescue with BDNF. Nat Neurosci 2011; 14: 1507–9. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Graybeal C, Bachu M, Mozhui K, Saksida LM, Bussey TJ, Sagalyn E, et al. Strains and stressors: an analysis of touchscreen learning in genetically diverse mouse strains. PLoS One 2014; 9: e87745. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Grone BP, Marchese M, Hamling KR, Kumar MG, Krasniak CS, Sicca F, et al. Epilepsy, behavioral abnormalities, and physiological comorbidities in syntaxin-binding protein 1 (STXBP1) mutant Zebrafish. PLoS One 2016; 11: e0151148. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Guacci A, Chetta M, Rizzo F, Marchese G, De Filippo MR, Giurato G, et al. Phenytoin neurotoxicity in a child carrying new STXBP1 and CYP2C9 gene mutations. Seizure 2016; 34: 26–8. [DOI] [PubMed] [Google Scholar]
- Hager T, Maroteaux G, du Pont P, Julsing J, van Vliet R, Stiedl O. Munc18-1 haploinsufficiency results in enhanced anxiety-like behavior as determined by heart rate responses in mice. Behav Brain Res 2014; 260: 44–52. [DOI] [PubMed] [Google Scholar]
- Hamdan FF, Piton A, Gauthier J, Lortie A, Dubeau F, Dobrzeniecka S, et al. De novo STXBP1 mutations in mental retardation and nonsyndromic epilepsy. Ann Neurol 2009; 65: 748–53. [DOI] [PubMed] [Google Scholar]
- Hamdan FF, Gauthier J, Dobrzeniecka S, Lortie A, Mottron L, Vanasse M, et al. Intellectual disability without epilepsy associated with STXBP1 disruption. Eur J Hum Genet 2011; 19: 607–9. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Han S, Tai C, Westenbroek RE, Yu FH, Cheah CS, Potter GB, et al. Autistic-like behaviour in Scn1a+/- mice and rescue by enhanced GABA-mediated neurotransmission. Nature 2012; 489: 385–90. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Harrison FE, Hosseini AH, McDonald MP. Endogenous anxiety and stress responses in water maze and Barnes maze spatial memory tasks. Behav Brain Res 2009; 198: 247–51. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Harrison SD, Broadie K, van de Goor J, Rubin GM. Mutations in the Drosophila Rop gene suggest a function in general secretion and synaptic transmission. Neuron 1994; 13: 555–66. [DOI] [PubMed] [Google Scholar]
- Hata Y, Südhof TC. A novel ubiquitous form of Munc-18 interacts with multiple syntaxins. Use of the yeast two-hybrid system to study interactions between proteins involved in membrane traffic. J Biol Chem 1995; 270: 13022–8. [DOI] [PubMed] [Google Scholar]
- Heeroma JH, Roelandse M, Wierda K, van Aerde KI, Toonen RF, Hensbroek RA, et al. Trophic support delays but does not prevent cell-intrinsic degeneration of neurons deficient for munc18-1. Eur J Neurosci 2004; 20: 623–34. [DOI] [PubMed] [Google Scholar]
- Herrera DG, Robertson HA. Activation of c-fos in the brain [Review]. Prog Neurobiol 1996; 50: 83–107. [DOI] [PubMed] [Google Scholar]
- Holmes A, Wrenn CC, Harris AP, Thayer KE, Crawley JN. Behavioral profiles of inbred strains on novel olfactory, spatial and emotional tests for reference memory in mice [Review]. Genes Brain Behav 2002; 1: 55–69. [DOI] [PubMed] [Google Scholar]
- Homanics GE, Quinlan JJ, Firestone LL. Pharmacologic and behavioral responses of inbred C57BL/6J and strain 129/SvJ mouse lines. Pharmacol Biochem Behav 1999; 63: 21–6. [DOI] [PubMed] [Google Scholar]
- Kataoka M, Yamamori S, Suzuki E, Watanabe S, Sato T, Miyaoka H, et al. A single amino acid mutation in SNAP-25 induces anxiety-related behavior in mouse. PLoS One 2011; 6: e25158. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kitamura K, Itou Y, Yanazawa M, Ohsawa M, Suzuki-Migishima R, Umeki Y, et al. Three human ARX mutations cause the lissencephaly-like and mental retardation with epilepsy-like pleiotropic phenotypes in mice. Hum Mol Genet 2009; 18: 3708–24. [DOI] [PubMed] [Google Scholar]
- Keogh MJ, Daud D, Pyle A, Duff J, Griffin H, He L, et al. A novel de novo STXBP1 mutation is associated with mitochondrial complex I deficiency and late-onset juvenile-onset parkinsonism. Neurogenetics 2015; 16: 65–7. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lek M, Karczewski KJ, Minikel EV, Samocha KE, Banks E, Fennell T, et al. Analysis of protein-coding genetic variation in 60,706 humans. Nature 2016; 536: 285–91. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Li L, Chin LS, Shupliakov O, Brodin L, Sihra TS, Hvalby O, et al. Impairment of synaptic vesicle clustering and of synaptic transmission, and increased seizure propensity, in synapsin I-deficient mice. Proc Natl Acad Sci USA 1995; 92: 9235–9. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Loos M, Koopmans B, Aarts E, Maroteaux G, van der Sluis S, Neuro-BSIK Mouse Phenomics Consortium, et al. Sheltering behavior and locomotor activity in 11 genetically diverse common inbred mouse strains using home-cage monitoring. PLoS One 2014; 9: e108563. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Löscher W, Hönack D. Profile of ucb L059, a novel anticonvulsant drug, in models of partial and generalized epilepsy in mice and rats. Eur J Pharmacol 1993; 232: 147–58. [DOI] [PubMed] [Google Scholar]
- Lynch BA, Lambeng N, Nocka K, Kensel-Hammes P, Bajjalieh SM, Matagne A, et al. The synaptic vesicle protein SV2A is the binding site for the antiepileptic drug levetiracetam. Proc Natl Acad Sci USA 2004; 101: 9861–6. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Maroteaux G, Loos M, van der Sluis S, Koopmans B, Aarts E, van Gassen K, et al. High-throughput phenotyping of avoidance learning in mice discriminates different genotypes and identifies a novel gene. Genes Brain Behav 2012; 11: 772–84. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Martin S, Papadopulos A, Tomatis VM, Sierecki E, Malintan NT, Gormal RS, et al. Increased polyubiquitination and proteasomal degradation of a Munc18-1 disease-linked mutant causes temperature-sensitive defect in exocytosis. Cell Rep 2014; 9: 206–18. [DOI] [PubMed] [Google Scholar]
- Meijer M, Burkhardt P, de Wit H, Toonen RF, Fasshauer D, Verhage M. Munc18-1 mutations that strongly impair SNARE-complex binding support normal synaptic transmission. EMBO J 2012; 31: 2156–68. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Mignot C, Moutard ML, Trouillard O, Gourfinkel-An I, Jacquette A, Arveiler B, et al. STXBP1-related encephalopathy presenting as infantile spasms and generalized tremor in three patients. Epilepsia 2011; 52: 1820–7. [DOI] [PubMed] [Google Scholar]
- Milh M, Villeneuve N, Chouchane M, Kaminska A, Laroche C, Barthez MA, et al. Epileptic and nonepileptic features in patients with early onset epileptic encephalopathy and STXBP1 mutations. Epilepsia 2011; 52: 1828–34. [DOI] [PubMed] [Google Scholar]
- Misura KM, Scheller RH, Weis WI. Three-dimensional structure of the neuronal-Sec1-syntaxin 1a complex. Nature 2000; 404: 355–62. [DOI] [PubMed] [Google Scholar]
- Nguyen HT, Bryois J, Kim A, Dobbyn A, Huckins LM, Munoz-Manchado A, et al. Integrated Bayesian analysis of rare exonic variants to identify risk genes for schizophrenia and neurodevelopmental disorders. Genome Med 2017; 9: 114 doi: 10.1101/135293. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Onat FY, van Luijtelaar G, Nehlig A, Snead OC 3rd. The involvement of limbic structures in typical and atypical absence epilepsy. Epilepsy Res 2013; 103: 111–23. [DOI] [PubMed] [Google Scholar]
- Otsuka M, Oguni H, Liang JS, Ikeda H, Imai K, Hirasawa K, et al. STXBP1 mutations cause not only Ohtahara syndrome but also West syndrome—result of Japanese cohort study. Epilepsia 2010; 51: 2449–52. [DOI] [PubMed] [Google Scholar]
- Paxinos G, Franklin K. The mouse brain in stereotaxic coordinates. San Diego: Academic Press; 2012. [Google Scholar]
- Peñagarikano O, Abrahams BS, Herman EI, Winden KD, Gdalyahu A, Dong H, et al. Absence of CNTNAP2 leads to epilepsy, neuronal migration abnormalities, and core autism-related deficits. Cell 2011; 147: 235–46. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Pitsch J, Becker AJ, Schoch S, Müller JA, de Curtis M, Gnatkovsky V. Circadian clustering of spontaneous epileptic seizures emerges after pilocarpine-induced status epilepticus. Epilepsia 2017; 58: 1159–71. [DOI] [PubMed] [Google Scholar]
- Remmelink E, Smit AB, Verhage M, Loos M. Measuring discrimination- and reversal learning in mouse models within 4 days and without prior food deprivation. Learn Mem 2016; 23: 660–7. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Rohena L, Neidich J, Truitt Cho M, Gonzalez KD, Tang S, Devinsky O, Chung WK. Mutation in SNAP25 as a novel genetic cause of epilepsy and intellectual disability. Rare Dis 2013; 1: e26314. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Rothwell PE, Fuccillo MV, Maxeiner S, Hayton SJ, Gokce O, Lim BK, et al. Autism-associated neuroligin-3 mutations commonly impair striatal circuits to boost repetitive behaviors. Cell 2014; 158: 198–212. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ryan MD, King AM, Thomas GP. Cleavage of foot-and-mouth disease virus polyprotein is mediated by residues located within a 19 amino acid sequence. J Gen Virol 1991; 72 (Pt 11): 2727–32. [DOI] [PubMed] [Google Scholar]
- Saitsu H, Kato M, Mizuguchi T, Hamada K, Osaka H, Tohyama J, et al. De novo mutations in the gene encoding STXBP1 (MUNC18-1) cause early infantile epileptic encephalopathy. Nat Genet 2008; 40: 782–8. [DOI] [PubMed] [Google Scholar]
- Saitsu H, Kato M, Okada I, Orii KE, Higuchi T, Hoshino H, et al. STXBP1 mutations in early infantile epileptic encephalopathy with suppression-burst pattern. Epilepsia 2010; 51: 2397–405. [DOI] [PubMed] [Google Scholar]
- Santos TC, Wierda K, Broeke JH, Toonen RF, Verhage M. Early golgi abnormalities and neurodegeneration upon loss of presynaptic proteins Munc18-1, Syntaxin-1, or SNAP-25. J Neurosci 2017; 37: 4525–39. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Schmitz SK, Hjorth JJ, Joemai RM, Wijntjes R, Eijgenraam S, de Bruijn P, et al. Automated analysis of neuronal morphology, synapse number and synaptic recruitment. J Neurosci Methods 2011; 195: 185–93. [DOI] [PubMed] [Google Scholar]
- Schmitz SK, King C, Kortleven C, Huson V, Kroon T, Kevenaar JT, et al. Presynaptic inhibition upon CB1 or mGlu2/3 receptor activation requires ERK/MAPK phosphorylation of Munc18-1. EMBO J 2016; 35: 1236–50. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Schubert J, Siekierska A, Langlois M, May P, Huneau C, Becker F, et al. Mutations in STX1B, encoding a presynaptic protein, cause fever-associated epilepsy syndromes. Nat Genet 2014; 46: 1327–32. [DOI] [PubMed] [Google Scholar]
- Stamberger H, Nikanorova M, Willemsen MH, Accorsi P, Angriman M, Baier H, et al. STXBP1 encephalopathy: a neurodevelopmental disorder including epilepsy. Neurology 2016; 86: 954–62. [DOI] [PubMed] [Google Scholar]
- Toonen RF, Wierda K, Sons MS, de Wit H, Cornelisse LN, Brussaard A, et al. Munc18-1 expression levels control synapse recovery by regulating readily releasable pool size. Proc Natl Acad Sci USA 2006; 103: 18332–7. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Toonen RF, de Vries KJ, Zalm R, Südhof TC, Verhage M. Munc18-1 stabilizes syntaxin 1, but is not essential for syntaxin 1 targeting and SNARE complex formation. J Neurochem 2005; 93: 1393–400. [DOI] [PubMed] [Google Scholar]
- Toonen RF, Verhage M. Munc18-1 in secretion: lonely Munc joins SNARE team and takes control. Trends Neurosci 2007; 30: 564–72. [DOI] [PubMed] [Google Scholar]
- Vanden Berghe T, Hulpiau P, Martens L, Vandenbroucke RE, Van Wonterghem E, Perry SW, et al. Passenger mutations confound interpretation of all genetically modified congenic mice. Immunity 2015; 43: 200–9. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Vatta M, Tennison MB, Aylsworth AS, Turcott CM, Guerra MP, Eng CM, et al. A novel STXBP1 mutation causes focal seizures with neonatal onset. J Child Neurol 2012; 27: 811–4. [DOI] [PubMed] [Google Scholar]
- Verhage M, Maia AS, Plomp JJ, Brussaard AB, Heeroma JH, Vermeer H, et al. Synaptic assembly of the brain in the absence of neurotransmitter secretion. Science 2000; 287: 864–9. [DOI] [PubMed] [Google Scholar]
- Verhoeven KJF, Simonsen KL, McIntyre LM. Implementing false discovery rate control: increasing your power. OIKOS 2005; 108: 643–47. [Google Scholar]
- Voets T, Toonen RF, Brian EC, de Wit H, Moser T, Rettig J, et al. Munc18-1 promotes large dense-core vesicle docking. Neuron 2001; 31: 581–91. [DOI] [PubMed] [Google Scholar]
- Wagnon JL, Korn MJ, Parent R, Tarpey TA, Jones JM, Hammer MF, et al. Convulsive seizures and SUDEP in a mouse model of SCN8A epileptic encephalopathy. Hum Mol Genet 2015; 24: 506–15. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Watanabe S, Yamamori S, Otsuka S, Saito M, Suzuki E, Kataoka M, et al. Epileptogenesis and epileptic maturation in phosphorylation site-specific SNAP-25 mutant mice. Epilepsy Res 2015; 115: 30–44. [DOI] [PubMed] [Google Scholar]
- Weckhuysen S, Holmgren P, Hendrickx R, Jansen AC, Hasaerts D, Dielman C, et al. Reduction of seizure frequency after epilepsy surgery in a patient with STXBP1 encephalopathy and clinical description of six novel mutation carriers. Epilepsia 2013; 54: e74–80. [DOI] [PubMed] [Google Scholar]
- Wierda KD, Toonen RF, de Wit H, Brussaard AB, Verhage M. Interdependence of PKC-dependent and PKC-independent pathways for Presynaptic plasticity. Neuron 2007; 54: 275–90. [DOI] [PubMed] [Google Scholar]
- Wolfer DP, Crusio WE, Lipp HP. Knockout mice: simple solutions to the problems of genetic background and flanking genes [Review]. Trends Neurosci 2002; 25: 336–40. [DOI] [PubMed] [Google Scholar]
- Yamamoto T, Shimojima K, Yano T, Ueda Y, Takayama R, Ikeda H, et al. Loss-of-function mutations of STXBP1 in patients with epileptic encephalopathy. Brain Dev 2016; 38: 280–4. [DOI] [PubMed] [Google Scholar]
- Yamashita S, Chiyonobu T, Yoshida M, Maeda H, Zuiki M, Kidowaki S, et al. Mislocalization of syntaxin-1 and impaired neurite growth observed in a human iPSC model for STXBP1-related epileptic encephalopathy. Epilepsia 2016; 57: e81–6. [DOI] [PubMed] [Google Scholar]
- Yu FH, Mantegazza M, Westenbroek RE, Robbins CA, Kalume F, Burton KA, et al. Reduced sodium current in GABAergic interneurons in a mouse model of severe myoclonic epilepsy in infancy. Nat Neurosci 2006; 9: 1142–9. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.