

Expression of the neuronal kinase DCLK3 is reduced in Huntington’s disease. Galvan et al. report that DCLK3 silencing in the mouse striatum exacerbates the toxicity of mutant huntingtin, whereas DCLK3 overexpression is neuroprotective, and show that DCLK3 regulates the expression of many genes involved in transcription regulation and chromatin remodelling.
Keywords: Huntington, kinase, neurons, transcription, chromatin
Abstract
The neurobiological functions of a number of kinases expressed in the brain are unknown. Here, we report new findings on DCLK3 (doublecortin like kinase 3), which is preferentially expressed in neurons in the striatum and dentate gyrus. Its function has never been investigated. DCLK3 expression is markedly reduced in Huntington’s disease. Recent data obtained in studies related to cancer suggest DCLK3 could have an anti-apoptotic effect. Thus, we hypothesized that early loss of DCLK3 in Huntington’s disease may render striatal neurons more susceptible to mutant huntingtin (mHtt). We discovered that DCLK3 silencing in the striatum of mice exacerbated the toxicity of an N-terminal fragment of mHtt. Conversely, overexpression of DCLK3 reduced neurodegeneration produced by mHtt. DCLK3 also produced beneficial effects on motor symptoms in a knock-in mouse model of Huntington’s disease. Using different mutants of DCLK3, we found that the kinase activity of the protein plays a key role in neuroprotection. To investigate the potential mechanisms underlying DCLK3 effects, we studied the transcriptional changes produced by the kinase domain in human striatal neurons in culture. Results show that DCLK3 regulates in a kinase-dependent manner the expression of many genes involved in transcription regulation and nucleosome/chromatin remodelling. Consistent with this, histological evaluation showed DCLK3 is present in the nucleus of striatal neurons and, protein-protein interaction experiments suggested that the kinase domain interacts with zinc finger proteins, including the transcriptional activator adaptor TADA3, a core component of the Spt-ada-Gcn5 acetyltransferase (SAGA) complex which links histone acetylation to the transcription machinery. Our novel findings suggest that the presence of DCLK3 in striatal neurons may play a key role in transcription regulation and chromatin remodelling in these brain cells, and show that reduced expression of the kinase in Huntington’s disease could render the striatum highly vulnerable to neurodegeneration.
Introduction
Huntington’s disease is an inherited neurodegenerative disorder caused by a mutation of the huntingtin (HTT) gene. The main hallmark of Huntington’s disease is the early loss of medium-sized spiny neurons from the striatum. This neuronal loss leads to striatal atrophy, followed by massive progressive neurodegeneration throughout the brain. Huntington’s disease is caused by an abnormal CAG repeat expansion in the HTT gene (The Huntington’s Disease Collaborative Research Group, 1993). Mutant huntingtin (mHtt) is neurotoxic to neurons, particularly medium-sized spiny neurons, through both gain- and loss-of-function mechanisms (Zuccato and Cattaneo, 2014). Mutant Htt causes transcriptional anomalies affecting the expression of a large range of genes (Seredenina and Luthi-Carter, 2012). It also causes changes in the cytoskeleton-mediated transport machinery (Hinckelmann et al., 2013), and defects in signalling pathways involved in cell survival (Roze et al., 2008). However, none of these mechanisms can account for the preferential damage to the striatum observed early in the disease.
It has been suggested that factors selectively expressed in the striatum may render GABAergic projection neurons (medium-sized spiny neurons) particularly susceptible to degeneration (Brouillet et al., 2005; Han et al., 2010; Francelle et al., 2014). We previously performed a transcriptome SAGE (serial analysis of gene expression) analysis of different regions of the mouse brain, which identified about 120 genes preferentially expressed in the striatum (de Chaldee et al., 2003; Brochier et al., 2008). Most of the products of these genes were confirmed to be ‘striatal-enriched’ by checking against a freely available transcriptomic database (e.g. Allen Brain Atlas). In our initial screen, some of these newly identified striatal gene products displayed significantly lower than normal levels of expression in the brains of Huntington’s disease mouse models (Brochier et al., 2008) and patients (Kuhn et al., 2007). More recent studies confirmed these findings, showing that a subset of approximately 70 striatal gene products (e.g. ADORA2A, DCLK3, GPR88, RGS2, RGS9, SCN4B) are downregulated in knock-in Huntington’s disease mice (Langfelder et al., 2016). However, the functions of these striatal gene products are frequently unknown, and their potential roles in the vulnerability of the striatum in Huntington’s disease remain a matter of speculation.
For a few of these striatal genes, the effects of their expression levels and/or inactivation have been assessed experimentally against the toxicity of mHtt in mice. Results suggest that in some cases, the reduced expression of the striatal marker found in the Huntington’s disease striatum could lead to a loss of function rendering striatal cells more vulnerable [e.g. the long non-coding RNA Abhd11os or Crym (Francelle et al., 2015a, c)] while in others, it might have no major impact on neurodegeneration (e.g. capucin) (Galvan et al., 2012). In some instances, although other striatal markers have reduced expression in Huntington’s disease, they are considered as risk factors, since the knock down of their genes or blockade of their function produces neuroprotection against HTT toxicity. For example, dopamine and its subtype 2 membrane receptors (D2R), PDE10a, and the small GTPase Rhes have been shown to be preferentially expressed in the striatum, have early reduction of their expression in Huntington’s disease patients and Huntington’s disease models and have been shown to render striatal neurons highly vulnerable to mHtt (Charvin et al., 2005, 2008; Benchoua et al., 2008; Subramaniam et al., 2009; Giampa et al., 2010).
In the present study, we focused on a kinase of unknown function, DCLK3. DCLK3 is the third member of the doublecortin-like kinase (DCLK) family (Ohmae et al., 2006b). The DCLK1/2 proteins have a conserved C-terminal ‘serine/threonine kinase’ domain similar to that of Ca2+/calmodulin kinase IV (CaMKIV) and an N-terminal putative doublecortin (DCX) domain mediating their anchorage to microtubules. DCLK1 and DCLK2 are involved in neural development and the maturation of neuronal architecture, including the organization of dendritic spines in particular (Kerjan et al., 2009; Shin et al., 2013) and play an important role in axonal regeneration mainly through their DCX domain (Nawabi et al., 2015). DCLK3 functions are likely not similar to those of DCLK1/2. The putative DCX domain in the N-terminal part of DCLK1/2 is not present in DCLK3. In addition, DCLK1 and DCLK2 have only 56–54% identities in the amino acid primary sequence of DCLK3, with differences in and around the kinase domain in the C-terminal part of the proteins. In line with this hypothesis, DCLK3 displays a more neuron-specific pattern of expression than DCLK1/2, which are also expressed in astrocytes (http://web.stanford.edu/group/barres_lab/brain_rnaseq.html) (Zhang et al., 2014). DCLK3 is mainly expressed in the adult and not in the developing forebrain. Intriguingly, a recent study pointed to DCLK3 as an oncogenic factor in colorectal cancer. High expression of DCLK3 (resulting from at-distance effects of a single polymorphism in the promoter of the MLH1 gene) in cancer cells was shown to be associated with increased malignancy. This suggested that DCLK3 has biological functions that may modulate cell survival (Liu et al., 2017).
We hypothesized that, if DCLK3 displays biological activity favouring survival in neurons, then the loss of this protein from neurons may contribute to striatal degeneration in Huntington’s disease. We investigated whether the modulation of DCLK3 levels in mouse models of Huntington’s disease affected mHtt toxicity in vivo. The function of DCLK3 is unknown. Thus we performed different types of experiments to identify the signalling pathways or mechanisms through which the kinase activity of DCLK3 could impact striatal neurons.
Materials and methods
Animal experiments
Mice
Mice were housed (five/cage in enriched environment) in a controlled-temperature room maintained on a 12-h light/dark cycle. Food and water were available ad libitum. All animal studies were conducted in accordance with French regulations (EU Directive 86/609 – French Act Rural Code R 214-87 to 131). The animal facility was approved by veterinary inspectors (authorization no. A 92-032-02) and complies with the Standards for Humane Care and Use of Laboratory Animals of the Office of Laboratory Animal Welfare (OLAW – no. #A5826-01). All procedures were approved by the ethics committee and the Research Ministry [no. 2015060417243726vl (APAFIS#770)]. Adult male C57BL/6J mice (each weighing 25 g; Charles River) were used for lentiviral infections.
We also studied knock-in mice expressing a chimeric mouse/human exon 1 containing 140 CAG repeats inserted into the murine Htt gene (KI140CAG) and their control littermates. The KI140CAG mouse colony was maintained by crossing heterozygous KI140CAG males and females (Menalled et al., 2003). Mice were N3 (B6) on a 129 Sv × C57BL/6J background. The resulting homozygous mice were used for the study. Genotyping was performed by PCR on tail DNA obtained from 10–15-day-old KI140CAG mice.
Lentiviral and AAV vector construction, production and administration
DNA sequences encoding green fluorescent protein (GFP) and C-terminally haemagglutinin (HA)-tagged mouse short and long forms of DCLK3 were inserted into the SIN-W-PGK lentiviral vector (de Almeida et al., 2002) to generate LV-GFP, LV-S-rDCLK3-HA and LV-L-rDCLK3-HA, respectively. The following PCR primers were used to generate S-rDCLK3-HA: 5′ CACCATGGGCAAAGAGCCGCTGAC 3′ and 5′ CTAGGAGGCGTAGTCAGGCACGTCGTATGGGTAGGCACTGTTGGGGGACTCCTC 3′.
We also cloned the C-terminal domain of DCLK3 (from amino acid 330 to the C-terminal end) for expression of the kinase domain alone, in the vector LV-Kin-rDCLK3-HA. The LV-L-rDCLK3-HA and LV-Kin-rDCLK3 vectors were cloned and produced by GeneArt based on the RNA sequence in the GENE database. The HA-tag sequence was inserted just before the stop codon.
We inserted short hairpin RNAs (shRNAs) targeting mouse DCLK3 (target sequence: 5′ GAGAAGTGTAAGAGAGAAA 3’) and luciferase (target sequence: 5′ CGTACGCGGAATACTTCGA 3′) into a bicistronic lentiviral vector (Drouet et al., 2009), such that the infected cells expressed the reporter protein GFP. The resulting constructs were designated LV-DCLK3-shRNA and LV-Luc-shRNA, respectively. The lentiviral vectors expressing a wild-type Htt fragment (LV-Htt171-18Q), a mutant Htt fragment (LV-Htt171-82Q) or beta-galactosidase (LV-βgal) have been described elsewhere (Diguet et al., 2009; Faideau et al., 2010). Lentiviral particles were produced as previously described (Hottinger et al., 2000). The particle content of the viral batches was determined by ELISA for the p24 antigen (Gentaur).
For adeno-associated virus (AAV) production, the mouse L-DCLK3 (rDCLK3) sequence was inserted into a single-stranded, rAAV2-based shuttle vector under the control of the mouse PGK promoter, by LR recombination, with Gateway® technology (Invitrogen, Life Technologies). All constructs were packaged into AAVrh10 capsids by the MIRCen viral production platform, as previously described (Berger et al., 2015). Viral particles were produced by the transient co-transfection of HEK-293 cells with an adenovirus helper plasmid (pXX6-80), an AAV packaging plasmid carrying the rep2 and cap10 genes, and the AAV2 transfer vector containing the expression cassettes described above. Virions were purified 72 h after transfection and concentrated from the cell lysate and supernatant by ultracentrifugation on an iodixaniol density gradient, followed by dialysis against PBSMK [0.5 mM MgCl2 and 1.25 mM KCl in phosphate-buffered saline (PBS)]. The concentration of the vector stocks was estimated by quantitative PCR as described by Aurnhammer et al. (2012) and expressed as the number of viral genomes per ml of concentrated stocks (vg/ml).
LV-Htt171-82Q was used at a concentration of 150 ng/µl of p24; LV-S-rDCLK3-HA, LV-L-rDCLK3-HA, LV-Kin-rDCLK3-HA, LV-DCLK3-shRNA, LV-Luc-shRNA and LV-βgal were used at a concentration of 100 ng/µl of p24, and LV-GFP at a concentration of 50 ng/µl of p24. AAV2/rh10-GFP and AAV2/rh10-rDCLK3 were used at a dose of 2 × 109 vg per site.
For intracerebral infections, animal were anaesthetized (100 mg/kg ketamine and 10 mg/kg xylazine). Local analgesia included subcutaneous lidocaine (5 mg/kg). A volume of 2 µl of lentiviral or AAV suspension was injected into the mouse striatum, as described (Faideau et al., 2010), at the following stereotaxic coordinates: 1.0 mm anterior and 2.0 mm lateral to bregma, at a depth of 2.7 mm from the dura, with the tooth bar set at 0.0 mm. The mice were then left for 1–2 h in a heated (30°C) ventilated box, until they had woken up and recovered fully from anaesthesia. Post-surgery analgesia included acetaminophen (Doliprane®) in drinking water for 48 h (1.6 mg/ml).
Behavioural tests on KI140CAG mice
KI140CAG mice receiving injections of AAV encoding GFP or L-rDCLK3 at 6 months of age underwent behavioural tests at 12 months of age: the rotarod test, the CatWalk and grip strength tests. We tested Huntington’s disease mice during the light phase of the light/dark cycle. All the mice were killed at the age of 13 months.
Rotarod
The rotarod test was used to evaluate the motor coordination and strength of the mice. Eight-month-old mice were trained on a rotarod (MED-Associates Rota-Rod) at 4 rpm for 2 min. They were then tested in three consecutive trials, each lasting 5 min, in which the speed of the rod was increased from 4 to 40 rpm, with 45 min between trials. This sequence was repeated on three consecutive days and values were averaged across all trials.
Grip strength
A validated grip strength test meter (BIO-GS3 BIOSEB; EB Instruments) was used to measure forelimb grip strength. The mice were handled by their tails and placed over the grid until they grasped the grid. The tail was then pulled horizontally until the mouse released its grip fully. Three separate readings were recorded and the results, in grams, were averaged for analysis.
CatWalk
The CatWalk XT gait analysis system (Noldus IT) used here consisted of an enclosed walkway constructed from plate glass and a camera for speed recording. Gait performance was assessed with recording and analysis software. On the first 2 days, the mice were allowed to get used to the apparatus for 10 min. This was considered to have been achieved if the mouse crossed the walkway. The following day, free runs across the walkway were recorded. A correct run was defined as one complete (60 cm) uninterrupted crossing of the walkway and a minimum of nine step sequences. We selected five correct runs per animal for analysis. Body speed variation (%) was calculated by dividing the absolute difference between body speed and the mean speed of a run by the mean speed. This parameter reflects the smoothness of mouse displacement whilst crossing the walkway. The number of twitches captured in the acquisition window during the run was determined. These twitches corresponded to brief, sudden jerks of one of the paws, resulting in a missing recording or a smaller paw print, and an abnormal paw base.
Histological studies and image analysis
Monkey tissue preparation
Serial coronal sections (40-µm thick) obtained from control male adult Macaca fascicularis monkeys were fixed in 4% paraformaldehyde (PFA) and used for histological studies. For DCLK3 immunohistochemistry, paraffin-embedded brain sections were used.
Mouse tissue preparation
Mice were deeply anaesthetized by the intraperitoneal injection of sodium pentobarbital solution (50 µg/g body weight). They were then transcardially perfused with 100 ml 4% PFA in phosphate buffer at a flow rate of 8 ml/min. The brains of the animals were removed, post-fixed overnight in the same solution, then cryoprotected by immersion in 30% sucrose for 36 h. Free-floating 30-µm serial coronal sections from throughout the striatum were collected with a freezing sliding microtome (SM2400; Leica Microsystems). Brain slices were placed in a storage solution (30% glycerol, 30% ethylene glycol in 0.1 M phosphate buffer) and stored at −20°C before use.
Immunohistochemistry
Monkeys
Brain slices were subjected to pretreatment with 0.3% hydrogen peroxide for 1 h, washed three times in PBS, blocked in PBS containing 4.5% normal goat serum (NGS) for 1 h, then incubated overnight at 4°C in PBS containing 3% NGS, 0.2% Triton™ X-100 and anti-DCLK3 antibody (1:1000, rabbit, 21890002, Novus). The brain slices were then rinsed and incubated with biotinylated secondary antibodies (Vector Laboratories) for 1 h at room temperature. Finally, slices were incubated with the Vectastain Elite ABC Kit (Vector Laboratories) and antibody binding was detected with DAB (Vector Laboratories).
Mice
Free-floating sections were subjected to pretreatment with 0.3% hydrogen peroxide for 1 h, washed three times in PBS, blocked in PBS containing 4.5% NGS for 1 h, then incubated overnight at 4°C in PBS containing 3% NGS, 0.2% Triton™ X-100 and one of the following antibodies: anti-DARPP32 (1:1000, rabbit, Santa Cruz Biotechnology), anti-NeuN (1:2000, mouse, Millipore), anti-ubiquitin (1:1000, rabbit, Wako Chemicals), or anti-HA (1:500, mouse, clone 11 Covance). Sections were rinsed three times in PBS and then incubated with the appropriate anti-IgG biotinylated antibody (Vector Laboratories) at a dilution of 1:5000, for 1 h. Staining was visualized by adding avidin-biotinylated peroxidase and incubating with DAB or VIP substrate (Vector Laboratories) for 1 min. For NeuN immunostaining, we used the MOM immunodetection kit (Vector Laboratories). Stained sections were mounted on microscopy slides.
Cytochrome c oxidase histochemistry
Brain slices were incubated in 0.1 M phosphate buffer, 4% sucrose, 0.05% DAB (Vector Laboratories), and 0.03% cytochrome c (Sigma-Aldrich) for 6 h at room temperature. Slices were washed three times in PBS, dehydrated and mounted on microscopy slides.
Immunofluorescence
Monkeys
PFA-fixed coronal sections of the striatum were treated with 0.5% citrate buffer in a 75°C water bath for 20 min to maximize antibody penetration into the tissue. Non-specific Fc-binding sites were blocked by incubation with 4.5% NGS for 1 h and sections were then incubated overnight at 4°C with anti-NeuN antibody (1:1000, mouse, Millipore). The sections were rinsed three times in PBS and incubated with anti-DCLK3 antibody (1:1000, rabbit, 21890002, Novus). Both primary antibodies were diluted in PBS supplemented with 3% NGS and 0.2% Triton™ X-100. We used the Tyramide Signal Amplification Kit (Life Technologies), according to the manufacturer’s protocol, for high-density DCLK3 labelling. For NeuN labelling, sections were rinsed three times in PBS and incubated for 1 h with Alexa Fluor® 488-conjugated goat anti-mouse secondary antibody (1:1000, Life Technologies). They were then rinsed again and incubated in 4′,6-diamidino-2-phenylindole (DAPI) diluted 1:2000 in PBS for 10 mins, before a final rinse in PBS and mounting.
Mice
For the detection of DCLK3 by immunofluorescence after infection with LV-rDCLK3-HA in mouse striatum, free-floating sections were prepared as described above. These sections were blocked by incubation with 4.5% NGS in PBS for 1 h, then incubated overnight at 4°C in PBS supplemented with 3% NGS, 0.2% Triton™ X-100 and one of the following antibodies: anti-HA (1:500, mouse, clone 11, Covance) or anti-DCLK3 (1:1000, rabbit, 21890002, Novus). Sections were rinsed three times in PBS and incubated with Alexa Fluor® 488-labelled anti-mouse IgG or Alexa Fluor® 594-labelled anti-rabbit IgG (Invitrogen) at room temperature for 1 h. The sections were rinsed in PBS and the nuclei were counterstained with DAPI (dilution 1:10 000, Wako) for 3 mins.
Confocal microscopy
Immunofluorescent labelling was analysed with a confocal laser-scanning microscope (SP8 Leica).
Image analysis of lesion area
The area covered by striatal lesions resulting from LV-Htt171-82Q infection was delineated manually by identifying the border of the lesion on each coronal brain section. The corresponding area was calculated with MCID image analysis software (InterFocus Imaging). The volume of the striatal lesion was determined by the Cavalieri method and the number of ubiquitin-positive inclusions was determined as previously described (Diguet et al., 2009; Francelle et al., 2015b, d) with an intersection distance of 210 µm (i.e. we used one in every seven sections). A 10× objective was used on an Axioplan 2 imaging microscope (Carl Zeiss). Objects with an apparent cross-sectional area exceeding 5 µm2 were reliably detected with this method.
Real-time quantitative RT-PCR and northern blot analysis
Adult mice were deeply anaesthetized by the intraperitoneal injection of sodium pentobarbital solution (50 µg/g of body weight) and decapitated. The brains were immediately removed and positioned in a coronal brain matrix (Ted Pella). For DCLK3 mRNA determinations in knockdown experiments, we examined 1-mm brain sections under a fluorescence microscope (DM6000 M; Leica Microsystems) and sampled the striatal area expressing the GFP reporter protein with a 1.5-mm diameter hole-punch (Ted Pella). We quantified DCLK3 levels by dissecting out the striatum of the mice on 1-mm thick sections, with a scalpel. Total RNA was extracted with TRIzol® (Invitrogen) and treated with RQ1 DNase (Promega). cDNA was synthesized with the SuperScript™Vilo™ cDNA synthesis kit (Invitrogen), and qPCR was performed with the Platinum Taq enzyme (SYBR® Green PCR kit, Invitrogen) on a Master cycle Realplex system (Eppendorf) using the primers 5′ TGGGCGGCAGGTGTGAT 3′ and 5′ GCTCGTCTTGGTCCCTCTCAG 3′ for DCLK3.
For northern blot analysis, a 5-mm thick block containing the entire striatum (between grooves 3 and 8 of the matrix, groove 1 being the most anterior) was removed from each brain and processed for mRNA extraction (Brochier et al., 2008). We denatured 3 µg of poly(A)+ RNA in glyoxal-dimethylsulfoxide solution and subjected it to electrophoresis in a 1% agarose gel. The bands were then transferred to a Hybond™ -XL nylon membrane (GE Healthcare) and hybridization was carried out at 65°C in Rapid-Hyb buffer (GE Healthcare) with an [α-32P]dCTP random primed (Prime-It® II; Agilent Technologies) 788-bp PstI fragment of the DCLK3 cDNA sequence as a probe (Fig. 1A).
Figure 1.
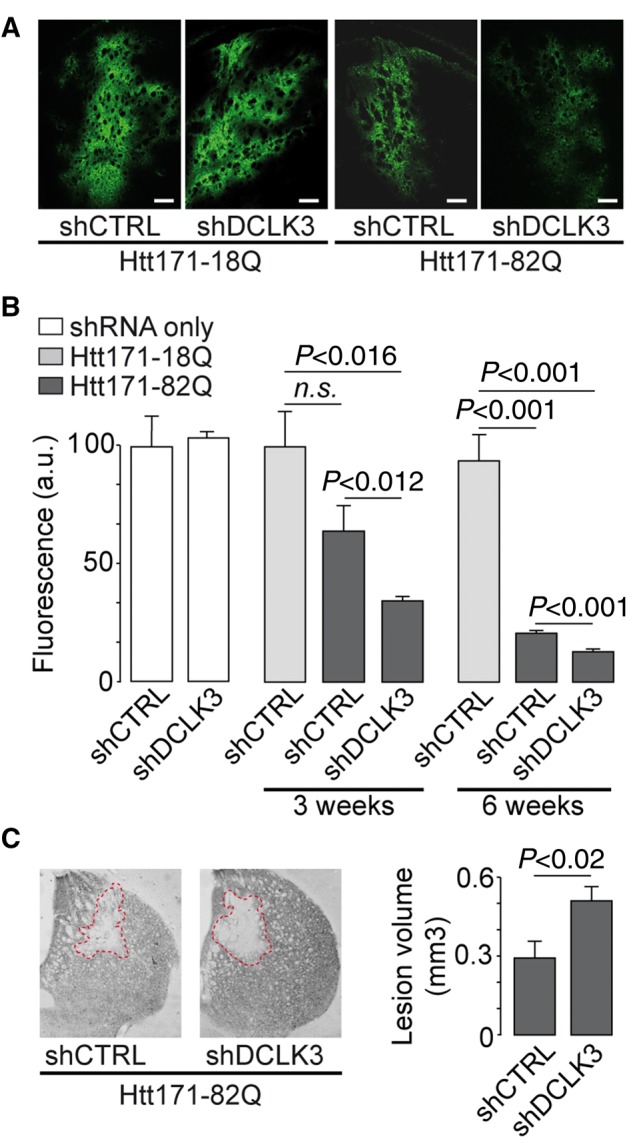
DCLK3 knockdown increases the toxicity of mHtt in vivo. Mice received lentiviral vectors encoding a shRNA targeting either DCLK3 (shDCLK3) or luciferase (shCTRL, the control). The shRNA-encoding constructs also contained the coding sequence for GFP. (A) Representative images of GFP expression in striatal sections from mice infected with LV-Htt171-82Q or LV-Htt171-18Q with LV-shDCLK3 or its control (LV-shCTRL) (3 weeks post-infection). (B) Quantification of fluorescence as an index of neuronal integrity in mice infected with LV-Htt171-18Q mixed with LV-shCTRL (control levels of GFP) or LV-Htt171-82Q mixed with LV-shDCLK3 or LV-shCTRL. Quantification was performed 3 and 6 weeks after infection. The Htt171-82Q-induced loss of GFP was exacerbated by LV-shDCLK3. (C) Brains were processed for histological evaluation 6 weeks after infection, with DARPP32 histochemistry used to detect Htt171-82Q toxicity. Left: Typical coronal sections of mouse brain, with areas of staining depletion observed in the presence of Htt171-82Q. Right: Quantitative determination of the size of the striatal lesions in the two groups. Infection with shDCLK3 significantly leads to increased lesion volume, as shown with DARPP32. Results are expressed as means (n = 7–10/group) ± SEM. Mann-Whitney and Kruskall-Wallis tests in B and paired Student’s t-test in C. Scale bars in A = 0.25 mm; B = 0.5 mm.
Tissue processing and western blotting
Primate tissue and fractionation experiments
For biochemical studies, we used control frozen macaque (M. fascicularis) brain samples, as previously described (Francelle et al., 2015a). All procedures conformed to European (European Union Directive 86/609/EEC) and French (French Act Rural Code R 214-87-131) regulations. The animal facility was approved by veterinary inspectors (Authorization 92-032-02) and complied with the Standards for Humane Care and Use of Laboratory Animals of the Office of Laboratory Animal Welfare (A5826-01). For fractionation experiments, a frozen piece of tissue (∼100 mg) was placed in a loose-fitting Dounce homogenizer with 2.0 ml of buffer A [10 mM HEPES pH 7.9, 150 mM NaCl, 1 mM EDTA, 10 μl/ml protease inhibitors (Roche), 10 μl/ml phosphatase inhibitors (Sigma-Aldrich)] and homogenized with 10 up-and-down passes. We added 100 μl of 10% NP-40 (0.5% final concentration) and the mixture was fully homogenized by 15 up-and-down passes. All these steps were performed on ice. The homogenate was transferred to an Eppendorf tube and incubated on ice for 10 min. We used 1 ml of homogenate as the total fraction. The rest was centrifuged for 10 min at 100g at 4°C. The supernatant was retained as the cytosolic fraction. The pellet was homogeneously suspended in 500 μl buffer A and centrifuged at 100g for 10 min at 4°C. The supernatant was discarded. The pellet was suspended in 250 μl buffer B [20 mM HEPES pH 7.9, 420 mM NaCl, 1.2 mM MgCl2, 0.2 mM EDTA, 25% glycerol, 0.5 mM dithiothreitol (DTT), 10 μl/ml protease inhibitors, 10 μl/ml phosphatase inhibitors] and vortexed to constitute the nuclear fraction. Each fraction was sonicated for 20 s at 20% amplitude. The nuclear fraction was vortexed at 5-min intervals over a 20-min period. Each fraction was then centrifuged at 16 000g for 40 min at 4°C. Protein concentration was determined by the Bradford method (Thermo Scientific).
Mouse tissue
Mice were killed with an overdose of sodium pentobarbital, and their brains were rapidly collected and sliced into 1-mm thick coronal sections with a brain matrix. The striatum was dissected out and homogenized with a glass homogenizer in 50 mM Tris-HCl, pH 7.4, 100 mM NaCl, and 1% SDS, with protease (Roche) and phosphatase inhibitor cocktails (Sigma-Aldrich). Protein concentration was determined by the BCA method (Thermo Scientific).
Western blot
Protein samples were diluted in NuPAGE® LDS sample buffer with NuPAGE Sample Reducing agent (Life Technologies), boiled for 5 min, loaded on a 4–12% Bis-Tris gel (Life Technologies), and subjected to SDS-PAGE. The bands obtained were then transferred to a nitrocellulose membrane with an iBlot® transfer device (Life Technologies). Membranes were incubated overnight at 4°C with antibodies against HA (1:2000, mouse, Covance), DCLK3 (1:1000, 21890002, rabbit, Novus), or beta-tubulin (1:2000, mouse, Sigma-Aldrich) diluted in Tris-buffered saline supplemented with 0.1% Tween 20 and 5% bovine serum albumin (BSA). The membranes were rinsed and incubated with horseradish peroxidase-conjugated secondary antibodies for 1 h (1:4000, GE HealthCare). Antibody binding was detected with a Fusion FX7 camera system (Thermo Fisher Scientific) after incubation with the Clarity enhanced chemiluminescence substrate (Bio-Rad).
In vitro experiments
Directed mutagenesis of DCLK3
S-DCLK3-HA was subjected to in vitro mutagenesis with the QuikChange® Site-Directed Mutagenesis Kit (Stratagene), to replace the threonine 286 residue (i.e. threonine 457 in L-DCLK3) with an alanine residue (T286A), with the following primers: T286A-F: 5′ CCACAGCTACCTAGAGCCCGAGGGGAGGAG 3′ and T286-R: 5′ CTCCTCCCCTCGGGCTCTAGGTAGCTGTGG 3′. The lysine 543 residue of L-DCLK3-HA and Kin-DCLK3 was mutated to a methionine (K543M) with the following primers: K543M-F: 5′ GACAAAGCAGGCATATGCCATGATGATGATTGACAAGAGC 3′ and K543M-R: 5′ GCTCTTGTCAATCATCATCATGGCATATGCCTGCTTTGTC 3′. A silent mutation was also added to introduce an NdeI site, to facilitate sequence verification.
Primary culture of neurons and transfection/infection
Primary cultures of striatal neurons were set up and maintained as previously described (Benchoua et al., 2008). These neurons were electroporated with the mouse striatal neuron Nucleofector® kit according to the instructions supplied (Amaxa, Biosystem).
Primary cortical neurons were prepared from embryonic Day 17 rat embryos. Briefly, cortices were dissected in cold Hank’s Balanced Salt Solution (HBSS) supplemented with 10 mM HEPES. Tissue digestion was performed with a 10-min incubation in a 0.025% trypsin solution. Enzymatic action of the trypsin was inactivated by addition of 1 ml of foetal calf serum. After 3 washes in 5 ml HBSS supplemented with 10 mM HEPES, cortices were mechanically dissociated by 10 up-and-down movements. Neurons were plated on glass coverslips coated with poly-l-lysine (1 mg/ml) in Neurobasal® + 2% B27 + 1% penicillin/streptomycin. Cells were maintained in 5% CO2 and humidity atmosphere.
HEK 293T cells and transfection
HEK293T cells were cultured in Dulbecco’s modified Eagle medium (DMEM, Wako) supplemented with 10% foetal bovine serum and 1% phenylalanine-streptomycin (Sigma-Aldrich). Cells were grown at 37°C in a damp incubator, under an atmosphere containing 5% CO2/95% air. HEK293T cells were plated at a density of 7.5 × 105 cells per well in six-well plates or 10 × 104 cells per well in 24-well plates. After 24 h of culture, cells were transfected by the calcium phosphate (CaPO4) transfection method, with a maximum of 5 μg of plasmid DNA. They were incubated for 4 to 6 h in DMEM supplemented with 10% foetal bovine serum and 1% phenylalanine-streptomycin, and the medium was then replaced. Cells were processed 48 h after transfection to ensure optimal protein expression.
Immunofluorescence of DCLK3 and rDCLK3-HA in HEK293T cells and neurons
HEK293T
Transfected cells were cultured on poly-l-lysine-coated 12 mm-diameter coverslips and treated with 100% ice-cold methanol and 5 mM EGTA for 3 min to preserve the microtubular network. The fixed cells were rinsed with TBS (0.02 M Tris-HCl pH 7.4, 0.15 M NaCl), and permeabilized by incubation with 0.1% Triton™ X-100 in blocking solution [TBS, 1% bovine serum albumin (BSA)] for 30 min. The cells were then incubated with anti-HA antibody (1:1000, mouse clone11, Covance) or anti-α-tubulin antibody (1:1000, rabbit, Millipore) diluted in 1% BSA in TBS-0.1% Triton™ X-100, at room temperature for 1 h. The cells were then incubated with Alexa Fluor® 594-labelled anti-mouse IgG or Alexa Fluor® 488-labelled anti-rabbit IgG (Invitrogen), respectively, at room temperature for 30 min. They were rinsed in TBS and treated with DAPI (1:10 000, Wako) diluted in TBS, at room temperature for 3 min, and were then observed under a confocal laser-scanning microscope (SPE and SP8 Leica).
Neuronal cultures
Cortical rat primary neurons were fixed with cold methanol during 5 min at −20°C then washed twice with PBS. Similar treatment was used for human neurons except that cells were fixed with PFA 4%. Blocking was performed with PBS + 3% NGS + 0.1% Triton™ X-100. Primary antibodies (anti-Dclk3 1/500 and anti-HA 1/1000) were incubated overnight at 4°C in the blocking buffer followed by three washes in the same buffer. Secondary antibodies conjugated with Alexa fluorochromes (anti-rabbit Alexa Fluor® 488 and anti-mouse Alexa Fluor® 594, 1/500) prepared in the blocking buffer were incubated during 1 h at room temperature. After two washes in the blocking buffer, a staining with Hoechst 33258 (1 µg/ml in PBS) was performed during 5 min at room temperature followed by two washes in PBS. Coverslips were mounted in Mowiol® before confocal microscopy.
DCLK3 antibody pre-blocking experiment
For the DCLK3 blocking peptide experiment, the DCLK3 antibody (Ab2189, Novus) was incubated (ratio of 1:5 with either H2O or the recombinant human DCLK3 protein) (ProteoGenix) overnight at 4°C under agitation. This step was performed in tubes previously coated with BSA (5 min incubation with 1 mg/ml of BSA in Tris 50 mM and NaCl 150 mM followed by three washes with H2O). Immunostaining was performed as described above.
Immunoprecipitation for the autophosphorylation assay
Cells were collected 48 h after transfection and lysed in modified-RIPA buffer [10 mM Tris–HCl (pH 8.0), 1% NP-40, 150 mM NaCl, 1 mM EDTA, 10 μg/ml protease inhibitors (Mini-Complete, Roche) and 1% phosphatase inhibitor cocktail 2 (Sigma-Aldrich)]. The homogenates were centrifuged at 16 000g for 20 min, and the supernatants were collected. Protein levels were determined by the BCA method.
We used the IP/co-IP kit, with magnetic beads coupled to A/G protein (Thermo Fisher). We rinsed 50 µl of magnetic beads and incubated them for 2 h with antibodies [0.5 μg of anti-HA antibody or anti-TADA3 antibody (NBP1-90243, rabbit, Novus)] at 4°C. We mixed 500 µg of total protein extract with the antibody-bead complex and incubated the mixture at 4°C for 3 h.
For immunoprecipitation for the autophosphorylation assay, the beads were rinsed in kinase assay buffer (25 mM HEPES pH 7.4; 25 mM, MgCl2) supplemented with inhibitors (0.1 mM orthovanadate, 25 mM β-glycerophosphate) and 2 mM DTT. For autophosphorylation assays, we added 20 µM unlabelled ATP. For protein kinase activity determination, we added 6000 Ci/mmol [γ-32P]ATP to the beads and incubated at 20°C for 30 min (or eluted with SDS-PAGE sample buffer for SDS-PAGE analysis). The beads were rinsed several times with the kinase assay buffer before incubation in 20 µl NuPAGE® LDS Sample Buffer (4×) at 80°C for 5 min. The supernatant was subjected to electrophoresis in a precast gel (NuPAGE® Novex 4–12% Bis-Tris Protein gel, Invitrogen). The gel was dried at 60°C for 3 h and then placed against a MultiSensitive phosphor screen (Perkin Elmer) for 2 h in the dark. This sensitive screen was used to measure radioactivity levels, with the high-resolution filmless Cyclone Plus Phosphor Imager autoradiography system (PerkinElmer).
For immunoprecipitation of kinase assays, the beads were rinsed with kinase assay buffer supplemented with inhibitors. For kinase assays with myelin basic protein (MBP) as a universal kinase substrate, we incubated beads in 200 µM unlabelled ATP and 5 mg/ml MBP at 30°C for 60 min. The reaction was stopped by adding NuPAGE® LDS Sample Buffer (4×) incubating at 80°C for 5 min. The supernatant was run on a precast gel (NuPAGE® Novex 4-12% Bis-Tris Protein gel, Invitrogen).
For immunoprecipitation for the TADA3-rDCLK3 interaction study, we used a two-step immunoprecipitation protocol. The beads were incubated with antibodies and protein extracts from cells transfected with plasmids coding mouse Kin-rDCLK3 or human TADA3 and then rinsed several times. We then incubated the beads for 2 h with protein extracts from cells transfected with TADA3 or rDCLK3, respectively. The beads were rinsed and the proteins eluted by boiling the beads in NuPAGE® LDS Sample Buffer (4×) at 80°C for 5 min.
Western blotting
SDS–PAGE was performed on NuPAGE® Novex® 4-12% Bis-Tris pre-cast polyacrylamide gels (Invitrogen). We used iBlot® Transfer Stacks and the iBlot® Gel Transfer Device (Thermo Fisher Scientific) for protein transfer. The 7-min transfer programme efficiently transferred most of the proteins.
Yeast two-hybrid analysis
Yeast two-hybrid screening was performed by Hybrigenics Services (http://www.hybrigenics-services.com). The coding sequence for human DCLK3 (GenBank accession number gi: 149589020) was amplified by PCR and inserted into pB29 as an N-terminal fusion to LexA (N-DCLK3-LexA-C). The construct was checked by sequencing the entire insert and used as bait for screening a random-primed human adult brain cDNA library constructed in pP6. The pB29 and pP6 constructs were derived from the original pBTM116 (Vojtek and Hollenberg, 1995; Beranger et al., 1997) and pGADGH (Bartel et al., 1993) plasmids, respectively.
We screened 110 million clones (11-fold coverage) by a mating approach, with YHGX13 (Y187 ade2-101::loxP-kanMX-loxP, matα) and L40ΔGal4 (mata) yeast strains, as previously described (Fromont-Racine et al., 1997). We selected 36 His+ colonies on a medium lacking tryptophan, leucine and histidine. The prey fragments of the positive clones were amplified by PCR and their 5′ and 3′ junctions were sequenced. The resulting sequences were used to identify the corresponding interacting proteins in the GenBank database (NCBI) by a fully automated procedure. A confidence score (predicted biological score) was attributed to each interaction, as previously described (Formstecher et al., 2005).
The protein interactions from this publication have been submitted to the IMEx (http://www.imexconsortium.org) consortium through IntAct (Orchard et al., 2014) and assigned the identifier IM-26152.
Human embryonic stem cell-derived neuronal samples and transcriptomic analyses
RC9 human embryonic stem cells (XY, passage 20–40; Roslin Cells) were maintained on CELLstart™ (Invitrogen) and cultured in STEMPRO medium (Invitrogen; cultures were fed daily and manually passaged every 4–5 days).
Striatal neuron progenitors were derived from human embryonic stem cells as previously described (Nicoleau et al., 2013; Arber et al., 2015). Briefly, human embryonic stem cell colonies were manually dissociated and suspended for 6 h in neural induction medium consisting of DMEM/F12, Neurobasal®, N2 and B27 supplement, β-mercaptoethanol and penicillin/streptomycin (N2B27 medium) supplemented with 0.1 µM LDN-193189 (Miltenyi Biotec), 20 µM SB-431542 (Tocris), 1 µM XAV-939 (Cellagen Technology) and 10 µM ROCK inhibitor (Y27632, Calbiochem). On Day 1, human pluripotent stem cell-aggregates were transferred to polyornithine laminin-coated dishes without Y27632. On Day 10, LDN-193189 and SB-431542 were removed and cells were cultured for a further 10 days in N2B27 supplemented with 50 ng/ml activin A (R&D Systems). On Day 21, striatal progenitors were passaged with Accutase® for 10–20 min at 37°C and plated on polyornithine laminin-coated dishes at 80–100 thousand cells per square centimetre in terminal specification medium consisting of N2B27 supplemented with 20 ng/ml BDNF (R&D Systems), 0.5 mM dbcAMP (Sigma–Aldrich), 0.5 mM valpromide (Lancaster Synthesis) and 50 ng/ml activin A (R&D Systems) for 24 additional days of culture.
After 15 days of neuronal differentiation, post-mitotic neuronal cultures were transduced with 24 ng of lentiviral vectors encoding GFP, wild-type Kin-rDCLK3 or rDCLK3-K543M. These infected neuronal cultures were processed for total RNA extraction 10 days after infection, with the RNeasy® Plus Micro Kit (Qiagen). For the large scale RNA study, each of the 16 samples, were reverse-transcribed (50 ng of total RNA) with the Ion AmpliSeq Transcriptome Human Gene Expression kit (Revision A.0) according to the manufacturer’s protocol (Life Technologies). The cDNA libraries were amplified and barcoded with the Ion AmpliSeq Transcriptome Human Gene Expression core panel and the Ion Xpress Barcode Adapter (Life Technologies). The amplicons were quantified with the Agilent High Sensitivity DNA kit and the samples were then pooled in sets of six. Emulsion PCR and Enrichment and loading onto the Ion Chef Instrument were performed with the Ion PI IC 200 Kit (Life Technologies). Samples were loaded on an Ion PI v2 BC Chip and sequenced on the Ion Proton System by Ion PI IC 200 Kit chemistry (200-bp read length; Life Technologies). The Ion Proton reads (FASTQ files) were imported into the RNA-seq pipeline of Partek Flow software (v 4.0, Partek Inc), with hg19 as the reference genome. We identified genes differentially expressed between groups, by quantifying mapped reads with the Partek E/M algorithm (the resulting counts represent the expression levels of more than 20 800 different genes present in the AmpliSeq Human Gene Expression panel) and applying the Partek Gene Specific Analysis (GSA) algorithm. The data of this experiment have been deposited on GEO under accession number GSE104091. For the RT-PCR study of human striatal neurons infected with LV-Htt171-82Q, the protocol for RNA extraction and analysis was similar to that described above.
The first analysis involved identifying the genes differentially expressed between Kin-rDCLK3WT and GFP for ontology analysis. Relatively stringent criteria were used [fold-change >1.20 and adjusted P < 0.0005, false discovery rate (FDR) < 0.05].
The second analysis involved identifying the genes for which expression was specifically regulated by the activity of the kinase domain. We searched for genes displaying expression changes triggered by kin-rDCLK3WT and not Kin-rDCLK3K543M. We used a lower stringency to identify genes displaying differences in expression between Kin-rDCLK3WT and GFP conditions [Fold Change (FC) > 1.1, adjusted P < 0.0009, FDR < 0.05, 275 genes, Supplementary Table 3] and between Kin-rDCLK3K543M and Kin-rDCLK3WT conditions (FC > 1.1, adjusted P < 0.002, FDR < 0.05; 371 genes, Supplementary Table 4).
Biological interpretations of the list of differentially expressed genes were obtained with dedicated software [Partek Genomics Suite (v6.6), Gene Set Enrichment Analysis (GSEA) software from the Broad Institute (http://www.broadinstitute.org/gsea) or online with DAVID (Huang da et al., 2009) and GREAT (McLean et al., 2010)].
Statistical analysis
All data are expressed as means ± standard error of the mean (SEM). Normality of data distribution was tested using the Shapiro-Wilk test and homogeneity of variance was tested with Levene’s test using the commercially available software (Statistica, 13.0). When normality and homogeneity of variance were met (on raw or log transformed data), unpaired Student’s t-tests were used for pairwise comparisons between groups. For comparisons of more than two groups, one-way ANOVA for multiple comparisons was carried out, with Fisher’s post hoc partial least-squares difference (PLSD) test. In the cases where assumption of normality and/or homogeneity of variance are not met, non-parametric tests were applied: Mann-Whitney and Kruskall-Wallis for comparison of two and more groups, respectively.
Results
DCLK3 downregulation exacerbates mHtt toxicity whereas overexpression is neuroprotective in vivo
The mouse Dclk3 gene sequence has two open reading frames corresponding to a large (L-DCLK3, 790 aa) and a short (S-DCLK3, 619 aa) form of DCLK3 (Supplementary Fig. 1). Both forms (L-DCLK3 and S-DCLK3) contain the full conserved kinase sequence, but only L-DCLK3 has a putative DCX domain (Supplementary Fig. 1A). Macaques and humans have only one open reading frame, encoding a 648 aa protein displaying high levels of similarity to the mouse S-DCLK3. Northern blot analysis with mRNA from adult mouse brains showed that L-DCLK3 was more strongly expressed than S-DCLK3 (Supplementary Fig. 1B). Dclk3 mRNA levels were much higher in the striatum than in other regions of the brain, consistent with previous findings (Brochier et al., 2008) and available transcriptomic datasets (http://mouse.brain-map.org/experiment/show/72079921). We also checked that DCLK3 is an active kinase. Recombinant mouse long- and short-forms of DCLK3 with a HA-tag (termed hereafter L- or S-rDCLK3-HA) were produced in HEK293 cells, purified by immunoprecipitation and tested in an autophosphorylation assay with [γ-32P]ATP. The autophosphorylation of proteins with apparent molecular weight corresponding to those of full-length S-rDCLK3 and L-rDCLK3-HA, respectively, was easily detected (Supplementary Fig. 1C).
First, we analysed the role of each mouse DCLK3 form, by using lentiviral vectors. As DCLK3 levels are low in Huntington’s disease patients and mouse models (Kuhn et al., 2007; Brochier et al., 2008; Langfelder et al., 2016), we investigated whether the silencing of the endogenous mouse protein using a lentivirus coding a shRNA targeting Dclk3 mRNA (LV-shDCLK3) aggravated mHtt toxicity in vivo. As a control, we used a shRNA targeting luciferase (shCTRL), a non-mammalian gene. Both vectors contained a GFP reporter gene (Supplementary Fig. 2A) and no difference in GFP fluorescence was observed in the striatum at 4 weeks post-infection when the two lentiviral shRNAs were compared (Fig. 1A and B). Thus, transduction and expression levels may therefore be considered to have been similar for both lentiviruses (Fig. 1B). RT-qPCR showed that shDCLK3 decreased endogenous Dclk3 mRNA levels significantly, by 75%, in the infected striatal region (Supplementary Fig. 2D).
We used lentiviruses expressing the N-terminal part of HTT with a pathological expansion (Htt171-82Q) that we have previously shown to model Huntington’s disease and to produce striatal lesions in a consistent manner (Galvan et al., 2012; Ruiz and Deglon, 2012; Francelle et al., 2015b, d). ShCTRL or shDCLK3 was co-expressed with Htt171-82Q in the mouse striatum. We used GFP expression levels as readout of neuronal integrity at 3 and 6 weeks post-infection. For each mouse, 3D determination of striatal GFP levels was performed by direct measurement of fluorescence levels using serially cut adjacent brain sections encompassing the entire striatum. GFP levels in the striatum were low 3 and 6 weeks after infection with the LV-Htt171-82Q, while in the shCTRL group (without mHtt) fluorescence levels remained high. The loss of GFP (∼30%) was only a trend at 3 weeks post-infection in the mHtt/shCTRL group as compared to shCTRL (Fig. 1A and B). In the group expressing mHtt and shDCLK3, the loss at 3 weeks post-infection was more profound and reached statistical significance as compared to control (shCTRL without mHtt). At 3 and 6 weeks post-infection, the mHtt-induced loss of fluorescence in the striata expressing shDCLK3 was significantly more pronounced than that seen in striata expressing the shCTRL. These results indicated that mHtt-induced loss of fluorescence, an index of neurodegeneration, was accelerated by the expression of shDCLK3 in the mouse striatum (Fig. 1A and B). Tri-dimensional measurement of the volume of the striatal lesions using DARPP32 immunohistochemistry confirmed that Htt171-82Q-induced disease was more pronounced in striata expressing shDCLK3 than in striata expressing shCTRL (Fig. 1C).
We then investigated whether the expression of rDCLK3s in striatal neurons modified the toxicity associated with mHtt production in vivo. The role of each mouse DCLK3 form was investigated with lentivirus coding for mouse recombinant L-DCLK3 and S-DCLK3 with a HA-tag at the C-terminus (LV-L-rDCLK3-HA and LV-S-rDCLK3-HA, respectively, Supplementary Fig. 2A). We injected LV-LacZ (control vector), LV-S-rDCLK3-HA or LV-L-rDCLK3-HA with LV-Htt171-82Q into the mouse striatum. In infected animals, the expression of the S- and L-rDCLK3 proteins in the striatum at 4 weeks post-infection could be checked by HA-tag immunohistochemistry (Fig. 2A) and western blot analysis (Supplementary Fig. 2E). Six weeks after infection, pathological features, characterized by the loss of the medium-sized spiny neuron marker DARPP32, neuronal marker (NeuN), and cytochrome c oxidase (COX), were observed in the striatum, in the control group (Fig. 2B, D and F). The area of lesions seen in the rostro-caudal extension of the striatum in serially cut coronal brain sections (five to eight sections depending on the animals) were quantified and used to determine the volume of the lesions with the Cavalieri method. The volume of the lesions measured using DARPP32 staining in mice co-expressing Htt171-82Q with L- or S-rDCLK3-HA was similar to that seen in mice co-expressing Htt171-82Q with the control protein (Fig. 2B and C). However, in mice co-expressing Htt171-82Q with either L- or S-rDCLK3-HA, the lesions detected using NeuN and COX staining were significantly attenuated with respect to the control group (Fig. 2D–G). The number of ubiquitin-positive aggregates in the entire striatum was also quantified by image analysis in five to eight serially-cut sections in all the mice infected with LV-171-82Q (Fig. 2H and I). No significant difference was found between groups, suggesting no major impact of rDCLK3 on mHtt accumulation.
Figure 2.

Effect of mouse recombinant DCLK3-HA overexpression on the toxicity of mHtt. Adult male mice received a bilateral intrastriatal injection of a mixture of two lentiviral vectors (LV): LV-Htt171-82Q with LV-CTRL (LV-LacZ, control), LV-S-rDCLK3-HA or LV-L-rDCLK3-HA. Six weeks after infection, brains were processed for histological evaluation by immunostaining for the HA-tag, DARPP32, NeuN, and ubiquitin, and histochemistry for cytochrome c oxidase (COX) detection, to evaluate Htt171-82Q neurotoxicity. Coronal sections of mouse brains displaying a representative area with rDCLK3-HA overexpression (A) or depletion of staining, for the different groups (B, D, F and H). (C, E, G and I) Quantitative determination of the volume of the striatal lesions in the different groups using serial sections for each animal. Results are expressed as means (n = 7–10/group) ± the SEM. One-way ANOVA and Fisher’s post hoc PLSD test. Scale bars = AH (A), 0.2 mm; NeuN (B), 0.1 mm; Cox (D), 0.5 mm; ubiquitin (F), 0.125 mm.
Thus, DCLK3 knockdown exacerbates mHtt-induced degeneration whereas DCLK3 overexpression is neuroprotective against mHtt toxicity in vivo.
DCLK3 improves motor signs in knock-in 140CAG mice
As we observed that DCLK3 could produce neuroprotection (i.e. blockade of cell death) without total rescue of the molecular phenotype of medium-sized spiny neurons (loss of DARPP32), we asked whether DCLK3 could produce beneficial effects on the function of striatal neurons in the KI140CAG mouse model of Huntington’s disease (Menalled et al., 2003). In heterozygous and homozygous knock-in 140CAG Huntington’s disease mice at 12 months of age, we found that striatal Dclk3 mRNA levels were significantly lower than in control littermates (Supplementary Fig. 3) as found by others (Langfelder et al., 2016). We therefore investigated whether the restoration of DCLK3 expression in the striatum of these mice could improve their well-characterized Huntington’s disease behavioural phenotype (Menalled et al., 2003; Pepin et al., 2016). We used the AAV2/rh10 vector (AAV-L-rDCLK3 and AAV-GFP as control) to produce L-rDCLK3-HA throughout much of the striatum (Fig. 3A, B). This led to markedly higher levels of mRNA production than for the endogenous mouse Dclk3 (Fig. 3C). Homozygous KI140CAG mice underwent bilateral injections before the onset of symptoms, at the age of 6 months (Fig. 3D).
Figure 3.

Functional effects of striatal overexpression of L-rDCLK3, using AAV2/rh10 in homozygous KI140 Huntington’s disease mice. (A) Schematic representation of the adeno-associated-virus (AAV) constructs used to overexpress GFP (control) or L-rDCLK3 in mice (with AAV2/rh10). (B) Typical image of AAV–mediated transduction of the mouse striatum with AAV-GFP. (C) DCLK3 levels in striatum infected with AAV-L-rDCLK3-HA in KI140 mice, as assessed by RT-qPCR. (D) Time course and experimental design: mice received injections of AAV-GFP or AAV-L-rDCLK3 (n = 7 for the GFP group and n = 8 for the L-DCLK3 group) at the age of 6 months. At 12 months of age, mice performed rotarod, grip strength and CatWalk tests. (E) Accelerated rotarod test: the results of the three tests (t1, 2, 3) on the last training day are shown. (F) Grip strength was measured in triplicate for all animals. (G) The CatWalk parameter ‘mean body speed variation’ was measured to evaluate the smoothness of displacement. (H) Number of twitches captured in the acquisition window during the run (short and sudden jerking of one of the paws). (I) Body weight of the KI140 mice after striatal infection. Results are expressed as means (n = 7/group) ± SEM. P-values indicate significance levels: ANOVA for repeated measures over the three tests in E; one-way ANOVA factorial analysis in F, G and H, followed by Fisher’s post hoc PLSD test).
Mice were studied in the rotarod motor test. The time for which the animals were able to remain on the rotarod was significantly shorter in uninjected Huntington’s disease mice and GFP-expressing Huntington’s disease mice than in littermate controls at the age of 12 months in accelerating speed trials (Fig. 3E). Rotarod performance of L-DCLK3-expressing Huntington’s disease mice was not significantly different from that of control littermates and significantly better than the performance of uninjected and GFP-expressing Huntington’s disease mice.
Consistent with the findings of the rotarod test, GFP–expressing Huntington’s disease mice also performed significantly less well in forelimb grip strength tests than GFP-expressing wild-type littermates. By contrast, the long-term overexpression of L-rDCLK3 alleviated the grip strength defects of Huntington’s disease mice at 12 months of age. Indeed, there was a significant difference between KI140CAG Huntington’s disease mice receiving AAV-GFP injections and wild-type mice, whereas Huntington’s disease mice receiving injections of AAV-L-rDCLK3 did not differ from wild-type mice in terms of grip strength (Fig. 3F).
We also used the CatWalk system to investigate gait deficits in the Huntington’s disease mice. Body speed was significantly increased in Huntington’s disease mice injection with AAV-GFP when compared to wild-type mice. Body speed varied significantly less in Huntington’s disease mice overexpressing L-rDCLK3 than in Huntington’s disease mice expressing GFP (Fig. 3G), indicating a more homogeneous displacement. Consistent with this finding, we also observed fewer twitches in the gait of Huntington’s disease mice overexpressing DCLK3 than in that of those expressing GFP (Fig. 3H).
Moreover, the body weight of Huntington’s disease mice overexpressing L-rDCLK3 increased over the 6 months of the experiment, whereas that of Huntington’s disease mice expressing GFP remained stable over time after infection (Fig. 3I).
Thus, the restoration of DCLK3 expression improves motor and health signs in KI140CAG mice.
Neuroprotection is mediated by the kinase domain of DCLK3
The tandem DCX domains of DCLK1 and DCLK2 play a major role in the known functions of these proteins in neurodevelopment and neuroprotection, through interaction with cytoskeletal elements (Ohmae et al., 2006a; Shin et al., 2013; Nawabi et al., 2015). The tandem DCX domains are not conserved in human DCLK3 and mouse S-DCLK3. Mouse L-DCLK3 has only one DCX domain. We therefore hypothesized that the kinase domain underlies key functions of DCLK3, including its neuroprotective effect against mHtt. To address this question, we generated different rDCLK3 mutants.
We first generated a truncated mouse DCLK3 protein containing only the kinase domain [aa 159–619], by deleting the N-terminal DCX domain (Kin-rDCLK3-HA) (Fig. 4A). Wild-type mice were co-infected with LV-Htt171-82Q together with either LV-CTRL (coding LacZ) or LV-rKin-DCLK3. Histological analyses of staining for NeuN and COX showed that the volumes of striatal lesions were significantly decreased by the overexpression of Kin-rDCLK3 (Fig. 4B and C). Thus, overexpression of the rDCLK3 kinase domain is sufficient for neuroprotection against mHtt toxicity.
Figure 4.

The active kinase domain of rDCLK3 is sufficient to protect against mHtt in the striatum in vivo. (A) Schematic illustration of DCLK3 and Kin-rDCLK3-HA constructs with and without the K543M mutation. (B) Mice received bilateral injection of LV-Htt171-82Q mixed with either LV-LacZ (control) or a lentivirus encoding the kinase domain of DCLK3 (LV-Kin-rDCLK3). Six weeks after infection, brains were processed for histological evaluation (B) of NeuN, HA and COX (not shown) levels. HA staining (B) shows expression of Kin-rDCLK3-HA in the striatum. (C) Quantification of the volumes of the Htt171-82Q-induced striatal lesions by staining for NeuN, COX and ubiquitin. (D) Autophosphorylation experiments with ATP [γ-32P] after the co-immunoprecipitation of wild type or mutant Kin-rDCLK3 with anti-HA antibody followed by SDS-PAGE, showing the incorporation of radioactive 32P into the protein. Colours indicate the radioactivity levels from white (no signal) to red (highest). (E) Assays of kinase activity, with MBP used as a kinase substrate after the of Kin-rDCLK3-HA, wild-type (WT) and mutant K543M forms (K > M) and detection of phosphorylated MBP (p-MBP) by western blotting. T = input (total protein extract before immunoprecipitation), S = sample test; n.t. = non-transfected cells (control). (F and H) Photomicrographs showing striatal lesions in mice injected with LV-Htt171-82Q mixed with LV-LacZ (control, CTRL), LV-Kin-rDCLK3 (WT) or lentivirus encoding the mutant Kin-DCLK3 form with the K543M (K>M) substitution. (G and I) Histograms representing the quantification of the volumes of Htt171-82Q-induced lesions using NeuN (G) and COX (I) staining. Scale bars = 0.2 mm. Results are expressed as means (n = 7–11/group) ± SEM. P-values indicate the level of significance (unpaired Student’s t-test, control versus wild-type).
We then investigated whether the disruption of kinase activation abolished the neuroprotective efficacy of the protein against mHtt. We generated a T286>A (T286A) substitution in S-rDCLK3 (similar to Th289 in human DCLK3) within a conserved motif of charged amino acids similar to that close to threonine 286 in CaMKII, which is known to play a major role in activation. We investigated the effect of this mutation on the toxicity of mHtt in cultured striatal neurons. Cell death was evaluated by counting transfected neurons with fragmented nuclei. The overexpression of mHtt (Htt171-82Q) led to neuron degeneration in primary striatal cultures, with an increase in the number of fragmented nuclei. Wild-type S-rDCLK3 was neuroprotective against mHtt toxicity, whereas the S-rDCLK3T286A mutant was not (Supplementary Fig. 4).
We checked that kinase activation was a critical event for the neuroprotective function of DCLK3 by generating ‘dead kinase’ mutants of L-rDCLK3 and Kin-rDCLK3 (Fig. 4A). We identified a highly conserved lysine residue (K543) in the ATP-binding pocket as the target (Supplemental Supplementary Fig. 5). Substitutions of this residue have been shown to abolish kinase activity in other kinases (e.g. AKT1, AMPK). We generated L-rDCLK3K543M and Kin-rDCLK3K543M mutants (K>M). As expected, these mutants displayed much lower levels of autophosphorylation than the wild-type forms in an assay with [γ-32P]-ATP (Fig. 4D). Consistent with this finding, L-rDCLK3K543M and Kin-rDCLK3K543M phosphorylated the pan-kinase substrate MBP only inefficiently when compared to the wild-type form of Kin-rDCLK3 (Fig. 4E). These experiments validate these mutants as ‘dead DCLK3 kinase’ proteins.
We then investigated the effects of the ‘efficient kinase’ DCLK3 (Kin-rDCLK3) and the ‘dead kinase’ DCLK3 (Kin-rDCLK3K543M) on the toxicity of mHtt (Htt171-82Q) in mouse striatum in vivo. The detection of the HA-tag by immunohistochemistry in vivo 6 weeks after lentiviral injection showed that expression levels were similar for Kin-rDCLK3K543M and the wild-type form (not shown). The volume of the mHtt-induced striatal lesions was measured using NeuN and COX staining. It was significantly reduced by the co-expression of Kin-rDCLK3 in striatal neurons (Fig. 4F–I). By contrast, striatal neurons were not rescued by Kin-rDCLK3K543M co-expression (Fig. 4F–I).
Thus, the kinase function of DCLK3 is critical for neuroprotection against mHtt.
DCLK3 behaves as a regulator of chromatin biology and transcription
Following the demonstration that the DCLK3 kinase domain was required for neuroprotection, we looked for downstream effectors, by identifying potential interactors. The human Kin-DCLK3 domain (aa 162–632) (Fig. 5A) was used as a bait in a yeast two-hybrid screen to identify potential partners from a human brain library. We identified seven interactors: TADA3, BTBD9, TRIM39, SALL1, ZNF12, ZNF292 and ZNF366 (Fig. 5B and Supplementary Table 1). The protein interactions from this screen have been submitted to the IMEx (http://www.imexconsortium.org) consortium through IntAct (Orchard et al., 2014) and assigned the identifier IM-26152. All the interactors identified have zinc finger domains. Bioinformatics-based searches for potential functions (based on similarity) suggested that these DCLK3 interactors were involved in transcription. TADA3 appeared to be the most relevant of the interactors identified, as the level of confidence for interaction with DCLK3 was highest for this molecule. Furthermore, an analysis of the expression of the genes encoding these seven interactors in the brain based on previously reported mouse SAGE data (Brochier et al., 2008), or more recent region- and cell type-specific transcriptomic databases (http://mouse.brain-map.org/; http://web.stanford.edu/group/barres_lab/brain_rnaseq.html) (Supplementary Table 1) revealed that TADA3 was more strongly expressed in the striatum than the other DCLK3 interactors. It was strongly expressed in neurons and other brain cell types. We obtained evidence supporting DCLK3–TADA3 interaction, by performing co-transfection followed by co-immunoprecipitation. HA-tagged rDCLK3 and a V5-tagged form of one of its potential partners, TADA3, were expressed in HEK293T cells. Co-immunoprecipitation experiments were conducted with whole-cell lysates prepared from the co-transfected cells. TADA3-V5 co-immunoprecipitated with Kin-rDCLK3-HA. Kin-rDCLK3-HA was also co-immunoprecipitated with TADA3-V5 (Fig. 5C).
Figure 5.
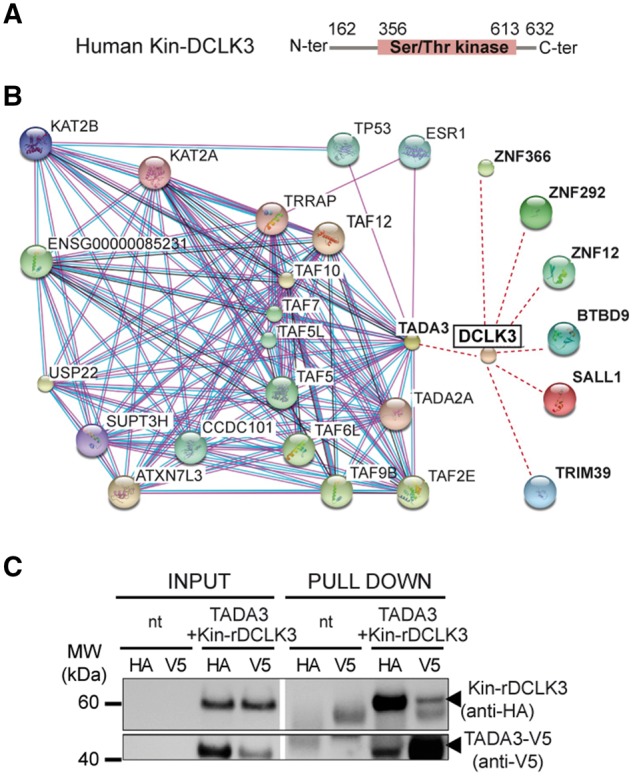
Yeast two-hybrid screen of interactors of the DCLK3 kinase domain. (A) The C-terminal domain of human DCLK3 (Kin-DCLK3) was used as bait to screen a human brain mRNA library. (B) The seven proteins (in bold) were found to interact with DCLK3. String 10.0 bioinformatics software (http://string-db.org) was used to search for TADA3 (Transcriptional adapter 3) interactors (human only) in public databases and for the schematic representation of the network of functional interactions for TADA3. (C) Interaction between Kin-DCLK3 and TADA3 was checked in double pull-down experiments after the transfection of HEK cells.
The TADA3 protein (transcriptional adapter 3, also called ADA3) is a component of the Spt-ada-Gcn5 acetyltransferase (SAGA) complex. This complex regulates transcription by modulating histone post-translational modifications, including histone acetylation (Wang et al., 2008). TADA3 has a large number of known partners, including TADA2, several TATA box-binding protein-associated factors (TAFs), the histone acetyltransferases KAT2B and KAT2A, and the transcription initiation protein SPT3 homolog SUPT3H (Fig. 5B), providing strong support for the hypothesis that DCLK3 could regulate transcription through SAGA-induced histone modifications.
We asked whether the DCLK3 kinase domain could modulate transcriptomic profile in neurons. We overexpressed human Kin-rDCLK3WT, Kin-rDCLK3K543M or GFP (as a control) in striatal neurons derived from wild-type human embryonic stem cells (Fig. 6A) using lentiviral vectors. After 10 days, we performed transcriptome analyses by ultrahigh-multiplex PCR (Ion AmpliSeq technology). A relatively stringent analysis of differential gene expression (fold-change 1.2; P < 0.0005, FDR, 0.05) between cells expressing Kin-rDCLK3WT and cells expressing GFP showed that 88 genes were upregulated and 73 downregulated in the presence of Kin-rDCLK3WT (Fig. 6B and Supplementary Table 2; GEO dataset GSE104091). Kin-rDCLK3 modified the expression levels of many genes encoding histones and transcription-related proteins. Gene ontology analyses with DAVID (Huang da et al., 2009) and GREAT (McLean et al., 2010) indicated that the upregulated genes were significantly enriched in functions and pathways linked to chromatin biology and nucleosomes, whereas the downregulated genes showed no significant enrichment in any specific function or pathway (Fig. 6 and Supplementary Fig. 6). Specifically, we observed an upregulation of genes encoding histones (HIST) and proteins modulating chromatin state and transcription dynamics, including MLL5 [KMT2E lysine (K)-specific methyltransferase 2E], TAF7 (transcription initiation factor TFIID 55 kDa subunit) and SUPT3H (SPT3 homologue). Like TADA3, SUPT3H is a core component of the SAGA complex (Wang et al., 2008).
Figure 6.

Effect of rDCLK3 kinase domain in human striatal neurons. (A) Schematic representation of the lentiviral (LV) constructs used to transduce human striatal neurons derived from neuronal stem cells for transcriptomic analysis of the effects of the rDCLK3 kinase domain by AmpliSeq at 12 days post-infection. (B) Gene ontology analysis of genes (n = 88) significantly upregulated by Kin-rDCLK3WT using DAVID and GREAT identified functions related to nucleosome and chromatin/DNA remodelling. (C) Genes with significant changes in expression dependent on the catalytic activity of Kin-rDCLK3 (i.e. genes displaying significant differences in expression (FDR < 0.05) between Kin-rDCLK3WT and GFP and between Kin-rDCLK3K543M and Kin-rDCLK3WT). (D) Expression levels of a subset of genes identified in C in human neurons expressing Htt171-82Q and either GFP, Kin-rDCLK3WT or Kin-rDCLK3K543M. Expression levels where determined by RT-qPCR. Results are expressed as means (C, n = 4 wells per group; D, n = 5–6 wells per group) ± SEM. One-way ANOVA factorial analysis in D followed by Fisher’s post hoc PLSD test. *P < 0.05.
We then identified genes for which expression levels were selectively modified by the catalytic activity of the kinase domain. We took the set of genes for which expression was significantly modified by Kin-rDCLK3WT (Supplementary Table 3), and identified those that also displayed significant differences in expression between Kin-rDCLK3WT and Kin-rDCLK3K543M conditions (Supplementary Table 4). There were 53 genes present on both lists (i.e. 9-fold enrichment) (Fig. 6C, Supplementary Tables 4 and 5). Many of these genes whose expression was regulated by DCLK3 in a kinase-dependent manner are involved in transcription regulation. They included the genes encoding the demethylase ZIC3 and the zinc finger domain-containing BTBD9 protein identified as a potential partner of human Kin-DCLK3 in the yeast two-hybrid screen described above. Eight of these genes encoded histones (HIST). Most of these genes (64%) were upregulated. Approximately 40% of these genes were found also to be significantly deregulated in knock-in mouse models of Huntington’s disease (Langfelder et al., 2016), suggesting a link between decreased DCLK3 and altered regulation of genes implicated in chromatin biology and transcription. To further investigate this relationship, using lentiviral vectors, we overexpressed a truncated version of mHtt, together with Kin-rDCLK3WT or Kin-rDCLK3K543M or GFP (as a control), in striatal neurons derived from wild-type human embryonic stem cells (Fig. 6D). After 10 days, we performed expression analyses, using qRT-PCR. More specifically, we analysed 24 genes of the above list of 53 genes (whose expression, in the absence of mHtt was changed both in cells expressing Kin-rDCLK3WT versus cells expressing GFP and in cells expressing Kin-rDCLK3K543M versus cells expressing Kin-rDCLK3WT cells). Subsets of these genes, including BTBD9 and histones, were also modulated by Kin-rDCLK3 in the context of the Huntington’s disease mutation (Fig. 6D), through a kinase-dependent manner, suggesting that the protective effect of DCLK3 in neurons expressing mHtt involves epigenetic and/or transcriptional mechanisms. Additionally, we examined the effect of rDCLK3 overexpression on the striatal marker genes, PPP1R1B (DARPP32), TAC1 and PDE10A, which are decreased in the Huntington’s disease pathology. Expression of PPP1R1B/DARPP32 and TAC1 was not markedly changed by Kin-rDCLK3WT or by Kin-rDCLK3K543M in Huntington’s disease-like human cells, a result that is consistent with results found in our lentiviral mouse models. In contrast, Kin-rDCLK3WT, and not Kin-rDCLK3K543M increased the expression of PDE10A, an important regulator of striatal physiology, further suggesting that restoring DCLK3 signalling is beneficial in Huntington’s disease-like striatal neurons.
Since these data suggested that DCLK3 may act in the nucleus, we studied the subcellular localization of endogenous DCLK3 and rDCLK3. The endogenous levels of mouse DCLK3 is low and could not be easily detected with the commercially available human anti-DCLK3 antibodies. For this reason, we first investigated whether DCLK3 could be detected by immunofluorescence in primary culture of rat neurons. We used an antibody directed against the internal N-terminal part of human DCLK3 (Ab2189, Novus) (Fig. 7A). Confocal imaging showed a relatively weak immunofluorescent signal in the cytoplasm and nucleus of neurons (Fig. 7B). When the primary Ab2189 antibody was omitted and when the antibody was pre-blocked with the recombinant human DCLK3 protein this signal markedly decreased (Supplementary Fig. 7A). When neurons were infected with LV-S-rDCLK3-HA, immunofluorescence signal related to the antibody Ab2189 increased in the cytoplasm and the nucleus as compared to not-infected cells (Fig. 7B). We could also detect Kin-rDCLK3 protein by immunofluorescence in the cytoplasm and in the nucleus of human striatal neurons expressing mHtt (Supplementary Fig. 8). L-rDCLK3-HA could be detected by the anti-HA antibody in LV-infected mice in vivo. The HA-tag was mainly found in the cytoplasm of striatal neurons. Cytoplasmic rDCLK3 related fluorescence was also detected with Ab 2189, which cross reacts with the mouse protein when overexpressed (not shown). Interestingly, signal was also found in the nucleus of transduced striatal neurons (Fig. 7C). This suggested that a cleaved product of DCLK3 lacking its C-terminal part (therefore lacking the HA-tag) accumulated in the nucleus in vivo. This finding is reminiscent of the reported migration of DCLK2 to the nucleus after the cleavage of a short C-terminal domain (Nagamine et al., 2014).
Figure 7.

Nuclear localization of DCLK3 in striatal neurons. DCLK3/rDCLK3 proteins were detected with an anti-HA antibody (green) and an anti-DCLK3 antibody (Ab2189; red) recognizing the internal domain of the protein. (A) Schematic representation of the immunodetection of endogenous and recombinant DCLK3. (B) Endogenous rat DCLK3 (top) and mouse rDCLK3 expressed with a lentiviral vector (bottom) in primary culture of rat cortical neurons. Note DCLK3 is present in both the cytoplasm and nucleus. (C) Cellular distribution of DCLK3 after the injection of LV-L-rDCLK3-HA into the mouse striatum, as determined with confocal images indicate the presence of DCLK3-HA in the cytoplasm (green) and of a form of DCLK3 lacking the C-terminal HA tag in the nucleus of the neurons (red). (D) Immunohistochemical detection of endogenous DCLK3 in the non-human primate caudate nucleus with an antibody recognizing the internal domain of the protein (Ab2189). Arrowheads indicate cells with essentially nuclear distribution (dark staining) of DCLK3. (E) Western blot analysis of DCLK3 levels in fractions (total homogenates = T; cytosol = Cyt; Nuclear = Nu) prepared from non-human primate caudate/putamen, showing the full-length (f.l.) protein and the putative cleaved product of the protein (cleaved). DCLK3 is both in the cytoplasm and nucleus. (F) Confocal images of endogenous DCLK3 (red), obtained with the Ab2189 antibody along with NeuN (green) and nuclear DNA (blue) in the non-human primate caudate nucleus. (G) Enlarged view of the two fields of view (1 and 2) in the image in B in the three fluorescence channels. The perimeter of the nucleus is delineated (white dotted lines). Scale bars in B and D = 10 µm, C and G = 5 µm.
To check that the nuclear localization of DCLK3 in rat and mouse neurons was not an artefact related to its overexpression after lentiviral-mediated transduction, we analysed the distribution of endogenous DCLK3 in non-human primate brains with the anti-DCLK3 antibody (Ab 2189). In the caudate-putamen prepared from paraffin-embedded section, immunohistochemical analysis of the striatum showed DCLK3 to be present in the nucleus of neurons (Fig. 7D). Subcellular fractionation of homogenates prepared from non-human primate striatum showed the non-human primate DCLK3 protein (∼74 kDa) to be present in both the cytoplasm and the nucleus (Fig. 7E). We performed immunofluorescence detection of DCLK3 on floating sections. Confocal microscopy showed DCLK3 to be present in the nucleus of many cells that were positive for the neuronal marker NeuN, consistent with the expression of the protein in medium-sized spiny neurons in this brain region (Fig. 7F and G). DCLK3 immunofluorescence decorates the chromatin in the vicinity of the nuclear membrane, consistent with nucleosomal localization, as suggested from a bioinformatics search (e.g. http://www.genecards.org/cgi-bin/carddisp.pl?gene=DCLK3). DCLK3 was detected both in the cytoplasm and the nucleus of striatal neurons and in large neurons (layers V) of the cerebral cortex (Supplementary Fig. 7B and C).
Together with the yeast two-hybrid screen and cellular localization data, transcriptomic results support the view that the DCLK3 in striatal neurons has a nuclear function linked to chromatin biology and transcription regulation.
Discussion
This study identifies for the first time DCLK3 as a key signalling kinase in the neurons likely to play a role in the susceptibility of striatal neurons to mHtt. Our results also provide evidence for a role of DCLK3 in a nuclear function linked to chromatin and transcription regulation, a function that is altered early in Huntington’s disease.
DCLK3 levels are low in Huntington’s disease patients and animal models (Kuhn et al., 2007; Langfelder et al., 2016). We confirmed this observation using qRT-PCR in our KI140CAG Huntington’s disease mice. Our results indicate that DCLK3 is neuroprotective against mHtt in murine medium-sized spiny neurons in vivo and that DCLK3 downregulation exacerbates mHtt toxicity. Intriguingly, expression of rDCLK3 did not restore DARPP32 levels while it rescued NeuN and COX levels in our in vivo models. Consistent with this, in human striatal neurons expressing mHtt, rDCLK3 did not change DARPP32 levels while levels of PDE10A, another well established marker of striatal neurons were increased in a kinase-dependent manner. The total number of ubiquitin-positive inclusions was not changed by rDCLK3 while in the striatal volume infected expressing mHtt the number of surviving neurons was likely increased by the kinase. Thus it is possible that the net number of inclusions per neurons was reduced, suggesting rDCLK3 may slightly diminish mHtt aggregation. Whether this could contribute to the neuroprotective effects of rDCLK3 awaits further studies. Thus, early decreases in DCLK3 levels in the striatum in Huntington’s disease may render medium-sized spiny neurons more susceptible to complex disease processes leading eventually to neuronal cell death. The neuroprotection conferred by rDCLK3 was observed in an acute in vitro model of Huntington’s disease, and in a relatively ‘aggressive’ mouse model of Huntington’s disease based on the overexpression of a short N-terminal fragment of mHtt, leading to severe neuronal marker loss and cell death within a few weeks. We also investigated the neuroprotective potential of rDCLK3 in a highly progressive ‘mild’ mouse model of Huntington’s disease, KI140CAG mice. We found that rDCLK3 overexpression improved mouse motor performance. Thus our results suggest that increased DCLK3 signalling in the striatum can improve neuronal dysfunction, supporting the hypothesis that the loss of DCLK3 function in KI140 Huntington’s disease mice could play a role in the motor signs in these animals. These observations highlight the probable role of early DCLK3 loss in precipitating both striatal dysfunction and associated motor symptoms in Huntington’s disease.
‘Striatal enriched’ gene products other than DCLK3 have been identified as potentially involved in the selective degeneration of the striatum in Huntington’s disease (for a review see Francelle et al., 2014). These gene products include for example the Ras-related protein Rhes (Subramaniam et al., 2009), the thyroid hormone binding protein CRYM (Francelle et al., 2015b), and the non-coding RNA Abhd11os (Francelle et al., 2015d). Thus, the additive and/or synergistic loss of several striatal markers, including DCLK3, may render the striatum particularly sensitive to mHtt toxicity in Huntington’s disease.
The mechanisms underlying the neuroprotection produced by rDCLK3 are unknown. High expression levels of DCLK3 have been recently shown to be a key risk factor in colorectal cancer through the modification of the expression of genes (associated to epithelial-to-mesenchymal transition) that drive tumour cells into a more aggressive behaviour (Liu et al., 2017). Thus high DCLK3 levels might be associated with anti-apoptotic pathways, which is consistent with its neuroprotective effects we found in the present study. However, nothing is known of the roles of DCLK3 in adult neurons. The present study provides novel results that may help understanding these probably complex mechanisms. We showed that the expression of a mouse truncated protein containing only the kinase domain of DCLK3 conferred neuroprotection against mHtt. We also showed that a ‘dead’ kinase form of DCLK3 was unable to mediate such neuroprotection, thereby demonstrating that the neuroprotective effect of DCLK3 against mHtt requires the catalytic kinase activity of the protein. These findings suggest that the function of the kinase domain of DCLK3 may be specific. DCLK1 and DCLK2 are also neuroprotective and their overexpression can improve neuroregeneration after axonal injury (Nawabi et al., 2015). The role of the kinase domain of DCLK1/2 in these effects is unlikely, because the overexpression of the DCX domains and of the adjacent S/P-rich domain in the absence of the kinase domain seems to be sufficient to improve regeneration. Similarly, DCLK1/2 can regulate the spindle orientation of neuronal precursors during mitosis and cortical development independently of kinase activity (Shu et al., 2006). In addition, DCLK3 displays a more neuron-specific pattern of expression than DCLK1/2, which are also expressed in astrocytes (http://web.stanford.edu/group/barres_lab/brain_rnaseq.html) (Zhang et al., 2014). Bioinformatics tools to compare DCLK3 and the other two members of the DCLKs family indicate that while the structure/organization of the kinase domain is conserved, many amino acids are different in the kinase domain and in regions surrounding the kinase domain (Supplementary Fig. 9). Thus it is tempting to speculate that interactors of Kin-rDCLK3 are different from those of the kinase domain of DCLK1/2, leading to a specific signalling in neurons. However, it cannot be totally ruled out that the three members of the family have interactors in common. The cell- and region-specific expression of DCLK3 and the key role of its kinase domain in neuroprotection highlight differences in neurological functions of this kinase and of DCLK1/2.
The present findings also suggest that DCLK3 has nuclear functions. Following its overexpression in mouse striatum, DCLK3 was detected in both the cytoplasm and the nucleus. Fractionation experiments and confocal imaging on non-human primate striatum indicated that DCLK3 was preferentially localized in the nucleus, despite the detection of some DCLK3 in the cytoplasm. This finding is consistent with the observation that DCLK2 enters the nucleus after the cleavage of its C-terminus (a probable ‘regulatory domain’) (Nagamine et al., 2014). Translocation from the cytoplasm to the nucleus has also been suggested for DCLK1 (Burgess and Reiner, 2001; Kuribara et al., 2011). Autophosphorylation assays suggested that a DCLK3 protein with an apparent molecular weight close to that of the full-length form had the highest level of catalytic activity. The nuclear location of DCLK3 may, therefore, be an important aspect of its neuronal function.
The yeast two-hybrid screen revealed that the proteins interacting with the DCLK3 kinase domain were related to transcriptional regulation and/or chromatin remodelling. One of these interactors, the transcriptional activator adaptor TADA3, is a component of the SAGA complex linking histone acetylation to the transcription machinery and is of particular interest in this context. SAGA plays a key role in transcription, regulating the transcription of almost all genes in yeast and human cells (Bonnet et al., 2014). Its importance in neurodevelopment and possibly neurodegenerative diseases has been recently underscored (Wang and Dent, 2014). Expression of the DCLK3 kinase domain in human striatal-differentiated embryonic stem cells leads to the upregulation of genes encoding histones and proteins modulating chromatin state and transcription dynamics, suggesting that DCLK3 may play a direct role in chromatin remodelling and transcription regulation. Our results in human neurons expressing mHtt fragment suggest that this function of DCLK3 is partially restored upon re-expression of rDCLK3, at least in part through a kinase-dependent manner. Whether this contributes to increased expression of subsets of striatal markers (e.g. PDE10A) will need to be further investigated. The precise mechanisms of SAGA regulation to generate cell-specific transcriptional signatures are unknown. The interaction of DCLK3 with TADA3 may provide us with a novel means of imprinting a SAGA-dependent transcriptional signature in medium-sized spiny neurons. Given the neuron-specific expression of DCLK3, this might constitute a novel pathway for regulating transcription in response to synaptic stimuli, for example. Further studies are needed to validate this hypothesis.
The deregulation of transcription in Huntington’s disease has been studied in detail over the past 15 years (Seredenina and Luthi-Carter, 2012). Transcriptomic studies of the striatum in mouse models of Huntington’s disease and Huntington’s disease patients have identified a functional signature associated with downregulated genes (Francelle et al., 2017). This signature is characterized by enrichment in genes associated with biological functions linked to neuronal activity (Kuhn et al., 2007; Seredenina and Luthi-Carter, 2012; Achour et al., 2015). The genes downregulated in Huntington’s disease striatum have been shown to be regulated by super-enhancers (Achour et al., 2015), a category of enhancers controlling cell-type specific identity through epigenetic mechanisms (Whyte et al., 2013). Super-enhancers were also shown to be selectively impaired in the striatum of mice with Huntington’s disease, due to low levels of histone acetylation (Achour et al., 2015). DCLK3 is one of the genes downregulated by the loss-of-function of ‘super-enhancers’ (Achour et al., 2015). Thus, DCLK3 loss of function may be part of a vicious cycle leading to major transcription defects and detrimental epigenetic changes underlying early neuronal dysfunction and eventually cell death. The validation of this hypothesis awaits further studies.
Supplementary Material
Acknowledgements
We thank Julien Mitja, Diane Houitte, and Carole Malgorn for their technical assistance in animal experiments and biochemical analyses. We also thank Alexandre Fayel for his help in the analysis of DCLK3 target genes. We are also very grateful for the fruitful scientific discussions with Dr Jocelyne Caboche.
Funding
The present research is supported by recurrent funds from C.E.A. and C.N.R.S.
The research generating these results received funding from the European Community's Seventh Framework Programme FP7/2007-2013 under grant agreement no. HEALTH-F5-2008-222925. L.G. was supported by the Neuropôle de Recherche Francilien and the Fondation pour la Recherche Médicale. L. de L. is the recipient of a PhD grant from Fondation pour la Recherche Medicale (n° PLP2015103462). L.F. was supported by the French Research Ministry. This work benefited from support from the national ‘Infrastructure de recherche’ NeurATRIS (Translational Research Infrastructure in Neurosciences, ‘Investissement d'Avenir’, ANR-11-INBS-0011).
Supplementary material
Supplementary material is available at Brain online.
Glossary
Abbreviation
- mHtt
mutant huntingtin
References
- Achour M, Le Gras S, Keime C, Parmentier F, Lejeune FX, Boutillier AL, et al. Neuronal identity genes regulated by super-enhancers are preferentially down-regulated in the striatum of Huntington's disease mice. Hum Mol Genet 2015; 24: 3481–96. [DOI] [PubMed] [Google Scholar]
- Arber C, Precious SV, Cambray S, Risner-Janiczek JR, Kelly C, Noakes Z, et al. Activin A directs striatal projection neuron differentiation of human pluripotent stem cells. Development 2015; 142: 1375–86. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Aurnhammer C, Haase M, Muether N, Hausl M, Rauschhuber C, Huber I, et al. Universal real-time PCR for the detection and quantification of adeno-associated virus serotype 2-derived inverted terminal repeat sequences. Hum Gene Ther Methods 2012; 23: 18–28. [DOI] [PubMed] [Google Scholar]
- Bartel L, Chien C-T, Sternglanz R, Fields S. Using the two-hybrid system to detect protein-protein interactions. In: Hartley DA, editor. Cellular interactions in development: a practical approach. Oxford: Oxford University Press; 1993. p. 153–79. [Google Scholar]
- Benchoua A, Trioulier Y, Diguet E, Malgorn C, Gaillard MC, Dufour N, et al. Dopamine determines the vulnerability of striatal neurons to the N-terminal fragment of mutant huntingtin through the regulation of mitochondrial complex II. Hum Mol Genet 2008; 17: 1446–56. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Beranger F, Aresta S, de Gunzburg J, Camonis J. Getting more from the two-hybrid system: N-terminal fusions to LexA are efficient and sensitive baits for two-hybrid studies. Nucleic Acids Res 1997; 25: 2035–6. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Berger A, Lorain S, Josephine C, Desrosiers M, Peccate C, Voit T, et al. Repair of rhodopsin mRNA by spliceosome-mediated RNA trans-splicing: a new approach for autosomal dominant retinitis pigmentosa. Mol Ther 2015; 23: 918–30. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bonnet J, Wang CY, Baptista T, Vincent SD, Hsiao WC, Stierle M, et al. The SAGA coactivator complex acts on the whole transcribed genome and is required for RNA polymerase II transcription. Genes Dev 2014; 28: 1999–2012. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Brochier C, Gaillard M-C, Diguet E, Caudy N, Dossat C, Ségurens B, et al. Quantitative gene expression profiling of mouse brain regions reveals differential transcripts conserved in human and affected in disease models. Physiol Genomics 2008; 33: 170–9. [DOI] [PubMed] [Google Scholar]
- Brouillet E, Jacquard C, Bizat N, Blum D. 3-Nitropropionic acid: a mitochondrial toxin to uncover physiopathological mechanisms underlying striatal degeneration in Huntington's disease. J Neurochem 2005; 95: 1521–40. [DOI] [PubMed] [Google Scholar]
- Burgess HA, Reiner O. Cleavage of doublecortin-like kinase by calpain releases an active kinase fragment from a microtubule anchorage domain. J Biol Chem 2001; 276: 36397–403. [DOI] [PubMed] [Google Scholar]
- Charvin D, Roze E, Perrin V, Deyts C, Betuing S, Pages C, et al. Haloperidol protects striatal neurons from dysfunction induced by mutated huntingtin in vivo. Neurobiol Dis 2008; 29: 22–9. [DOI] [PubMed] [Google Scholar]
- Charvin D, Vanhoutte P, Pages C, Borrelli E, Caboche J. Unraveling a role for dopamine in Huntington's disease: the dual role of reactive oxygen species and D2 receptor stimulation. Proc Natl Acad Sci USA 2005; 102: 12218–23. [DOI] [PMC free article] [PubMed] [Google Scholar]
- de Almeida LP, Ross CA, Zala D, Aebischer P, Deglon N. Lentiviral-mediated delivery of mutant huntingtin in the striatum of rats induces a selective neuropathology modulated by polyglutamine repeat size, huntingtin expression levels, and protein length. J Neurosci 2002; 22: 3473–83. [DOI] [PMC free article] [PubMed] [Google Scholar]
- de Chaldee M, Gaillard MC, Bizat N, Buhler JM, Manzoni O, Bockaert J, et al. Quantitative assessment of transcriptome differences between brain territories. Genome Res 2003; 13: 1646–53. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Diguet E, Petit F, Escartin C, Cambon K, Bizat N, Dufour N, et al. Normal aging modulates the neurotoxicity of mutant huntingtin. PLoS One 2009; 4: e4637. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Drouet V, Perrin V, Hassig R, Dufour N, Auregan G, Alves S, et al. Sustained effects of nonallele-specific Huntingtin silencing. Ann Neurol 2009; 65: 276–85. [DOI] [PubMed] [Google Scholar]
- Faideau M, Kim J, Cormier K, Gilmore R, Welch M, Auregan G, et al. In vivo expression of polyglutamine-expanded huntingtin by mouse striatal astrocytes impairs glutamate transport: a correlation with Huntington's disease subjects. Hum Mol Genet 2010; 19: 3053–67. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Formstecher E, Aresta S, Collura V, Hamburger A, Meil A, Trehin A, et al. Protein interaction mapping: a Drosophila case study. Genome Res 2005; 15: 376–84. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Francelle L, Galvan L, Brouillet E. Possible involvement of self-defense mechanisms in the preferential vulnerability of the striatum in Huntington's disease. Front Cell Neurosci 2014; 8: 295–9. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Francelle L, Galvan L, Gaillard MC, Guillermier M, Houitte D, Bonvento G, et al. Loss of the thyroid hormone-binding protein Crym renders striatal neurons more vulnerable to mutant huntingtin in Huntington's disease. Hum Mol Genet 2015a; 24: 1563–73. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Francelle L, Galvan L, Gaillard MC, Guillermier M, Houitte D, Bonvento G, et al. Loss of the thyroid hormone-binding protein Crym renders striatal neurons more vulnerable to mutant huntingtin in Huntington's disease. Hum Mol Genet 2015b; 24: 1563–73. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Francelle L, Galvan L, Gaillard MC, Petit F, Bernay B, Guillermier M, et al. The striatal long noncoding RNA Abhd11os is neuroprotective against an N-terminal fragment of mutant huntingtin in vivo. Neurobiol Aging 2015c; 36. [DOI] [PubMed] [Google Scholar]
- Francelle L, Galvan L, Gaillard MC, Petit F, Bernay B, Guillermier M, et al. The striatal long noncoding RNA Abhd11os is neuroprotective against an N-terminal fragment of mutant huntingtin in vivo. Neurobiol Aging 2015d; 36: 1601.e7–16. [DOI] [PubMed] [Google Scholar]
- Francelle L, Lotz C, Outeiro T, Brouillet E, Merienne K. Contribution of neuroepigenetics to Huntington's disease. Front Hum Neurosci 2017; 11: 17. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Fromont-Racine M, Rain JC, Legrain P. Toward a functional analysis of the yeast genome through exhaustive two-hybrid screens. Nat Genet 1997; 16: 277–82. [DOI] [PubMed] [Google Scholar]
- Galvan L, Lepejova N, Gaillard MC, Malgorn C, Guillermier M, Houitte D, et al. Capucin does not modify the toxicity of a mutant Huntingtin fragment in vivo. Neurobiol Aging 2012; 33: 1845.e5–6. [DOI] [PubMed] [Google Scholar]
- Giampa C, Laurenti D, Anzilotti S, Bernardi G, Menniti FS, Fusco FR. Inhibition of the striatal specific phosphodiesterase PDE10A ameliorates striatal and cortical pathology in R6/2 mouse model of Huntington's disease. PLoS One 2010; 5: e13417. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Han I, You Y, Kordower JH, Brady ST, Morfini GA. Differential vulnerability of neurons in Huntington's disease: the role of cell type-specific features. J Neurochem 2010; 113: 1073–91. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hinckelmann MV, Zala D, Saudou F. Releasing the brake: restoring fast axonal transport in neurodegenerative disorders. Trends Cell Biol 2013; 23: 634–43. [DOI] [PubMed] [Google Scholar]
- Hottinger AF, Azzouz M, Deglon N, Aebischer P, Zurn AD. Complete and long-term rescue of lesioned adult motoneurons by lentiviral-mediated expression of glial cell line-derived neurotrophic factor in the facial nucleus. J Neurosci 2000; 20: 5587–93. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Huang da W, Sherman BT, Lempicki RA. Systematic and integrative analysis of large gene lists using DAVID bioinformatics resources. Nat Protoc 2009; 4: 44–57. [DOI] [PubMed] [Google Scholar]
- Kerjan G, Koizumi H, Han EB, Dube CM, Djakovic SN, Patrick GN, et al. Mice lacking doublecortin and doublecortin-like kinase 2 display altered hippocampal neuronal maturation and spontaneous seizures. Proc Natl Acad Sci USA 2009; 106: 6766–71. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kuhn A, Goldstein DR, Hodges A, Strand AD, Sengstag T, Kooperberg C, et al. Mutant huntingtin's effects on striatal gene expression in mice recapitulate changes observed in human Huntington's disease brain and do not differ with mutant huntingtin length or wild-type huntingtin dosage. Hum Mol Genet 2007; 16: 1845–61. [DOI] [PubMed] [Google Scholar]
- Kuribara M, Jenks BG, Dijkmans TF, de Gouw D, Ouwens DT, Roubos EW, et al. ERK-regulated double cortin-like kinase (DCLK)-short phosphorylation and nuclear translocation stimulate POMC gene expression in endocrine melanotrope cells. Endocrinology 2011; 152: 2321–9. [DOI] [PubMed] [Google Scholar]
- Langfelder P, Cantle JP, Chatzopoulou D, Wang N, Gao F, Al-Ramahi I, et al. Integrated genomics and proteomics define huntingtin CAG length-dependent networks in mice. Nat Neurosci 2016; 19: 623–33. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Liu NQ, Ter Huurne M, Nguyen LN, Peng T, Wang SY, Studd JB, et al. The non-coding variant rs1800734 enhances DCLK3 expression through long-range interaction and promotes colorectal cancer progression. Nat Commun 2017; 8: 14418. [DOI] [PMC free article] [PubMed] [Google Scholar]
- McLean CY, Bristor D, Hiller M, Clarke SL, Schaar BT, Lowe CB, et al. GREAT improves functional interpretation of cis-regulatory regions. Nat Biotechnol 2010; 28: 495–501. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Menalled LB, Sison JD, Dragatsis I, Zeitlin S, Chesselet MF. Time course of early motor and neuropathological anomalies in a knock-in mouse model of Huntington's disease with 140 CAG repeats. J Compar Neurol 2003; 465: 11–26. [DOI] [PubMed] [Google Scholar]
- Nagamine T, Nomada S, Onouchi T, Kameshita I, Sueyoshi N. Nuclear translocation of doublecortin-like protein kinase and phosphorylation of a transcription factor JDP2. Biochem Biophys Res Commun 2014; 446: 73–8. [DOI] [PubMed] [Google Scholar]
- Nawabi H, Belin S, Cartoni R, Williams PR, Wang C, Latremoliere A, et al. Doublecortin-like kinases promote neuronal survival and induce growth cone reformation via distinct mechanisms. Neuron 2015; 88: 704–19. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Nicoleau C, Varela C, Bonnefond C, Maury Y, Bugi A, Aubry L, et al. Embryonic stem cells neural differentiation qualifies the role of Wnt/beta-Catenin signals in human telencephalic specification and regionalization. Stem Cells 2013; 31: 1763–74. [DOI] [PubMed] [Google Scholar]
- Ohmae E, Ouchi Y, Oda M, Suzuki T, Nobesawa S, Kanno T, et al. Cerebral hemodynamics evaluation by near-infrared time-resolved spectroscopy: correlation with simultaneous positron emission tomography measurements. Neuroimage 2006a; 29: 697–705. [DOI] [PubMed] [Google Scholar]
- Ohmae S, Takemoto-Kimura S, Okamura M, Adachi-Morishima A, Nonaka M, Fuse T, et al. Molecular identification and characterization of a family of kinases with homology to Ca2+/calmodulin-dependent protein kinases I/IV. J Biol Chem 2006b; 281: 20427–39. [DOI] [PubMed] [Google Scholar]
- Orchard S, Ammari M, Aranda B, Breuza L, Briganti L, Broackes-Carter F, et al. The MIntAct project–IntAct as a common curation platform for 11 molecular interaction databases. Nucleic Acids Res 2014; 42: D358–63. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Pepin J, Francelle L, Carrillo-de Sauvage MA, de Longprez L, Gipchtein P, Cambon K, et al. In vivo imaging of brain glutamate defects in a knock-in mouse model of Huntington's disease. Neuroimage 2016; 139: 53–64. [DOI] [PubMed] [Google Scholar]
- Roze E, Saudou F, Caboche J. Pathophysiology of Huntington's disease: from huntingtin functions to potential treatments. Curr Opin Neurol 2008; 21: 497–503. [DOI] [PubMed] [Google Scholar]
- Ruiz M, Deglon N. Viral-mediated overexpression of mutant huntingtin to model HD in various species. Neurobiol Dis 2012; 48: 202–11. [DOI] [PubMed] [Google Scholar]
- Seredenina T, Luthi-Carter R. What have we learned from gene expression profiles in Huntington's disease? Neurobiol Dis 2012; 45: 83–98. [DOI] [PubMed] [Google Scholar]
- Shin E, Kashiwagi Y, Kuriu T, Iwasaki H, Tanaka T, Koizumi H, et al. Doublecortin-like kinase enhances dendritic remodelling and negatively regulates synapse maturation. Nat Commun 2013; 4: 1440. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Shu T, Tseng HC, Sapir T, Stern P, Zhou Y, Sanada K, et al. Doublecortin-like kinase controls neurogenesis by regulating mitotic spindles and M phase progression. Neuron 2006; 49: 25–39. [DOI] [PubMed] [Google Scholar]
- Subramaniam S, Sixt KM, Barrow R, Snyder SH. Rhes, a striatal specific protein, mediates mutant-huntingtin cytotoxicity. Science 2009; 324: 1327–30. [DOI] [PMC free article] [PubMed] [Google Scholar]
- The Huntington's Disease Collaborative Research Group. A novel gene containing a trinucleotide repeat that is expanded and unstable on Huntington's disease chromosomes. The Huntington's Disease Collaborative Research Group. Cell 1993; 72: 971–83. [DOI] [PubMed] [Google Scholar]
- Vojtek AB, Hollenberg SM. Ras-Raf interaction: two-hybrid analysis. Methods Enzymol 1995; 255: 331–42. [DOI] [PubMed] [Google Scholar]
- Wang L, Dent SY. Functions of SAGA in development and disease. Epigenomics 2014; 6: 329–39. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wang YL, Faiola F, Xu M, Pan S, Martinez E. Human ATAC is a GCN5/PCAF-containing acetylase complex with a novel NC2-like histone fold module that interacts with the TATA-binding protein. J Biol Chem 2008; 283: 33808–15. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Whyte WA, Orlando DA, Hnisz D, Abraham BJ, Lin CY, Kagey MH, et al. Master transcription factors and mediator establish super-enhancers at key cell identity genes. Cell 2013; 153: 307–19. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zhang Y, Chen K, Sloan SA, Bennett ML, Scholze AR, O'Keeffe S, et al. An RNA-sequencing transcriptome and splicing database of glia, neurons, and vascular cells of the cerebral cortex. J Neurosci 2014; 34: 11929–47. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zuccato C, Cattaneo E. Huntington's disease. Handb Exp Pharmacol 2014; 220: 357–409. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.