

Abstract
The advent of human induced pluripotent stem cell (hiPSC) technology has revitalized much of the efforts within the past decade to more fully realize the potential of human embryonic stem cells (hESCs). Adding to the possibility of generating unlimited supplies of any cell types of interest, the hiPSC technology now enables the derivation of cells with patient-specific phenotypes. With the Precision Medicine Initiative, it is clear that the hiPSC technology will play a vital role in the advancement of cardiovascular research and medicine. This review summarizes the tremendous and continuing progress that has been made in the field of hiPSC technology, with particular emphasis on cardiovascular disease modeling and drug development. Wherever appropriate, the growing roles of hiPSC technology in the practice of precision medicine will be specifically discussed.
Keywords: precision medicine, induced pluripotent stem cells, cardiovascular diseases, disease modeling, drug development
1. Introduction
In the past few decades, we have seen an explosion of research activities aiming at understanding the fundamental molecular underpinnings of human diseases. However, our comprehension of clinical diseases has lagged behind. Indeed, much of the “molecular data” thought to be crucial in connecting various disease processes still have largely unknown clinical implications. For these reasons, the Precision Medicine Initiative was launched with efforts to both link molecular data with the clinical disease phenotypes and to precisely identify patient subpopulations that differ in their disease susceptibility, progression, and prognosis so that patient-specific treatments can be implemented.1, 2 Among many promising techniques for this mission, the human induced pluripotent stem cell (hiPSC) technology offers a unique platform by which human diseases with their underlying genetic and epigenetic contributions can be modeled in cell culture (Figure 1).3-7 Using this platform, drugs can also be screened against patients’ complex genetic backgrounds so as to yield information regarding patient-specific cardiotoxicity.8-10 Here we first describe how hiPSCs can play an important role in precision medicine that will shape the future of clinical practice. Following a concise overview of the technical advances in hiPSC research, we then discuss in depth various clinically relevant applications of hiPSCs, with a particular emphasis on disease modeling and drug discovery. We expect that with further refinement, the hiPSC technology will greatly shape the future of cardiovascular research and contribute to the clinical practice of precision cardiovascular medicine.
Figure 1.
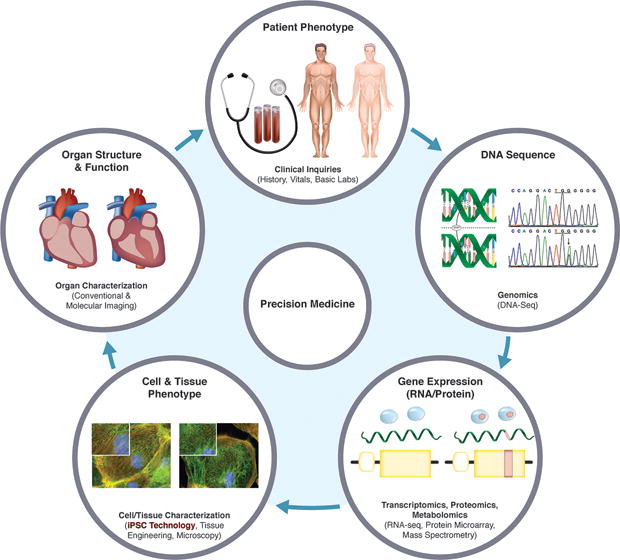
Role of hiPSC technology in precision medicine. Differences in patients’ clinical phenotypes are the results of their unique DNA sequences being acted on by varying environmental influences, leading to differential gene expression patterns, cellular/tissue phenotypes, and organ structures/functions. DNA sequences alone do not entirely predict the clinical phenotypes. Patient-specific hiPSC derivatives recapitulate the phenotypes of their in vivo counterparts. In precision medicine, the patients’ disease risks, prognoses, and treatment responses can be predicted based on the behaviors of their hiPSC derivatives in cell culture.
2. Roles of hiPSCs in Precision Medicine
The fundamental goal of the Precision Medicine Initiative is to develop prevention and treatment strategies that take into account individual variability. The underlying assumption of this approach is that differences in patients’ genetic makeup and environmental exposure contribute to their differential clinical outcomes. Indeed, a growing body of research has shown that differences at the genetic level can be characterized by genome sequencing and be exploited to guide clinical decisions. As a prime example, Nicholas Volker, a 4-year-old boy survived a life-threatening gut inflammation after his doctors found a mutation known to cause immune dysregulation by whole-exome sequencing and performed a cord blood transplant accordingly to save his life.11 The strong push for a more wide-spread use of whole-genome sequencing makes practical sense, as both the rate of increase in the speed of genome sequencing and the rate of decline in the genome sequencing cost in recent years easily surpasses the Moore’s law—a projection in the computer industry describing the doubling of growth (e.g., number of transistors in an integrated circuit) every 2 years.12 However, does DNA alone predict disease?
Studies from monozygotic twins have shown that despite similar height and appearance, they do not always develop or die from the same diseases.13 Numerous studies have found that genetics alone may not be better than traditional risk factors for predicting a person’s risk of developing most diseases, especially for those complex and polygenic in nature.14 It is also well known that epigenetic modulation of gene expression as a result of varying environmental exposure can influence disease risks.15 Numerous post-translational mechanisms in response to environmental influences have also been implicated in cardiovascular diseases.16 Short of cloning a replica of the patient or his heart, the primary cardiovascular cells (e.g., cardiomyocytes, endothelial cells, smooth muscle cells) containing the same genetic landscape and the environmental exposure as the patient arguably may serve as the next-best predictive model of the patient’s risks of developing diseases. However, the procurement of primary cardiovascular cells, especially adult cardiomyocytes, requires invasive maneuvers that carry nontrivial risks. Furthermore, the long-term maintenance of quality primary cells in culture is not feasible to allow prolonged in vitro investigation. For these reasons, the hiPSC technology is an attractive tool because it holds the key to generating unlimited amount of patient-specific cardiovascular cells that closely mimic the endogenous counterparts.
Besides mimicking primary cardiovascular cells, the hiPSC-derived cardiovascular cells play the role of an “integrator” in precision medicine. For example, when exposed to environmental perturbation in cell culture, the hiPSC-derived cardiovascular cells integrate the patient’s genomic disease susceptibility with the environmental influence to produce a disease phenotype simulating the patient’s condition. Therefore, one can imagine the use of hiPSC-derived cardiomyocytes (hiPSC-CMs) in a patient with unknown cardiomyopathy or life-threatening arrhythmia to understand whether a variant of unknown significance (VUS) on genetic testing is disease-causing. The same can be done to understand why a patient with familial dilated cardiomyopathy has a much more severe clinical phenotype than his or her sibling who has the same genetic mutation in the cardiac troponin T gene but exhibits only mild clinical phenotype. It is also possible to envision the use of hiPSC-CMs in a patient with familial cardiomyopathy to predict whether exposure to certain antipsychotic medications would trigger drug-induced life-threatening arrhythmia. The hiPSC-CMs in this case can be challenged with adrenergic stress to further elicit the disease phenotypes. The potential applications for hiPSCs in precision medicine are therefore enormous. We believe the findings obtained from hiPSC-based interrogation can complement other existing clinical diagnostic tools to best guide the practice of precision medicine.
3. Concise Overview of hiPSC Research
Before describing the various exciting applications of hiPSCs for cardiovascular research, we will first present a concise overview of the technical advances that have been made in the field of hiPSCs, including refined protocols for hiPSC reprogramming and hiPSC differentiation into various cardiovascular cell types (i.e., cardiomyocytes, endothelial cells, and smooth muscle cells).7, 17-19 These protocols have opened the door for many exciting hiPSC applications, including disease modeling, drug screening, and regenerative therapy. In this review, we will focus on the first two applications, and the readers are referred to excellent review articles written elsewhere on hiPSC-based regenerative therapy.20,21
The hiPSC technology has grown rapidly after the initial discovery by Yamanaka et al. that human fibroblasts can be reprogramed by overexpressing 4 different transcription factors: OCT4, SOX2, c-MYC, and KLF4.22 This reprogramming technique was also reproduced by other groups using either the same or different variants of the Yamanaka factors.23-25 Similar to human embryonic stem cells (hESCs), hiPSCs derived from somatic cell sources exhibit unlimited self-renewal and pluripotency as characterized by the capacity to differentiate into cells of all 3 germ layers. But, unlike hESCs, making hiPSCs does not require human embryos and therefore raises fewer ethical concerns, which have hindered the progress of hESC research in the past. Furthermore, hiPSC derivatives from different patients confer patient-specific phenotypes due to their unique genomic landscapes. The patient-specific hiPSC derivatives therefore serve as ideal platforms not only for modeling monogenic/polygenic diseases in cell culture, but also for conducting drug discovery on a real-world patient population with diverse genetic backgrounds.
3.1 Reprogramming Methods
Since Dr. Yamanaka’s seminal research on mouse iPSCs26 and hiPSCs22 for which he received the Nobel Prize in Medicine or Physiology in 2012, there have been continuing efforts to make the reprogramming process safer and more efficient. For instance, integrating retroviral/lentiviral vectors that were initially used for reprogramming could cause insertional mutagenesis,27 and the retained expression or reactivation of a reprogramming factor such as c-MYC could be tumorigenic.28 Due to these concerns, non-integrating viral vectors (e.g., Sendai virus,29 adenovirus,30 Epstein-Barr virus23), nonviral vectors (e.g., plasmids,31 minicircles,32 CoMiP33), excisable integrating vectors (e.g., Cre-recombinase-based,34 piggyBac transposon-based35), synthetic modified mRNAs,36 and pluripotency proteins37 have all been explored as safer alternatives to generating hiPSCs. The choice of reprogramming strategies, however, depends on the downstream applications desired and should be carefully made to balance factors such as reprogramming efficiency, amounts of residual transgene retention, rates of aneuploidy, cost, and required workload.38 It is important to keep in mind that although the reprogramming process can potentially introduce genetic variation to produce undesirable phenotypes for the hiPSCs or their derivatives, most of the genetic variations found in the hiPSC derivatives can be traced to those variations already existing in the somatic source cells.39 Similarly, the epigenetic variations found in hiPSCs can be more attributed to the genetic background of the source cells than the reprogramming process, as demonstrated by a recent study using Sendai viral vector-mediated reprogramming.40
3.2. Cardiomyocyte Differentiation
The protocols for generating hiPSC derivatives have been gleaned from early hESC research. Initial efforts to differentiate hiPSCs into cardiomyocytes focused mainly on the use of embryoid bodies (EBs). Inductive strategies using co-cultures of hESCs and mouse visceral endodermal-like (END-2) cells were also explored but were found to be relatively inefficient.41 For EB-mediated differentiation, the hESCs grown on irradiated mouse embryonic fibroblasts (MEFs) were dispersed and cultured in suspension, where spontaneous cell aggregates (EBs) formed.42, 43 Following plating, cardiomyocytes from contracting EBs could be separated out and used for subsequent analysis and manipulation. The EB-derived cardiomyocytes resembled early-stage cardiomyocytes in terms of myofibrillar organization, gene expression pattern, and chronotropic responses to catecholamines. Similar differentiation protocols were later validated for hiPSC-CMs, which were structurally and electrophysiologically indistinguishable from those of hESCs (hESC-CMs).44 The major problems with early EB-based protocols were their low efficiency and significant line-to-line variability as a result of heterogeneous EB sizes. For these reasons, EB-based protocols were modified to include “forced aggregation” strategies.45 In these modified protocols, a defined number of enzymatically adapted hESCs/hiPSCs were placed in “U” or “V-bottomed” shaped wells and then subjected to centrifugation. These maneuvers led to aggregates of uniform sizes more conducive to consistent cardiac differentiation. The precise modulation of mesodermal induction, followed by early cardiac differentiation, was achieved by the timely introduction and removal of growth factors that influenced 4 major important pathways in the normal cardiac development: (1) the bone morphogenetic protein (BMP), (2) transforming growth factor-β (TGFβ)/activin/NODAL, (3) WNT, and (4) fibroblast growth factor (FGF) pathways.17 Using an EB-based approach with sequential addition of multiple growth factors (i.e., BMP4, activin A, FGF2, vascular endothelial growth factor, and Dickkopf-1) followed by flow cytometry-based purification against a novel cardiac-specific cell surface maker, signal-regulatory protein alpha (SIRPA), Dubois et al. were able to differentiate hESCs into cardiomyocytes of up to 98% purity, as assessed by both cardiac troponin T (cTnT) positivity on flow cytometry and confirmatory expression of cardiac markers including NK2 homoeobox 5 (NKX2.5), myosin heavy chains 6 (MYH6), myosin heavy chain 7 (MYH7), and myosin light chain 7 (MYL7).46 However, because EB-based methods were both technically complex and labor intensive, the monolayer-based cardiac differentiation method has become more popular.
Laflamme et al. cultured hESCs as monolayers and then induced cardiac differentiation with sequential addition and subsequent removal of activin A and BMP4, such that they were able to obtain cultures of up to 30% pure cardiomyocytes.47 Subsequently, other versions of the monolayer approach using additional growth factors such as WNT3A, DKK1, FGF2, or WNT inhibitors were found to be more effective.48-50 Lian et al. further demonstrated that small molecule GSK3B and WNT inhibitors could be used to temporally modulate the canonical WNT signaling pathway and produce up to 87 % pure cardiomyocytes from multiple hESC/hiPSC lines without any purification step, as determined by flow cytometry of cTnT- and MF20 (myosin heavy chain)-positive cells.51 More recently, Burridge et al. refined the monolayer/small molecule approach by using a simplified formulation containing the basal RPMI 1640 medium, L-ascorbic acid 2-phosphate, and rice-derived recombinant human albumin in conjunction with GSK3B and WNT inhibitors to produce contractile sheets of greater than 95% pure cardiomyocytes from 11 hiPSC lines.52 Collectively, these studies show that chemically-defined protocols are both efficient and cost-effective for large-scale de novo production of functional cardiomyocytes.
3.3. Endothelial Cell and Smooth Muscle Cell Differentiation
For vascular biology research, efficient protocols for differentiating hESCs/hiPSCs into endothelial cells (ECs) and smooth muscle cells (SMCs) have also been validated. Levenberg et al. demonstrated the feasibility of spontaneously differentiating hESCs into endothelial cells via EB intermediates. Platelet endothelial cell adhesion molecule 1-positive (PECAM1+) cells isolated from days 13-15 EBs expressed endothelial markers (e.g., CD34, Flk-1) that were comparable to those of human umbilical vein endothelial cells.53 Orlova et al., later established a protocol for simultaneously deriving ECs and pericytes from monolayers of hESCs and hiPSCs grown on Matrigel and exposed to timely introduction of differentiation growth factors in B(P)EL media.54 The differentiation efficiency for either cell type was as high as 30% by day 10 of differentiation. More importantly, these cells formed primary vascular plexus upon co-culture. Ferreira et al. also showed the possibility of isolating CD34 vascular progenitor cells from EB suspension and differentiating them into both endothelial cells using endothelial growth medium-2 (EGM-2) supplemented with VEGF, or SMCs using EGM-2 supplemented with platelet-derived growth factor (PDGF).55 The resultant cells expressed characteristic cell type-specific markers and functionally contributed to the formation of microvasculature following implantation into mice. A more recent refinement of this protocol used co-cultures of hESCs and OP9 stroma cells in media supplemented with VEGF, FGF2, and BMP4 to generate CD31+CD34+ vascular progenitor cells, which were then further differentiated into ECs using EGM-2 media supplemented with VEGF, or into SMCs using SMC proliferation media supplemented with PDGF followed by SMC differentiation media supplemented with TGFβ1.56 The resultant yields of ECs and SMCs were 10-100-fold greater than those obtainable from the previous version of the protocol. As with cardiomyocyte differentiation, further optimization of these protocols will likely include the use of monolayers of hiPSCs in serum-free, chemically defined media supplemented with small molecules so as to achieve simplicity, convenience, cost-effectiveness, and efficiency.
4. Disease Modeling Using Patient-Specific hiPSC Derivatives
In order to practice precision cardiovascular medicine, we need to be able to answer questions such as why some patients with certain genetic mutations or single nucleotide polymorphisms are at higher risks of developing complications, and why patients with the same genetic alteration respond differently to the same clinical treatment. These fundamental questions can only be fully answered if we have a clear understanding of the different diseases at the molecular/cellular level. The hiPSC technology has allowed us to efficiently produce human cells with specific patient and disease characteristics. Extensive efforts are now devoted to modeling various cardiovascular diseases using hiPSC derivatives with the hope of understanding mechanistically how patients’ genotypes are related to their cellular phenotypes. Table 1 lists most of the hiPSC models of cardiac diseases to date.57-97
Table 1.
Selected hiPSC models of cardiac diseases. LQT, long QT Syndrome; JLNS, Jervell and Lange-Nielsen Syndrome; CPVT, catecholaminergic polymorphic ventricular tachycardia; DCM, dilated cardiomyopathy; HCM, hypertrophic cardiomyopathy; ARVD/C, arrhythmogenic right ventricular dysplasia/cardiomyopathy; DMD, Duchenne muscular dystrophy; CM, cardiomyopathy; ALHD, aldehyde dehydrogenase; HLHS, hypoplastic left heart syndrome; RV, retrovirus; LV, lentivirus; SV, Sendai virus; EPS, episomal; S-mRNA, synthetic modified mRNA; O, OCT4; S, SOX2; K, KLF4; C, c-MYC; N, NANOG; L, LIN28; EB (SP), embryoid bodies (suspension); EB (FA) embryoid bodies (force aggregation); ML, monolayer; END-2, mouse endodermal-like cell line co-culture; GF, growth factors; SM, small molecules; VPA, valproic acid; ↑, increased; ↓, decreased; APD, action potential duration; FPD, field potential duration; EAD, early afterdepolarization; DAD, delayed afterdepolarization; hERG, human Ether-a-go-go-related gene; ALLN, (N-[N-(N-acetyl-L-leucyl)-L-leucyl]-L-norleucine); CaMKII, Ca2+/calmodulin-dependent protein kinase II; PDE2A/PDE3A, phosphodiesterase 2A/3A; DES, desmin; TnT, troponin T; ANP, atrial natriuretic peptide; cMyBP-C, myosin binding protein C; ETA-b, endothelin receptor type A blocker; NFATC4, nuclear factor of activated T-cells, cytoplasmic 4; PKP2, plakophilin 2; GSK3β, glycogen synthase kinase 3 beta; PPARα/PPARγ, peroxisome proliferator-activated receptor α/γ; ROS, reactive oxygen species; TAZ, tafazzin; GAA, acid alpha-glucosidase; Ikr, rapidly activating delayed rectifier potassium current; Iks, slowly activating delayed rectifier potassium current; INaL, late sodium current; iPSC-CM, induced pluripotent stem cell-derived cardiomyocyte.
| Class | Disorder | Gene | Mutation | Reprog. Method | Diff. Method | iPSC-CM Phenotypes | Ref. |
|---|---|---|---|---|---|---|---|
| Arrhythmia Syndromes | LQT1 | KCNQ1 | R190Q | RV (OSKC) | EB (SP) | ↑ APD, ↓ IKs current, catecholamine-induced arrhythmia; phenotypes attenuated by β-blockers. | [57] |
| 1893delC | RV (OSKC) | EB (SP) | ↑ FPD, trafficking defect of Iks channels; phenotypes aggravated by Ikr blockers. | [58] | |||
| Exon 7 del | RV (OSKC) | ML (+SM) | ↑ APD, ↓ Iks, ↓ full-length Kv7.1 protein; phenotypes partially reversed with Kv7.1 activator. | [59] | |||
| JLNS | c.478-2A>T, c.1781G>A | SV (OSKC) | ML (+SM) EB (S) (+GF) | ↑ APD with adrenergic stimulation, ↓ or absent IKs, ↑ sensitivity to proarrhythmic drugs. | [60] | ||
| LQT2 | KCNH2 | A561T | LV (OSNL) | EB (FA) | ↑ APD/FPD, ↓ IKr, EADs with Ikr blockers; phenotypes improved with Ikr enhancers. | [61] | |
| A614V | RV (OSK + VPA) | EB (SP) | ↑ APD, ↓ IKr, EADs; phenotypes ameliorated by Ca2+-channel blockers or KATP-channel openers. | [62] | |||
| R176W | RV (OSK) | END-2 | ↑ APD/FPD, ↓ IKr, and increased sensitivity to pro-arrhythmic drugs. | [63] | |||
| A561V | EPS (OSK+SV40L, OSKCNL) | EB (SP) | ↑ APD/FPD, ↓ IKr, defective hERG trafficking; phenotypes reversed with ALLN. | [64] | |||
| A561P | EPS (OSKCNL+ SV40LT) | ML (+GF) | ↑ APD, ↓ IKr current, defective hERG trafficking. | [65] | |||
| LQT3 | SCN5A | F1473C | LV (OSKC) | EB (S) (+GF) | ↑ INaL, ↑ APD; phenotypes improved with increased pacing rate. | [66] | |
| V1763M | S-mRNA (OSKCL) | EB (SP) | ↑ INaL, ↑ APD, faster recovery from INaL inactivation; phenotype reversed by NaV1.5 blockade. | [67] | |||
| V240M, R535Q | RV (OSKC) | END-2 | Insignificant ↑ APD, delayed time to peak INa or time to 90% INa inactivation. | [68] | |||
| CPVT1 | RYR2 | F2483I | RV (OSKC) | END-2 | Ca2+ release from SR long after repolarization, frequent DADs with adrenergic stimulation. | [69] | |
| RV (OSKC) | END-2 | Aberrant unitary Ca2+-signaling, ↓ Ca2+-stores, and sensitized adrenergic regulation. | [70] | ||||
| S406L | RV (OSKC) | EB (SP) | ↑ frequency/duration of Ca2+ sparks with adrenergic stimulation; phenotypes restored with dantrolene. | [71] | |||
| M4109R | RV (OSK + VPA) | EB (SP) | ↑ frequency/amplitude of DADs with adrenergic stimulation, phenotypes reversed by β-blockers. | [72] | |||
| P2328S | RV (OSKC) | EB (SP) | ↑ EADs and DADs with adrenergic stimulation, but not during pacing. | [73] | |||
| Q2311D | LV (OSNL) | EB (SP) | DADs at rest and during adrenergic stimulation; phenotypes rescued by CaMKII inhibitors. | [74] | |||
| R420Q | LV/SV (OSKC) | EB (SP) | Immature ultrastructural features, lack of norepinephrine-induced inotropic or lusitropic effects. | [75] | |||
| CPVT2 | CASQ2 | D307H | LV (OSKC) | EB (SP) | Catecholamine-induced DADs, oscillatory arrhythmic prepotentials, and diastolic [Ca2+]i rise. | [76] | |
| Timothy Syndrome | CACNA1C | G406R | RV (OSKC) | EB (SP) | ↑ APD, abnormal calcium transients; phenotypes rescued by Roscovitine, which inactivates Cav1.2. | [77] | |
| Cardiomyopathies | DCM | TNNT2 | R173W | LV (OSKC) | EB (SP) | Abnormal Ca2+ handling, ↓ contractility, myofibrillar disarray; adrenergic challenge worsens phenotypes. | [78] |
| LV (OSKC) | ML (+SM) | ↑ PDE2A/PDE3A leading to compromised β-adrenergic signaling and contractile dysfunction. | [79] | ||||
| LMNA | R225X | LV (OSKC) | EB (SP) | ↑ nuclear blebbing, ↑apoptosis with electrical stimulation; phenotypes rescued by MEK1/2 inhibitors. | [80] | ||
| DES | A285V | RV (OSKC) | END-2 | Abnormal DES aggregations, abnormal calcium handling, abnormal response to inotropic stress. | [81] | ||
| HCM | MYH7 | R663H | LV (OSKC) | EB (SP) | ↑ cell size, abnormal calcium handling, ↑ [Ca2+]i; all phenotypes reversed with verapamil. | [82] | |
| R442G | RV (OSKC) | EBs (+GF) | Sarcomere disorganization, ↑ APD, irregular contraction, ↓ calcium transients in response to caffeine. | [83] | |||
| MYBPC3 | G999‐Q1004del | SV (OSKC) | EB (SP) | ↑ cell size, myofibrillar disarray, ↑ TnT/ANP, ↓ cMyBP‐C; phenotypes rescued by ETA-b. | [84] | ||
| ARVD | PKP2 | A324fs335X | RV (OSK + VPA) | EB (SP) | ↓ PKP2, ↑ PPAR-γ, ↓ plakoglobin, lipid accumulation; phenotypes corrected by GSK3β inhibition. | [85] | |
| c.2484C>T, c.2013delC | EPS (OSKCL) | EB (SP) | Abnormal plakoglobin nuclear translocation and ↓ β-catenin activity; exaggerated lipogenesis and apoptosis with coactivation of PPARα and PPARγ pathways. | [86] | |||
| c.1841T>C | RV (OSKC) | EB (FA) | ↓ PKP2 and plakoglobin expression, ↑ cell size, ↑ lipid content. | [87] | |||
| DMD | DMD | exon 47(48)-50del | LV (OSNL) | EB (SP) (+GF) | ↓ dystrophin expression partially restored by antisense oligonucleotide-mediated exon skipping. | [88] | |
| exon 45-52del | RV (OSKC)/ LV (OSNL) | EB (SP) (+GF) | Dystrophin deficiency, ↑ resting Ca2+, ↑ apoptosis; phenotypes attenuated by membrane sealant. | [89] | |||
| Diabetic CM | N/A | N/A | RV (OSKC) | Not described | Myofibrillar disarray, ↑ lipid accumulation, ↑ oxidative stress, ↓ calcium transient. | [90] | |
| Metabolic Disorders | ALHD Deficiency | ALDH2 | E487K | LV (OSNC) | ML (+GF) | ↑ ROS, ↑ toxic aldehydes, cell cycle arrest, ↑ apoptosis during ischemic injury. | [91] |
| Barth Syndrome | TAZ | c.517delG, c.517ins | RV (OSKC), S-mRNA (OSKML) | EB (SP) | ↑ ROS, abnormal sarcomere assembly, impaired contractility; phenotypes improved with wild-type TAZ or a mitochondria-targeted antioxidant. | [92] | |
| Pompe Disease | GAA | D645E, Y354X | RV (OSKC) | ML (+SM) | Low GAA activity, high glycogen content; phenotypes reversed with recombinant GAA or L-carnitine. | [93] | |
| exon 18del, 1441delT, c.G2237A | LV (OSKCNL) | ML (+SM) | Undetectable GAA, characteristic glycogen-filled lysosomes, impaired Golgi-based glycosylation. | [94] | |||
| c.796C>T, c.1316T>A | SV (OSKC) | EB (SP) | Weak GAA expression, ↑glycogen content; phenotypes improved with GAA gene therapy. | [95] | |||
| HLHS | UKN | UKN | LV (OSKC) | EB (SP) (+GF) | ↓ cardiac differentiation, ↑ fetal gene expression, myofibrillar disarray, abnormal calcium handling. | [96] | |
| Myocarditis | N/A | N/A | LV/SV (OSKC) | ML (+SM) | Irregular/ceased beating, ↑ cell death; phenotypes improved with antiviral therapy. | [97] |
4.1 Arrhythmic Syndromes/Channelopathies
Early attempts at using hiPSC-CMs to model cardiovascular diseases began with looking at arrhythmic syndromes/channelopathies because of their clear, distinct, and easily assessable phenotypes both clinically and in cell culture. Of these, familial long QT syndrome (LQTS) and catecholaminergic polymorphic ventricular tachycardia (CPVT) were the most commonly modeled diseases, aside from the very rare conditions of Jervell and Lange-Nielsen syndrome (JLNS)60 and Timothy syndrome.77
4.1.1. Familial Long QT Syndromes
Familial LQTS is an arrhythmic condition in which mutations in either the ion channel proteins or other proteins that participate in the formation of the cardiomyocyte’s action potential cause prolongation of action potential duration (APD) and corresponding QT interval on a surface electrocardiogram (ECG).98 The latter is associated with an increased risk of life-threatening arrhythmia (i.e., Torsade de Pointes; TdP). Using the hiPSC-CMs, investigators have modeled LQTS based on the premises that neither the traditional heterologous expression systems nor transgenic animal models can fully simulate diseased human cardiomyocytes due to species-dependent differences in the electrophysiological (EP) characteristics of cardiomyocytes.99
Moretti et al. first demonstrated the feasibility of modeling type 1 LQTS (LQT1) using hiPSC-CMs from a patient with a missense mutation (R190Q) in the KCNQ1 gene.57 Such a mutation was found to cause a trafficking defect in the Kv7.1 channel protein, leading to 70% reduction in the slowly activating delayed rectifier potassium current (Iks), prolongation of APD, and an increase in arrhythmia with β-adrenergic agonists in the diseased hiPSC-CMs. Ma et al. subsequently showed that hiPSC-CMs carrying a different KCNQ1 mutation (exon 7 del) not only faithfully recapitulated patients’ clinical phenotypes but also responded to ML277, a novel drug under development for arrhythmia suppression.59 Egashira et al. performed EP testing on hiPSC-CMs derived from a young survivor of sudden cardiac death (SCD) with clinical suspicion of LQT1.58 They found a novel KCNQ1 mutation (1893delC) associated with a trafficking defect in Kv7.1 that was thought to be the culprit of the patient’s progressive clinical course. These findings demonstrated the diagnostic and predictive potential of patient-specific hiPSC-CMs despite their relatively immature ion channel profile. Although Iks is significantly reduced in hiPSC-CMs compared to adult cardiomyocytes, these cells exhibit repolarization reserve, an important concept explaining why some Iks inhibitors alone do not cause appreciable APD prolongation unless Ikr is also compromised.100 With further improvement in cardiomyocyte maturation protocols (e.g., pacing), we are hopeful that these cells will become even more robust for modeling LQT1.101
Type 2 LQTS (LQT2) is the second most common LQTS following LQT1 characterized by mutations in the KCNH2 gene that lead to disruption of the human ether-a-go-go related gene (hERG) potassium channel and consequent reduction in the rapidly activating delayed rectifier potassium current (Iks).98 Itzhaki et al. first reported that hiPSC-CMs derived from a patient carrying an A614V KCNH2 mutation exhibited reduced Ikr, increased APD, and early afterdepolarizations (EADs), leading to triggered activities that were consistent with the patient’s history of prolonged QT and TdP.62 Matsa et al. further demonstrated that hiPSC-CMs derived from an asymptomatic disease carrier of G1681A KCNH2 mutation exhibited only APD prolongation but no catecholamine-induced EADs, consistent with the patient’s clinical course.61 Furthermore, allele-specific knockdown of the mutated mRNAs that left intact wild-type mRNAs was able to rescue the LQT2 phenotype at the cellular level, further confirming the dominant negative effect of this particular mutation that is thought to impair hERG glycosylation, maturation, trafficking, and degradation.102 These findings emphasized the importance of understanding the functional ramifications of different genetic defects at the molecular/cellular level so that novel, targeted therapeutics can be developed accordingly in a personalized fashion.
Type 3 LQTS (LQT3) comprises 1-2% of all familial LQTs and is caused by gain-of-function mutations in the SCN5A gene, which encodes for the alpha subunit of the Nav1.5 sodium channel.98, 103 Compared to modeling of LQT1 and LQT2, LQT3 modeling has met with more difficulties in correlating hiPSC-CM and clinical phenotypes. In a LQT3 patient with both a de novo F1473C SCN5A mutation and a K897T KCNH2 gene polymorphism, the APDs of LQT3 hiPSC-CMs via a current clamp were not measurable because of a relatively depolarized diastolic membrane potential in these immature hiPSC-CMs.66 Fatima et al. likewise had difficulty showing increased ADP in LQT3 hiPSC-CMs derived from patients carrying different SCN5A mutations (p.V240M and p.R535Q) because of variations in the EP parameters among individual hiPSC-CMs.68 These findings underscored the current limitations of hiPSC models, including the inability to accurately recapitulate all clinical disease thus far.
4.1.2. Catecholaminergic Polymorphic Ventricular Tachycardia
CPVT is an inherited arrhythmic disorder with genetic defects in calcium handling proteins. Clinically, CPVT often manifests as life-threatening arrhythmia triggered by conditions of increased catecholamine levels (e.g., physical exertion and emotional stress).98 CPVT1 has causal mutations in the ryanodine receptor isoform 2 (RYR2), whereas CPVT2 has causal mutations in the calsequestrin isoform 2, the major luminal calcium binding protein in the sarcoplasmic reticulum (SR). Early studies on hiPSC modeling of CPVT1 showed that hiPSC-CMs with a mutation in the FK506-binding protein 12.6 (FKBP12.6)-binding domain of RYR2 (e.g., F2483I) exhibited characteristic delayed afterdepolarizations (DADs) upon catecholamine stimulation.69 The same mutation was found to cause decreased Ca2+ storage in the SR, as well as increased gain in the calcium-induced calcium release mechanisms.70 These changes were consistent with an increased propensity for abnormal Ca2+ release during diastole which contributed to the development of DADs via the sodium-calcium exchanger. Itzhaki et al. further contributed to the understanding of the pathogenesis of CPVT1 by showing that hiPSC-CMs from a patient with a M4109R RYR2 mutation had a significantly lower threshold for store-overload-induced Ca2+ release (SOICR), which made the patient more susceptible to ventricular arrhythmia as observed clinically.72 Jung et al. also demonstrated that dantrolene could effectively reverse the CPVT phenotypes by stabilizing the N-terminus of RYR2, whose interaction with the central region modulates the closure of RYR2 or Ca2+ leakage during diastole.71
Collectively, these findings underscored the need to fully understand the functional consequences of a given mutation so as to design effective therapies. This point is highly pertinent to CPVT and precision medicine because both of its subtypes (CPVT1 and CPVT2) clinically present themselves similarly, and yet their underlying Ca2+ handling disturbances were different due to their distinct mutations. Indeed, CPVT2 hiPSC-CMs had significantly decreased Ca2+ buffering capacity than CPVT1 hiPSC-CMs (or caffeine-mediated Ca2+ release) due to their mutated CASQ2, whereas the latter hiPSC-CMs had more altered sensitivity to ryanodine due to their mutated RYR2 proteins.75 These functional differences are important and suggest that the treatments for patients with different CPVT subtypes may need to be tailored.
4.2 Familial Cardiomyopathies
Modeling of cardiomyopathies using hiPSC-CMs is by far easier with familial cardiomyopathies than acquired cardiomyopathies because the disease-causing gene mutations are known in most circumstances. Familial cardiomyopathies including dilated cardiomyopathy (DCM), hypertrophic cardiomyopathy (HCM), and arrhythmogenic right ventricular dysplasia/ cardiomyopathy (ARVD/C) will be specifically discussed in this section.
4.2.1. Dilated Cardiomyopathy
DCMs are the most common cardiomyopathies, with familial DCMs making up approximately half of the cases.104 In a pioneering study, Sun et al. used hiPSC-CMs to model a family of 7 patients spanning 3 generations, some of whom had a heterozygous point mutation (R173W) in the troponin T type 2 (TNNT2) gene, while others served as controls.78 Compared to control iPSC-CMs, the diseased hiPSC-CMs had impaired calcium handling and reduced contractility. Despite the immaturity of the derived hiPSC-CMs, this study paved the way for a subsequent study by Wu et al., which showed more mechanistically that the mutated TNNT2 caused the cardiomyopathic phenotypes by nuclear translocation, followed by epigenetic activation of the phosphodiesterase 2A/3A (PDE2A/PDE3A) genes leading to contractile dysfunction.79 These studies demonstrated the ability of hiPSC technology to enable mechanistic studies on patient-specific cardiomyocytes.
Familial DCMs caused by mutations of other genes such as lamin A/C and desmin have also been successfully modeled using hiPSC-CMs.80, 81 Most recently, DCMs due to mutations that truncate the massive sarcomere protein titin (TTNtvs) were modeled using hiPSC-derived 3-dimensional cardiac microtissues (CMTs), which had the advantage of yielding more mature cardiac phenotypes than a monolayer of hiPSC-CMs.105 In this study, patient-derived CMTs with TTNtvs developed sarcomere insufficiency, impaired contractile response to mechanical or beta-adrenergic stress, as well as attenuated growth factor signaling activation. Interestingly, CMTs with I-band TTNtvs experienced less contractile deficits than those with A-band TTNtvs because of differential splicing favoring partial omission of the mutated I-band exons, thus providing an explanation for why DCM patients with I-band TTNtvs tend to fare better clinically than those with A-band TTNtvs. Here, the hiPSC technology enables prognostication of titin-related DCMs, an accomplishment that is in sync with the goals of precision medicine.
The hiPSC-based studies of DCM so far serve as a great starting point for further investigation of this disease in a biologically relevant human cell culture model. It is exciting to find that the diseased hiPSC-CMs recapitulate some clinical phenotypes of the patients from which the cells were derived, despite their relative immaturity. Furthermore, the findings obtained from these cells should lead to more testable hypotheses. Further studies will be needed to confirm whether these cells fully recapitulate every aspect of human DCM, as well as to make the full maturation of hiPSC-CMs possible so that they can best simulate adult-onset cardiomyopathies.
4.2.2. Hypertrophic Cardiomyopathy
HCM manifests clinically as asymmetrical left ventricular hypertrophy with increased risks of heart failure and life-threating arrhythmia. In a seminal study, Lan et al. first showed the feasibility of modeling HCM using hiPSC-CMs derived from a family of 10 individuals with an autosomal dominant missense β-myosin heavy chain (MYH7) mutation (A663H).82 The diseased hiPSC-CMs were found to recapitulate numerous HCM phenotypes including cellular enlargement, sarcomere disarray, and arrhythmia secondary to impaired Ca2+ handling abnormalities. Verapamil, a calcium channel blocker, was found capable of preventing the HCM phenotypes by improving Ca2+ regulation. A subsequent study modeling a different missense MYH7 mutation (R442G) confirmed Ca2+ handling abnormalities and arrhythmic potential in the diseased hiPSC-CMs and added whole transcriptome sequencing results to implicate the potential roles of WNT1, FGF, and Notch signaling pathways in the pathogenesis of HCM, some of which could serve as potential therapeutic targets for the treatments of HCM beyond beta-blockers and calcium channel blockers.83
4.2.3. Arrhythmogenic Right Ventricular Dysplasia/Cardiomyopathy
ARVD/C is an inherited heart disease characterized by both fibrofatty infiltration and cardiomyocyte loss predominantly in the right ventricle and is associated with an increased risk of SCD.104 Modeling of ARVD/C is challenging because of its adult-onset phenotype. This issue was particularly addressed in a pioneering study by Kim et al., which showed that hiPSC-CMs from ARVD/C patients (homozygous c.2484C->T mutation in the desmosomal plakophilin 2 (PKP2) gene) had abnormal plakoglobin nuclear translocation and decreased β-catenin activity in baseline cardiogenic conditions, but failed to recapitulate the pathological phenotypes of ARVD/C (e.g., exaggerated lipogenesis, cardiomyocyte apoptosis, and significant EP disturbances) unless the diseased hiPSC-CMs were metabolically matured using a lipogenic cocktail consisting of insulin, dexamethasone, and 3-isobutyl-1-methilxantine with rosiglitazone and indomethacin added.86 Ma et al., further demonstrated that hiPSC-CMs derived from patients with a c.1841T>C PKP2 mutation needed to be exposed to adipogenic differentiation medium in order to develop significant lipogenesis.87 More recently, Asimaki et al. showed that iPSC-CMs from plakophilin-2 mutation patients can be used to complement a zebrafish model for high throughput screening of drugs that modulate aberrant trafficking of intercalated disc proteins.106 These studies affirmed the challenges of modeling adult-onset diseases using immature hiPSC-CMs and called for further efforts to structurally and metabolically mature the hiPSC-CMs.
4.3. Cardiometabolic Disorders
hiPSC-CMs have been used to model metabolic disorders with cardiac complications, including Pompe disease,93-95 mitochondrial aldehyde dehydrogenase deficiency,91 diabetic cardiomyopathy,90 Barth syndrome,92 and Dannon disease.107 In the following sections, we will discuss a sampling of these cardiometabolic disorders.
4.3.1. Pompe Disease
Pompe disease is a glycogen storage disease with autosomal recessive mutations in the lysosomal acid α-glucosidase (GAA) gene.108 Patients with this condition have insufficient GAA activity, leading to increased accumulation of glycogen in the nerves and the heart, with the latter typically manifesting as cardiomegaly, cardiomyopathy, and arrhythmia. This disease is found more frequently but less severely in the infantile-onset form than the adult-onset form. Huang et al. first showed that hiPSC-CMs derived from patients with the infantile form of Pompe disease had reduced GAA level, leading to increased glycogen content, swollen mitochondrial cristae, increased autophagosome-like structures, and mitochondrial dysfunction manifested as lower oxygen consumption rate and extracellular acidification rate.93 Raval et al. introduced an additional mechanistic angle by showing that hiPSC-CMs from the infantile-onset form had deficiency of Golgi-based glycosylation defects, which by speculation could increase membrane Ca2+ permeability and stimulate downstream hypertrophic signaling.94 Importantly, these hiPSC-based disease models also enabled the screening of drugs (e.g., Carnitine) that could serve as effective adjuncts or alternatives to enzyme replacement therapy for Pompe disease.93
4.3.2. Mitochondrial Aldehyde Dehydrogenase 2 Deficiency
Mitochondrial aldehyde dehydrogenase 2 (ALDH2) deficiency is commonly seen in patients of Asian descents. This disease can be easily appreciated in social encounters that involve alcohol ingestion. Patients with ALDH2*2 polymorphism (E487K) have reduced ALDH2 enzyme activity, which leads to facial flushing upon alcohol ingestion.91 This polymorphism also has been linked to increased risk of CAD109 and complications from diabetes.110 Ebert et al. first used hiPSC-CMs derived from a cohort of sex-matched East Asian individuals carrying this polymorphism and showed that these cells experienced more oxidative stress and apoptosis secondary to toxic aldehyde buildup during ischemic injury.91 These effects were mediated by reactive oxygen species (ROS)-mediated upregulation of JNK, such that administration of JNK inhibitor (JIP) restored ROS level as well as viability post-ischemia. This study was the first hiPSC study to investigate a genetic polymorphism with strong ethnic prevalence. Studies such as this allow for a better elucidation of the roles of ethnicity in the development and progression of cardiovascular diseases.
4.3.3. Diabetic Cardiomyopathy
Diabetic cardiomyopathy, a long-term complication in patients with diabetes, was recently modeled by Drawnel et al.90 In this study, hiPSC-CMs from healthy donors were specifically metabolically matured with insulin and fatty acid supplementation in the absence of glucose to simulate adult metabolism. Following exposure to a diabeto-mimetic medium, the hiPSC-CMs then developed phenotypes of diabetic cardiomyopathy characterized by cellular hypertrophy, upregulation of hypertrophic genes, increased BNP release, myofilament disarray, and lipid accumulation/peroxidation. By contrast, the hiPSC-CMs derived from patients with type 2 diabetes were able to develop similar pathological phenotypes in the absence of the diabeto-mimetic medium. These results clearly showed a genetic component to the development of diabetic cardiomyopathy because the diseased hiPSC-CMs behaved differently at baseline from those of normal hiPSC-CMs. Conceivably, many other cardiomyopathies thought to be acquired could also have a genetic component and are worth studying using hiPSC-CMs (e.g., chemotherapy-induced cardiomyopathy,111 alcohol cardiomyopathy, viral cardiomyopathy81).
4.4. Diseases of Endothelial Cells or Vascular Smooth Muscle Cells
In addition to diseases affecting cardiomyocytes, many diseases that affect ECs or vascular SMCs are also ideal targets for hiPSC modeling for the following reasons.112 First, there are species-dependent differences in EC/SMC characteristics and functions, which make animal models alone insufficient for fully capturing disease phenotypes. Secondly, there is limited access to primary ECs/SMCs because the isolation procedures are invasive. Third, primary ECs/SMCs lose primary phenotypes after long-term cell culture. For these reasons, the limitless supply of patient- and disease-specific ECs/SMCs greatly facilitates the studying of EC/SMC-related diseases in cell culture.
4.4.1. Williams-Beuren Syndrome / Supravalvular Aortic Stenosis
Williams-Beuren syndrome (WBS) is a rare primary genetic vascular disorder characterized clinically by progressive arterial stenoses.113 Patients with this condition have a 1.5-1.8 Mb microdeletion on chromosome 7q11.23 encompassing 26-28 contiguous genes encompassing the elastin (ELN) gene. Patients with congenital supravalvular aortic stenosis (SVAS) have mutations only in the ELN gene but have cardiovascular lesions that are identical to those accompanying WBS, which has other additional craniofacial or neurobehavioral features.114 In both conditions, ELN mutations are thought to disinhibit SMC proliferation and cause arterial stenosis.
Ge et al. first derived hiPSC-SMCs from patients with a 4-nucleotide insertion mutation in exon 9 and showed that these cells had fewer organized networks of smooth muscle alpha actin filament bundles, conferring a less mature phenotype and increased proliferation rates.115 These pathological features were rescued by elastin recombinant protein, thus confirming elastin haploinsufficiency as the cause. Subsequently, hiPSC-SMCs derived from patients with WBS were found to have the same pathological features that were later confirmed by Kinnear et al.116 The use of hiPSC platform was a significant breakthrough, as hemizygous transgenic mouse models of WBS failed to exhibit supravalvular aortic stenosis.117 Importantly, the authors found the drug rapamycin, an antiproliferative agent, to be effective at reversing the pathological phenotypes, demonstrating the potential use of this FDA-approved drug for the prevention of arterial stenosis following surgical correction.
4.4.2 Calcific Aortic Valve Disease in Association with Congenital Bicuspid Valve
Calcific aortic valve disease is responsible for more than 100,000 valve transplants every year in the United States. About 50% of aortic stenosis leading to aortic valve replacement in adults is due to congenital bicuspid valve, which occurs in 1-2% of the population.118 It is long recognized that in this disease condition, the aortic side of the valve is associated with reduced shear stress, increased calcification, and downregulation of NOTCH1 signaling, whereas the ventricular side of the valve experiences the exact opposite trends.119-121 In an elegant study, Theodoris et al. derived hiPSC-ECs from 3 patients with calcific aortic valve disease secondary to congenital bicuspid valve. These patients have a nonsense heterozygous mutation (N1) leading to NOTCH1 haploinsufficiency.122 Under shear stress, this NOTCH signaling defect led to epigenetic dysregulation and subsequent aberrant upregulation of pro-osteogenic and inflammatory signaling in these cells, consistent with the clinical observation of more calcification and inflammation on the aortic side of the valve. This study demonstrated the ability of hiPSC-ECs to recapitulate the disease clinical phenotype and emphasized the need to evaluate endothelial biology under shear stress, which is important for eliciting disease phenotypes.
4.4.3. Hutchinson-Gilford Progeria Syndrome
Hutchinson-Gilford progeria syndrome (HGPS) is a genetic condition characterized by premature aging beginning in childhood.123 A single point mutation in the LMNA gene leading to the formation of progerin, a truncated splicing mutant of the nuclear protein lamin A, is thought to underlie this condition. Indeed, hiPSC-SMCs taken from patients with HGPS had appearance of premature senescence phenotypes associated with vascular aging, specifically characterized by increased senescence-associated-β-Gal staining, reduced telomere length, reduced number of Ki67-positive cells, and compromised cell proliferation.124 Progerin was shown to downregulate DNA-dependent protein kinase catalytic subunit (DNAPKcs) expression, which served as a marker for premature vascular aging. Another study showed that hiPSC-derived mesenchymal stem cells (hiPSC-MSCs) survived less well than control hiPSC-MSCs under starvation and hypoxia, suggesting that perhaps progerin exerted its pathological effects through inhibiting the stem cell pool in the vascular compartment.125 These results highlighted the utility of differentiating hiPSCs into multiple cell types within the vasculature so that the contribution from each cell type to the vascular condition can be precisely isolated.
4.5. Genome-Editing Strategies for Proper Isogenic Control and Creation of Mutation
Most of the aforementioned studies used hiPSC derivatives from healthy patients or non-afflicted relatives of the diseased study patients as negative controls. However, the genetic landscapes of the control hiPSCs and the diseased hiPSCs are not equivalent beyond the mutations of interest, and these differences could easily muddle the study findings. For these reasons, genome-editing strategies that employ zinc-finger nucleases (ZFNs), transcription activator-like effector nucleases (TALENs), or clustered regulatory interspaced short palindromic repeat (CRISPR)/Cas-based RNA-guided DNA endonucleases have been employed to create isogenic controls from the diseased lines.126, 127 Alternatively, a desired mutation can be introduced into a normal hiPSC line to create a diseased line in cell culture, bypassing entirely the need to recruit patients with the same mutation—a task that can be very challenging for rare genetic disorders. Phenotypic rescue following correction of the mutation can then help to confirm the specificity of the mutation in contributing to the diseased phenotype.
Karakikes et al. recently demonstrated the use of TALEN-mediated homology-directed repair in hiPSC-CMs derived from a patient with p.R14del mutation in the phospholamban (PLN) gene.128 Upon differentiation, the DCM phenotypes characterized by calcium handling abnormalities and abnormal cytoplasmic distribution of PLN were evident in the patient’s hiPSC-CMs. These phenotypes were ameliorated in the TALEN-corrected hiPSC-CMs, further confirming the causal relationship between the mutation and the DCM phenotypes. In a different application, Wang et al. successfully used ZFNs to create diseased hiPSC-CMs carrying LQT1 (KCNQ1) and LQT2 (KCNH2) mutations from wild-type control hiPSC-CMs. These mutated lines had prolonged APDs compared to isogenic controls, consistent with the expected LQTS phenotypes.129 Such an approach for studying the contributions of a specific gene mutation is expected to become more popular due to the difficulties gaining access to patients with rare genetic diseases. Ultimately, knowing exactly what each gene mutation or polymorphism can do for each patient clinically will be the first step towards developing tailored therapies in the practice of precision medicine.
5. Drug Discovery Employing hiPSC Platforms
The advent of hiPSC technology has made it possible to pursue preclinical drug testing and discovery in a manner that significantly deviates from traditional platforms, namely, animal disease models and heterologous expression systems. This shift of paradigm has been supported by both researchers and pharmaceutical companies, who have been frustrated with the inability to deliver new, safe, and efficacious medications to address unmet medical needs. As the hiPSC technology continues to mature, many hiPSC platforms are now being incorporated into the standards for drug safety testing and major pharmaceutical drug development pipelines.
5.1. hiPSC-Based Platforms for Cardiotoxicity Testing
Cardiotoxicity such as drug-induced QT prolongation can cause life-threatening arrhythmia (e.g., TdP) and is the most common reason for late-stage drug attrition and market withdrawal.130 For these reasons, the International Conference on Harmonization (ICH) in 2005 issued a guidance document (E14), which mandated all new drugs to undergo a dedicated ECG study in healthy volunteers called “the Thorough QT/QTc (TQT) study.”131 Since its implementation, no drug with negative TQT results has been associated with TdP. However, the TQT study is costly and has been criticized for its low specificity, leading to the withdrawal of many efficacious QT-prolonging drugs that were actually non-proarrhythmic.
The ICH in its S7B guidance document likewise required nonclinical assessment of drugs’ arrhythmogenic potential using both the hERG assay and a non-rodent QT interval study.131 The former assesses a compound’s ability to block the hERG current (Ikr) in transformed cell lines that overexpress hERG, namely Chinese hamster ovary (CHO) cells or human embryonic kidney (HEK) cells, even though they have largely different ion channel profiles than adult human cardiomyocytes. In an effort to make cardiotoxicity testing more biologically relevant, the FDA-sponsored Cardiac Safety Research Consortium and the non-profit Health and Environmental Sciences Institute (HESI) have recommended as a part of the Comprehensive in vitro Proarrhythmia Assay (CiPA) initiative to include both in silico computational modeling and hiPSC-CMs into the non-clinical drug cardiotoxicity assessment.132 Computer simulations that incorporate experimentally-determined behaviors of multiple ion channels have been shown to predict clinical risks of arrhythmia better than the hERG assay.133 Likewise, hiPSC-CMs have been validated for their ability to recapitulate the electrophysiological phenotypes of many genetic arrhythmic disorders.98 Compared to in silico modeling, hiPSC-CMs can provide information on proarrhythmia potential in addition to the long QT phenotype.
For drug toxicity testing, multielectrode array (MEA) has been validated for detecting drug-induced arrhythmia on hiPSC-CMs based on field potential recordings.134 The impedance-based platforms also offer assessment of cardiotoxicity by characterizing parameters related to the beating of cardiomyocytes.135 Guo et al. applied one such platform to assess the beating rate and amplitude of hiPSC-CMs exposed to different classes of cardioactive compounds and found that these cells developed the expected responses based on their known pharmacological actions.136 Interestingly, arrhythmia induced by terfenadine, a drug known for torsadogenicity, was observed only after extended drug treatment and monitoring. Importantly, this platform was able to differentiate torsadogenic drugs from those that prolonged QT but did not cause TdP. A human cardiomyocyte arrhythmic risk (hCAR) model based on this platform, incorporating parameters such as drug-induced arrhythmic beating and change in beating rate, and the known effective serum therapeutic concentration of each drug, provided better predictability for torsadogenicity in humans than either the hERG assay or clinically observed QT prolongation.137
In contrast to the electrode-based techniques, Sirenko et al. used time-lapsed fluorescence imaging in conjunction with fluorescent dyes to screen drugs for their effects on both viability and Ca2+ influx patterns that are characteristic of known ion channel blockers.138 A scaled up version of this system incorporating the Toxicological Prioritization Index for data analysis enabled the effective ranking of numerous drugs in terms of cardiotoxicity.139 However, the use of fluorescent labels in this platform raised the potential for both phototoxicity and undesirable interactions between the labels and the test compounds, and these effects needed to be carefully excluded before cardiotoxicity could be fairly interpreted.
Most recently, a label-free video-based platform that captures and analyzes the beating patterns of cardiomyocytes has been developed to assess cardiotoxicity in terms of changes in beat rate, beat duration, and contractile arrhythmia.140 The major advantages of this system are its compatibility with standard multiwell plates, sterility, and and its label-free noninvasive nature. Furthermore, drug effects can be easily interrogated longitudinally without any compromise to cell integrity. The system is limited by the time lag between video capture from one well to the next, which makes studying fast drug responses challenging. Further optimization of this platform should include a microfluidic drug delivery system to allow the analysis of fast-drug responses. Ultimately, a multimodal platform that can simultaneously assess morphology, contractility, arrhythmia, biosynthetic function, and metabolism will be the ideal platform for drug developers (Figure 2).
Figure 2.
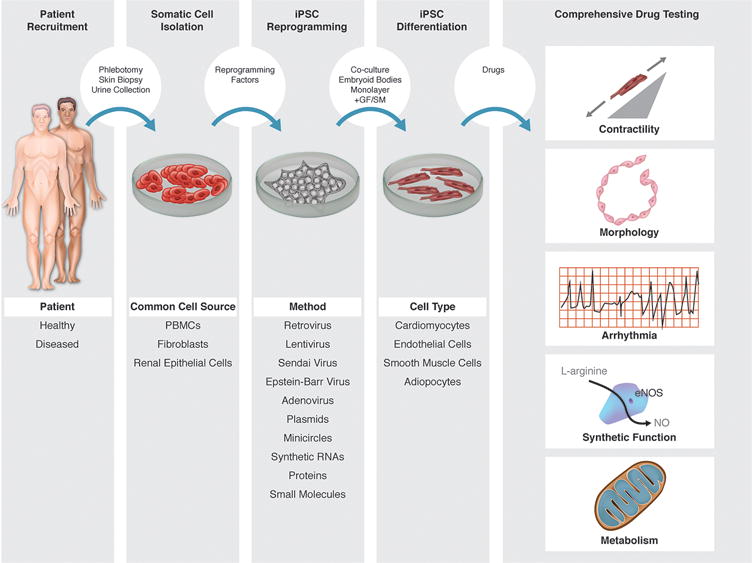
hiPSC-based platforms for drug development. Various cell types can be derived from patients’ somatic cells using different techniques in multiple steps: somatic cell isolation, hiPSC reprogramming, and hiPSC differentiation. The resultant hiPSC-derived cells can be subjected to comprehensive drug testing not limited to assessments of contractility, arrhythmia potential, metabolism, cell morphology, and synthetic function.
Compared to the high costs associated with animal experiments,141 hiPSC-based drug discovery platform can produce high-quality drugs at a relatively low cost. It may be possible to save up to $100 million per drug development process and reduce the current 10% attrition rate.142 Coupled with the use of chemically defined media, instead of commercial media, the cost of hiPSC culture and cardiac differentiation can be further reduced by as much as 75 %.143 Thus, from a budgetary standpoint, there is also a strong financial impetus to develop a hiPSC-based platform for drug development.
5.2. Predicting Clinical Drug Response Using hiPSC Derivatives
For hiPSC technology to deliver its great promises for precision medicine, we also need to look at how well patient-specific hiPSC derivatives can recapitulate the patients’ clinical disease phenotypes, including their responses to administered drugs. The hiPSC-CMs in most studies have demonstrated predictable responses to most cardioactive drugs based on their pharmacological mechanisms of action, despite concerns over their immature phenotypes. If the patients’ own hiPSC-CMs responded to administered drugs in a similar fashion and to the same degree as the patients taking the drugs themselves, however, then the hiPSC technology would become an even more effective and powerful tool in the practice of precision medicine.
Stillitano et al. recently conducted a prospective study to investigate drug-induced LQTS in 126 healthy patients, who individually underwent a pharmacological challenge of a single oral dose of sotalol 80 mg.144 The hiPSC-CMs derived from 17 patients with extreme clinical responses to sotalol in terms of QT prolongation also had corresponding extreme measures of APD. LQT-related arrhythmia were observed in hiPSC-CMs derived from 5 of 7 patients with the most significant responses to sotalol clinically. These results provided some initial evidence that hiPSC-CMs could accurately predict clinical drug response and called for more incorporation of hiPSC-CMs into the early stages of clinical trials so that more data regarding their predictability of cardiotoxicity could be gathered.10
Since the initial discovery of hiPSC technology, some drugs that have been developed through the hiPSC platform are now making their appearance in clinical trials, mainly in the area of neurological diseases.145 Indeed, in the field of Alzheimer’s disease, BMS-986168, a tau-specific antibody is now undergoing phase 1 toxicity testing in healthy volunteers after initial development through the use of hiPSC-derived cortical neurons.146 Retigabine, an FDA-approved anticonvulsant drug capable of suppressing the hyperexcitable phenotypes of different hiPSC models of amyotrophic lateral sclerosis is undergoing phase 2 trial, bypassing further animal model validations.147 These trials offer valuable insights into how well the hiPSC derivatives can predict patient-specific drug responses. These studies also highlighted an important niche for the hiPSC-based platforms in drug repurposing.
5.3. hiPSC-Based Tissue Engineering Platforms for Drug Development
Advances in tissue engineering and microfabrication have provided additional avenues for advancing hiPSC-based strategies for drug development. Various groups have demonstrated the utility of a 3D hiPSC culture to better simulate adult cardiac tissue than a 2D monolayer of hiPSCs. In a pioneering study, Eschenhagen et al. developed a 3D model of contracting heart tissue by culturing embryonic chick cardiomyocytes in a collagen matrix.148 Later refinement of this technique included changing collagen to fibrin for faster polymerization and more uniform cell distribution, introduction of elastic silicone posts to allow physiological auxotonic contraction, and adaptation to a standard 24-well format suitable for drug screening.149 These 3D cardiac tissues display well-organized sarcomeres, express adult contractile proteins, display Frank-Starling mechanism, respond well to adrenergic stimulation, and can be readily adapted for hESC-CM/hiPSC-CM-based applications.150, 151 Zhang et al. also showed that 3D patches made of hESC-CMs casted in fibrinogen/Matrigel mixtures exhibited matured conduction velocities, sarcomere length, and gene expression associated with contractile function compared to their 2D counterparts.152 Nunes et al. demonstrated that aligned 3D cardiac tissue constructs (biowires) made of hiPSC-CMs mixed with collagen could be electrically stimulated to improve sarcomeric organization, conduction velocity, as well as electrical and Ca2+ handling properties.153 Most recently, a microphysiological system (MPS) made of an aligned 3D cardiac tissue construct flanked by microchannels to simulate capillary exchange was found to yield drug responses that are more comparable to human tissue than a 2D counterpart.154 Coupled with high content imaging and multimodal phenotypic readouts, such a robust platform, if made more widely available, can help accelerate the drug discovery process already aided by the hiPSC technology.155
5.4. Comparison of hiPSC and Traditional Models of Arrhythmia Risk Assessment
Prior to the development of hiPSC technology, numerous cellular, multicellular, isolated perfused heart, and animal models have been used to predict drug-induced arrhythmia risks in humans. There has not been an all-purpose “swiss knife” equivalent for proarrhythmia risk assessment because each model has its own advantages and disadvantages, and each captures only a partial aspect of proarrhythmia in humans. For example, the hERG assay has been popular with most early drug development programs because of their simplicity, low cost, high sensitivity, and potential for medium-to-high throughput when coupled with automated electrophysiological or noninvasive fluorescence-based platforms.156 However, they are known for limited specificity, as some drugs can modulate arrhythmia risks by interacting with ion channels besides hERG, thereby generating either false positive (e.g., verapamil) or false negative (e.g., afluzosin) results.9, 157 The hERG assay also suffers from a lack of standardization in that the half maximal inhibitory concentration (IC50) determination often varies significantly from lab to lab, depending on protocol-related parameters such as temperature, cell type, and the actual bath drug concentration.157 Furthermore, due to its single-cell nature, the hERG assay misses tissue-level contribution to TdP (e.g., spatial dispersion of repolarization) which can be better interrogated with multicellular preparations such as the arterial perfused wedge preparations. However, these tissue preparations are themselves low throughput and vulnerable to selection bias, depending on where in the heart the tissue slab was prepared from.158 This issue can be addressed using a Langendorf perfused heart model, which allows interrogation of monophasic action potential measurements both transmurally and around the ventricles.159 Moreover, repolarization abnormalities such as monophasic action potential duration prolongation, triangulation, instability, and reverse use dependence can serve as additional markers for proarrhythmia.160 The common drawback of the intro models discussed above is the exclusion of neurohormonal and metabolic influences, which are known to modulate arrhythmia risks and can only be studied in intact animal models.
Animal models including rodents, rabbits, dogs, and pigs have all been used to predict drug-induced arrhythmia. Rodent models are generally less relevant than large animal models due to their different molecular and cellular electrophysiology (differences in heart rate, action potential duration, ion channel expression profile, and calcium-handling properties), similar aspects of which are more commonly shared by large animals and humans.98 Non-rodent models, however, incur greater costs and have their own well-described limitations.161 For example, the methoxamine-sensitized anesthetized rabbit model relies on the use of α1-adrenergic stimulation to produce an increase in intracellular Ca2+ concentration, which predisposes the myocardium to drug-induced arrhythmia. Although sensitive, the need for anesthesia alters autonomic tone and influences myocardial electrophysiology. By contrast, the chronic AV block canine (or non-human primate) model does not require anesthesia but relies on the chronic myocardial adaption to artificially introduced third-degree AV block to generate susceptibility to arrhythmia. However, because most patients with drug-induced TdP do not have third-degree AV block, such a model, although more accurate in predicting the effects of many known torsadogens, may have limited predictability for patients with other underlying disease conditions.
The main advantages of using the hiPSC-based cell model for assessing proarrhythmia include the following. First, compared to the CHO and HEK cells used in the conventional hERG assay, hiPSC-CMs retain the complexity of human action potential generation by expressing important ion channels that modulate proarrhythmia risks beyond just hERG. Second, hiPSC-CMs can be derived from patients of varying underlying disease conditions, thus enabling the investigation of disease-specific susceptibility to drug-induced arrhythmia. Finally, genome editing of hiPSC-CMs with specific polymorphisms or mutations can be readily performed to examine their precise contribution to arrhythmia risks. As drug-induced arrhythmia is influenced by multiple risk factors (e.g., genetic predisposition, female gender, and presence of structural heart disease),130 hiPSC-based platforms allow many of these risk factors to be mechanistically investigated at the cellular level. However, it should be noted that hiPSC-CMs can only be as good as a cellular model. Mechanisms or modulating factors that can be better interrogated at the tissue, organ, or organism levels (e.g., spatial dispersion of repolarization, autonomic/metabolic/adrenergic modulation, etc.) cannot be captured with hiPSC-based platforms. Further studies will be needed to determine how they can complement large animal models to constitute a complete assessment of proarrhythmia prior to human cardiotoxicity testing.
6. Challenges and Future Directions
In this review, we have alluded on several occasions to one of the major limitations of the hiPSC technology—the immaturity of hiPSC derivatives. For instance, compared to adult cardiomyocytes, hiPSC-CMs appear rounder and have fewer mitochondria and less organized sarcomeres.162, 163 The gene expression profiles, especially those of contractile proteins, simulate fetal cardiomyocytes.164 Furthermore, the hiPSC-CMs have poorly developed SR and altered calcium handling at early stages of differentiation,165 nonexistent t-tubules,166 automaticity,167 and preference for glucose metabolism over fatty acid metabolism,168 which are all consistent with immature phenotypes. Although various maturation techniques (e.g., electromechanical stimulation,153, 169 overexpression of cardiac genes101 or miR-499,170 exposure to fatty acid supplementation,90 and incorporation of cardiomyocytes into 3D tissue constructs105) have all been tried with varying degrees of success, it is unclear whether the most mature hiPSC-CMs developed in an artificial cell culture environment can achieve the same level of maturity as adult cardiomyocytes in vivo. However, whether hiPSC-CMs can play a significant role in precision medicine depends on their as yet undetermined ability to accurately predict the clinical phenotypes in some or all cases that other existing animal or cellular models cannot. The fact that most hiPSC-based models of genetic disorders seem to exhibit clinically expected characteristics is encouraging. Nevertheless, more focused studies incorporating hiPSCs in clinical trials to correlate patients’ hiPSC-CMs and clinical phenotypes will be necessary to improve confidence into this technology.10
A second major issue concerning hiPSC research is the heterogeneity of hiPSC lines and the cells derived from them. Due to the clonal nature and low efficiency of hiPSC derivation, significant clone-to-clone variations may exist because of varying genetic and epigenetic influences from individual somatic donor cells.39 Furthermore, aneuploidy or subchromosomal variations accumulated during the reprogramming process can lead to varying differentiation capacity and resultant cellular phenotypes.39 Even after differentiation, the cell types derived are often a mixture of cells (e.g., atrial, ventricular, nodal for cardiomyocytes) which can further complicate the analysis. As most of the hiPSC studies thus far included only limited numbers of patient-derived lines, the resultant conclusions, although mostly encouraging, needed to be interpreted with caution as the variations associated with the reprogramming and differentiation processes could be significant. Selection of donor cells with the least accumulated mutations for reprogramming (young versus old),39 use of non-integrating vectors for reprogramming to avoid retention/reactivation of transgenes or insertional mutagenesis,38 employment of differentiation protocols that promote hiPSC homogeneity (EB with forced aggregation),45 and purification for the desired cell types (glucose deprivation for cardiomyocytes)168 are just some examples of efforts to reduce the heterogeneity inherent in the hiPSC platform. Ultimately, even with these efforts, the hiPSC lines could behave heterogeneously because of patient-specific differences in their genomics or epigenomics. Thus, for an hiPSC-based investigation to be truly considered “a trial in the dish”, hundreds of hiPSC lines will need to be generated in order to fully assess cardiotoxicity over diverse genetic backgrounds. In this regard, the establishment of hiPSC repositories at different institutions to store patient-derived hiPSCs along with the patients’ clinical data will be crucial for carrying out meaningful large-scale hiPSC-based drug testing.171
Lastly, for both precision medicine and large-scale drug screening in the pharmaceutical industry, there remains the challenge of how best to scale up the hiPSC platform in order to test tens to hundreds of hiPSC lines and drugs. In this regard, the use of dynamic suspension cultures represents a feasible solution able to accomplish the derivation, expansion, and even differentiation of hiPSCs into beating cardiomyocytes in either Erlenmeyer or spinner flasks.172, 173 The hiPSC yield can be augmented by 25-fold in 10 days, with retention of karyotype stability and pluripotency.174 Coupled with the use of chemically defined media and small molecules instead of recombinant growth factors, such a platform can also reduce the cost dramatically, which is lower on a per cell basis compared to adherent cell cultures.175 On an even larger scale, an automatic, modular robotic platform can be used for the high-throughput derivation, characterization, and differentiation of induced pluripotent stem cells.176 Such a platform can reprogram, expand, and characterize several hundred hiPSC lines per month. More importantly, the system can reduce reagent cost by 5-6-fold and increase productivity by 10-12-fold compared to previous automated systems that worked with a small number of lines at a time. Compared to manually selected hiPSC clones, the pooled polyclonal hiPSC lines derived from this automation had significantly less line-to-line variation that is known to occur with manual hiPSC clone selection. Thus, with further refinement such as system will not only enable large-scale production, characterization, and manipulations of iPSC lines, but will also make the experimental results less prone to technical errors, which is necessary for the objective comparison of a large number of iPSC lines for both drug screen and phenotypic assessment in precision medicine.
7. Conclusion
The Precision Medicine Initiative has created an emphasis for deeper investigations into the functional significances of the patients’ genetic backgrounds. To this end, the hiPSC technology has a unique role from its ability to provide an unlimited supply of various cell types from patients, which otherwise would not be as easily obtained, cultured, or studied. Among other achievements thus far, patient-specific hiPSC derivatives have enabled 1) the successful modeling of a number of cardiovascular disorders that led to valuable new mechanistic insights, 2) the comprehensive cardiotoxicity testing of drugs focusing on proarrhythmic potential instead of QT/APD prolongation alone, and 3) the development of drugs that can be tailored based on patient-specific differences at the cellular level.
As with any other experimental technique, there are fundamental limitations with hiPSCs which may prevent the technology from completely replacing the existing experimental models or drug discovery methods, including clinical trials. As such, the predictability of hiPSC-based platforms will need to be better tested clinically so that a better appreciation of its strengths and weaknesses can be obtained. Ultimately, if used in concert with the other existing approaches, we expect that the hiPSC-based platforms will have a unique and prominent role in advancing the practice of precision medicine.
Acknowledgments
We thank Blake Wu for his assistance with manuscript preparation and Amy Thomas for her artistic rendition of the figures included in this manuscript. Due to space limitation, we are unable to include all of the important papers relevant to hiPSC research, and we apologize to those investigators who have otherwise contributed significantly to this field. This work is supported by research grants from the National Institute of Health T32 training grant (IYC), American Heart Association 13EIA14420025, Burroughs Wellcome Foundation, NIH R01 HL123968, NIH HL130020, NIH R01 HL128170, and NIH R01 HL126527 (JCW).
Contributor Information
Ian Y. Chen, Department of Medicine, Division of Cardiovascular Medicine, Stanford University School of Medicine, Stanford, California
Elena Matsa, Stanford Cardiovascular Institute, Stanford University School of Medicine, Stanford, California
Joseph C. Wu, Stanford Cardiovascular Institute, Stanford University School of Medicine, Stanford, California Department of Medicine, Division of Cardiovascular Medicine, Stanford University School of Medicine, Stanford, California; Department of Radiology, Stanford University School of Medicine, Stanford, California.
References
- 1.Collins FS, Varmus H. A new initiative on precision medicine. N Engl J Med. 2015;372:793–5. doi: 10.1056/NEJMp1500523. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Jaffe S. Planning for US Precision Medicine Initiative underway. Lancet. 2015;385:2448–9. doi: 10.1016/S0140-6736(15)61124-2. [DOI] [PubMed] [Google Scholar]
- 3.Matsa E, Burridge PW, Wu JC. Human stem cells for modeling heart disease and for drug discovery. Sci Transl Med. 2014;6:239ps6. doi: 10.1126/scitranslmed.3008921. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Karakikes I, Ameen M, Termglinchan V, Wu JC. Human induced pluripotent stem cell-derived cardiomyocytes: insights into molecular, cellular, and functional phenotypes. Circ Res. 2015;117:80–8. doi: 10.1161/CIRCRESAHA.117.305365. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Wilson KD, Wu JC. Induced pluripotent stem cells. JAMA. 2015;313:1613–4. doi: 10.1001/jama.2015.1846. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Eschenhagen T, Mummery C, Knollmann BC. Modelling sarcomeric cardiomyopathies in the dish: from human heart samples to iPSC cardiomyocytes. Cardiovasc Res. 2015;105:424–38. doi: 10.1093/cvr/cvv017. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Yamanaka S. Induced pluripotent stem cells: past, present, and future. Cell Stem Cell. 2012;10:678–84. doi: 10.1016/j.stem.2012.05.005. [DOI] [PubMed] [Google Scholar]
- 8.Mercola M, Colas A, Willems E. Induced pluripotent stem cells in cardiovascular drug discovery. Circ Res. 2013;112:534–48. doi: 10.1161/CIRCRESAHA.111.250266. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Liang P, et al. Drug screening using a library of human induced pluripotent stem cell-derived cardiomyocytes reveals disease-specific patterns of cardiotoxicity. Circulation. 2013;127:1677–91. doi: 10.1161/CIRCULATIONAHA.113.001883. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Engle SJ, Puppala D. Integrating human pluripotent stem cells into drug development. Cell Stem Cell. 2013;12:669–77. doi: 10.1016/j.stem.2013.05.011. [DOI] [PubMed] [Google Scholar]
- 11.Katsnelson A. Momentum grows to make ‘personalized’ medicine more ‘precise’. Nat Med. 2013;19:249. doi: 10.1038/nm0313-249. [DOI] [PubMed] [Google Scholar]
- 12.Hayden EC. Technology: The $1,000 genome. Nature. 2014;507:294–5. doi: 10.1038/507294a. [DOI] [PubMed] [Google Scholar]
- 13.Wong AH, Gottesman II, Petronis A. Phenotypic differences in genetically identical organisms: the epigenetic perspective. Hum Mol Genet. 2005;14(1):R11–8. doi: 10.1093/hmg/ddi116. [DOI] [PubMed] [Google Scholar]
- 14.Janssens AC, van Duijn CM. Genome-based prediction of common diseases: advances and prospects. Hum Mol Genet. 2008;17:R166–73. doi: 10.1093/hmg/ddn250. [DOI] [PubMed] [Google Scholar]
- 15.Loscalzo J, Handy DE. Epigenetic modifications: basic mechanisms and role in cardiovascular disease (2013 Grover Conference series) Pulm Circ. 2014;4:169–74. doi: 10.1086/675979. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Smith LE, White MY. The role of post-translational modifications in acute and chronic cardiovascular disease. Proteomics Clin Appl. 2014;8:506–21. doi: 10.1002/prca.201400052. [DOI] [PubMed] [Google Scholar]
- 17.Mummery CL, et al. Differentiation of human embryonic stem cells and induced pluripotent stem cells to cardiomyocytes: a methods overview. Circ Res. 2012;111:344–58. doi: 10.1161/CIRCRESAHA.110.227512. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Yoder MC. Differentiation of pluripotent stem cells into endothelial cells. Curr Opin Hematol. 2015;22:252–7. doi: 10.1097/MOH.0000000000000140. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Mordwinkin NM, Lee AS, Wu JC. Patient-specific stem cells and cardiovascular drug discovery. JAMA. 2013;310:2039–40. doi: 10.1001/jama.2013.282409. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Neofytou E, O’Brien CG, Couture LA, Wu JC. Hurdles to clinical translation of human induced pluripotent stem cells. J Clin Invest. 2015;125:2551–7. doi: 10.1172/JCI80575. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Zhao T, Zhang ZN, Rong Z, Xu Y. Immunogenicity of induced pluripotent stem cells. Nature. 2011;474:212–5. doi: 10.1038/nature10135. [DOI] [PubMed] [Google Scholar]
- 22.Takahashi K, et al. Induction of pluripotent stem cells from adult human fibroblasts by defined factors. Cell. 2007;131:861–72. doi: 10.1016/j.cell.2007.11.019. [DOI] [PubMed] [Google Scholar]
- 23.Yu J, et al. Induced pluripotent stem cell lines derived from human somatic cells. Science. 2007;318:1917–20. doi: 10.1126/science.1151526. [DOI] [PubMed] [Google Scholar]
- 24.Park IH, Lerou PH, Zhao R, Huo H, Daley GQ. Generation of human-induced pluripotent stem cells. Nat Protoc. 2008;3:1180–6. doi: 10.1038/nprot.2008.92. [DOI] [PubMed] [Google Scholar]
- 25.Lowry WE, et al. Generation of human induced pluripotent stem cells from dermal fibroblasts. Proc Natl Acad Sci U S A. 2008;105:2883–8. doi: 10.1073/pnas.0711983105. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Takahashi K, Yamanaka S. Induction of pluripotent stem cells from mouse embryonic and adult fibroblast cultures by defined factors. Cell. 2006;126:663–76. doi: 10.1016/j.cell.2006.07.024. [DOI] [PubMed] [Google Scholar]
- 27.Hawley RG. Does retroviral insertional mutagenesis play a role in the generation of induced pluripotent stem cells? Mol Ther. 2008;16:1354–5. doi: 10.1038/mt.2008.142. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Lee AS, Tang C, Rao MS, Weissman IL, Wu JC. Tumorigenicity as a clinical hurdle for pluripotent stem cell therapies. Nat Med. 2013;19:998–1004. doi: 10.1038/nm.3267. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Ban H, et al. Efficient generation of transgene-free human induced pluripotent stem cells (iPSCs) by temperature-sensitive Sendai virus vectors. Proc Natl Acad Sci U S A. 2011;108:14234–9. doi: 10.1073/pnas.1103509108. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Stadtfeld M, Nagaya M, Utikal J, Weir G, Hochedlinger K. Induced pluripotent stem cells generated without viral integration. Science. 2008;322:945–9. doi: 10.1126/science.1162494. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Okita K, et al. A more efficient method to generate integration-free human iPS cells. Nat Methods. 2011;8:409–12. doi: 10.1038/nmeth.1591. [DOI] [PubMed] [Google Scholar]
- 32.Jia F, et al. A nonviral minicircle vector for deriving human iPS cells. Nat Methods. 2010;7:197–9. doi: 10.1038/nmeth.1426. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Diecke S, et al. Novel codon-optimized mini-intronic plasmid for efficient, inexpensive, and xeno-free induction of pluripotency. Sci Rep. 2015;5:8081. doi: 10.1038/srep08081. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Soldner F, et al. Parkinson’s disease patient-derived induced pluripotent stem cells free of viral reprogramming factors. Cell. 2009;136:964–77. doi: 10.1016/j.cell.2009.02.013. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Woltjen K, et al. piggyBac transposition reprograms fibroblasts to induced pluripotent stem cells. Nature. 2009;458:766–70. doi: 10.1038/nature07863. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Warren L, et al. Highly efficient reprogramming to pluripotency and directed differentiation of human cells with synthetic modified mRNA. Cell Stem Cell. 2010;7:618–30. doi: 10.1016/j.stem.2010.08.012. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Kim D, et al. Generation of human induced pluripotent stem cells by direct delivery of reprogramming proteins. Cell Stem Cell. 2009;4:472–6. doi: 10.1016/j.stem.2009.05.005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Schlaeger TM, et al. A comparison of non-integrating reprogramming methods. Nat Biotechnol. 2015;33:58–63. doi: 10.1038/nbt.3070. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Liang G, Zhang Y. Genetic and epigenetic variations in iPSCs: potential causes and implications for application. Cell Stem Cell. 2013;13:149–59. doi: 10.1016/j.stem.2013.07.001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Choi J, et al. A comparison of genetically matched cell lines reveals the equivalence of human iPSCs and ESCs. Nat Biotechnol. 2015;33:1173–81. doi: 10.1038/nbt.3388. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.Mummery C, et al. Differentiation of human embryonic stem cells to cardiomyocytes: role of coculture with visceral endoderm-like cells. Circulation. 2003;107:2733–40. doi: 10.1161/01.CIR.0000068356.38592.68. [DOI] [PubMed] [Google Scholar]
- 42.Kehat I, et al. Human embryonic stem cells can differentiate into myocytes with structural and functional properties of cardiomyocytes. J Clin Invest. 2001;108:407–14. doi: 10.1172/JCI12131. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43.Zwi L, et al. Cardiomyocyte differentiation of human induced pluripotent stem cells. Circulation. 2009;120:1513–23. doi: 10.1161/CIRCULATIONAHA.109.868885. [DOI] [PubMed] [Google Scholar]
- 44.Zhang J, et al. Functional cardiomyocytes derived from human induced pluripotent stem cells. Circ Res. 2009;104:e30–41. doi: 10.1161/CIRCRESAHA.108.192237. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45.Burridge PW, et al. Improved human embryonic stem cell embryoid body homogeneity and cardiomyocyte differentiation from a novel V-96 plate aggregation system highlights interline variability. Stem Cells. 2007;25:929–38. doi: 10.1634/stemcells.2006-0598. [DOI] [PubMed] [Google Scholar]
- 46.Dubois NC, et al. SIRPA is a specific cell-surface marker for isolating cardiomyocytes derived from human pluripotent stem cells. Nat Biotechnol. 2011;29:1011–8. doi: 10.1038/nbt.2005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47.Laflamme MA, et al. Cardiomyocytes derived from human embryonic stem cells in pro-survival factors enhance function of infarcted rat hearts. Nat Biotechnol. 2007;25:1015–24. doi: 10.1038/nbt1327. [DOI] [PubMed] [Google Scholar]
- 48.Zhang Q, et al. Direct differentiation of atrial and ventricular myocytes from human embryonic stem cells by alternating retinoid signals. Cell Res. 2011;21:579–87. doi: 10.1038/cr.2010.163. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49.Elliott DA, et al. NKX2-5(eGFP/w) hESCs for isolation of human cardiac progenitors and cardiomyocytes. Nat Methods. 2011;8:1037–40. doi: 10.1038/nmeth.1740. [DOI] [PubMed] [Google Scholar]
- 50.Hudson J, Titmarsh D, Hidalgo A, Wolvetang E, Cooper-White J. Primitive cardiac cells from human embryonic stem cells. Stem Cells Dev. 2012;21:1513–23. doi: 10.1089/scd.2011.0254. [DOI] [PubMed] [Google Scholar]
- 51.Lian X, et al. Robust cardiomyocyte differentiation from human pluripotent stem cells via temporal modulation of canonical Wnt signaling. Proc Natl Acad Sci U S A. 2012;109:E1848–57. doi: 10.1073/pnas.1200250109. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 52.Burridge PW, et al. Chemically defined generation of human cardiomyocytes. Nat Methods. 2014;11:855–60. doi: 10.1038/nmeth.2999. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 53.Levenberg S, Ferreira LS, Chen-Konak L, Kraehenbuehl TP, Langer R. Isolation, differentiation and characterization of vascular cells derived from human embryonic stem cells. Nat Protoc. 2010;5:1115–26. doi: 10.1038/nprot.2010.31. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 54.Orlova VV, et al. Generation, expansion and functional analysis of endothelial cells and pericytes derived from human pluripotent stem cells. Nat Protoc. 2014;9:1514–31. doi: 10.1038/nprot.2014.102. [DOI] [PubMed] [Google Scholar]
- 55.Ferreira LS, et al. Vascular progenitor cells isolated from human embryonic stem cells give rise to endothelial and smooth muscle like cells and form vascular networks in vivo. Circ Res. 2007;101:286–94. doi: 10.1161/CIRCRESAHA.107.150201. [DOI] [PubMed] [Google Scholar]
- 56.Marchand M, et al. Concurrent generation of functional smooth muscle and endothelial cells via a vascular progenitor. Stem Cells Transl Med. 2014;3:91–7. doi: 10.5966/sctm.2013-0124. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 57.Moretti A, et al. Patient-specific induced pluripotent stem-cell models for long-QT syndrome. N Engl J Med. 2010;363:1397–409. doi: 10.1056/NEJMoa0908679. [DOI] [PubMed] [Google Scholar]
- 58.Egashira T, et al. Disease characterization using LQTS-specific induced pluripotent stem cells. Cardiovasc Res. 2012;95:419–29. doi: 10.1093/cvr/cvs206. [DOI] [PubMed] [Google Scholar]
- 59.Ma D, et al. Characterization of a novel KCNQ1 mutation for type 1 long QT syndrome and assessment of the therapeutic potential of a novel IKs activator using patient-specific induced pluripotent stem cell-derived cardiomyocytes. Stem Cell Res Ther. 2015;6:39. doi: 10.1186/s13287-015-0027-z. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 60.Zhang M, et al. Recessive cardiac phenotypes in induced pluripotent stem cell models of Jervell and Lange-Nielsen syndrome: disease mechanisms and pharmacological rescue. Proc Natl Acad Sci U S A. 2014;111:E5383–92. doi: 10.1073/pnas.1419553111. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 61.Matsa E, et al. Drug evaluation in cardiomyocytes derived from human induced pluripotent stem cells carrying a long QT syndrome type 2 mutation. Eur Heart J. 2011;32:952–62. doi: 10.1093/eurheartj/ehr073. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 62.Itzhaki I, et al. Modelling the long QT syndrome with induced pluripotent stem cells. Nature. 2011;471:225–9. doi: 10.1038/nature09747. [DOI] [PubMed] [Google Scholar]
- 63.Lahti AL, et al. Model for long QT syndrome type 2 using human iPS cells demonstrates arrhythmogenic characteristics in cell culture. Dis Model Mech. 2012;5:220–30. doi: 10.1242/dmm.008409. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 64.Mehta A, et al. Re-trafficking of hERG reverses long QT syndrome 2 phenotype in human iPS-derived cardiomyocytes. Cardiovasc Res. 2014;102:497–506. doi: 10.1093/cvr/cvu060. [DOI] [PubMed] [Google Scholar]
- 65.Jouni M, et al. Toward personalized medicine: using cardiomyocytes differentiated from urine-derived pluripotent stem cells to recapitulate electrophysiological characteristics of type 2 long QT syndrome. J Am Heart Assoc. 2015;4 doi: 10.1161/JAHA.115.002159. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 66.Terrenoire C, et al. Induced pluripotent stem cells used to reveal drug actions in a long QT syndrome family with complex genetics. J Gen Physiol. 2013;141:61–72. doi: 10.1085/jgp.201210899. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 67.Ma D, et al. Modeling type 3 long QT syndrome with cardiomyocytes derived from patient-specific induced pluripotent stem cells. Int J Cardiol. 2013;168:5277–86. doi: 10.1016/j.ijcard.2013.08.015. [DOI] [PubMed] [Google Scholar]
- 68.Fatima A, et al. The disease-specific phenotype in cardiomyocytes derived from induced pluripotent stem cells of two long QT syndrome type 3 patients. PLoS One. 2013;8:e83005. doi: 10.1371/journal.pone.0083005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 69.Fatima A, et al. In vitro modeling of ryanodine receptor 2 dysfunction using human induced pluripotent stem cells. Cell Physiol Biochem. 2011;28:579–92. doi: 10.1159/000335753. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 70.Zhang XH, et al. Ca2+ signaling in human induced pluripotent stem cell-derived cardiomyocytes (iPS-CM) from normal and catecholaminergic polymorphic ventricular tachycardia (CPVT)-afflicted subjects. Cell Calcium. 2013;54:57–70. doi: 10.1016/j.ceca.2013.04.004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 71.Jung CB, et al. Dantrolene rescues arrhythmogenic RYR2 defect in a patient-specific stem cell model of catecholaminergic polymorphic ventricular tachycardia. EMBO Mol Med. 2012;4:180–91. doi: 10.1002/emmm.201100194. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 72.Itzhaki I, et al. Modeling of catecholaminergic polymorphic ventricular tachycardia with patient-specific human-induced pluripotent stem cells. J Am Coll Cardiol. 2012;60:990–1000. doi: 10.1016/j.jacc.2012.02.066. [DOI] [PubMed] [Google Scholar]
- 73.Kujala K, et al. Cell model of catecholaminergic polymorphic ventricular tachycardia reveals early and delayed afterdepolarizations. PLoS One. 2012;7:e44660. doi: 10.1371/journal.pone.0044660. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 74.Di Pasquale E, et al. CaMKII inhibition rectifies arrhythmic phenotype in a patient-specific model of catecholaminergic polymorphic ventricular tachycardia. Cell Death Dis. 2013;4:e843. doi: 10.1038/cddis.2013.369. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 75.Novak A, et al. Functional abnormalities in iPSC-derived cardiomyocytes generated from CPVT1 and CPVT2 patients carrying ryanodine or calsequestrin mutations. J Cell Mol Med. 2015;19:2006–18. doi: 10.1111/jcmm.12581. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 76.Novak A, et al. Cardiomyocytes generated from CPVTD307H patients are arrhythmogenic in response to beta-adrenergic stimulation. J Cell Mol Med. 2012;16:468–82. doi: 10.1111/j.1582-4934.2011.01476.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 77.Yazawa M, et al. Using induced pluripotent stem cells to investigate cardiac phenotypes in Timothy syndrome. Nature. 2011;471:230–4. doi: 10.1038/nature09855. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 78.Sun N, et al. Patient-specific induced pluripotent stem cells as a model for familial dilated cardiomyopathy. Sci Transl Med. 2012;4:130ra47. doi: 10.1126/scitranslmed.3003552. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 79.Wu H, et al. Epigenetic regulation of phosphodiesterases 2A and 3A underlies compromised beta-adrenergic signaling in an iPSC model of dilated cardiomyopathy. Cell Stem Cell. 2015;17:89–100. doi: 10.1016/j.stem.2015.04.020. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 80.Siu CW, et al. Modeling of lamin A/C mutation premature cardiac aging using patient-specific induced pluripotent stem cells. Aging. 2012;4:803–822. doi: 10.18632/aging.100503. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 81.Tse HF, et al. Patient-specific induced-pluripotent stem cells-derived cardiomyocytes recapitulate the pathogenic phenotypes of dilated cardiomyopathy due to a novel DES mutation identified by whole exome sequencing. Hum Mol Genet. 2013;22:1395–403. doi: 10.1093/hmg/dds556. [DOI] [PubMed] [Google Scholar]
- 82.Lan F, et al. Abnormal calcium handling properties underlie familial hypertrophic cardiomyopathy pathology in patient-specific induced pluripotent stem cells. Cell Stem Cell. 2013;12:101–13. doi: 10.1016/j.stem.2012.10.010. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 83.Han L, et al. Study familial hypertrophic cardiomyopathy using patient-specific induced pluripotent stem cells. Cardiovasc Res. 2014;104:258–69. doi: 10.1093/cvr/cvu205. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 84.Tanaka A, et al. Endothelin-1 induces myofibrillar disarray and contractile vector variability in hypertrophic cardiomyopathy-induced pluripotent stem cell-derived cardiomyocytes. J Am Heart Assoc. 2014;3:e001263. doi: 10.1161/JAHA.114.001263. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 85.Caspi O, et al. Modeling of arrhythmogenic right ventricular cardiomyopathy with human induced pluripotent stem cells. Circ Cardiovasc Genet. 2013;6:557–68. doi: 10.1161/CIRCGENETICS.113.000188. [DOI] [PubMed] [Google Scholar]
- 86.Kim C, et al. Studying arrhythmogenic right ventricular dysplasia with patient-specific iPSCs. Nature. 2013;494:105–10. doi: 10.1038/nature11799. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 87.Ma D, et al. Generation of patient-specific induced pluripotent stem cell-derived cardiomyocytes as a cellular model of arrhythmogenic right ventricular cardiomyopathy. Eur Heart J. 2013;34:1122–33. doi: 10.1093/eurheartj/ehs226. [DOI] [PubMed] [Google Scholar]
- 88.Dick E, et al. Exon skipping and gene transfer restore dystrophin expression in human induced pluripotent stem cells-cardiomyocytes harboring DMD mutations. Stem Cells Dev. 2013;22:2714–24. doi: 10.1089/scd.2013.0135. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 89.Lin B, et al. Modeling and study of the mechanism of dilated cardiomyopathy using induced pluripotent stem cells derived from individuals with Duchenne muscular dystrophy. Dis Model Mech. 2015;8:457–66. doi: 10.1242/dmm.019505. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 90.Drawnel FM, et al. Disease modeling and phenotypic drug screening for diabetic cardiomyopathy using human induced pluripotent stem cells. Cell Rep. 2014;9:810–21. doi: 10.1016/j.celrep.2014.09.055. [DOI] [PubMed] [Google Scholar]
- 91.Ebert AD, et al. Characterization of the molecular mechanisms underlying increased ischemic damage in the aldehyde dehydrogenase 2 genetic polymorphism using a human induced pluripotent stem cell model system. Sci Transl Med. 2014;6:255ra130. doi: 10.1126/scitranslmed.3009027. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 92.Wang G, et al. Modeling the mitochondrial cardiomyopathy of Barth syndrome with induced pluripotent stem cell and heart-on-chip technologies. Nat Med. 2014;20:616–23. doi: 10.1038/nm.3545. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 93.Huang HP, et al. Human Pompe disease-induced pluripotent stem cells for pathogenesis modeling, drug testing and disease marker identification. Hum Mol Genet. 2011;20:4851–64. doi: 10.1093/hmg/ddr424. [DOI] [PubMed] [Google Scholar]
- 94.Raval KK, et al. Pompe disease results in a Golgi-based glycosylation deficit in human induced pluripotent stem cell-derived cardiomyocytes. J Biol Chem. 2015;290:3121–36. doi: 10.1074/jbc.M114.628628. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 95.Sato Y, et al. Disease modeling and lentiviral gene transfer in patient-specific induced pluripotent stem cells from late-onset Pompe disease patient. Mol Ther Methods Clin Dev. 2015;2:15023. doi: 10.1038/mtm.2015.23. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 96.Jiang Y, et al. An induced pluripotent stem cell model of hypoplastic left heart syndrome (HLHS) reveals multiple expression and functional differences in HLHS-derived cardiac myocytes. Stem Cells Transl Med. 2014;3:416–23. doi: 10.5966/sctm.2013-0105. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 97.Sharma A, et al. Human induced pluripotent stem cell-derived cardiomyocytes as an in vitro model for coxsackievirus B3-induced myocarditis and antiviral drug screening platform. Circ Res. 2014;115:556–66. doi: 10.1161/CIRCRESAHA.115.303810. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 98.Sallam K, Li Y, Sager PT, Houser SR, Wu JC. Finding the rhythm of sudden cardiac death: new opportunities using induced pluripotent stem cell-derived cardiomyocytes. Circ Res. 2015;116:1989–2004. doi: 10.1161/CIRCRESAHA.116.304494. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 99.O’Hara T, Rudy Y. Quantitative comparison of cardiac ventricular myocyte electrophysiology and response to drugs in human and nonhuman species. Am J Physiol Heart Circ Physiol. 2012;302:H1023–30. doi: 10.1152/ajpheart.00785.2011. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 100.Braam SR, et al. Repolarization reserve determines drug responses in human pluripotent stem cell derived cardiomyocytes. Stem Cell Res. 2013;10:48–56. doi: 10.1016/j.scr.2012.08.007. [DOI] [PubMed] [Google Scholar]
- 101.Lieu DK, et al. Mechanism-based facilitated maturation of human pluripotent stem cell-derived cardiomyocytes. Circ Arrhythm Electrophysiol. 2013;6:191–201. doi: 10.1161/CIRCEP.111.973420. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 102.Matsa E, et al. Allele-specific RNA interference rescues the long-QT syndrome phenotype in human-induced pluripotency stem cell cardiomyocytes. Eur Heart J. 2014;35:1078–87. doi: 10.1093/eurheartj/eht067. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 103.Lei M, Huang CL, Zhang Y. Genetic Na+ channelopathies and sinus node dysfunction. Prog Biophys Mol Biol. 2008;98:171–8. doi: 10.1016/j.pbiomolbio.2008.10.003. [DOI] [PubMed] [Google Scholar]
- 104.Sallam K, Kodo K, Wu JC. Modeling inherited cardiac disorders. Circ J. 2014;78:784–94. doi: 10.1253/circj.cj-14-0182. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 105.Hinson JT, et al. Titin mutations in iPS cells define sarcomere insufficiency as a cause of dilated cardiomyopathy. Science. 2015;349:982–6. doi: 10.1126/science.aaa5458. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 106.Asimaki A, et al. Identification of a new modulator of the intercalated disc in a zebrafish model of arrhythmogenic cardiomyopathy. Sci Transl Med. 2014;6:240ra74. doi: 10.1126/scitranslmed.3008008. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 107.Hashem SI, et al. Brief report: oxidative stress mediates cardiomyocyte apoptosis in a human model of Danon disease and heart failure. Stem Cells. 2015;33:2343–50. doi: 10.1002/stem.2015. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 108.Nakamura K, Hirano K, Wu SM. iPS cell modeling of cardiometabolic diseases. J Cardiovasc Transl Res. 2013;6:46–53. doi: 10.1007/s12265-012-9413-4. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 109.Guo YJ, et al. The ALDH2 Glu504Lys polymorphism is associated with coronary artery disease in Han Chinese: Relation with endothelial ADMA levels. Atherosclerosis. 2010;211:545–50. doi: 10.1016/j.atherosclerosis.2010.03.030. [DOI] [PubMed] [Google Scholar]
- 110.Xu F, et al. ALDH2 genetic polymorphism and the risk of type II diabetes mellitus in CAD patients. Hypertens Res. 2010;33:49–55. doi: 10.1038/hr.2009.178. [DOI] [PubMed] [Google Scholar]
- 111.Dhingra R, et al. Bnip3 mediates doxorubicin-induced cardiac myocyte necrosis and mortality through changes in mitochondrial signaling. Proc Natl Acad Sci U S A. 2014;111:E5537–44. doi: 10.1073/pnas.1414665111. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 112.Clayton ZE, Sadeghipour S, Patel S. Generating induced pluripotent stem cell derived endothelial cells and induced endothelial cells for cardiovascular disease modelling and therapeutic angiogenesis. Int J Cardiol. 2015;197:116–22. doi: 10.1016/j.ijcard.2015.06.038. [DOI] [PubMed] [Google Scholar]
- 113.Pober BR. Williams-Beuren syndrome. N Engl J Med. 2010;362:239–52. doi: 10.1056/NEJMra0903074. [DOI] [PubMed] [Google Scholar]
- 114.Merla G, Brunetti-Pierri N, Piccolo P, Micale L, Loviglio MN. Supravalvular aortic stenosis: elastin arteriopathy. Circ Cardiovasc Genet. 2012;5:692–6. doi: 10.1161/CIRCGENETICS.112.962860. [DOI] [PubMed] [Google Scholar]
- 115.Ge X, et al. Modeling supravalvular aortic stenosis syndrome with human induced pluripotent stem cells. Circulation. 2012;126:1695–704. doi: 10.1161/CIRCULATIONAHA.112.116996. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 116.Kinnear C, et al. Modeling and rescue of the vascular phenotype of Williams-Beuren syndrome in patient induced pluripotent stem cells. Stem Cells Transl Med. 2013;2:2–15. doi: 10.5966/sctm.2012-0054. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 117.Goergen CJ, Li HH, Francke U, Taylor CA. Induced chromosome deletion in a Williams-Beuren syndrome mouse model causes cardiovascular abnormalities. J Vasc Res. 2011;48:119–29. doi: 10.1159/000316808. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 118.Roberts WC, Ko JM. Frequency by decades of unicuspid, bicuspid, and tricuspid aortic valves in adults having isolated aortic valve replacement for aortic stenosis, with or without associated aortic regurgitation. Circulation. 2005;111:920–5. doi: 10.1161/01.CIR.0000155623.48408.C5. [DOI] [PubMed] [Google Scholar]
- 119.Weinberg EJ, Mack PJ, Schoen FJ, Garcia-Cardena G, Kaazempur Mofrad MR. Hemodynamic environments from opposing sides of human aortic valve leaflets evoke distinct endothelial phenotypes in vitro. Cardiovasc Eng. 2010;10:5–11. doi: 10.1007/s10558-009-9089-9. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 120.Combs MD, Yutzey KE. Heart valve development: regulatory networks in development and disease. Circ Res. 2009;105:408–21. doi: 10.1161/CIRCRESAHA.109.201566. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 121.Masumura T, Yamamoto K, Shimizu N, Obi S, Ando J. Shear stress increases expression of the arterial endothelial marker ephrinB2 in murine ES cells via the VEGF-Notch signaling pathways. Arterioscler Thromb Vasc Biol. 2009;29:2125–31. doi: 10.1161/ATVBAHA.109.193185. [DOI] [PubMed] [Google Scholar]
- 122.Theodoris CV, et al. Human disease modeling reveals integrated transcriptional and epigenetic mechanisms of NOTCH1 haploinsufficiency. Cell. 2015;160:1072–86. doi: 10.1016/j.cell.2015.02.035. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 123.Pollex RL, Hegele RA. Hutchinson-Gilford progeria syndrome. Clin Genet. 2004;66:375–81. doi: 10.1111/j.1399-0004.2004.00315.x. [DOI] [PubMed] [Google Scholar]
- 124.Liu GH, et al. Recapitulation of premature ageing with iPSCs from Hutchinson-Gilford progeria syndrome. Nature. 2011;472:221–5. doi: 10.1038/nature09879. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 125.Zhang J, et al. A human iPSC model of Hutchinson Gilford Progeria reveals vascular smooth muscle and mesenchymal stem cell defects. Cell Stem Cell. 2011;8:31–45. doi: 10.1016/j.stem.2010.12.002. [DOI] [PubMed] [Google Scholar]
- 126.Kim H, Kim JS. A guide to genome engineering with programmable nucleases. Nat Rev Genet. 2014;15:321–34. doi: 10.1038/nrg3686. [DOI] [PubMed] [Google Scholar]
- 127.Hsu PD, Lander ES, Zhang F. Development and applications of CRISPR-Cas9 for genome engineering. Cell. 2014;157:1262–78. doi: 10.1016/j.cell.2014.05.010. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 128.Karakikes I, et al. Correction of human phospholamban R14del mutation associated with cardiomyopathy using targeted nucleases and combination therapy. Nat Commun. 2015;6:6955. doi: 10.1038/ncomms7955. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 129.Wang Y, et al. Genome editing of isogenic human induced pluripotent stem cells recapitulates long QT phenotype for drug testing. J Am Coll Cardiol. 2014;64:451–9. doi: 10.1016/j.jacc.2014.04.057. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 130.Frommeyer G, Eckardt L. Drug-induced proarrhythmia: risk factors and electrophysiological mechanisms. Nat Rev Cardiol. 2016;13:36–47. doi: 10.1038/nrcardio.2015.110. [DOI] [PubMed] [Google Scholar]
- 131.Darpo B, et al. Cardiac Safety Research Consortium: can the thorough QT/QTc study be replaced by early QT assessment in routine clinical pharmacology studies? Scientific update and a research proposal for a path forward. Am Heart J. 2014;168:262–72. doi: 10.1016/j.ahj.2014.06.003. [DOI] [PubMed] [Google Scholar]
- 132.Sager PT, Gintant G, Turner JR, Pettit S, Stockbridge N. Rechanneling the cardiac proarrhythmia safety paradigm: a meeting report from the Cardiac Safety Research Consortium. Am Heart J. 2014;167:292–300. doi: 10.1016/j.ahj.2013.11.004. [DOI] [PubMed] [Google Scholar]
- 133.Mirams GR, et al. Simulation of multiple ion channel block provides improved early prediction of compounds’ clinical torsadogenic risk. Cardiovasc Res. 2011;91:53–61. doi: 10.1093/cvr/cvr044. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 134.Navarrete EG, et al. Screening drug-induced arrhythmia [corrected] using human induced pluripotent stem cell-derived cardiomyocytes and low-impedance microelectrode arrays. Circulation. 2013;128:S3–13. doi: 10.1161/CIRCULATIONAHA.112.000570. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 135.Jonsson MK, Wang QD, Becker B. Impedance-based detection of beating rhythm and proarrhythmic effects of compounds on stem cell-derived cardiomyocytes. Assay Drug Dev Technol. 2011;9:589–99. doi: 10.1089/adt.2011.0396. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 136.Guo L, et al. Estimating the risk of drug-induced proarrhythmia using human induced pluripotent stem cell-derived cardiomyocytes. Toxicol Sci. 2011;123:281–9. doi: 10.1093/toxsci/kfr158. [DOI] [PubMed] [Google Scholar]
- 137.Guo L, et al. Refining the human iPSC-cardiomyocyte arrhythmic risk assessment model. Toxicol Sci. 2013;136:581–94. doi: 10.1093/toxsci/kft205. [DOI] [PubMed] [Google Scholar]
- 138.Sirenko O, et al. Multiparameter in vitro assessment of compound effects on cardiomyocyte physiology using iPSC cells. J Biomol Screen. 2013;18:39–53. doi: 10.1177/1087057112457590. [DOI] [PubMed] [Google Scholar]
- 139.Sirenko O, et al. Assessment of beating parameters in human induced pluripotent stem cells enables quantitative in vitro screening for cardiotoxicity. Toxicol Appl Pharmacol. 2013;273:500–7. doi: 10.1016/j.taap.2013.09.017. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 140.Maddah M, et al. A non-invasive platform for functional characterization of stem-cell-derived cardiomyocytes with applications in cardiotoxicity testing. Stem Cell Reports. 2015;4:621–31. doi: 10.1016/j.stemcr.2015.02.007. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 141.Doke SK, Dhawale SC. Alternatives to animal testing: A review. Saudi Pharm J. 2015;23:223–9. doi: 10.1016/j.jsps.2013.11.002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 142.Giri S, Bader A. A low-cost, high-quality new drug discovery process using patient-derived induced pluripotent stem cells. Drug Discov Today. 2015;20:37–49. doi: 10.1016/j.drudis.2014.10.011. [DOI] [PubMed] [Google Scholar]
- 143.Burridge PW, Holmstrom A, Wu JC. Chemically defined culture and cardiomyocyte differentiation of human pluripotent stem cells. Curr Protoc Hum Genet. 2015;87:21 3 1–21 3 15. doi: 10.1002/0471142905.hg2103s87. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 144.Stillitano F, et al. Modeling drug-induced long QT syndrome with patient-specific induced pluripotent stem cell-derived cardiomyocytes. Circ. 2014;130:A18442. [Google Scholar]
- 145.Mullard A. Stem-cell discovery platforms yield first clinical candidates. Nat Rev Drug Discov. 2015;14:589–91. doi: 10.1038/nrd4708. [DOI] [PubMed] [Google Scholar]
- 146.Bright J, et al. Human secreted tau increases amyloid-beta production. Neurobiol Aging. 2015;36:693–709. doi: 10.1016/j.neurobiolaging.2014.09.007. [DOI] [PubMed] [Google Scholar]
- 147.Wainger BJ, et al. Intrinsic membrane hyperexcitability of amyotrophic lateral sclerosis patient-derived motor neurons. Cell Rep. 2014;7:1–11. doi: 10.1016/j.celrep.2014.03.019. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 148.Eschenhagen T, et al. Three-dimensional reconstitution of embryonic cardiomyocytes in a collagen matrix: a new heart muscle model system. FASEB J. 1997;11:683–94. doi: 10.1096/fasebj.11.8.9240969. [DOI] [PubMed] [Google Scholar]
- 149.Eder A, Vollert I, Hansen A, Eschenhagen T. Human engineered heart tissue as a model system for drug testing. Adv Drug Deliv Rev. 2016;96:214–224. doi: 10.1016/j.addr.2015.05.010. [DOI] [PubMed] [Google Scholar]
- 150.Schaaf S, et al. Human engineered heart tissue as a versatile tool in basic research and preclinical toxicology. PLoS One. 2011;6:e26397. doi: 10.1371/journal.pone.0026397. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 151.Stoehr A, et al. Automated analysis of contractile force and Ca2+ transients in engineered heart tissue. Am J Physiol Heart Circ Physiol. 2014;306:H1353–63. doi: 10.1152/ajpheart.00705.2013. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 152.Zhang D, et al. Tissue-engineered cardiac patch for advanced functional maturation of human ESC-derived cardiomyocytes. Biomaterials. 2013;34:5813–20. doi: 10.1016/j.biomaterials.2013.04.026. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 153.Nunes SS, et al. Biowire: a platform for maturation of human pluripotent stem cell-derived cardiomyocytes. Nat Methods. 2013;10:781–7. doi: 10.1038/nmeth.2524. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 154.Mathur A, et al. Human iPSC-based cardiac microphysiological system for drug screening applications. Sci Rep. 2015;5:8883. doi: 10.1038/srep08883. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 155.Tzatzalos E, Abilez OJ, Shukla P, Wu JC. Engineered heart tissues and induced pluripotent stem cells: Macro- and microstructures for disease modeling, drug screening, and translational studies. Adv Drug Deliv Rev. 2016;96:234–44. doi: 10.1016/j.addr.2015.09.010. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 156.Priest BT, Bell IM, Garcia ML. Role of hERG potassium channel assays in drug development. Channels. 2008;2:87–93. doi: 10.4161/chan.2.2.6004. [DOI] [PubMed] [Google Scholar]
- 157.Gintant GA, Su Z, Martin RL, Cox BF. Utility of hERG assays as surrogate markers of delayed cardiac repolarization and QT safety. Toxicol Pathol. 2006;34:81–90. doi: 10.1080/01926230500431376. [DOI] [PubMed] [Google Scholar]
- 158.Chen X, Cordes JS, Bradley JA, Sun Z, Zhou J. Use of arterially perfused rabbit ventricular wedge in predicting arrhythmogenic potentials of drugs. J Pharmacol Toxicol Methods. 2006;54:261–72. doi: 10.1016/j.vascn.2006.02.005. [DOI] [PubMed] [Google Scholar]
- 159.Milberg P, et al. Transmural dispersion of repolarization as a key factor of arrhythmogenicity in a novel intact heart model of LQT3. Cardiovasc Res. 2005;65:397–404. doi: 10.1016/j.cardiores.2004.10.016. [DOI] [PubMed] [Google Scholar]
- 160.Lawrence CL, Bridgland-Taylor MH, Pollard CE, Hammond TG, Valentin JP. A rabbit Langendorff heart proarrhythmia model: predictive value for clinical identification of Torsades de Pointes. Br J Pharmacol. 2006;149:845–60. doi: 10.1038/sj.bjp.0706894. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 161.Sugiyama A. Sensitive and reliable proarrhythmia in vivo animal models for predicting drug-induced torsades de pointes in patients with remodelled hearts. Br J Pharmacol. 2008;154:1528–37. doi: 10.1038/bjp.2008.240. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 162.Lundy SD, Zhu WZ, Regnier M, Laflamme MA. Structural and functional maturation of cardiomyocytes derived from human pluripotent stem cells. Stem Cells Dev. 2013;22:1991–2002. doi: 10.1089/scd.2012.0490. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 163.Robertson C, Tran DD, George SC. Concise review: maturation phases of human pluripotent stem cell-derived cardiomyocytes. Stem Cells. 2013;31:829–37. doi: 10.1002/stem.1331. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 164.Xu XQ, Soo SY, Sun W, Zweigerdt R. Global expression profile of highly enriched cardiomyocytes derived from human embryonic stem cells. Stem Cells. 2009;27:2163–74. doi: 10.1002/stem.166. [DOI] [PubMed] [Google Scholar]
- 165.Hwang HS, et al. Comparable calcium handling of human iPSC-derived cardiomyocytes generated by multiple laboratories. J Mol Cell Cardiol. 2015;85:79–88. doi: 10.1016/j.yjmcc.2015.05.003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 166.Lieu DK, et al. Absence of transverse tubules contributes to non-uniform Ca(2+) wavefronts in mouse and human embryonic stem cell-derived cardiomyocytes. Stem Cells Dev. 2009;18:1493–500. doi: 10.1089/scd.2009.0052. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 167.Kim JJ, et al. Mechanism of automaticity in cardiomyocytes derived from human induced pluripotent stem cells. J Mol Cell Cardiol. 2015;81:81–93. doi: 10.1016/j.yjmcc.2015.01.013. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 168.Tohyama S, et al. Distinct metabolic flow enables large-scale purification of mouse and human pluripotent stem cell-derived cardiomyocytes. Cell Stem Cell. 2013;12:127–37. doi: 10.1016/j.stem.2012.09.013. [DOI] [PubMed] [Google Scholar]
- 169.Zhu R, et al. Physical developmental cues for the maturation of human pluripotent stem cell-derived cardiomyocytes. Stem Cell Res Ther. 2014;5:117. doi: 10.1186/scrt507. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 170.Wilson KD, et al. Dynamic microRNA expression programs during cardiac differentiation of human embryonic stem cells: role for miR-499. Circ Cardiovasc Genet. 2010;3:426–35. doi: 10.1161/CIRCGENETICS.109.934281. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 171.Marx V. Stem cells: disease models that show and tell. Nature Methods. 2015;12:111–114. doi: 10.1038/nmeth.3263. [DOI] [PubMed] [Google Scholar]
- 172.Fluri DA, et al. Derivation, expansion and differentiation of induced pluripotent stem cells in continuous suspension cultures. Nat Methods. 2012;9:509–16. doi: 10.1038/nmeth.1939. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 173.Chen VC, et al. Development of a scalable suspension culture for cardiac differentiation from human pluripotent stem cells. Stem Cell Res. 2015;15:365–75. doi: 10.1016/j.scr.2015.08.002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 174.Amit M, et al. Dynamic suspension culture for scalable expansion of undifferentiated human pluripotent stem cells. Nat Protoc. 2011;6:572–9. doi: 10.1038/nprot.2011.325. [DOI] [PubMed] [Google Scholar]
- 175.Kirouac DC, Zandstra PW. The systematic production of cells for cell therapies. Cell Stem Cell. 2008;3:369–81. doi: 10.1016/j.stem.2008.09.001. [DOI] [PubMed] [Google Scholar]
- 176.Paull D, et al. Automated, high-throughput derivation, characterization and differentiation of induced pluripotent stem cells. Nat Methods. 2015;12:885–92. doi: 10.1038/nmeth.3507. [DOI] [PubMed] [Google Scholar]